

Abstract
Disruption of neuronal Ca2+ homeostasis contributes to neurodegenerative diseases through mechanisms that are not fully understood. A polymorphism in CALHM1, a recently described ion channel that regulates intracellular Ca2+ levels, is a possible risk factor for late-onset Alzheimer's disease. Since there are six potentially redundant CALHM family members in humans, the physiological and pathophysiological consequences of CALHM1 function in vivo remain unclear. The nematode Caenorhabditis elegans expresses a single CALHM1 homolog, CLHM-1. Here we find that CLHM-1 is expressed at the plasma membrane of sensory neurons and muscles. Like human CALHM1, C. elegans CLHM-1 is a Ca2+-permeable ion channel regulated by voltage and extracellular Ca2+. Loss of clhm-1 in the body-wall muscles disrupts locomotory kinematics and biomechanics, demonstrating that CLHM-1 has a physiologically significant role in vivo. The motility defects observed in clhm-1 mutant animals can be rescued by muscle-specific expression of either C. elegans CLHM-1 or human CALHM1, suggesting that the function of these proteins is conserved in vivo. Overexpression of either C. elegans CLHM-1 or human CALHM1 in neurons is toxic, causing degeneration through a necrotic-like mechanism that is partially Ca2+ dependent. Our data show that CLHM-1 is a functionally conserved ion channel that plays an important but potentially toxic role in excitable cell function.
Introduction
Ca2+ acts as an intracellular messenger required for essential processes including neurotransmitter release and muscle contraction. Multiple active and passive mechanisms are used to precisely regulate cytoplasmic Ca2+ including Ca2+ pumps and cotransporters, voltage-gated Ca2+ channels (VGCCs), transient receptor potential (TRP) channels, store-operated Ca2+ entry (SOCE) channels, hemichannels, inositol 1,4,5-triphosphate (InsP3) receptors, and ryanodine receptors (Berridge et al., 1998). Disruption of neuronal Ca2+ homeostasis, either through inappropriate entry of extracellular Ca2+ (Cao2+) or unregulated release of Ca2+ from intracellular stores, has been linked to neurodegenerative diseases such as Alzheimer's disease (AD) (Bezprozvanny and Mattson, 2008). However, the molecular mechanisms that underlie alterations in cellular Ca2+ handling and AD pathogenesis are poorly understood.
Human CALHM1 is a previously described pore-forming subunit of an ion channel that modulates cellular Ca2+ homeostasis (Dreses-Werringloer et al., 2008; Ma et al., 2012). Heterologous expression of CALHM1 in Xenopus oocytes and N2A cells gives rise to a voltage and Cao2+-sensitive, outwardly rectifying, Ca2+-permeable current (Ma et al., 2012). CALHM1 is predicted to contain four transmembrane (tm) domains and exhibits structural and functional similarities with connexins, pannexins, and innexins, but does not form gap junctions or exhibit significant sequence similarity with known ion channels (Siebert et al., 2013). Human genetic studies suggest that a polymorphism in CALHM1 [proline at residue 86 replaced by leucine (P86L)] may be associated with age of onset of late-onset Alzheimer's disease (LOAD) (Dreses-Werringloer et al., 2008; Boada et al., 2010; Lambert et al., 2010), although existing genetic data supporting a role for CALHM1 in LOAD pathogenesis remain controversial (Bertram et al., 2008; Beecham et al., 2009; Minster et al., 2009). While these genetic data indicate a potential role for mutant CALHM1 in disease, the role of CALHM1 in regulating Ca2+ homeostasis in vivo is unknown. Thus, it is essential to determine physiological and pathophysiological functions of normal and mutant CALHM1 in an in vivo system to understand the role of CALHM1 in neuronal Ca2+ homeostasis and its relationship to LOAD.
Calhm1 is one of six members in the Calhm gene family in humans. Both genetic redundancy and the fact that CALHM proteins form a multimeric structure complicate the study of mammalian CALHM1 in vivo (Dreses-Werringloer et al., 2008; Siebert et al., 2013). Thus, analysis of a Calhm homolog in a simpler organism might yield insights into CALHM1 function. While Calhm1 homologs are absent in Drosophila and yeast, Caenorhabditis elegans expresses a single homolog, clhm-1. We found that C. elegans CLHM-1 is an ion channel that shares biophysical and pharmacological properties with human CALHM1. C. elegans CLHM-1 is expressed at the plasma membrane of excitable cells, is required in body-wall muscles for coordinated locomotion, and causes cell death when overexpressed. Furthermore, expression of either C. elegans CLHM-1 or human CALHM1 in body-wall muscles can rescue C. elegans clhm-1 mutant motility defects, suggesting that these two proteins may play a similar role in vivo. Our work establishes Calhm family members as a new, evolutionarily conserved class of physiologically significant and potentially toxic ion channels.
Materials and Methods
Nematode culture.
C. elegans hermaphrodites were grown at 20° under standard conditions unless noted otherwise. Double-mutant strains were constructed using standard genetic techniques (Brenner, 1974), and all genotypes were confirmed by PCR and/or sequencing. The wild-type strain was Bristol N2; mutants used were clhm-1(tm4071), clhm-1(ok3617), cnx-1(ok2234), crt-1(ok948), mec-4(e1611), and unc-119(ed3). Both clhm-1 mutants were backcrossed four times to wild-type to remove background mutations.
Electrophysiology in Xenopus oocytes.
The C. elegans clhm-1 cDNA was inserted into the pBF oocyte expression vector; the CLHM-1 D125A mutation was created using the QuikChange II site-directed mutagenesis kit (Agilent Technologies). cRNA was synthesized from linearized plasmids with SP6 RNA polymerase (mMessage mMachine kit; Ambion). Five nanograms of clhm-1 cRNA along with 80 ng of Xenopus connexin 38 (Cx38) antisense oligonucleotide (Ma et al., 2012) were injected into stage IV–VI Xenopus oocytes that had been isolated from female Xenopus laevis and defolliculated with type IV collagenase (Worthington Biochemical). Oocytes were incubated 1–3 d at 16°C in ND96 medium (96 mm NaCl, 2 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, 2.5 mm Na-pyruvate, 1× penicillin–streptomycin, pH 7.6). Oocytes injected with clhm-1::gfp cRNA were imaged with a Zeiss LSM 510 confocal microscope 48 h after injection.
Oocytes used for two-electrode voltage-clamp experiments were injected with 50 nl of a 20 mm BAPTA, 10 mm Ca2+ solution at least 30 min before recording to prevent activation of endogenous Ca2+-activated Cl− currents (Ma et al., 2012). Standard bath solutions contained 100 mm Na+, 100 mm Cl−, 2 mm K+, and 10 mm HEPES, and various concentrations of Ca2+ and Mg2+ as indicated. EGTA (0.5 mm) and EDTA (0.5 mm) were added to the standard bath solution lacking divalent cations to create a divalent cation-free solution. All solutions were pH 7.2, adjusted with methanesulfonic acid. Data were acquired with an OC-725C amplifier (Warner Instrument) at 1 kHz with a 16 bit analog-to-digital converter (Instrutech ITC-16). Electrodes were made from thin-walled TW100F-6 glass (World Precision Instruments) using a micropipette puller (model P-87; Sutter Instrument) and were filled with 3 m KCl. All recordings were analyzed with Igor Pro.
Relative permeabilities.
For ion permeability experiments, sucrose was used as substitute for NaCl or divalent cations in solutions to maintain constant osmolality while varying ion concentrations. KCl (3 m) agar bridges connected ground electrodes to bath solutions. Reversal potentials (Erev) obtained from instantaneous I–V recordings were used in the Goldman–Hodgkin–Katz (GHK) constant field equation to estimate relative K+, Na+, and Cl− permeabilities (Hille, 2001). Estimations of relative Ca2+ and Mg2+ permeabilities were calculated using the extended GHK equation (Jan and Jan, 1976; Ma et al., 2012).
Pharmacology.
The standard bath solution for pharmacology experiments contained 100 mm Na+, 100 mm Cl−, and 10 mm HEPES in the presence or absence of 5 Ca2+. Stock solutions of all potential inhibitors (Sigma-Aldrich) were made as follows: 200 mm MK801, 90 mm niflumic acid, 1.5 mm thapsigargin, and 120 mm nimodipine in DMSO; 450 mm 2-aminoethoxydiphenyl borate (2-APB) in MeOH; 770 mm octanol in EtOH; and 40 mm Ruthenium red, 50 mm SKF96365, 25 mm ZnCl2, 100 mm GdCl3, and 163 mm carbenoxolone in H2O. Stock solutions were added to 0 mm Ca2+ pharmacology bath solution to final concentrations as indicated in Figure 2.
Figure 2.

CLHM-1 exhibits unique pharmacological properties. A, Shown are representative traces of CLHM-1 current inhibition by 100 μm Gd3+ (black dotted line) and 100 μm Zn2+ (gray solid line) and activation by 1 mm octanol (gray dotted line) compared with current in an oocyte maintained in 0 mm Ca2+ bath solution (black solid line). CLHM-1 currents were activated by depolarization to +60 mV, with tail currents measured at −80 mV. This was repeated ≥50 times at 10 s intervals for each oocyte, with solution changes as indicated. B, Summary of pharmacological responses, with currents measured at 350 s normalized to currents immediately before inhibitor application; n = 4–6 oocytes (mean ± SE). MK801 (100 μm), n = 4; nimodipine (10 μm), n = 4; 2-APB (1 mm[scap]), n = 5; Ruthenium red (80 μm), n = 4; SKF96365 (50 μm), n = 4; Zn2+ (100 μm), n = 4; Gd3+ (100 μm), n = 5; niflumic acid (200 μm), n = 4; thapsigargin (100 nm), n = 4; octanol (1 mm), n = 6; and carbenoxolone (200 μm), n = 6. **p < 0.01; ***p < 0.001.
