

Abstract
The quantification of residual plasmatic ADAMTS13 activity in congenital thrombotic thrombocytopenic purpura (TTP) patients is constrained by limitations in sensitivity and reproducibility of commonly used assays at low levels of ADAMTS13 activity, blunting efforts to establish genotype-phenotype correlations. In the present study, the residual plasmatic activity of ADAMTS13 was measured centrally by surface-enhanced laser desorption/ionization time-of-flight mass spectrometry (limit of detection = 0.5%) in 29 congenital TTP patients. The results were used to study correlations among ADAMTS13 genotype, residual plasmatic activity, and clinical phenotype severity. An ADAMTS13 activity above 0.5% was measured in 26 (90%) patients and lower levels of activity were associated with earlier age at first TTP episode requiring plasma infusion, more frequent recurrences, and prescription of fresh-frozen plasma prophylaxis. Receiver operating characteristic curve analysis showed that activity levels of less than 2.74% and 1.61% were discriminative of age at first TTP episode requiring plasma infusion < 18 years, annual rate of TTP episodes > 1, and use of prophylaxis. Mutations affecting the highly conserved N-terminal domains of the protein were associated with lower residual ADAMTS13 activity and a more severe phenotype in an allelic-dose dependent manner. The results of the present study show that residual ADAMTS13 activity is associated with the severity of clinical phenotype in congenital TTP and provide insights into genotype-phenotype correlations.
Introduction
Congenital thrombotic thrombocytopenic purpura (TTP; also known as Upshaw-Schulman syndrome, OMIM #274150) is a rare, recessively inherited thrombotic microangiopathy. The disease is characterized by the severe congenital deficiency of ADAMTS13 plasmatic activity caused by mutations in the ADAMTS13 gene.1–6 The phenotype of congenital TTP is variable in its clinical severity. Some patients present with the disease in the neonatal period, whereas others have an adult onset. Moreover, patients may experience only a few isolated episodes of TTP, whereas others have frequent recurrences leading to the prescription of fresh-frozen plasma (FFP) prophylaxis.5–8 A recent review of the approximately 100 published cases of congenital TTP showed that patients carrying the same ADAMTS13 gene mutations develop their first disease episode at a similar age.9 This observation suggests that different ADAMTS13 mutations may influence the severity of clinical phenotype, probably by determining different levels of residual plasmatic activity of ADAMTS13. However, the quantification of residual ADAMTS13 activity in congenital TTP patients is blunted by limitations in the analytical sensitivity and performance in the lower end of the ADAMTS13 activity distribution (ie, activity below 6%) of the commonly used ADAMTS13 activity assays.10–13 A recently described method that is based on surface-enhanced laser desorption/ionization time-of-flight (SELDI-TOF) mass spectrometry, is able to accurately measure ADAMTS13 plasmatic activity with high analytical sensitivity (limit of detection = 0.5%, 5- to 10-fold higher than most commercially available assays).14,15
In the present study, we measured the residual activity of ADAMTS13 by SELDI-TOF mass spectrometry in a cohort of 29 patients with congenital TTP and studied the relationships among ADAMTS13 genotype, residual plasmatic activity, and clinical phenotype of the disease.
Methods
Patients
Patients registered between 2000 and 2010 in 4 European TTP registries, the Milan TTP registry (Milan, Italy),16,17 the United Kingdom TTP Registry (London, United Kingdom),18 the International Registry for HUS and TTP (Bergamo, Italy),19 and the TMA Registry of the French Reference Center for the management of thrombotic microangiopathies (Paris, France),20 were evaluated for study eligibility.
Inclusion criteria were: (1) history of at least 1 episode of TTP (defined by the presence of thrombocytopenia and microangiopathic Coombs-negative hemolytic anemia with signs of RBC fragmentation); (2) severe deficiency of ADAMTS13 with activity < 6% as measured by the collagen-binding assay14 or fluorescence resonance energy transfer15; (3) absence of anti-ADAMTS13 autoantibodies as shown by Western blotting21 and/or ELISA17–20; (4) documented mutations of ADAMTS13 after sequencing of the protein coding area of the gene by PCR and Sanger sequencing; and (5) availability of 100 μL of citrated plasma collected during remission at least 20 days from the last infusion of FFP or any other blood-derived products. A total of 29 patients were included in the study (Milan, n = 12; United Kingdom, n = 7; Bergamo, n = 5; France, n = 5). Sixteen of these patients have been described previously,9,20–28 whereas 13 are presented here for the first time.
For each patient, phenotypic data were retrieved, including: age at last followup, sex, ethnicity, age at first TTP episode requiring FFP, lifetime and annual rate of TTP episodes, use of regular FFP prophylaxis, history of neonatal jaundice and/or thrombocytopenia, and presence of renal damage (defined as the presence of chronic renal failure with glomerular filtration rate below 60 mL/min/m2) and/or neurologic damage, (ie, persistence after disease remission of motor deficit, sensory deficit, or focal neurologic signs). Detailed individual clinical and genetic information are reported in supplemental Table 1 (available on the Blood Web site; see the Supplemental Materials link at the top of the online article). This study was approved by the institutional review boards of the participating centers and all subjects gave informed consent in accordance with the Declaration of Helsinki.
SELDI-TOF mass spectrometry–based measurement of ADAMTS13
All samples were shipped in dry ice to the Wu laboratory at the Department of Pathology (The Ohio State University, Columbus, OH) for centralized measurement. ADAMTS13 activity was determined in this central laboratory using a SELDI-TOF mass spectrometry–based method.14 The measuring laboratory personnel were blind to the clinical features of the congenital TTP patients.
ADAMTS13 activity was determined by mixing patient plasma with the enzyme substrate VWF73 containing a 6X His tag. The cleavage product was enriched by IMAC ProteinChip and then quantified using a SELDI-TOF mass spectrometer.14,15 An internal control was generated by cleaving recombinant-6X His-tagged human VWF73 by PreScission Protease (Amersham Biosciences) as described previously.14 Briefly, 10 μL of patient plasma was mixed with 30 μL of buffer (5mM Tris HCl, 5mM NaCl, 1mM BaCl2, pH 7.5) containing 2.5 μg of 6X His–tagged human VWF73 (D1596-R1668). The cleavage reaction was performed for 16 hours at 37°C and then terminated by heating the samples at 95°C for 2 minutes. Each experiment included a standard curve performed under identical conditions except that the plasma sample was replaced by pooled normal plasma diluted at 7.5%, 5%, 3.5%, 2.5%, 1.5%, 1%, and 0.5% in 100mM NaCl containing 0.1% BSA (Figure 1). After the cleavage reaction, 40 μL of internal control (0.01 μg/μL) was mixed with 35 μL of reaction sample from each patient on the corresponding IMAC ProteinChip spot. After incubation for 30 minutes at room temperature with constant shaking, each spot was washed 5 times with 200 μL of washing buffer. This was followed by 1 quick wash with 1mM HEPES, pH 7.0. Finally, 1 μL of energy-absorbing molecule solution (100% saturated sinapinic acid in 50% acetonitrile and 0.5% trifluoroacetic acid) was added to each spot. The cleavage products on the IMAC Protein Chips were analyzed with the PCS4000 SELDI-TOF mass spectrometer (Vermillion). To evaluate the test reproducibility in the lower analytical range, 2 external controls (2 TTP patients with their plasma samples aliquoted and frozen at −80°C) were tested repeatedly over time and by different operators. The ADAMTS13 activity levels obtained (means ± 2 SD) for these 2 patients were 1.8% ± 0.2% and 4.8% ± 0.8%, respectively, indicating good test reproducibility. The coefficients of variation for those 2 controls were 6.8% and 7.8%, respectively.
Figure 1.