Sequence analysis.
The multiple sequence alignment (Fig. 1) was created by the Clustal W method using MegAlign (Lasergene). The hypothetical model of C. elegans CLHM-1 was constructed using the transmembrane domain predictions from the TMPred (http://www.ch.embnet.org/software/TMPRED_form.html) and TMHMM (http://www.cbs.dtu.dk/services/TMHMM/) servers. The accession numbers of the sequences used were as follows: C. elegans CLHM-1, NP_495403; human CALHM1, NP_001001412.
Figure 1.

C. elegans CLHM-1 is a Ca2+ permeable ion channel. A, CLHM-1::GFP localized to the plasma membrane (arrow) of Xenopus oocytes. B, Currents observed in oocytes expressing C. elegans CLHM-1 in standard bath solution containing 2 mm Ca2+ in response to voltage pulses from −80 to +80 mV with a holding potential of −40 mV. C, Using the same pulse protocol as in B, currents were not observed in H2O-injected control oocytes. All oocytes were injected with an antisense Xenopus Cx38 oligonucleotide and BAPTA to inhibit endogenous currents (Ma et al., 2012). D–F, Reversal potentials (Erev) determined by activating CLHM-1 at +60 mV and then measuring the instantaneous I–V relationship after stepping to different voltages over a range of NaCl concentrations in the absence (black) and presence (gray) of Cao2+ (D), Mg2+ activities (E), and Ca2+ activities (F). Solid lines show fit as determined by the extended GHK equation, used to calculate all relative permeability ratios. D, PNa:PK:PCl = 1:1.09 ± 0.02:0.49 ± 0.05 in the absence of Cao2+, and PNa:PK:PCl = 1:1.06 ± 0.02:0.55 ± 0.05 in the presence of 2 mm Cao2+. E, PNa:PK:PCl:PMg = 1:1.15 ± 0.01:0.49 ± 0.02:1.74 ± 0.09. F, PNa:PK:PCl:PCa = 1:1.09 ± 0.02:0.51 ± 0.03:3.63 ± 0.21 for wild-type CLHM-1 (black), and PNa:PK:PCl:PCa = 1:1.02 ± 0.06:0.90 ± 0.03:1.83 ± 0.13 for CLHM-1(D125A) (gray). n ≥ 5 oocytes for each condition. Error bars indicate SE. **p < 0.01. G, Predicted topology of C. elegans CLHM-1, with Asp125 located on the extracellular side of TM3. H, Alignment of the TM3 region. The star indicates the position of the conserved Asp residue.
Transgenes.
The transgene used to examine the clhm-1 expression pattern was generated by directly injecting a 3 kb clhm-1 promoter::gfp::unc-54 3′ UTR PCR product (50 ng/μl) amplified from the plasmid pJT46 with pRol-6 (100 ng/μl) into wild-type animals, using the standard germline transformation technique (Mello et al., 1991; Etchberger and Hobert, 2008). The 3 kb clhm-1 promoter::clhm-1::gfp::unc-54 3′UTR construct pJT44 was injected and chromosomally integrated using Mos1 single-copy insertion (Frøkjaer-Jensen et al., 2008) to create the transgene drSi33 to examine CLHM-1 localization. To further study CLHM-1 localization in the body-wall muscles, the myo-3 promoter::clhm-1::gfp::unc-54 3′UTR construct pJT19 was injected at 20 ng/μl with myo-3 promoter::DsRed2 (pJer1; 50 ng/μl) and the coinjection marker pRol-6 (100 ng/μl).
Body-wall muscle cell-specific expression construct pJT48, created by inserting the clhm-1 genomic sequence directly in frame with the myo-3 promoter, was chromosomally integrated with the Mos1 insertion technique (Frøkjaer-Jensen et al., 2008) to generate the single copy insertion (SCI) transgene drSi34 used for rescue experiments (see Fig. 5). The body-wall muscle cell-specific expression construct pJT41 was created by inserting the human Calhm1 cDNA directly in frame with the myo-3 promoter. This construct was chromosomally integrated with the Mos1 insertion technique (Frøkjaer-Jensen et al., 2012) to generate transgene drSi51 used for rescue experiments (see Fig. 5).
Figure 5.

CLHM-1 is required for swimming locomotion. A, Intron–exon structure for C. elegans clhm-1; locations of the tm4071 and ok3617 deletions as well as the transmembrane domain coding region are indicated. B, Representative body curvature plots for wild-type and clhm-1(tm4071) mutant C. elegans. Average wave speed (speed at which the body bend propagates from head to tail) ± SD is significantly reduced in the clhm-1 mutant. p < 0.01. C, Average head amplitude exhibited no significant differences. p > 0.05. D–F Forward speed, mechanical power, and propulsive force are significantly decreased in clhm-1 mutants. **p < 0.01; ***p < 0.001 compared to wild-type worms. Expression of either C. elegans clhm-1 or human Calhm1 in the body-wall muscles of the clhm-1(tm4071) mutant using the single-copy transgene method rescued speed, power, and force defects. p > 0.05 compared to wild-type. For C–F, n ≥ 10 animals per genotype. Error bars indicate SD. All strains were recorded and analyzed blind to genotype at least two independent times; data presented are from a representative experiment.
For body-wall muscle overexpression experiments, the myo-3 promoter::clhm-1::unc-54 3′UTR was amplified off of pJT48 and injected at 2 or 20 ng/μl, with pJer1 at 50 ng/μl as a coinjection marker. The clhm-1 promoter::clhm-1::clhm-1 3′ UTR sequence was amplified off of genomic DNA and injected at 100 ng/μl with pJer1 at 50 ng/μl. Control lines were isolated by injecting pJer1 alone at 50 ng/μl. A multicopy array created by injection of pJT48 at 20 ng/μl with pJer1 at 50 ng/μl was isolated and then integrated by growing injected animals and subsequent generations on clhm-1 RNAi (Source Biosource) to create drIs22.
To overexpress clhm-1 in the touch neurons, an mec-4 promoter::clhm-1::unc-54 3′UTR PCR product was amplified from pJT47 and injected at 2 ng/μl (unless noted otherwise) with an mec-4 promoter::mCherry::unc-54 3′UTR PCR product (10 ng/μl) and the pRol-6 co-injection marker (100 ng/μl). Wild-type, cnx-1(ok2234), and crt-1(ok948) animals that had been crossed with zdIs5 (an integrated touch neuron marker, mec-4p::GFP) were injected on the same day, with the same injection mix and needle. The mec-4 promoter::hCalhm1 cDNA::unc-54 3′UTR PCR product was amplified from pJT51, the mec-4 promoter::hCalhm1 P86L cDNA::unc-54 3′UTR PCR product was amplified from pJT52, and the mec-4 promoter::mec-4(d) PCR product was amplified from a construct supplied by M. Driscoll (Rutgers University, Piscatawa, NJ). These PCR products were injected at 2 ng/μl with mec-4 promoter::mCherry::unc-54 3′UTR and pRol-6 as detailed above for clhm-1. Control animals were injected with the mec-4 promoter::mCherry::unc-54 3′UTR PCR product (12 ng/μl) and pRol6 (100 ng/μl). At least five independent lines were analyzed for each genotype; animals containing mosaic neurons (GFP+, mCherry−) were excluded from the analysis.
Immunocytochemical staining.
Adult wild-type and myo-3 promoter::hCalhm1 cDNA::unc-54 3′ UTR SCI C. elegans were prepared for staining using the previously described freeze-crack protocol, followed by a methanol/acetone fixation (Duerr et al., 1999; Gendrel et al., 2009). Staining was performed following the method used by Gendrel et al. (2009), with anti-CALHM1 antibodies (Sdix) at a 1:400 dilution and an Alexa-594-labeled donkey anti-rabbit IgG secondary antibody (Invitrogen) at 1:1000.
Fluorescence microscopy.
C. elegans were immobilized with 10 mm levamisole (Sigma) on 3% agar pads. clhm-1 expression pattern images were obtained with a Zeiss LSM 510 confocal microscope. Animals with the clhm-1 promoter::gfp transgene were exposed to the lipophilic fluorescent dye DiI and analyzed with a Leica DMI4000B inverted microscope to identify sensory neurons that expressed clhm-1 (Tong and Bürglin, 2010). Images of touch neurons, CLHM-1::GFP localization, and human CALHM1 localization were obtained using a Leica DMI4000B inverted microscope (63× objective) with a Leica DFC340FX digital camera. The area and average intensity of CLHM-1::GFP fluorescence were determined using ImageJ software. Statistical significance was determined using one-way ANOVA with Dunnett's multiple comparison test.
Mechanics.
A MATLAB-based image analysis and biomechanics algorithm was used to quantify C. elegans motility (Sznitman et al., 2010; Krajacic et al., 2012). Groups of one to three young adult worms grown at 25°C were transferred into a 50 μl drop of M9 buffer in the recording chamber and, after 2 min of acclimation, covered with a cover glass (catalog #12-544-10; Fisher Scientific). Recordings of C. elegans swimming for 4–10 s in the chamber were obtained with a Leica MZ16FA microscope equipped with a Leica DFC 340 FX camera using standard bright-field microscopy at 15 frames per second. For each recording, worm curvature (envelope of motion) was captured and plotted over time. Worm speed, force, and power calculations were performed in MATLAB (Sznitman et al., 2010; Krajacic et al., 2012). All analyses were performed blind to genotype. Statistical significance was determined using one-way ANOVA with Dunnett's multiple comparison test.
Touch neuron analysis.