Linearity of SELDI-TOF–based measurements of plasmatic activity of ADAMTS13 at concentrations below 10%. The figure shows a representative example of the calibration curve that was prepared before each experiment to confirm the linear behavior of the assay and the detection of the cleavage product.
Mutation analysis and annotation
In keeping with Human Genome Variation Society (HGVS) instructions, nucleotide numbering for reported mutations refers to cDNA numbering with the A of the ATG codon numbered as +1. According to new instructions, stop codons were designed as “*” instead of “X.” NM_139025.3 was used as a reference for nucleotide changes. NP_620596.2 was used as a protein reference sequence. The review of ADAMTS13 mutations associated with congenital TTP was based on published articles found through searches on PubMed (updated to July 2011), using “ADAMTS13 mutation,” “ADAMTS13 genotype,” and “congenital thrombotic thrombocytopenic purpura” as queries. Polymorphisms of ADAMTS13 were searched for on dbSNP. As suggested by the HGVS, the accuracy of nucleotide changes for reported mutations was verified by Mutalyzer Version 2.0 software (http://www.mutalyzer.nl/2.0/). Annotation of genetic variants was performed on the dbSNP131 (http://www.ncbi.nlm.nih.gov/projects/SNP) and 1000 Genomes (www.1000genomes.org) databases (used as databases of common genetic variation) and Polyphen 2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.jcvi.org/) software (used to predict the effect of amino acid changes on protein function). A list of all of the proteins used in the SIFT alignment to determine amino acid conservation is provided in supplemental Table 2. Splicing mutations were annotated on NetGene2 and their effect on protein translation predicted by Expasy (http://web.expasy.org/translate/). Annotation was performed either manually or by automated submission of mutation batches with custom scripts written in the Perl programming language. Sequencing of ADAMTS13 in 99 control subjects free from TTP was performed in the framework of the Milan DVT Study, a study on deep vein thrombosis predisposing genetic variants. In these subjects, ADAMTS13 exons were sequenced by DNA target capture on NimbleGen Custom Human Sequence Capture 2.1M Array chips followed by next-generation DNA sequencing on ABI SOLiD 4 platforms as described previously.29 Sequencing was performed at the Human Genome Sequencing Center at the Baylor College of Medicine (Houston, TX). The ADAMTS13 sequence coverage was above an average of 20 times per base in each of the sequenced individuals, providing enough coverage for confident variant calls.
Construction of a genotype risk score
To study the relationship between mutation localization and clinical phenotype, we constructed a genotype score considering N-terminal mutations as a risk factor for more severe disease. The hypothesis that the N-terminal mutations could be associated with severe disease stems from several studies showing the importance of the N-terminal domain for ADAMTS13 function and TTP pathophysiology. N-terminal domains (signal peptide through spacer) were shown to be necessary for ADAMTS13-mediated cleavage of VWF in vitro under both static and in-flow conditions.30–32 Almost all patients with autoimmune TTP display anti-ADAMTS13 autoantibodies directed against N-terminal domains, whereas only some of these patients have autoantibodies against C-terminal domains.33 The N-terminal domains are highly conserved across species, whereas the C-terminal domains are less conserved.9 N-terminal mutations were herein defined as mutations affecting ADAMTS13 domains from signal peptide through spacer (amino acid residues 1-685),34 C-terminal mutations were mutations affecting domains from thrombospondin-1-like domain number 2 (TSP1-2) through complement C1r/C1s, Uegf, and Bmp1 domain number 2 (CUB2; amino acid residues 686-1427).34 One point was added to the genotype score for each missense mutation affecting the N-terminal of the ADAMTS13 protein. Mutations at the C-terminal (either missense or truncating mutations affecting C-terminal domains only) did not add points to the score, being treated in the analysis as “protective” from severe disease. Patients with splice-site or truncating mutations affecting both the N- and C-terminal domains were excluded from analysis. The resulting scores were: genotype score = 0 (ie, a “mild genotype”) for patients with homozygous or compound heterozygous mutations both affecting the C-terminal of ADAMTS13; genotype score = 1 (ie, a “moderate genotype”) for compound heterozygous patients with one mutation affecting the N-terminal and one affecting the C-terminal of ADAMTS13; genotype score = 2 (ie, a “severe genotype”) for patients with homozygous or compound heterozygous mutations both affecting the N-terminal of ADAMTS13; and “unclassified genotype” for patients who had at least one splice-site mutation or a truncating mutation affecting both N- and C-terminal domains of ADAMTS13. Mutation and genotype score classification data for all patients are provided in supplemental Table 1.
Statistical analysis
Descriptive statistics are presented as medians (interquartile range [IQR]) and percentages. Bivariate associations between categorical variables were tested using the Fisher exact test. Linear and logistic regression analyses were used to calculate estimates and 95% confidence intervals (95% CIs) of the associations among plasmatic ADAMTS13 activity, genotype risk score, and clinical outcomes. Receiver operating characteristic curve analysis was used to determine the ADAMTS13 activity levels that could discriminate patients who had severe clinical outcomes with the highest sum of sensitivity and specificity. All P values were 2-sided with the level of significance set at P = .05.
Results
Patient characteristics and SELDI-TOF–based measurement of ADAMTS13 activity
The features of the 29 patients with congenital TTP included in this study are presented in Table 1. Residual plasmatic activity of ADAMTS13 was measured in a central laboratory on samples collected during remission at least 20 days after the last infusion of FFP or other blood-derived products. Measurements were carried out in 2 duplicates of the same sample, the median difference in the measurement between the 2 duplicates being 0.13% (IQR, 0.43%). Raw data on duplicate measurement results are available in supplemental Table 1. A total of 26 patients (90%) had measurable residual activity of ADAMTS13, and 3 patients had activity < 0.5%. Activity was below 10% in all patients, confirming the finding of severe deficiency of ADAMTS13. The median plasmatic ADAMTS13 activity was 3.08% (IQR, 4.17%; range, < 0.5%-6.77%). In a few patients with multiple remission samples available for measurement, residual ADAMTS13 activity was similar in the different samples (supplemental Table 3).
Table 1.
Patient characteristics (N = 29)
| Parameter | Value |
|---|---|
| Median age at last follow-up, y (IQR, range) | 27 (24, 3-66) |
| Male sex, n (%) | 13 (45) |
| Ethnicity, n (%) | |
| White | 22 (76) |
| Arab | 4 (14) |
| Caribbean | 3 (10) |
| Median age at first TTP episode requiring FFP, y (IQR, range) | 11 (23, neonatal-32) |
| Neonatal jaundice and/or thrombocytopenia, n (%) | 11 (38) |
| Persistence of renal/neurological damage, n (%) | 3 (10) |
| Median total lifetime TTP episodes, n (IQR, range) | 5 (8, 1-60) |
| Multiple TTP episodes, n (%) | 23 (79) |
| Median annual rate of TTP episodes, n (IQR, range) | 0.28 (0.67, 0.03-4.00) |
| Regular FFP prophylaxis, n (%) | 12 (42) |
ADAMTS13 mutations
ADAMTS13 genetic analysis in the 29 congenital TTP patients revealed 31 mutations (2 mutations of the splice site, 7 indels, 3 nonsense, and 19 missense), of which 9 (1 splice site mutation and 8 missense) were novel. All identified mutations and their functional annotations are reported in Table 2. Support for the causal role of the newly identified ADAMTS13 mutations was sought for by annotation on databases of common genetic variation and sequencing of the ADAMTS13 gene in 99 healthy white subjects. None of the mutations was present in dbSNP131, in the 1000 Genomes database, or in 198 alleles from 99 controls, enabling us to exclude that the identified variants were common polymorphisms.
Table 2.