The numbers of visible PLM and ALM touch neurons were counted as GFP-positive cells every 3 h using a Leica DMI4000B inverted microscope (63× objective) during the L1 stage starting 14 h after the end of a 1 h pulse lay. Images of PLM touch neurons (see Fig. 6A–C) were taken 18–20 h after pulse lay. For animals at the fourth larval stage (L4), the presence/absence of each neuron was recorded. To determine whether EGTA or crt-1 or cnx-1 mutations could suppress the clhm-1 overexpression phenotype, animals which hatched from eggs laid in a 1 h pulse lay period were analyzed either 22 h later or as L4 larvae. Animals that did not contain the transgene in all touch neurons (i.e., those with GFP-positive touch neurons not expressing the mCherry marker) were not analyzed. Touch sensitivity was determined by response to an initial anterior touch. Statistical significance was determined using Student's t test.
Figure 6.
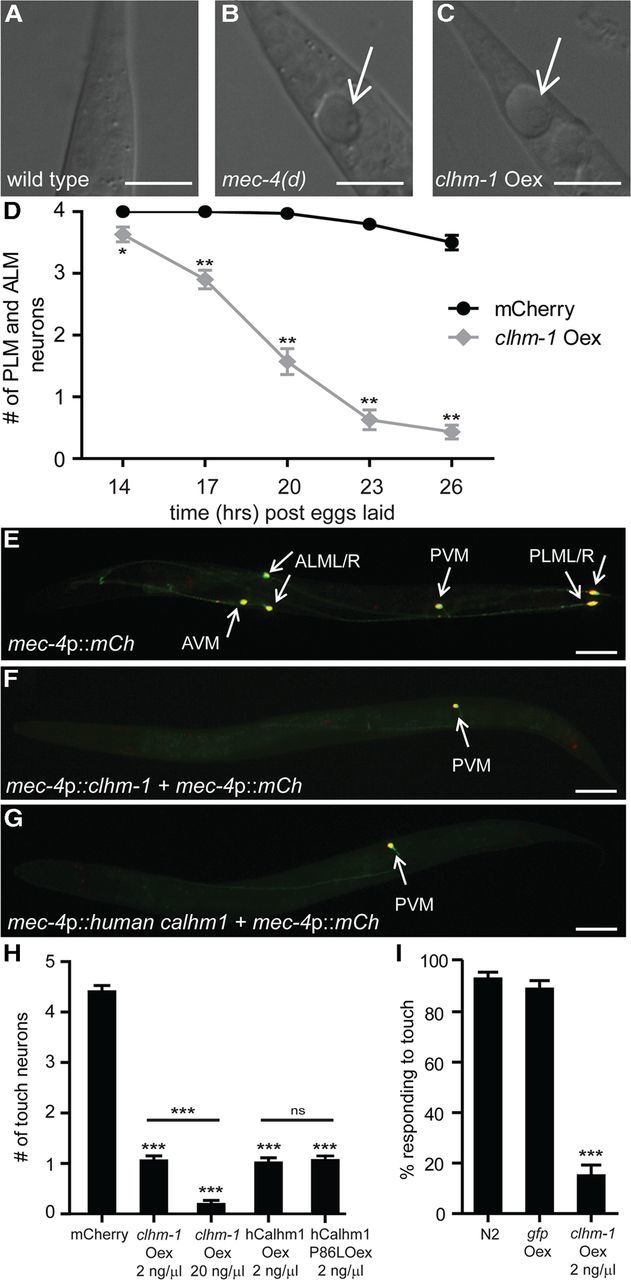
Overexpression of CALHM proteins is sufficient to cause neurodegeneration. A–C, Nomarski images of the C. elegans tail show that both the mec-4(d) mutation (B) and overexpression of CLHM-1 (C) caused swelling of a PLM neuron (arrow). D, Touch neuron overexpression of clhm-1 (gray), but not mCherry (mCh; black), resulted in progressive and nearly complete loss of PLM and ALM neurons by the end of the L1 stage (26 h after eggs laid). n = 30 animals per genotype (6 animals from 5 independent transgenic lines). E, L4 stage wild-type animals have six touch neurons (arrows): a pair of PLMs, a pair of ALMs, an AVM and a PVM. F, G, Overexpression of either C. elegans clhm-1 or human Calhm1 in the touch neurons caused degeneration of all touch neurons except the PVM (arrow). H, Quantification of touch neuron degeneration; n = 100 animals per genotype (20 animals from five independent lines); ns, indicates not significant. I, Overexpression of clhm-1, but not gfp caused loss of touch sensitivity as determined by response to an initial anterior touch; n = 100 animals per genotype. Scale bars: A–C, 10 μm; E–G, 50 μm. Error bars indicate SEM. ns, Not significant. *p < 0.05; **p < 0.01; ***p < 0.001.
Muscle toxicity analysis.
The viability of the myo-3 promoter::clhm-1, clhm-1 promoter::clhm-1, and myo-3 promoter::DsRed2 lines was determined by screening stable lines expressing DsRed2 in the F2 generation; a line was classified as nonviable if none of the animals expressing DsRed2 reached adulthood. A myo-3 promoter::clhm-1::unc-54 3′UTR extrachromosomal array line was integrated into the genome using UV bombardment and subsequently outcrossed four times to generate strain OG471 drIs22 [myo-3 promoter::clhm-1::unc-54 3′UTR; myo-3 promoter::DsRed2::unc-54 3′UTR]. This strain is only viable when grown on clhm-1 RNAi. To identify extragenic suppressors of toxicity, five animals grown on clhm-1 RNAi plates were transferred to empty vector, clhm-1, cnx-1, or crt-1 RNAi (Source Biosource) as L4 animals, and the time of flight (i.e., size) of the offspring of these animals was measured with a COPAS Biosort (Union Biometrica) 72 h later (Morton and Lamitina, 2010).
Results
C. elegans CLHM-1 exhibits functional ion channel properties similar to human CALHM1
Heterologously expressed human CALHM1 displays properties consistent with it being the pore-forming subunit of a novel ion channel (Ma et al., 2012). Since C. elegans CLHM-1 is only 16% identical/28% similar at the amino acid level to human CALHM1, it was unclear whether the two proteins would exhibit similar functional properties. To determine whether C. elegans CLHM-1 is an ion channel in the plasma membrane, we first analyzed CLHM-1::GFP expressed in Xenopus oocytes and observed localization of CLHM-1 at or near the plasma membrane (Fig. 1A). We then recorded whole-cell currents from Xenopus oocytes injected with C. elegans clhm-1 cRNA using protocols established previously by Ma et al. (2012). Membrane depolarization of CLHM-1-expressing oocytes in 2 mm Ca2+ solution produced large outward currents that deactivated upon stepping to hyperpolarized voltages (Fig. 1B). These currents were CLHM-1-dependent, since similar currents were not observed in control oocytes recorded under the same conditions (Fig. 1C). Thus, plasma membrane expression of C. elegans CLHM-1 in Xenopus oocytes produces voltage-dependent, outwardly rectifying currents.
To establish which ions are responsible for the currents observed in CLHM-1-expressing oocytes, we estimated relative permeabilities from reversal potential (Erev) measurements (for pulse protocol, see Fig. 3D). Changing bath NaCl concentration in nominally Ca2+-free solution caused a shift in Erev values that were used in the Goldman–Hodgkin–Katz equation to calculate relative permeabilities PNa:PK:PCl = 1:1.09:0.51 (Fig. 1D). Relative Na+, K+, and Cl− permeabilities were unchanged in the presence of 2 mm Cao2+, suggesting that Cao2+ does not alter CLHM-1 ion selectivity (Fig. 1D). Increasing Cao2+ or Mgo2+ in solutions containing constant NaCl shifted Erev to more depolarized voltages, with relative permeabilities PNa:PCa:PMg = 1:3.63:1.74 (Fig. 1E,F). Thus, like human CALHM1 (Ma et al., 2012), C. elegans CLHM-1 is permeable to monovalent cations, divalent cations, and anions with selectivity Ca2+ > Mg2+ > Na+ = K+ > Cl−.
Figure 3.

C. elegans CLHM-1 is regulated by voltage and extracellular divalent cations. A, Currents observed in oocytes expressing C. elegans CLHM-1 in response to voltage pulses from −120 to +60 mV. The holding potential was at the resting potential (ranged from −5 to −15 mV) in divalent-free bath solution containing 0.5 mm EGTA and 0.5 mm EDTA. B, C, Using the same protocol as in Figure 1B, currents observed in oocytes expressing C. elegans CLHM-1 in standard bath solution containing 5 mm Ca2+ (B) or 5 mm Mg2+ (C). All oocytes were injected with the Cx38 oligonucleotide and BAPTA to inhibit endogenous currents (Ma et al., 2012); currents were not observed in H2O-injected oocytes in the presence or absence of divalent cations (Fig. 1C; data not shown). D, Instantaneous current–voltage relationship in divalent-free (black) and 2 mm Ca2+ (gray) bath solutions. Tail currents observed after stepping to the different voltages were normalized to average current at the end of the activating +60 mV prepulse; solid lines show linear fit. E, CLHM-1 currents at −80 mV were measured after a series of voltage steps (as in A, B) to determine conductance–voltage relationships in the absence and presence of Cao2+. The currents in various Cao2+ solutions were normalized to Gmax determined by fitting 0 mm Cao2+ data with a Boltzmann function for each oocyte. All normalized data were then fit with Boltzmann functions (lines) as for human CALHM1 (Ma et al., 2012) with the assumption that Cao2+ does not affect Gmax. This fit gave rise to the apparent conductance–voltage relationship. Half-activation voltages are as follows: 0 mm Cao2+ (black squares), V0.5 = −6.8 ± 0.4 mV; 0.1 mm Cao2+ (light gray circles), V0.5 = 24.1 ± 0.4 mV; 0.5 mm Cao2+ (dark gray diamonds), V0.5 = 41.4 ± 0.3 mV; 2 mm Cao2+ (black circles), V0.5 = 66 ± 0.4 mV; 5 mm Cao2+ (light gray squares), V0.5 = 69.1 ± 0.5 mV. n ≥ 5 oocytes for each condition. Error bars show the SE of normalized data.
C. elegans clhm-1 encodes an ion channel
Although currents were observed only in CLHM-1-expressing oocytes and not in control oocytes, CLHM-1 could either be an ion channel or activate endogenous Xenopus ion channels. To distinguish between these possibilities, we mutated potential pore-lining residues and determined relative Ca2+ permeability. A change in ion selectivity for any of the mutant CLHM-1 proteins would be consistent with CLHM-1 being a pore-forming ion channel subunit. Acidic residues have been shown to influence ion selectivity of Ca2+-permeable ion channels (Catterall 1993; Keramidas et al., 2004). Charge neutralization of the conserved residue Asp125 to Ala (D125A) caused a change in relative Ca2+ and Cl− permeabilities from PCa:PNa:PK:PCl = 3.63:1:1.09:0.51 for wild-type CLHM-1 to PCa:PNa:PK:PCl = 1.83:1:1.02:0.90 for CLHM-1(D125A) (Fig. 1F–H). Mutation of the homologous Asp in human CALHM1 also altered ion selectivity (Ma et al., 2012), indicating that this residue plays an evolutionarily conserved role in Ca2+ permeability. As a second approach to show that CLHM-1 is an ion channel, we expressed C. elegans CLHM-1 in a different heterologous expression system, the Axolotyl oocyte. CLHM-1-expressing Axolotyl oocytes generated outwardly rectifying, voltage-dependent currents similar to those observed in CLHM-1-expressing Xenopus oocytes (data not shown). This further argues against the possibility that expression of CLHM-1 simply activates endogenous Xenopus conductances. Together, our results are consistent with CLHM-1 being a bona fide ion channel and suggest that a conserved Asp residue at the extracellular end of TM3 may line the channel pore.
CLHM-1 exhibits unique pharmacological properties
Since C. elegans CLHM-1 exhibits properties consistent with it being an ion channel, we sought to determine whether known channel inhibitors altered CLHM-1 currents. The relatively nonspecific blockers Gd3+ and Ruthenium red irreversibly inhibited CLHM-1 currents, while Zn2+ caused partially reversible inhibition (Fig. 2A,B,). The NMDA receptor blocker MK801 (Karp et al., 1993), L-type VGCC blocker nimodipine (Xu and Lipscombe, 2001), InsP3 receptor and SOCE inhibitor 2-APB (Maruyama et al., 1997; Bootman et al., 2002), TRP channel blocker SKF96365 (Boulay et al., 1997), Cl− channel blocker niflumic acid (White and Aylwin, 1990), and SERCA blocker thapsigargin (Lytton et al., 1991) did not affect CLHM-1 currents (Fig. 2B). These pharmacological properties are similar to those observed for human CALHM1 (Dreses-Werringloer et al., 2008; Ma et al., 2012). Strikingly, the connexin and pannexin hemichannel blockers octanol and carbenoxolone (Saez et al., 2003) caused a twofold increase in CLHM-1 currents (Fig. 2A,B). These pharmacological properties are distinct from other known channels, supporting the hypothesis that C. elegans CLHM-1 belongs to a unique family of ion channels.
CLHM-1 is regulated by extracellular Ca2+ and membrane voltage
Removal and subsequent add-back of Cao2+ has been shown to cause a dramatic increase in intracellular Ca2+ (Cai2+) levels in cell lines expressing human CALHM1 (Dreses-Werringloer et al., 2008; Ma et al., 2012). This likely occurs because lowering Cao2+ activates human CALHM1 channels at resting membrane potentials (Ma et al., 2012). To determine whether Cao2+ alters the gating of C. elegans CLHM-1, we recorded currents from CLHM-1 expressing oocytes in divalent-free (Fig. 3A), 5 mm Ca2+ (B), and 5 mm Mg2+ (C) bath solutions. Compared with CLHM-1 currents observed in the presence of either Cao2+ or Mgo2+, CLHM-1 currents activated much more rapidly in the absence of divalent cations (Fig. 3A–C). This regulation by Cao2+ and Mgo2+ was completely reversible (Fig. 2A; data not shown). These results indicate that CLHM-1 gating is regulated by both voltage and extracellular divalent cations.
While our results suggest that Cao2+ regulates CLHM-1 gating, the mechanism by which Cao2+ has this effect is unclear. Fast channel pore block that physically impedes ion flow leads to nonlinear instantaneous I–V relationships. To determine whether Cao2+ acts as a voltage-dependent fast pore blocker of CLHM-1, similar to the voltage-dependent pore block of NMDA receptors by Mgo2+ (Mayer et al., 1984; Nowak et al., 1984), we measured currents in the presence and absence of Cao2+ using an instantaneous I–V pulse protocol. The instantaneous I–V relationships were linear in both the presence and absence of Cao2+ (Fig. 3D). Therefore, Cao2+ does not regulate CLHM-1 gating through a voltage-dependent fast pore-block mechanism.
To further analyze the effects of Cao2+ and voltage on gating regulation, we determined the apparent conductance–voltage (G–V) relationships for CLHM-1 in both divalent-free and various Cao2+ containing solutions. Using Boltzmann equations to fit the apparent G–V relationships, we determined that 5 mm Cao2+ right-shifted the apparent G–V relationship by ∼75 mV from a half-activation voltage (V0.5) of −6.8 mV in 0 mm Cao2+ to +69.1 mV in 5 mm Cao2+ (Fig. 3E). Similarly, 5 mm Mgo2+ right-shifted the apparent G–V relationship by 62 mV (data not shown). The IC50 for Cao2+ was 184.3 ± 51.2 μm, with a Hill coefficient of 0.88 ± 0.2. Cao2+ did not appear to significantly affect the voltage-dependent slope (Fig. 3E), suggesting that Cao2+ may not directly affect the voltage-dependent gating. However, since the current was still in a rising phase at the end of the depolarizing pulses, our analysis of the apparent G–V relationship (Fig. 3E) does not exclude the possibility that Cao2+ could in fact affect voltage-dependent gating. Although using longer depolarizing pulses to address this problem was not possible due to instability of the oocytes and endogenous currents activated by such pulses, we note that our data are directly comparable to the apparent G–V relationship for human CALHM1, which was determined using the same protocol (Ma et al., 2012). In conclusion, our results indicate that Cao2+ regulates CLHM-1 gating by stabilizing CLHM-1 channels in the closed state at hyperpolarized voltages.
C. elegans CLHM-1 is expressed at the cell surface of excitable cells
RT-PCR has suggested that human Calhm1 is primarily expressed in the brain and spinal cord, and in situ hybridization showed Calhm1 expression in TRPM5-expressing taste cells (Dreses-Werringloer et al., 2008; Moyer et al., 2009; Taruno et al., 2013). Overexpression of human Calhm1 in CHO cells resulted in plasma membrane and endoplasmic reticulum localization (Dreses-Werringloer et al., 2008); however, an analysis of the expression pattern and subcellular localization of mammalian CALHM proteins has not been performed in vivo. To determine the expression pattern of clhm-1 in C. elegans, we generated transgenic worms expressing a transcriptional reporter consisting of a 3kb clhm-1 promoter region followed by gfp. We observed GFP expression in head and body-wall muscles; IL2, ASG, ASI, ASJ, PHA and PHB sensory neurons; and the spermatheca (Fig. 4A–C).
Figure 4.

C. elegans CLHM-1 is expressed in excitable cells. A, GFP fluorescence in animals carrying a clhm-1 promoter::gfp transgene. B, C, This reporter transgene was expressed in many cells, including head muscles, IL2 neurons (arrows), and other sensory neurons (lines; B), as well as the body-wall muscles (arrows; C). D, CLHM-1::GFP fluorescence localized to cilia of sensory neurons (arrow) in animals carrying a clhm-1 promoter::clhm-1::gfp single-copy transgene. E, F, Overexpression of DsRed2 and CLHM-1::GFP, respectively, in the body-wall muscles. G, Merge. Offset images on the right are magnifications of boxed areas in E–G; the lines indicate the location where DsRed2 and CLHM-1::GFP fluorescence appear to meet, but not overlap. H, Localization of human CALHM1 in animals carrying a rescuing myo-3 promoter::hCalhm1 transgene at single copy. Arrowheads point to muscle intercellular membranes, arrows point to muscle arms, and the double-headed arrow indicates the synaptic region. I, CALHM1 staining is not present in wild-type animals. Scale bars: A, 100 μm; B, C, 20 μm; D–I, 10 μm; offset images, 3 μm.
To examine the localization of the CLHM-1 protein, we inserted a single copy clhm-1 promoter::clhm-1::gfp translational fusion construct into the C. elegans genome. When expressed in Xenopus oocytes, CLHM-1::GFP produced an ion conductance indistinguishable from untagged CLHM-1, suggesting that the presence of a C-terminal GFP tag does not interfere with CLHM-1 channel localization or function (data not shown). CLHM-1::GFP at single copy localized to ciliary endings of sensory neurons and in a punctate pattern at or near the plasma membrane of muscles (Fig. 4D; data not shown). To further investigate the localization of CLHM-1 in muscles, we expressed DsRed2 in the cytoplasm of the body-wall muscles along with a multicopy myo-3 promoter::clhm-1::gfp translational fusion construct. The CLHM-1::GFP and DsRed2 fluorescence were not colocalized, further demonstrating CLHM-1 localization at or near the plasma membrane (Fig. 4E–G).