Mutations of ADAMTS13 found in patients included in this study and their functional annotations
| DNA | Location | Predicted effect | Domain | Polyphen 2 | SIFT | Reference |
|---|---|---|---|---|---|---|
| Splicing mutations | ||||||
| c.106-1G → C | Int 1 | p.(S36_C37delfs*102) | Novel | |||
| c.1309-?G → A | Int 11 | 36 | ||||
| Indels | ||||||
| c.82dupT | Ex 1 | p.W28Lfs*111 | Signal peptide | 23 | ||
| c.4143dupA | Ex 29 | p.E1382Rfs*6 | CUB-2 | 5 | ||
| c.291_319del | Ex 3 | p.E98Pfs*31 | Metalloprotease | 35 | ||
| c.825-?_?del | Int8_ex8 | 36 | ||||
| c.718_724del | Ex 7 | p.S240Afs*7 | Metalloprotease | 24 | ||
| c.2930_2935del | Ex 23 | p.C977_R979delinsW | TSP1-6 | 28 | ||
| c.3254-3255del | Ex 25 | p.S1085Cfs*12 | TSP1-8 | 36 | ||
| Nonsense | ||||||
| c.2728C → T | Ex 21 | p.R910* | TSP1-5 | 5 | ||
| c.3047G → A | Ex 24 | p.W1016* | TSP1-7 | 23 | ||
| c.3616C → T | Ex 26 | p.R1206* | CUB-1 | 8 | ||
| Missense | ||||||
| c.237C → G | Ex 3 | p.I79M | Metalloprotease | Ben | Tolerated | 36 |
| c.262G → A | Ex 3 | p.V88M | Metalloprotease | Prod | Affect | 27 |
| c.304C → T | Ex 3 | p.R102C | Metalloprotease | Prod | Tolerated | 2 |
| c.428T → C | Ex 5 | p.I143T | Metalloprotease | Prod | Affect | Novel |
| c.448T → C | Ex 5 | p.S150P | Metalloprotease | Prod | Affect | Novel |
| c.578G → A | Ex 6 | p.R193Q | Metalloprotease | Prod | Affect | Novel |
| c.607T → C | Ex 6 | p.S203P | Metalloprotease | Prod | Tolerated | 36 |
| c.703G → T | Ex 7 | p.D235Y | Metalloprotease | Prod | Affect | Novel |
| c.706G → T | Ex 7 | p.G236C | Metalloprotease | Prod | Affect | Novel |
| c.803G → C | Ex 7 | p.R268P | Metalloprotease | Ben | Tolerated | 3 |
| c.1308G → C | Ex 11 | p.Q436H | TSP1-1 | Prod | Tolerated | Novel |
| c.1520G → A | Ex 13 | p.R507Q | Cysteine-rich | Prod | Tolerated | 36 |
| c.1787C → T | Ex 16 | p.A596V | Spacer | Prod | Tolerated | 36 |
| c.2272T → C | Ex 19 | p.C758R | TSP1-3 | Prod | Affect | 36 |
| c.3178C → T | Ex 24 | p.R1060W | TSP1-7 | Prod | Affect | 25 |
| c.3251G → A | Ex 25 | p.C1084Y | TSP1-8 | Prod | Affect | Novel |
| c.3283C → T | Ex 25 | p.R1095W | TSP1-8 | Prod | Affect | Novel |
| c.3367C → T | Ex 25 | p.R1123C | TSP1-8 | Prod | Affect | 23 |
| c.3716G → T | Ex 27 | p.G1239V | CUB-1 | Prod | Affect | 27 |
None of the mutations was found in the dbSNP131 or 1000 Genomes study databases.
Int indicates intron; Ex, exon; Ben, predicted benign; and Prod, probably damaging.
All of the novel missense mutations were also annotated on SIFT and Polyphen 2 software for the prediction of the functional effect of protein changes (predicted to be damaging vs predicted to be benign; Table 2). As a critical test of the utility of these annotations, we determined their ability to distinguish common from disease-associated variants of ADAMTS13. To estimate the sensitivity and specificity of each software, we used as positive controls 55 missense mutations of ADAMTS13 found through a review of all congenital TTP cases reported in the literature and as negative controls 20 missense single nucleotide polymorphisms (SNPs) of ADAMTS13 reported by dbSNP. Polyphen 2 was the most sensitive software, with a sensitivity of 95% and a specificity of 60%. SIFT was the most specific software, with a sensitivity of 71% and a specificity of 95%. Annotation of all of the ADAMTS13 missense mutations and SNPs is presented in supplemental Table 4.
All 8 newly identified missense mutations were predicted to be damaging for protein function by Polyphen 2 software. Only p.Q436H was predicted by SIFT to be tolerated. This mutation is caused by a c.1308G → C nucleotide substitution located in the donor splice site of exon 11. In addition to the Q → H amino acid change, this nucleotide change was also predicted by NetGene 2 to result in a reduction of splicing efficiency, providing further support of its causal role.
The novel c.106-1G → C mutation, located at the acceptor splice site of in intron 1, was predicted by NetGene2 to result in a 2-bp shift of the splice site. This splice shift is in turn expected to determine a deletion of 2 amino acid residues (S36 and C37), with frameshift of protein translation and formation of premature stop codon after 102 amino acid residues.
Association between residual ADAMTS13 activity and the severity of clinical phenotype in congenital TTP
The associations between residual ADAMTS13 activity and the phenotypic features of congenital TTP are outlined in Table 3. A lower residual ADAMTS13 activity was associated with earlier age at first TTP episode requiring FFP (Figure 2A). Lower levels of plasmatic ADAMTS13 were also associated with a higher annual rate of TTP episodes (Figure 2B) and with higher odds of regular FFP prophylaxis prescription. Adjustment for FFP prophylaxis status did not affect the association between residual ADAMTS13 activity and the annual rate of TTP episodes (beta: −0.200; 95% CI, −0.391 to −0.010; R2, 0.456; P < .001). Association results remained unchanged after adjustment for family clustering (Table 3). We used receiver operating characteristic curve analysis to determine the levels of residual ADAMTS13 activity that best discriminated patients at risk for unfavorable clinical endpoints (ie, age at first TTP episode requiring FFP below 18 years, annual rate of TTP episodes greater than 1, and prescription of FFP prophylaxis). An ADAMTS13 activity < 2.74% discriminated patients who had a first TTP episode requiring FFP below 18 years and those who had prescription of regular FFP prophylaxis, whereas an activity < 1.61% discriminated patients who had an annual rate of TTP episodes greater than 1 (Table 4).
Table 3.
Association between residual ADAMTS13 activity and phenotypic outcomes
| Phenotype | ADAMTS13 activity unadjusted analysis, % | ADAMTS13 activity adjusted for family clustering, % |
|---|---|---|
| Age at first TTP episode requiring FFP, y* | ||
| beta (95% CI) | 3.242 (1.432 to 5.052) | 3.260 (1.535 to 4.986) |
| R2 | 0.334 | 0.419 |
| P | 0.001 | 0.001 |
| Total no. of lifetime TTP episodes* | ||
| beta (95% CI) | −2.081 (−4.442 to 0.281) | −2.082 (−4.443 to 0.279) |
| R2 | 0.112 | 0.150 |
| P | 0.082 | 0.081 |
| Annual rate of TTP episodes* | ||
| beta (95% CI) | −0.306 (−0.500 to −0.111) | −0.306 (−0.502 to −0.110) |
| R2 | 0.285 | 0.309 |
| P | 0.003 | 0.004 |
| Multiple TTP episodes† | ||
| OR (95% CI) | 0.798 (0.519 to 1.225)‡ | 0.795 (0.517 to 1.225) |
| P | 0.302 | 0.299 |
| Neonatal jaundice and/or thrombocytopenia† | ||
| OR (95% CI) | 0.879 (0.616 to 1.255)‡ | 0.878 (0.615 to 1.255) |
| P | 0.478 | 0.475 |
| Persistence of renal/neurological damage† | ||
| OR (95% CI) | 2.014 (0.863 to 4.699)‡ | 2.285 (0.805 to 6.485) |
| P | 0.105 | 0.120 |
| Regular FFP prophylaxis† | ||
| OR (95% CI) | 0.625 (0.410 to 0.954)‡ | 0.625 (0.409 to 0.955) |
| P | 0.030 | 0.030 |
On linear regression analysis.