Although the ion channel function of the CLHM-1::GFP fusion protein is indistinguishable from untagged CLHM-1, it is possible that the GFP tag alters the subcellular distribution of CLHM-1 in a way that forces its accumulation at or near the plasma membrane. A CLHM-1-specific antibody to localize endogenous CLHM-1 could distinguish between these possibilities, although our initial attempts at developing such a reagent were unsuccessful (data not shown). As an alternative approach, we expressed a functional human Calhm1 transgene (Fig. 5D–F) at single copy in the body-wall muscles and performed immunofluorescence to determine the CALHM1 localization pattern using a commercially available antibody. Consistent with the localization pattern observed with CLHM-1::GFP, CALHM1 localized to the plasma membrane at body-wall muscle intercellular junctions as well as in the muscle arms and synaptic regions (Fig. 4H–I). These data suggest that CALHM family members are primarily localized at or near the plasma membrane of excitable cells and represent the first in vivo description of the physiological expression pattern and localization of a CALHM family member.
CLHM-1 mutants exhibit uncoordinated locomotion
While genetic studies suggest that a polymorphism in human Calhm1 results in pathophysiological consequences (Dreses-Werringloer et al., 2008; Boada et al., 2010; Lambert et al., 2010), phenotypes associated with loss of Calhm1 expression have not been determined. To examine the physiological role(s) of CLHM-1 in C. elegans, we obtained two knock-out mutants, clhm-1(ok3617) and clhm-1(tm4071). Both deletions remove a significant section of the transmembrane domain region, resulting in predicted null alleles (Fig. 5A). Since a primary site of clhm-1 expression is the body-wall muscles and these cells control motility, we quantified the swimming gait of outcrossed clhm-1 mutants using a quantitative fluid mechanics approach (Sznitman et al., 2010; Krajacic et al., 2012). Loss of clhm-1 significantly disrupted some, but not all, kinematic parameters, including mean wave speed (Fig. 5B,C), suggesting that loss of clhm-1 affects specific aspects of the swim gait. The kinematic defects observed in both clhm-1 mutants caused reduced forward velocity and diminished muscle force and power production compared to wild-type worms (Fig. 5D–F).
To test whether the biomechanical defects were caused by loss of clhm-1 expression in muscle, we transgenically expressed C. elegans CLHM-1 in the body-wall muscles of the clhm-1 mutant and determined whether this could rescue the locomotion defects. Expression of a body-wall muscle promoter::clhm-1 single-copy transgene did not cause a significant biomechanical phenotype in the wild-type background (Fig. 5C–F), while expression in the clhm-1 mutant worms was sufficient to rescue the defects in forward velocity, force, and power production (Fig. 5D–F). Despite exhibiting only 16% sequence identity with C. elegans CLHM-1, single-copy muscle-specific expression of human CALHM1 was also sufficient to rescue the biomechanical defects of the clhm-1 mutant, demonstrating that worm CLHM-1 and human CALHM1 are functionally conserved (Fig. 5D–F). In summary, our results demonstrate that CLHM-1 acts in the body-wall muscles to maintain normal motility and show that the function of Calhm family members is evolutionarily conserved.
Overexpression of CLHM-1 in the C. elegans touch neurons induces neurodegeneration
Mutation of human Calhm1 has been associated with LOAD (Dreses-Werringloer et al., 2008; Boada et al., 2010; Lambert et al., 2010), which is characterized by neuronal dysfunction and degeneration, yet the consequences of overexpression of mammalian CALHM proteins in vivo have not been determined. We explored the effects of clhm-1 overexpression in neurons even though we have not yet identified a phenotype associated with loss of neuronal clhm-1 (data not shown). Although not a site of endogenous clhm-1 expression, the C. elegans touch neurons are a well-established model for studying the impact of toxic ion channels on neuron function and death (Driscoll and Chalfie, 1991; Chung et al., 2000; Xu et al., 2001; Syntichaki et al., 2002). Overexpression of clhm-1 in the touch neurons caused swelling and degeneration of the ALM and PLM neurons during the first larval (L1) stage (Fig. 6A–D), similar to the effect of dominant mutations in the DEG/ENaC channel mec-4(d) (Driscoll and Chalfie, 1991; Xu et al., 2001). This rapid neurodegeneration was not simply a general response to increased ion channel expression, since overexpression of wild-type mec-4 or another DEG/ENaC channel, mec-10(d), is not sufficient to cause touch insensitivity or touch neuron degeneration, respectively (Royal et al., 2005; Zhang et al., 2008). Thus, overexpression of clhm-1 specifically induces cell swelling and neurodegeneration characteristic of pathophysiologically induced necrotic cell death.
Overexpression of C. elegans CLHM-1, human CALHM1, or LOAD-associated P86L human CALHM1 caused completely penetrant degeneration of the AVM and pairs of PLM and ALM neurons by L4 (Fig. 6E–H). The PVM touch neurons, which are born at the end of the L1 stage and are not required for the touch response (Chalfie et al., 1985), were largely unaffected by overexpression of CALHM proteins (Fig. 6E–H). Higher expression of clhm-1 was sufficient to induce PVM degeneration, suggesting that CLHM-1 toxicity is dose dependent (Fig. 6H; data not shown). To test whether the touch neurons in the CLHM-1-overexpressing animals were indeed absent, we performed touch assays on the transgenic worms. Consistent with loss of the touch neurons, animals that overexpressed clhm-1 exhibited a touch-insensitive phenotype (Fig. 6I). Our results establish Calhm family members as conditionally toxic ion channels, capable of inducing necrotic-like neuronal death.
Alteration of Ca2+ levels delays CLHM-1-induced neurodegeneration without affecting CLHM-1 expression
To determine the mechanism by which clhm-1 overexpression leads to cell death, we looked for conditions that could suppress CLHM-1-induced neurodegeneration. Disruption of cellular Ca2+ balance by both chemical reagents and genetic mutations has been used to suppress the touch neuron necrosis observed in mec-4(d) mutants (Xu et al., 2001; Syntichaki et al., 2002). Growth of clhm-1 overexpression animals in the presence of the Ca2+ chelator EGTA to reduce Cao2+ partially suppressed the clhm-1 overexpression phenotype in the L1 stage, suggesting that Ca2+ plays a role in CLHM-1-induced cell death (Fig. 7A). To analyze the role of intracellular Ca2+ stores, we used a candidate gene approach and overexpressed clhm-1 in calreticulin (crt-1) and calnexin (cnx-1) mutants. Loss of crt-1 or cnx-1 caused significant suppression of CLHM-1-induced neurodegeneration in L1 animals, but the remaining PLMs and ALMs still degenerated by the L4 stage (Fig. 7A,B). Thus, crt-1 and cnx-1 mutations were only sufficient to delay CLHM-1-induced neurodegeneration. In contrast, loss of crt-1 or cnx-1 was sufficient to suppress touch neuron degeneration induced by overexpression of mec-4(d) in L4 animals (Fig. 7B). These results indicate that Ca2+ signaling plays a role in CLHM-1-induced neurodegeneration; however, other mechanisms, including overall disruption of ion homeostasis or excitotoxicity, may also be contributing factors.
Figure 7.

Reduction of extracellular Ca2+ and plasma membrane CLHM-1 expression delays CLHM-1-induced neurodegeneration. A, Pharmacological and genetic manipulations were used to alter cellular Ca2+ levels. Growth of worms on 50 mm EGTA plates to reduce Cao2+ concentration partially suppressed CLHM-1-induced loss of PLM and ALM neurons in the L1 stage. Mutations in crt-1 and cnx-1, which decrease release of Ca2+ from intracellular stores, also partially suppressed CLHM-1-induced neurodegeneration. B, Analysis of animals in the L4 stage indicated that mutations in crt-1 and cnx-1 were not sufficient to suppress CLHM-1-induced cell death, while mec-4(d) overexpression was fully suppressed by loss of crt-1 and partially suppressed by cnx-1. For A and B, n = 50 animals (controls) or n = 100 animals from five independent lines (20 animals from each line). C, D, Quantification of CLHM-1::GFP fluorescence area (in square nanometers) and intensity in cilia of L4 animals expressing a clhm-1 promoter::clhm-1::gfp single-copy transgene. n ≥ 30 animals per genotype. Error bars indicate SEM. ns, Not significant. **p < 0.01; ***p < 0.001.
Since crt-1 and cnx-1 have dual functions in regulating ER Ca2+ homeostasis and protein folding (Coe and Michalak, 2009), it is possible that these mutations change surface expression levels of CLHM-1. To determine whether CLHM-1 expression levels were altered under any of the conditions tested, we measured CLHM-1::GFP fluorescence levels in animals grown in the presence of EGTA as well as in crt-1 and cnx-1 mutants. A significant decrease in the area as well as intensity of CLHM-1::GFP fluorescence was observed in the cnx-1 mutant (Fig. 7C,D) indicating a decrease in CLHM-1 expression. The presence of EGTA and the crt-1 mutation did not have a significant effect on CLHM-1::GFP fluorescence, suggesting that these manipulations likely delay CLHM-1-induced neurodegeneration by altering Ca2+ levels and not CLHM-1 expression (Fig. 7C,D). In conclusion, factors that regulate cellular Ca2+ levels and CLHM-1 surface expression can significantly delay the toxic effects of clhm-1 overexpression.
Overexpression of CLHM-1 in a subset of endogenous cell types induces toxicity
While overexpression of CLHM-1 in the touch neurons demonstrates that this channel is capable of causing neurodegeneration, this result is based on expression of the channel in a nonnative cell type. We sought to determine whether CLHM-1 causes toxicity when overexpressed in a subset of cells where the endogenous channel is normally expressed. Overexpression of clhm-1 in the body-wall muscles caused completely penetrant embryonic/L1 lethality that was suppressed by growing the animals on clhm-1 RNAi, demonstrating that the toxicity was due to CLHM-1 (Fig. 8A–D). As observed with CLHM-1 overexpression in the touch neurons, the toxicity associated with overexpression in the body-wall muscles was dose dependent (Fig. 8E) and could be partially suppressed by loss of cnx-1 (Fig. 8F). Overexpression of CLHM-1 from its endogenous promoter was also sufficient to cause lethality in a small percentage of transgenic lines (Fig. 8E). Interestingly, when we overexpressed CLHM-1 in the IL2 neurons, another subset of cells that express the endogenous channel, we did not observe neurodegeneration (data not shown). This suggests that some of the cells that normally express CLHM-1 may have mechanisms to deal with toxicity associated with high expression of the channel. Thus, overexpression of CLHM-1 in some, but not all of the cells where it is normally expressed, results in toxicity.
Figure 8.
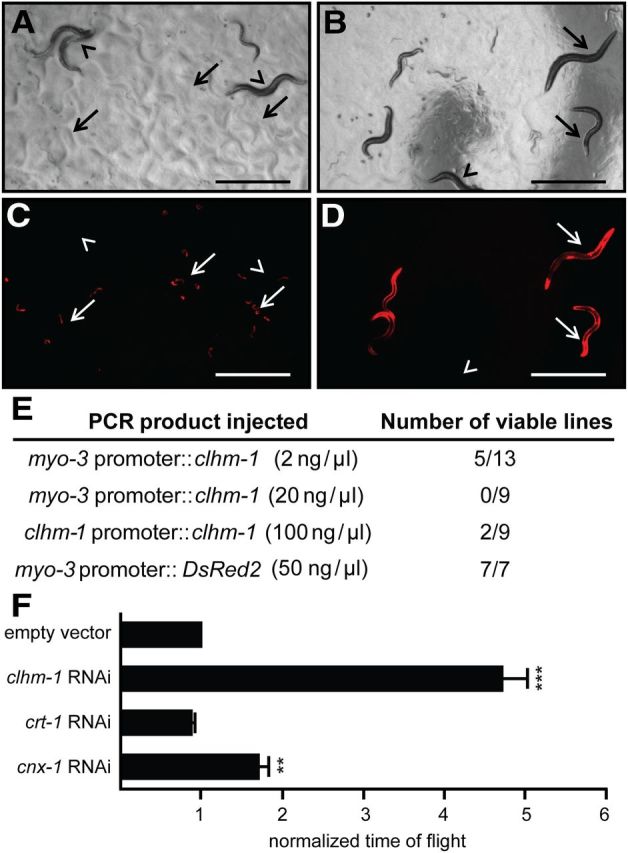
Overexpression of CLHM-1 in the body-wall muscles causes lethality. A–D, Bright-field (A, B) and fluorescent images (C, D) of transgenic animals, identified by DsRed2 expression, with clhm-1 overexpressed in the body-wall muscles. A, C, Animals grown on empty-vector RNAi. B, D, Animals grown on clhm-1 RNAi. Arrowheads indicate nontransgenic animals that lack the DsRed2 marker; arrows indicate transgenic animals. Scale bars: 1000 μm. E, Number of viable transgenic lines isolated when clhm-1 was expressed from the body-wall muscle myo-3 promoter and the endogenous clhm-1 promoter. F, Growth of myo-3 promoter::clhm-1-overexpressing worms on the indicated RNAi bacterial clones. Normalized time of flight is a measure of worm length that corresponds to developmental stage. n ≥ 3 independent experiments per RNAi clone; n > 1000 animals per experiment. Data shown are mean ± SEM. **p < 0.01; ***p < 0.001.
Discussion
In this work, we show that C. elegans CLHM-1 encodes an evolutionarily conserved ion channel, as human CALHM1 and C. elegans CLHM-1 exhibit similar biophysical properties when expressed in Xenopus oocytes and functional conservation when expressed in C. elegans. Based on our data demonstrating that CLHM-1 (1) is a bona fide ion channel, (2) exhibits a permeability preference for Ca2+, (3) is regulated by membrane voltage and extracellular Ca2+, and (4) is expressed at the surface of excitable cells, and that (5) CLHM-1 overexpression-induced neurodegeneration is delayed by lowering Ca2+ levels, we suggest that CALHM proteins are novel ion channels that sense and regulate Ca2+ signaling in vivo.
CLHM-1 acts as an extracellular Ca2+ sensor
At times of high excitatory activity at synaptic clefts, local Cao2+ transiently becomes depleted due to NMDA receptor and VGCC activation (Rusakov and Fine, 2003). The sensitivity of CLHM-1 to voltage and Cao2+ suggests that it may act as a Cao2+ sensor to amplify neurotransmission. An emerging number of neuronal channels including hemichannels, the sodium leak channel NALCN, and other unidentified nonselective cation channels are capable of sensing physiological changes in Cao2+ (Xiong et al., 1997; Gómez-Hernández et al., 2003; Saez et al., 2003; Smith et al., 2004; Lu et al., 2010). Most recently, Ma et al. (2012) showed that human CALHM1 plays a role in the electrophysiological responses of cortical neurons to changes in Cao2+. Compared with previously described channels that respond to Cao2+, C. elegans CLHM-1 and human CALHM1 channels exhibit unique regulatory mechanisms, permeability properties, and sequences (Ma et al., 2012). In particular, the unusual regulatory properties and the neurodegeneration phenotype associated with overexpression of Calhm family members suggest that these channels may contribute to neuronal death associated with excitotoxic synaptic activity (MacDonald et al., 2006).
Although it is clear that Cao2+ plays a fundamental role in CLHM-1 regulation, we do not know how voltage or Cao2+ is sensed by the channel. Despite remarkable similarities in biophysical properties, there are some differences in the gating of C. elegans CLHM-1 compared to that of human CALHM1. Removal of Cao2+ shifted CALHM1 and CLHM-1 voltage-dependent activation toward hyperpolarized voltages; however, the magnitude of this shift was different for the two proteins (Ma et al., 2012). In addition, Cao2+ affected the voltage-dependence (G–V slope) of human CALHM1 (Ma et al., 2012), but not C. elegans CLHM-1. Given that neither human CALHM1 nor C. elegans CLHM-1 contain known Ca2+-binding sequences or canonical voltage-sensing domains, defining the sequences underlying differences between the two proteins could reveal new molecular mechanisms for both voltage and Ca2+ sensing.
C. elegans CLHM-1 activity at the plasma membrane regulates excitable cell function
The power of the C. elegans model system has allowed us to address important unanswered questions concerning the subcellular localization and in vivo function of Calhm family members. For the first time, we defined the protein localization pattern of a CALHM family member in vivo. Comparison of the localization patterns of CLHM-1 and other proteins that regulate body-wall muscle function suggests that CLHM-1 does not localize to the same regions as the L-type VGCC EGL-19 (Kim et al. 2009), Shaker K+ channel SHK-1 (Fawcett et al., 2006), or BK channel SLO-1 (Carre-Pierrat et al., 2006). While AChRs and GABA receptors are strictly localized to the synapse (Bamber et al., 1999; Gendrel et al., 2009), CALHM family members exhibit both synaptic and extrasynaptic localization like the innexin UNC-9 (Liu et al., 2006). CALHM proteins are similar in oligomeric structure, though not sequence, to connexins and innexins, but human CALHM1 cannot form gap junctions (Siebert et al., 2013). Since human CALHM1 can rescue the C. elegans clhm-1 mutant biomechanical defects, our data suggest that the requirement for CLHM-1 in motility may depend more on its role as a plasma membrane ion channel than on another putative function(s).
Loss of clhm-1 in the body-wall muscles caused an uncoordinated locomotion phenotype, demonstrating that this ion channel plays a physiologically significant role in regulating behavior. However, we still do not understand why loss of clhm-1 results in locomotion defects. Biomechanical profiling (BMP) as conducted on clhm-1 mutants was performed previously on 21 mutants with neuromuscular structure or signaling defects (Krajacic et al., 2012). Utilizing a clustering analysis, the biomechanical profile of a mutant can be used to predict gene function (Krajacic et al., 2012). Interestingly, the clhm-1 mutant biomechanical profile clustered with the synaptic signaling proteins and not the dystrophin-associated glycoprotein complex proteins (data not shown). Currently, only a limited number of mutants have been tested with BMP. Determining whether the biomechanical profile of clhm-1 is similar to genes that regulate cell excitability, Ca2+ signaling, or electrical coupling between cells could help suggest a mechanism by which CLHM-1 regulates locomotion.
Coordinated muscle contraction and movement in C. elegans is regulated by a balance of synaptic acetylcholine and GABA signaling (Richmond and Jorgensen, 1999) as well as the electrical properties of the body-wall muscles (Liu et al., 2011a,b). Although C. elegans lack voltage-gated Na+ channels (Bargmann, 1998), action potentials are generated in the body-wall muscles by Ca2+ entry through the L-type VGCC EGL-19 (Jospin et al., 2002; Liu et al., 2011a). Under physiological conditions, activity of many ion channels including the K+ channels SHK-1 and SLO-2, the innexin UNC-9, the GABA receptor UNC-49, and subunits of the levamisole-sensitive AChR modulates action potentials in the muscles (Liu et al., 2011a,b). As loss of human CALHM1 in cortical neurons results in a decrease in the firing of action potentials (Ma et al., 2012), it is possible that loss of C. elegans CLHM-1 alters action potentials in the body-wall muscles, affecting locomotion.
While only a small number of channels function in the body-wall muscles under normal physiological circumstances, additional conductances are activated by more specific conditions (Santi et al., 2003). For example, SHL-1 and SHK-1 are responsible for most of the voltage-dependent K+ conductance under physiological conditions (Fawcett et al., 2006), while an increase in intracellular Cl− and Ca2+ activates the K+ channel SLO-2 (Santi et al. 2003). We still do not know when CLHM-1 is active in vivo, and, in fact, it may only be activated under specific conditions, such as low Cao2+. Future efforts will be directed toward determining whether loss of CLHM-1 under physiological or low Cao2+ conditions alters resting membrane potential, action potential firing, Ca2+ transients, and/or muscle morphology.