On logistical regression analysis.
Per 1% increase in ADAMTS13 activity.
OR indicates odds ratio.
Figure 2.
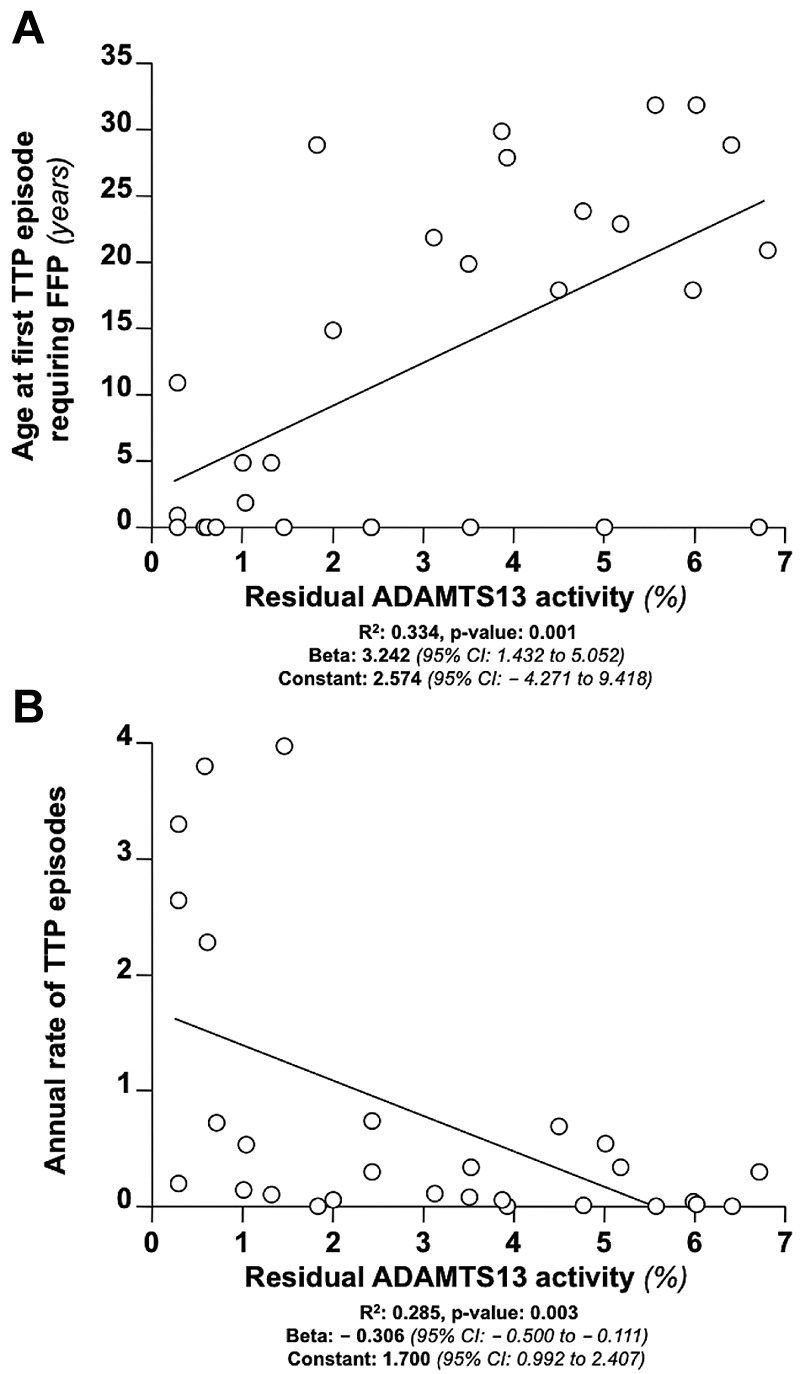
Relationship between residual ADAMTS13 activity and clinical features of congenital TTP. (A) Association between residual ADAMTS13 activity and age at first TTP episode requiring FFP in 29 patients with congenital TTP at regression analysis. (B) Association between residual ADAMTS13 activity and the annual rate of TTP episodes.
Table 4.
Receiver operating characteristic curve analysis
| Phenotype | Age at first TTP episode requiring FFP < 18 y | Annual rate of TTP episodes > 1 | Regular FFP prophylaxis |
|---|---|---|---|
| ADAMTS13 activity cutoff, % | < 2.74 | < 1.61 | < 2.74 |
| Area under the curve (95% CI) | 0.880 (0.746-1.013) | 0.930 (0.829-1.032) | 0.752 (0.564-0.941) |
| P | .001 | .003 | .023 |
| Sensitivity | 92.3% | 78.3% | 70.6% |
| Specificity | 81.3% | 100% | 75.0% |
| OR (95% CI) | 52.00 (4.74-570.53) | 1.615 × 109 (NC) | 7.20 (1.35-38.33) |
OR indicates odds ratio; and NC, noncalculable.
Relationships among ADAMTS13 mutations, residual plasmatic activity of ADAMTS13, and clinical outcomes of congenital TTP
Because patients with the same ADAMTS13 genotype were found to have similar age of onset,9 we hypothesized that ADAMTS13 gene mutations could influence the clinical phenotype of congenital TTP and therefore we determined levels of residual ADAMTS13 activity. We plotted ADAMTS13 genotypes that occurred more than once in the study cohort against the residual ADAMTS13 activity and the age at first TTP episode requiring FFP of the corresponding patients (Figure 3). A clustering of both age at first TTP episode requiring FFP and residual ADAMTS13 activity in patients with the same genotype was observed for both related and unrelated individuals. This suggests that ADAMTS13 mutations influence both the amount of residual ADAMTS13 activity and the clinical features of the disease.
Figure 3.
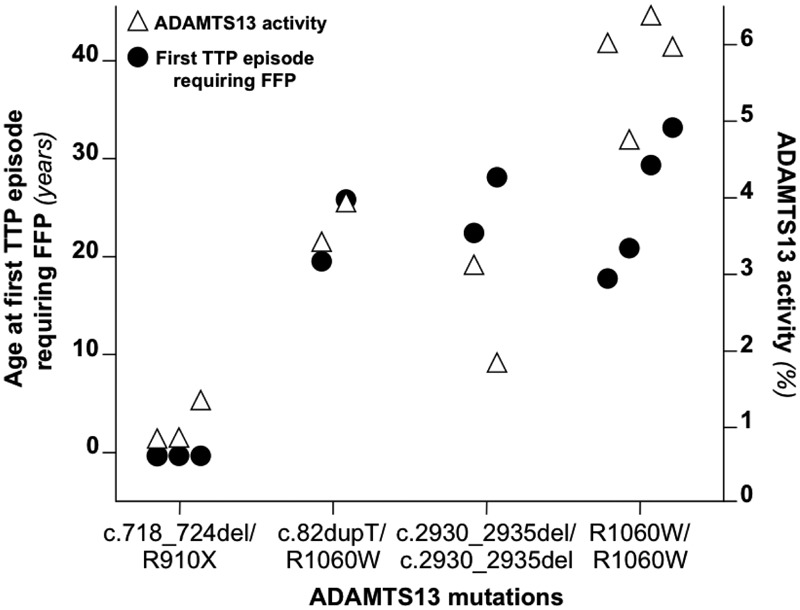
Relationship among ADAMTS13 genotype, residual activity, and age at first episode requiring FFP. Shown is the age at first TTP episode requiring FFP and residual ADAMTS13 activity in multiple patients carrying the same mutations of ADAMTS13. A clustering of both the age at first TTP episode requiring FFP and the plasmatic activity of ADAMTS13 can be observed for patients with a given ADAMTS13 genotype. The clustering was observed both for related patients (the first 3 genotypes) and for unrelated individuals (the homozygous p.R1060W genotype). For each patient, ADAMTS13 activity (▵) and age of the first TTP episode requiring FFP (●) are aligned.
To identify the relationships between specific ADAMTS13 mutations and the clinical features of congenital TTP, we studied the type and distribution of congenital-TTP–causing ADAMTS13 mutations on the ADAMTS13 protein domains.
Including the 9 novel mutations, 122 mutations of ADAMTS13 have been described to date in congenital TTP patients. Of these, 63 (51%) are missense. Notably, 71% (n = 45) of the missense mutations of ADAMTS13 described in TTP patients localize in the N-terminal of the protein (binomial probability, P = 9.05 × 10−5), which is the area with the highest degree of evolutionary conservation9 and where the domains necessary for ADAMTS13 catalytic activity are located.30–32,37,38 Conversely, missense SNPs of ADAMTS13 included in dbSNP131 do not show evidence of skewed distribution (P = .14).
We also evaluated the evolutionary conservation at the sites of all reported missense mutations of ADAMTS13 (only mutations of ADAMTS13 reported in association with congenital TTP were considered). Mutations of the C-terminal end of ADAMTS13 occurred almost exclusively at amino acid residues that are highly conserved across species (SIFT score below 0.05; mutations at highly conserved residues, 16 of 18, 89%), whereas causal mutations of congenital TTP in the N-terminal domains of ADAMTS13 were found at amino acid residues that are less conserved (mutations at highly conserved residues, 30 of 45, 67%; Figure 4A). All of these analyses indicated that mutations at the N-terminal domains of ADAMTS13 might be more severe than C-terminal domain mutations. To investigate whether mutations affecting different domains of ADAMTS13 were associated with variable degrees of clinical severity, we compared the residual plasmatic activity of study participants carrying mutations of the N-terminal domains of ADAMTS13 with that of patients with C-terminal domain mutations. Only patients with homozygous mutations were considered for analysis to avoid the confounding that derives from the coexistence of 2 different mutations in compound heterozygous patients. Patients with homozygous mutations at the N-terminal domains (n = 4) displayed lower residual activity of ADAMTS13 and earlier age at first TTP episode requiring FFP compared with those with homozygous C-terminal domain mutations (n = 8; Figure 4B). To further study the relationship between mutation localization and clinical phenotype of congenital TTP, a genotype score was constructed considering N-terminal mutations as a risk factor for more severe disease. After genotype score computation (see “Construction of a genotype score” for details), 9 patients had a mild genotype (ie, with homozygous or compound heterozygous mutations both affecting the C-terminal of ADAMTS13; score = 0), 5 had a moderate genotype (ie, compound heterozygous patients with 1 mutation affecting the N-terminal and 1 affecting the C-terminal of ADAMTS13; score = 1), 7 had a severe genotype (ie, with homozygous or compound heterozygous mutations both affecting the N-terminal of ADAMTS13; score = 2), and 8 patients were not classifiable in any of the groups (ie, they either had at least 1 splice-site mutation or a truncating mutation affecting both N- and C-terminal domains of ADAMTS13). On linear regression analysis, the residual plasmatic activity of ADAMTS13 decreased with increasing genotype severity (Figure 5A). On logistic regression analysis, patients with a moderate genotype were 2 times more likely (95% CI, 0.098-41.003) to receive regular FFP prophylaxis, whereas patients with a severe genotype were 10.667 times more likely (95% CI, 0.823-138.222) to receive regular FFP prophylaxis compared with patients with a mild genotype. The estimates were lower when the analysis was adjusted for residual ADAMTS13 activity (moderate genotype: odds ratio = 1.027; 95% CI, 0.035-30.545 and severe genotype: odds ratio = 4.757; 95% CI, 0.246-91.969 compared with the mild genotype). On linear regression analysis, there was a statistically significant negative association between the genotype severity score and age at first TTP episode requiring FFP (Figure 5B). Moreover, there was a positive association between genotype severity and the annual rate of TTP episodes (Figure 5C). Results remained unchanged when patients carrying truncating mutations at the C-terminal domains were excluded from analysis.
Figure 4.
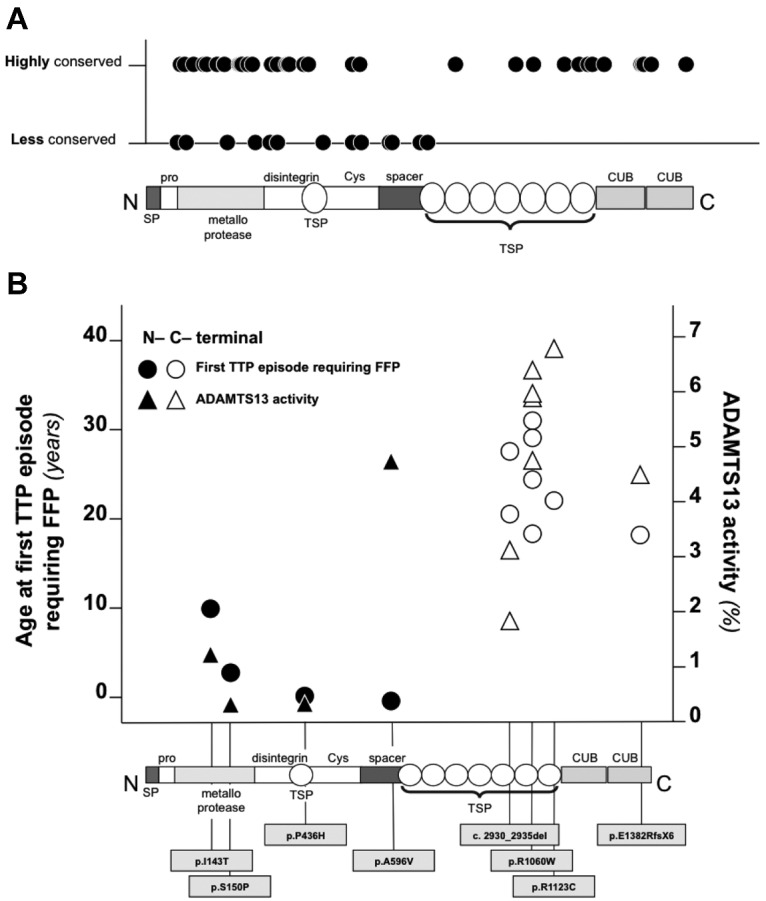
Evolutionary conservation, associated ADAMTS13 plasmatic activity, and age at first TTP episode requiring FFP of N- and C-terminal mutations of ADAMTS13. (A) Evolutionary conservation of amino acid residues affected by missense mutations of ADAMTS13 associated with congenital TTP in all cases reported in the literature. Conservation was estimated using SIFT. (B) Residual activity of ADAMTS13 and age at first TTP episode requiring FFP of patients with different C- and N-terminal mutations. Only mutations in homozygosis were considered.
Figure 5.