There are likely other phenotypes associated with clhm-1 loss of function. Our work has shown that C. elegans CLHM-1 is expressed in several sensory neurons, analogous to human Calhm1 expression in neurons and the sensory TRPM5-positive taste cells (Dreses-Werringloer et al., 2008; Moyer et al., 2009; Taruno et al., 2013). While the Ca2+ permeability of human CALHM1 has been well established (Dreses-Werringloer et al., 2008; Ma et al., 2012), CALHM1 was shown previously to act as a voltage-gated ATP release channel required for sweet, bitter, and umami tastes (Taruno et al., 2013). Although C. elegans does not have P2X receptors that are activated by extracellular ATP (Khakh and North 2006), worms may have an alternative receptor that can be activated by ATP or CLHM-1 may release different substances. Understanding how CLHM-1 regulates the activity of the sensory neurons could provide insight into CLHM-1 function in a native neuronal context.
Calhm family members are Ca2+-permeable, conditionally toxic ion channels
The P86L polymorphism in CALHM1 is likely a contributing, but not an independent, risk factor for LOAD (Lambert et al., 2010). While the P86L polymorphism has been shown to alter CALHM1-induced Ca2+ permeability and amyloid precursor protein processing in cell culture (Dreses-Werringloer et al., 2008), the mechanism by which aberrant CALHM1 signaling causes neuron dysfunction in vivo is unknown. Here we show that overexpression of Calhm family members in the C. elegans touch neurons is sufficient to induce necrotic-like neuronal death. C. elegans do not produce endogenous Aβ (Wu and Luo, 2005), suggesting that the mechanism by which neurodegeneration is induced is Aβ independent. Thus, it is possible that altered signaling through human CALHM1, which is regulated by and permeable to Ca2+, could disrupt neuronal Ca2+ homeostasis and cause necrotic neuron death via an Aβ independent mechanism.
The proteins that regulate CALHM1 trafficking and function in humans, as well as those that act downstream of CALHM1, are completely unknown. While we have shown that manipulations which reduce release of Ca2+ from intracellular stores significantly delay the toxic effects of clhm-1 overexpression, additional unidentified proteins and/or mechanisms likely also play a role since the neurons still die. Such mechanisms can be defined in C. elegans using unbiased forward genetic screens to identify mutants that suppress the toxicity associated with CLHM-1 overexpression. This genetic approach has the potential to identify novel and physiologically significant CALHM interactors in a way not currently possible in mammalian systems. Identification of CLHM-1 regulators in C. elegans will provide insights into the molecular mechanisms that control human CALHM1 localization and function as well as downstream proteins required for the conditionally toxic effects of CALHM signaling.
Footnotes
This work was supported by National Institutes of Health Grant R21NS072775 (T.L., J.K.F.), a summer research grant from Mount Desert Island Biological Laboratory (T.L.), and NIH Fellowship F32AR060128 and an American Heart Association postdoctoral fellowship (J.E.T.). Some nematode strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources. We also thank the National Bioresource Project for the tm4071 allele, Monica Driscoll for clones, and Adam Siebert and Akiyuki Taruno for helpful discussions.
The authors declare no competing financial interests.
References
- Bamber BA, Beg AA, Twyman RE, Jorgensen EM. The Caenorhabditis elegans unc-49 locus encodes multiple subunits of a heteromultimeric GABA receptor. J Neurosci. 1999;19:5348–5359. doi: 10.1523/JNEUROSCI.19-13-05348.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargmann CI. Neurobiology of the Caenorhabditis elegans genome. Science. 1998;282:2028–2033. doi: 10.1126/science.282.5396.2028. [DOI] [PubMed] [Google Scholar]
- Beecham GW, Schnetz-Boutaud N, Haines JL, Pericak-Vance MA. CALHM1 polymorphism is not associated with late-onset Alzheimer disease. Ann Hum Genet. 2009;73:379–381. doi: 10.1111/j.1469-1809.2009.00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Lipp P. Calcium—a life and death signal. Nature. 1998;395:645–648. doi: 10.1038/27094. [DOI] [PubMed] [Google Scholar]
- Bertram L, Schjeide BM, Hooli B, Mullin K, Hiltunen M, Soininen H, Ingelsson M, Lannfelt L, Blacker D, Tanzi RE. No association between CALHM1 and Alzheimer's disease risk. Cell. 2008;135:993–994. doi: 10.1016/j.cell.2008.11.030. author reply 994–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008;31:454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boada M, Antúnez C, López-Arrieta J, Galán JJ, Morón FJ, Hernández I, Marín J, Martínez-Lage P, Alegret M, Carrasco JM, Moreno C, Real LM, González-Pérez A, Tárraga L, Ruiz A. CALHM1 P86L polymorphism is associated with late-onset Alzheimer's disease in a recessive model. J Alzheimers Dis. 2010;20:247–251. doi: 10.3233/JAD-2010-1357. [DOI] [PubMed] [Google Scholar]
- Bootman MD, Collins TJ, Mackenzie L, Roderick HL, Berridge MJ, Peppiatt CM. 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J. 2002;16:1145–1150. doi: 10.1096/fj.02-0037rev. [DOI] [PubMed] [Google Scholar]
- Boulay G, Zhu X, Peyton M, Jiang M, Hurst R, Stefani E, Birnbaumer L. Cloning and expression of a novel mammalian homolog of Drosophila transient receptor potential (Trp) involved in calcium entry secondary to activation of receptors coupled by the Gq class of G protein. J Biol Chem. 1997;272:29672–29680. doi: 10.1074/jbc.272.47.29672. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carre-Pierrat M, Grisoni K, Gieseler K, Mariol MC, Martin E, Jospin M, Allard B, Ségalat L. The SLO-1 BK channel of Caenorhabditis elegans is critical for muscle function and is involved in dystrophin-dependent muscle dystrophy. J Mol Biol. 2006;358:387–395. doi: 10.1016/j.jmb.2006.02.037. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Structure and function of voltage-gated ion channels. Trends Neurosci. 1993;16:500–506. doi: 10.1016/0166-2236(93)90193-P. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Sulston JE, White JG, Southgate E, Thomson JN, Brenner S. The neural circuit for touch sensitivity in Caenorhabditis elegans. J Neurosci. 1985;5:956–964. doi: 10.1523/JNEUROSCI.05-04-00956.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S, Gumienny TL, Hengartner MO, Driscoll M. A common set of engulfment genes mediates removal of both apoptotic and necrotic cell corpses in C. elegans. Nat Cell Biol. 2000;2:931–937. doi: 10.1038/35046585. [DOI] [PubMed] [Google Scholar]
- Coe H, Michalak M. Calcium binding chaperones of the endoplasmic reticulum. Gen Physiol Biophys. 2009;28:F96–F103. [PubMed] [Google Scholar]
- Dreses-Werringloer U, Lambert JC, Vingtdeux V, Zhao H, Vais H, Siebert A, Jain A, Koppel J, Rovelet-Lecrux A, Hannequin D, Pasquier F, Galimberti D, Scarpini E, Mann D, Lendon C, Campion D, Amouyel P, Davies P, Foskett JK, Campagne F, et al. A polymorphism in CALHM1 influences Ca2+ homeostasis, Abeta levels, and Alzheimer's disease risk. Cell. 2008;133:1149–1161. doi: 10.1016/j.cell.2008.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll M, Chalfie M. The mec-4 gene is a member of a family of Caenorhabditis elegans genes that can mutate to induce neuronal degeneration. Nature. 1991;349:588–593. doi: 10.1038/349588a0. [DOI] [PubMed] [Google Scholar]
- Duerr JS, Frisby DL, Gaskin J, Duke A, Asermely K, Huddleston D, Eiden LE, Rand JB. The cat-1 gene of Caenorhabditis elegans encodes a vesicular monoamine transporter required for specific monoamine-dependent behaviors. J Neurosci. 1999;19:72–84. doi: 10.1523/JNEUROSCI.19-01-00072.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etchberger JF, Hobert O. Vector-free DNA constructs improve transgene expression in C. elegans. Nat Methods. 2008;5:3. doi: 10.1038/nmeth0108-3. [DOI] [PubMed] [Google Scholar]
- Fawcett GL, Santi CM, Butler A, Harris T, Covarrubias M, Salkoff L. Mutant analysis of the Shal (Kv4) voltage-gated fast transient K+ channel in Caenorhabditis elegans. J Biol Chem. 2006;281:30725–30735. doi: 10.1074/jbc.M605814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frøkjaer-Jensen C, Davis MW, Hopkins CE, Newman BJ, Thummel JM, Olesen SP, Grunnet M, Jorgensen EM. Single-copy insertion of transgenes in Caenorhabditis elegans. Nat Genet. 2008;40:1375–1383. doi: 10.1038/ng.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frøkjaer-Jensen C, Davis MW, Ailion M, Jorgensen EM. Improved Mos1-mediated transgenesis in C. elegans. Nat Methods. 2012;9:117–118. doi: 10.1038/nmeth.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendrel M, Rapti G, Richmond JE, Bessereau JL. A secreted complement-control-related protein ensures acetylcholine receptor clustering. Nature. 2009;461:992–996. doi: 10.1038/nature08430. [DOI] [PubMed] [Google Scholar]