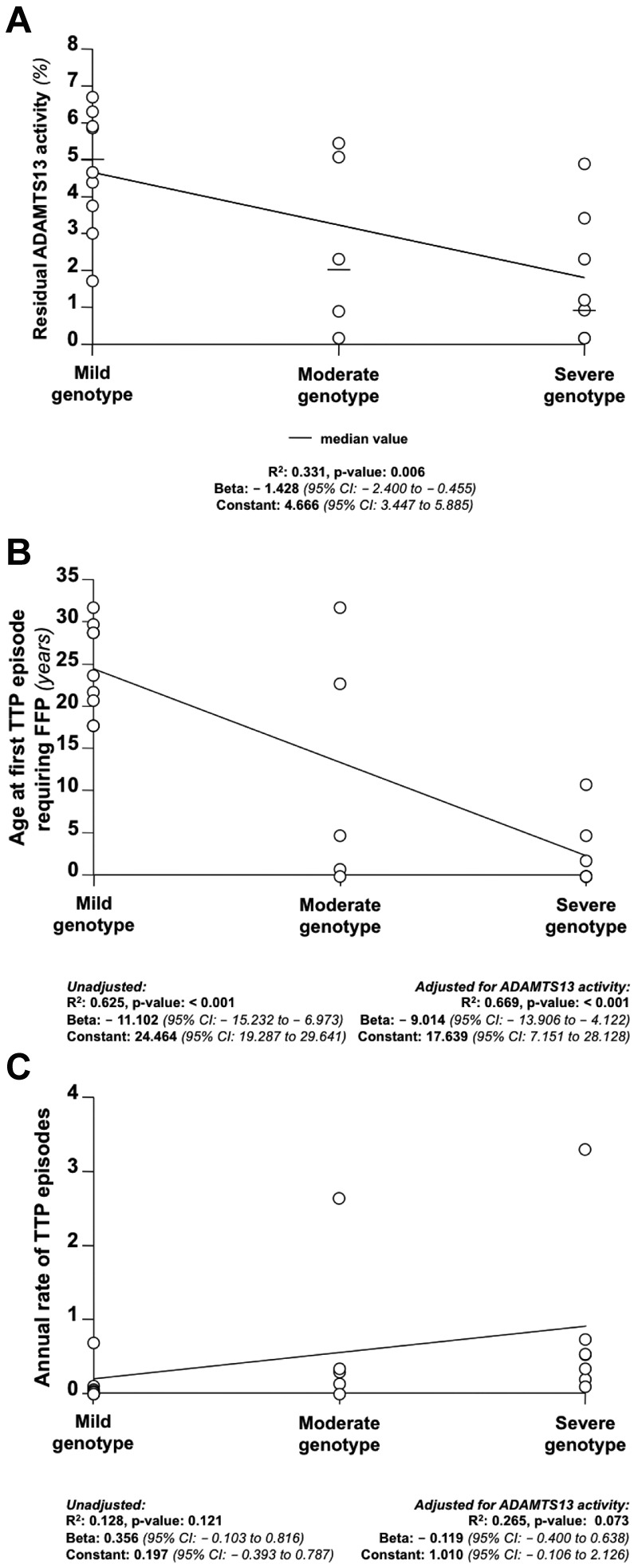
Association among genotype severity score, plasmatic activity of ADAMTS13, age at first TTP episode requiring FFP, and annual rate of TTP episodes. Increasing severity in the genotype score (ie, an increasing number of N-terminal mutations; mild genotype indicates 2 C-terminal mutations; moderate genotype, 1 C- and 1 N-terminal mutations; and severe genotype, 2 N-terminal mutations) was associated with lower plasmatic activity of ADAMTS13 (A), earlier age at first TTP episode requiring FFP (B), and a higher annual rate of TTP episodes (C).
Discussion
In the present study, a new SELDI-TOF mass spectrometry–based method was used to measure the residual plasmatic activity of ADAMTS13 in congenital TTP patients, with the aim of investigating the relationships among ADAMTS13 genotype, residual plasmatic activity, and the clinical phenotype of the disease. Our main study findings were: (1) a residual plasmatic activity of ADAMTS13 was measurable by SELDI-TOF mass-spectrometry in 90% of the study participants; (2) the amount of residual plasmatic activity of ADAMTS13 measured by SELDI-TOF mass spectrometry was inversely correlated with the clinical severity of the phenotype; (3) residual plasmatic activity levels below 2.74% and 1.61% discriminated patients with adverse clinical outcomes; and (4) ADAMTS13 mutations were associated with activity levels and clinical severity, with N-terminal domain mutations being associated with lower activity and severe disease in an allele-dosage–dependent way.
The existence of some degree of ADAMTS13 activity in the majority of congenital TTP patients was documented for the first time in the present study. Previous case reports and case series reported the detection of a residual activity in only a few patients. Because many of these studies adopted immunoblotting and collagen-binding assay for the identification of severe ADAMTS13 deficiency, this result is not surprising in light of the relatively high limit of detection and scarce reproducibility of these assays at low ADAMTS13 concentrations.12,13 More recently, Fujimura et al reported a large cohort of congenital TTP patients in whom ADAMTS13 was measured by an ELISA method with a reportedly low limit of detection (0.5%), in which residual activity was found in only a minority of patients. Because limits of detection are calculated against different calibration curves with different reference samples, a formal comparison of the limits of detection of that assay with ours cannot be made unless both assays are tested against a common “standard.” However, several proofs of the reliability and validity of our measurements are provided in the present study, arguing against random or biased results. First, a calibration curve using calibrators with concentrations of ADAMTS13 < 10% was calculated before each assay, showing the linearity of the method at very low concentrations (Figure 1). Second, repeated measurements on the same sample (including duplicate measurements and 2 external controls repeated over time by different operators) and multiple samples of the same patient consistently produced similar activity results. Third, the detection method of the assay used in this study (mass spectrometry) is considered to be the “gold standard” for protein detection, providing high analytical sensitivity and specificity for very tiny amounts of cleavage products.
Decreasing levels of residual ADAMTS13 activity were associated with earlier age at first TTP episode requiring FFP, more frequent disease recurrences, and prescription of FFP prophylaxis to prevent further recurrences. These results are consistent with the views that congenital TTP patients with a higher residual plasmatic activity of ADAMTS13 are protected from disease onset until they come across a strong challenge to their fragile ADAMTS13-VWF balance (eg, pregnancy, which is often a triggering factor of TTP in women with adult-onset disease), whereas patients with a low or with no residual activity are vulnerable to environmental challenges, developing early onset disease and frequent recurrences that prompt caring physicians to prescribe FFP prophylaxis. We identified 2.74% and 1.61% as the levels of ADAMTS13 activity predictive of clinical end points of disease severity. An activity of ADAMTS13 below these levels discriminated patients with early TTP requiring treatment with FFP, frequent recurrences, and the need for preventive FFP infusion with high specificity, suggesting that patients with low residual ADAMTS13 activity are unlikely to have mild disease. Individual exceptions to the observed associations occurred, with patients with relatively high residual activity having early disease episodes or frequent recurrences. This result likely reflects the multicausal nature of disease severity in congenital TTP. Phenotypic severity in congenital TTP is conceivably the result of multiple environmental and genetic factors acting in concert, so that factors other than residual ADAMTS13 activity might explain these apparently outlying results. Mutation-specific effects of the ADAMTS13 mutations, of other mutations genome wide, and of environmental modifiers (eg, infections, pregnancy, surgery, etc) may all be factors contributing to disease severity.
In the present study, we also reported 9 novel mutations of ADAMTS13 associated with congenital TTP. A detailed analysis of the annotation, functional consequences, and distribution on ADAMTS13 of these novel mutations and of previously reported mutations revealed that mutations in the highly conserved N-terminal domains of ADAMTS13, which have been shown to be required for the catalytic activity of this enzyme,30–32,37 are more likely to be associated with congenital TTP, lower residual ADAMTS13 activity, and earlier age at TTP requiring FFP treatment than those of C-terminal domains. The association of the ADAMTS13 genotype with the clinical phenotype was attenuated by adjusting for ADAMTS13 plasmatic activity, confirming that the ADAMTS13 genotype influences disease severity at least in part through residual plasmatic ADAMTS13 activity.