- Gómez-Hernández JM, de Miguel M, Larrosa B, González D, Barrio LC. Molecular basis of calcium regulation in connexin-32 hemichannels. Proc Natl Acad Sci U S A. 2003;100:16030–16035. doi: 10.1073/pnas.2530348100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ion channels of excitable membranes. Ed 3. Sunderland, MA: Sinauer; 2001. [Google Scholar]
- Jan LY, Jan YN. L-glutamate as an excitatory transmitter at the Drosophila larval neuromuscular junction. J Physiol. 1976;262:215–236. doi: 10.1113/jphysiol.1976.sp011593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jospin M, Jacquemond V, Mariol MC, Ségalat L, Allard B. The L-type voltage-dependent Ca2+ channel EGL-19 controls body wall muscle function in Caenorhabditis elegans. J Cell Biol. 2002;159:337–348. doi: 10.1083/jcb.200203055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp SJ, Masu M, Eki T, Ozawa K, Nakanishi S. Molecular cloning and chromosomal localization of the key subunit of the human N-methyl-D-aspartate receptor. J Biol Chem. 1993;268:3728–3733. [PubMed] [Google Scholar]
- Keramidas A, Moorhouse AJ, Schofield PR, Barry PH. Ligand-gated ion channels: mechanisms underlying ion selectivity. Prog Biophys Mol Biol. 2004;86:161–204. doi: 10.1016/j.pbiomolbio.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Khakh BS, North RA. P2X receptors as cell-surface ATP sensors in health and disease. Nature. 2006;442:527–532. doi: 10.1038/nature04886. [DOI] [PubMed] [Google Scholar]
- Kim H, Pierce-Shimomura JT, Oh HJ, Johnson BE, Goodman MB, McIntire SL. The dystrophin complex controls BK channel localization and muscle activity in Caenorhabditis elegans. PLoS Genet. 2009;5:e1000780. doi: 10.1371/journal.pgen.1000780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajacic P, Shen X, Purohit PK, Arratia P, Lamitina T. Biomechanical profiling of C. elegans motility. Genetics. 2012;191:1015–1021. doi: 10.1534/genetics.112.141176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Sleegers K, González-Pérez A, Ingelsson M, Beecham GW, Hiltunen M, Combarros O, Bullido MJ, Brouwers N, Bettens K, Berr C, Pasquier F, Richard F, Dekosky ST, Hannequin D, Haines JL, Tognoni G, Fiévet N, Dartigues JF, Tzourio C, et al. The CALHM1 P86L polymorphism is a genetic modifier of age at onset in Alzheimer's disease: a meta-analysis study. J Alzheimers Dis. 2010;22:247–255. doi: 10.3233/JAD-2010-100933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Ge Q, Chen B, Salkoff L, Kotlikoff MI, Wang ZW. Genetic dissection of ion currents underlying all-or-none action potentials in C. elegans body-wall muscle cells. J Physiol. 2011a;589:101–117. doi: 10.1113/jphysiol.2010.200683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Chen B, Wang ZW. Gap junctions synchronize action potentials and Ca2+ transients in Caenorhabditis elegans body wall muscle. J Biol Chem. 2011b;286:44285–44293. doi: 10.1074/jbc.M111.292078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Chen B, Gaier E, Joshi J, Wang ZW. Low conductance gap junctions mediate specific electrical coupling in body-wall muscle cells of Caenorhabditis elegans. J Biol Chem. 2006;281:7881–7889. doi: 10.1074/jbc.M512382200. [DOI] [PubMed] [Google Scholar]
- Lu B, Zhang Q, Wang H, Wang Y, Nakayama M, Ren D. Extracellular calcium controls background current and neuronal excitability via an UNC79-UNC80-NALCN cation channel complex. Neuron. 2010;68:488–499. doi: 10.1016/j.neuron.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytton J, Westlin M, Hanley MR. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J Biol Chem. 1991;266:17067–17071. [PubMed] [Google Scholar]
- Ma Z, Siebert AP, Cheung KH, Lee RJ, Johnson B, Cohen AS, Vingtdeux V, Marambaud P, Foskett JK. CALHM1 is the pore-forming subunit of an ion channel that mediates extracellular Ca2+ regulation of neuronal excitability. Proc Natl Acad Sci U S A Plus. 2012;109:E1963–E1971. doi: 10.1073/pnas.1204023109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JF, Xiong ZG, Jackson MF. Paradox of Ca2+ signaling, cell death and stroke. Trends Neurosci. 2006;29:75–81. doi: 10.1016/j.tins.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Maruyama T, Kanaji T, Nakade S, Kanno T, Mikoshiba K. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. Jpn J Biochem. 1997;122:498–505. doi: 10.1093/oxfordjournals.jbchem.a021780. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minster RL, Demirci FY, DeKosky ST, Kamboh MI. No association between CALHM1 variation and risk of Alzheimer disease. Hum Mutat. 2009;30:E566–E569. doi: 10.1002/humu.20989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton E, Lamitina T. A suite of MATLAB-based computational tools for automated analysis of COPAS Biosort data. Biotechniques. 2010;48:xxv–xxx. doi: 10.2144/000113427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer BD, Hevezi P, Gao N, Lu M, Kalabat D, Soto H, Echeverri F, Laita B, Yeh SA, Zoller M, Zlotnik A. Expression of genes encoding multi-transmembrane proteins in specific primate taste cell populations. PLoS One. 2009;4:e7682. doi: 10.1371/journal.pone.0007682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- Richmond JE, Jorgensen EM. One GABA and two acetylcholine receptors function at the C. elegans neuromuscular junction. Nat Neurosci. 1999;2:791–797. doi: 10.1038/12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royal DC, Bianchi L, Royal MA, Lizzio M, Jr, Mukherjee G, Nunez YO, Driscoll M. Temperature-sensitive mutant of the Caenorhabditis elegans neurotoxic MEC-4(d) DEG/ENaC channel identifies a site required for trafficking or surface maintenance. J Biol Chem. 2005;280:41976–41986. doi: 10.1074/jbc.M510732200. [DOI] [PubMed] [Google Scholar]
- Rusakov DA, Fine A. Extracellular Ca2+ depletion contributes to fast activity-dependent modulation of synaptic transmission in the brain. Neuron. 2003;37:287–297. doi: 10.1016/S0896-6273(03)00025-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83:1359–1400. doi: 10.1152/physrev.00007.2003. [DOI] [PubMed] [Google Scholar]
- Santi CM, Yuan A, Fawcett G, Wang ZW, Butler A, Nonet ML, Wei A, Rojas P, Salkoff L. Dissection of K+ currents in Caenorhabditis elegans muscle cells by genetics and RNA interference. Proc Natl Acad Sci U S A. 2003;100:14391–14396. doi: 10.1073/pnas.1935976100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebert AP, Ma Z, Grevet JD, Demuro A, Parker I, Foskett JK. Convergent structural and functional evolution of CALHM ion channel with connexins and pannexins-innexins. J Biol Chem. 2013;288:6140–6153. doi: 10.1074/jbc.M112.409789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Bergsman JB, Harata NC, Scheller RH, Tsien RW. Recordings from single neocortical nerve terminals reveal a nonselective cation channel activated by decreases in extracellular calcium. Neuron. 2004;41:243–256. doi: 10.1016/S0896-6273(03)00837-7. [DOI] [PubMed] [Google Scholar]
- Syntichaki P, Xu K, Driscoll M, Tavernarakis N. Specific aspartyl and calpain proteases are required for neurodegeneration in C. elegans. Nature. 2002;419:939–944. doi: 10.1038/nature01108. [DOI] [PubMed] [Google Scholar]
- Sznitman J, Purohit PK, Krajacic P, Lamitina T, Arratia PE. Material properties of Caenorhabditis elegans swimming at low Reynolds number. Biophys J. 2010;98:617–626. doi: 10.1016/j.bpj.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taruno A, Vingtdeux V, Ohmoto M, Ma Z, Dvoryanchikov G, Li A, Adrien L, Zhao H, Leung S, Abernethy M, Koppel J, Davies P, Civan MM, Chaudhari N, Matsumoto I, Hellekant G, Tordoff MG, Marambaud P, Foskett JK. CALHM1 ion channel mediates purinergic neurotransmission of sweet, bitter and umami tastes. Nature. 2013;495:223–226. doi: 10.1038/nature11906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong YG, Bürglin TR. Conditions for dye-filling of sensory neurons in Caenorhabditis elegans. J Neurosci Methods. 2010;188:58–61. doi: 10.1016/j.jneumeth.2010.02.003. [DOI] [PubMed] [Google Scholar]
- White MM, Aylwin M. Niflumic and flufenamic acids are potent reversible blockers of Ca2+-activated Cl-channels in Xenopus oocytes. Mol Pharmacol. 1990;37:720–724. [PubMed] [Google Scholar]
- Wu Y, Luo Y. Transgenic C. elegans as a model in Alzheimer's research. Curr Alzheimer Res. 2005;2:37–45. doi: 10.2174/1567205052772768. [DOI] [PubMed] [Google Scholar]
- Xiong Z, Lu W, MacDonald JF. Extracellular calcium sensed by a novel cation channel in hippocampal neurons. Proc Natl Acad Sci U S A. 1997;94:7012–7017. doi: 10.1073/pnas.94.13.7012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Tavernarakis N, Driscoll M. Necrotic cell death in C. elegans requires the function of calreticulin and regulators of Ca2+ release from the endoplasmic reticulum. Neuron. 2001;31:957–971. doi: 10.1016/S0896-6273(01)00432-9. [DOI] [PubMed] [Google Scholar]
- Xu W, Lipscombe D. Neuronal Ca(V)1.3alpha(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci. 2001;21:5944–5951. doi: 10.1523/JNEUROSCI.21-16-05944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Bianchi L, Lee WH, Wang Y, Israel S, Driscoll M. Intersubunit interactions between mutant DEG/ENaCs induce synthetic neurotoxicity. Cell Death Differ. 2008;15:1794–1803. doi: 10.1038/cdd.2008.114. [DOI] [PubMed] [Google Scholar]