Limitations of the present study include the fact that individual ADAMTS13 activity was measured once for the majority of patients and that clinical outcomes were ascertained retrospectively. The serial measurement of ADAMTS13, made difficult by the fact that included patients were from different regions of different continents, was beyond the scope of this explorative study. However, by measuring ADAMTS13 in more than 1 sample of a few representative patients, we showed that residual ADAMTS13 activity during remission is similar in a given patient. Because the prevalence of congenital TTP is estimated to be approximately 1 in 1 million,39 a prospective study would not have been feasible. In addition, the clinically relevant end points of the study (ie, age at first TTP episode requiring treatment with FFP, rate of episodes, and use of FFP prophylaxis) can be considered “hard” end points that are unlikely to be misclassified. To further minimize subjectivity in the definition of age of onset, we considered a clinically relevant disease manifestation (ie, acute episode of TTP requiring FFP treatment) as a more robust end point. If some misclassification persisted, it could have been in both directions. ADAMTS13 activity measurement was blind to phenotype severity.
In conclusion, in the present study we found that residual ADAMTS13 activity is correlated with the phenotype severity of congenital TTP and that mutations affecting the evolutionary conserved N-terminal domains of the protein are associated with more severe clinical phenotype. This study identifies a disease-severity biomarker of potential clinical relevance, residual plasmatic activity of ADAMTS13, and provides insights into genotype-phenotype correlations in this rare, life-threatening disease.
Supplementary Material
Acknowledgments
The authors thank Dr Luigi Flaminio Ghilardini for help with the figures and tables; Drs Roberta Palla, Andrea Cairo, and Carla Valsecchi for help with the experiments, organizing sample shipments, and collecting clinical information; Profs Armando Tripodi and Pier Mannuccio Mannucci for their valuable critical remarks; and Prof Sam J. Machin for help with setting up the study.
This study was supported by the Call per la Ricerca indipendente Regione Lombardia, decreto (grant 6716 del 1/7/2009) and by the Italian Ministry of Health (grant RF-2009-1530493); and Fondazione Cariplo (grant 2011-0524). The United Kingdom TTP Registry is sponsored by the Medical Research Council. L.A.L. was supported by the 2009 Bayer Hemophilia Clinical Training Award and is also the recipient of the Associazione Italiana Centri Emofilia De Biasi Prize.
Footnotes
There is an Inside Blood commentary on this article in this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: L.A.L. designed the research, carried out part of the analyses, interpreted the results, and wrote the manuscript; H.M.W., A.H., and S.Y. performed the ADAMTS13 measurement by SELDI-TOF mass spectrometry and critically reviewed the manuscript; H.M.W. participated in study design; I.J.M., M.A.S., M.N., A.V., G.R., P.C., R.L., R.D., C.L., I.G., L.A.L., and F.P. enrolled the patients, collected and standardized clinical and laboratory information, and critically reviewed the manuscript; R.A.G. performed the next-generation sequencing and mutation annotation and critically reviewed the manuscript; K.M.M. conducted the statistical analyses and wrote the manuscript; and F.P. designed the research, interpreted the results, and critically reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Luca A. Lotta, MD, PhD, Angelo Bianchi Bonomi Hemophilia and Thrombosis Center, U.O.S. Dipartimentale per la Diagnosi e la Terapia delle Coagulopatie, Fondazione Istituto di Ricovero e Cura a Carattere Scientifico Ca' Granda–Ospedale Maggiore Policlinico, Università degli Studi di Milano and Luigi Villa Foundation, Via Pace 9, 20122, Milan, Italy; e-mail: luca.lotta@unimi.it.
References
- 1.Schulman I, Pierce M, Lukens A, Currimbhoy Z. Studies on thrombopoiesis. I. A factor in normal human plasma required for platelet production; chronic thrombocytopenia due to its deficiency. Blood. 1960;16:943–957. [PubMed] [Google Scholar]
- 2.Levy GG, Nichols WC, Lian EC, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413(6855):488–494. doi: 10.1038/35097008. [DOI] [PubMed] [Google Scholar]
- 3.Kokame K, Matsumoto M, Soejima K, et al. Mutations and common polymorphisms in ADAMTS13 gene responsible for von Willebrand factor-cleaving protease activity. Proc Natl Acad Sci U S A. 2002;99(18):11902–11907. doi: 10.1073/pnas.172277399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsai HM. Pathophysiology of thrombotic thrombocytopenic purpura. Int J Hematol. 2010;91(1):1–19. doi: 10.1007/s12185-009-0476-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schneppenheim R, Budde U, Oyen F, et al. von Willebrand factor cleaving protease and ADAMTS13 mutations in childhood TTP. Blood. 2003;101(5):1845–1850. doi: 10.1182/blood-2002-08-2399. [DOI] [PubMed] [Google Scholar]
- 6.Fujimura Y, Matsumoto M, Isonishi A, et al. Natural history of Upshaw-Schulman syndrome based on ADAMTS13 gene analysis in Japan. J Thromb Haemost. 2011;9(Suppl 1):283–301. doi: 10.1111/j.1538-7836.2011.04341.x. [DOI] [PubMed] [Google Scholar]
- 7.Fujimura Y, Matsumoto M, Kokame K, et al. Pregnancy-induced thrombocytopenia and TTP, and the risk of fetal death, in Upshaw-Schulman syndrome: a series of 15 pregnancies in 9 genotyped patients. Br J Haematol. 2009;144(5):742–754. doi: 10.1111/j.1365-2141.2008.07515.x. [DOI] [PubMed] [Google Scholar]
- 8.Shibagaki Y, Matsumoto M, Kokame K, et al. Novel compound heterozygote mutations (H234Q/R1206X) of the ADAMTS13 gene in an adult patient with Upshaw-Schulman syndrome showing predominant episodes of repeated acute renal failure. Nephrol Dial Transplant. 2006;21(5):1289–1292. doi: 10.1093/ndt/gfk072. [DOI] [PubMed] [Google Scholar]
- 9.Lotta LA, Garagiola I, Palla R, Cairo A, Peyvandi F. ADAMTS13 mutations and polymorphisms in congenital thrombotic thrombocytopenic purpura. Hum Mutat. 2010;31(1):11–19. doi: 10.1002/humu.21143. [DOI] [PubMed] [Google Scholar]
- 10.Gerritsen HE, Turecek PL, Schwarz HP, Lammle B, Furlan M. Assay of von Willebrand factor (vWF)-cleaving protease based on decreased collagen binding affinity of degraded vWF: a tool for the diagnosis of thrombotic thrombocytopenic purpura (TTP). Thromb Haemost. 1999;82(5):1386–1389. [PubMed] [Google Scholar]
- 11.Kokame K, Nobe Y, Kokubo Y, Okayama A, Miyata T. FRETS-VWF73, a first fluorogenic substrate for ADAMTS13 assay. Br J Haematol. 2005;129(1):93–100. doi: 10.1111/j.1365-2141.2005.05420.x. [DOI] [PubMed] [Google Scholar]
- 12.Tripodi A, Chantarangkul V, Bohm M, et al. Measurement of von Willebrand factor cleaving protease (ADAMTS-13): results of an international collaborative study involving 11 methods testing the same set of coded plasmas. J Thromb Haemost. 2004;2(9):1601–1609. doi: 10.1111/j.1538-7836.2004.00879.x. [DOI] [PubMed] [Google Scholar]
- 13.Tripodi A, Peyvandi F, Chantarangkul V, et al. Second international collaborative study evaluating performance characteristics of methods measuring the von Willebrand factor cleaving protease (ADAMTS-13). J Thromb Haemost. 2008;6(9):1534–1541. doi: 10.1111/j.1538-7836.2008.03099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin M, Cataland S, Bissell M, Wu HM. A rapid test for the diagnosis of thrombotic thrombocytopenic purpura using surface enhanced laser desorption/ionization time-of-flight (SELDI-TOF)-mass spectrometry. J Thromb Haemost. 2006;4(2):333–338. doi: 10.1111/j.1538-7836.2006.01758.x. [DOI] [PubMed] [Google Scholar]
- 15.Jin M, Casper TC, Cataland SR, et al. Relationship between ADAMTS13 activity in clinical remission and the risk of TTP relapse. Br J Haematol. 2008;141(5):651–658. doi: 10.1111/j.1365-2141.2008.07107.x. [DOI] [PubMed] [Google Scholar]
- 16.Lotta LA, Mariani M, Consonni D, et al. Different clinical severity of first episodes and recurrences of thrombotic thrombocytopenic purpura. Br J Haematol. 2010;151(5):488–494. doi: 10.1111/j.1365-2141.2010.08385.x. [DOI] [PubMed] [Google Scholar]
- 17.Lotta LA, Lombardi R, Mariani M, et al. Platelet reactive conformation and multimeric pattern of von Willebrand factor in acquired thrombotic thrombocytopenic purpura during acute disease and remission. J Thromb Haemost. 2011;9(9):1744–1751. doi: 10.1111/j.1538-7836.2011.04428.x. [DOI] [PubMed] [Google Scholar]
- 18.Scully M, Yarranton H, Liesner R, et al. Regional UK TTP registry: correlation with laboratory ADAMTS 13 analysis and clinical features. Br J Haematol. 2008;142(5):819–826. doi: 10.1111/j.1365-2141.2008.07276.x. [DOI] [PubMed] [Google Scholar]
- 19.Caprioli J, Noris M, Brioschi S, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108(4):1267–1279. doi: 10.1182/blood-2005-10-007252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coppo P, Schwarzinger M, Buffet M, et al. Predictive features of severe acquired ADAMTS13 deficiency in idiopathic thrombotic microangiopathies: the French TMA reference center experience. PLoS One. 2010;5(4):e10208. doi: 10.1371/journal.pone.0010208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peyvandi F, Lavoretano S, Palla R, et al. ADAMTS13 and anti-ADAMTS13 antibodies as markers for recurrence of acquired thrombotic thrombocytopenic purpura during remission. Haematologica. 2008;93(2):232–239. doi: 10.3324/haematol.11739. [DOI] [PubMed] [Google Scholar]
- 22.Noris M, Bucchioni S, Galbusera M, et al. Complement factor H mutation in familial thrombotic thrombocytopenic purpura with ADAMTS13 deficiency and renal involvement. J Am Soc Nephrol. 2005;16(5):1177–1183. doi: 10.1681/ASN.2005010086. [DOI] [PubMed] [Google Scholar]
- 23.Donadelli R, Banterla F, Galbusera M, et al. In-vitro and in-vivo consequences of mutations in the von Willebrand factor cleaving protease ADAMTS13 in thrombotic thrombocytopenic purpura. Thromb Haemost. 2006;96(4):454–464. [PubMed] [Google Scholar]
- 24.Assink K, Schiphorst R, Allford S, et al. Mutation analysis and clinical implications of von Willebrand factor-cleaving protease deficiency. Kidney Int. 2003;63(6):1995–1999. doi: 10.1046/j.1523-1755.63.6s.1.x. [DOI] [PubMed] [Google Scholar]
- 25.Camilleri RS, Cohen H, Mackie IJ, et al. Prevalence of the ADAMTS-13 missense mutation R1060W in late onset adult thrombotic thrombocytopenic purpura. J Thromb Haemost. 2008;6(2):331–338. doi: 10.1111/j.1538-7836.2008.02846.x. [DOI] [PubMed] [Google Scholar]
- 26.Peyvandi F, Ferrari S, Lavoretano S, Canciani MT, Mannucci PM. von Willebrand factor cleaving protease (ADAMTS-13) and ADAMTS-13 neutralizing autoantibodies in 100 patients with thrombotic thrombocytopenic purpura. Br J Haematol. 2004;127(4):433–439. doi: 10.1111/j.1365-2141.2004.05217.x. [DOI] [PubMed] [Google Scholar]
- 27.Peyvandi F, Lavoretano S, Palla R, et al. Mechanisms of the interaction between two ADAMTS13 gene mutations leading to severe deficiency of enzymatic activity. Hum Mutat. 2006;27(4):330–336. doi: 10.1002/humu.20267. [DOI] [PubMed] [Google Scholar]
- 28.Palla R, Lavoretano S, Lombardi R, et al. The first deletion mutation in the TSP1-6 repeat domain of ADAMTS13 in a family with inherited thrombotic thrombocytopenic purpura. Haematologica. 2009;94(2):289–293. doi: 10.3324/haematol.13524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lotta LA, Wang M, Yu J, et al. Identification of genetic risk variants for deep vein thrombosis by multiplexed next-generation sequencing of 186 hemostatic/pro-inflammatory genes. BMC Med Genomics. 2012;5:7. doi: 10.1186/1755-8794-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soejima K, Matsumoto M, Kokame K, et al. ADAMTS-13 cysteine-rich/spacer domains are functionally essential for von Willebrand factor cleavage. Blood. 2003;102(9):3232–3237. doi: 10.1182/blood-2003-03-0908. [DOI] [PubMed] [Google Scholar]
- 31.Zheng X, Nishio K, Majerus EM, Sadler JE. Cleavage of von Willebrand factor requires the spacer domain of the metalloprotease ADAMTS13. J Biol Chem. 2003;278(32):30136–30141. doi: 10.1074/jbc.M305331200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tao Z, Wang Y, Choi H, et al. Cleavage of ultralarge multimers of von Willebrand factor by C-terminal-truncated mutants of ADAMTS-13 under flow. Blood. 2005;106(1):141–143. doi: 10.1182/blood-2004-11-4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng XL, Wu HM, Shang D, et al. Multiple domains of ADAMTS13 are targeted by autoantibodies against ADAMTS13 in patients with acquired idiopathic thrombotic thrombocytopenic purpura. Haematologica. 2010;95(9):1555–1562. doi: 10.3324/haematol.2009.019299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng X, Chung D, Takayama TK, Majerus EM, Sadler JE, Fujikawa K. Structure of von Willebrand factor-cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J Biol Chem. 2001;276(44):41059–41063. doi: 10.1074/jbc.C100515200. [DOI] [PubMed] [Google Scholar]
- 35.Garagiola I, Valsecchi C, Lavoretano S, Oren H, Bohm M, Peyvandi F. Nonsense-mediated mRNA decay in the ADAMTS13 gene caused by a 29-nucleotide deletion. Haematologica. 2008;93(11):1678–1685. doi: 10.3324/haematol.13102. [DOI] [PubMed] [Google Scholar]
- 36.Veyradier A, Lavergne JM, Ribba AS, et al. Ten candidate ADAMTS13 mutations in six French families with congenital thrombotic thrombocytopenic purpura (Upshaw-Schulman syndrome). J Thromb Haemost. 2004;2(3):424–429. doi: 10.1111/j.1538-7933.2004.00623.x. [DOI] [PubMed] [Google Scholar]
- 37.de Groot R, Bardhan A, Ramroop N, Lane DA, Crawley JT. Essential role of the disintegrin-like domain in ADAMTS13 function. Blood. 2009;113(22):5609–5616. doi: 10.1182/blood-2008-11-187914. [DOI] [PubMed] [Google Scholar]
- 38.Zhou Z, Yeh HC, Jing H, et al. Cysteine residues in CUB-1 domain are critical for ADAMTS13 secretion and stability. Thromb Haemost. 2011;105(1):21–30. doi: 10.1160/TH10-07-0446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kokame K, Kokubo Y, Miyata T. Polymorphisms and mutations of ADAMTS13 in the Japanese population and estimation of the number of patients with Upshaw-Schulman syndrome. J Thromb Haemost. 2011;9(8):1654–1656. doi: 10.1111/j.1538-7836.2011.04399.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.