

Abstract
Autophagy is a cell process that in normal conditions serves to recycle cytoplasmic components and aged or damaged organelles. The autophagic pathway has been implicated in many physiological and pathological situations, even during the course of infection by intracellular pathogens. Many compounds are currently used to positively or negatively modulate the autophagic response. Recently it was demonstrated that the polyamine spermidine is a physiological inducer of autophagy in eukaryotic cells. We have previously shown that the etiological agent of Chagas disease, the protozoan parasite Trypanosoma cruzi, interacts with autophagic compartments during host cell invasion and that preactivation of autophagy significantly increases host cell colonization by this parasite. In the present report we have analyzed the effect of polyamine depletion on the autophagic response of the host cell and on T. cruzi infectivity. Our data showed that depleting intracellular polyamines by inhibiting the biosynthetic enzyme ornithine decarboxylase with difluoromethylornithine (DFMO) suppressed the induction of autophagy in response to starvation or rapamycin treatment in two cell lines. This effect was associated with a decrease in the levels of LC3 and ATG5, two proteins required for autophagosome formation. As a consequence of inhibiting host cell autophagy, DFMO impaired T. cruzi colonization, indicating that polyamines and autophagy facilitate parasite infection. Thus, our results point to DFMO as a novel autophagy inhibitor. While other autophagy inhibitors such as wortmannin and 3-methyladenine are nonspecific and potentially toxic, DFMO is an FDA-approved drug that may have value in limiting autophagy and the spread of the infection in Chagas disease and possibly other pathological settings.
Keywords: autophagy, polyamines, Trypanosoma cruzi, spermidine, DFMO, autophagic response, LC3, ATG5
Introduction
Polyamines (PAs) are ubiquitous polycations that participate in multiple known and unknown biological processes. These low molecular weight basic substances are present in practically all living organisms and play essential roles in the biosynthesis of nucleic acids and proteins as well as cell proliferation and differentiation.1 PAs stabilize anionic macromolecules and modulate DNA:protein and protein:protein interactions.
The biosynthesis of PAs starts from the amino-acid L-ornithine, an intermediate of the urea cycle, which is converted to the diamine putrescine (Put) after decarboxylation catalyzed by ornithine decarboxylase 1 (ODC1). The sequential addition of two aminopropyl groups to Put by the aminopropyltransferases spermidine synthase and spermine synthase generates spermidine (Spd) and spermine (Spm), respectively. Both reactions require the previous decarboxylation of S-adenosylmethionine to generate the aminopropyl groups under a reaction catalyzed by S-adenosylmethionine decarboxylase 1 (AMD1).2
ODC1 and AMD1 are the rate-limiting enzymes in polyamine biosynthesis and their actions are highly controlled. ODC1 is an obligate homodimer with two shared active sites in the C-terminal domain of the second subunit, the Cys360 residue plays an essential role in the decarboxylation reaction.3 α-difluoromethylornithine (DFMO) acts as an enzyme-activated irreversible inhibitor of ODC1, forming a covalent adduct with Cys360.4
A particular feature of polyamines is that their concentration is increased in proliferating cells including cancer cells5-8 and in parasitic organisms.2 Targeting of the polyamine biosynthetic and catabolic enzymes is being evaluated in different neoplastic diseases such as nonmelanoma skin cancers,9 lung cancers10 and colorectal adenomas.11 ODC1 inhibition represents an important molecular target-based approach for preventing malignant diseases. In a randomized, double-blind, phase-III trial, DFMO treatment has achieved promising results in men and women with a previous history of skin cancer.12 Moreover, DFMO is an effective clinical agent against Trypanosoma brucei gambiense, one of the causative agents of African sleeping sickness;13 this effectiveness is based on a stable ODC1 activity plus a negligible polyamine uptake in T. brucei.13 In contrast, a direct action of DFMO on T. cruzi was discarded because of the well-demonstrated lack of ODC1 enzymatic activity in this parasite.13-15 Among polyamines, Spd is considered an universal anti-aging drug because its exogenous supply increases the life span of several organisms: yeast (Saccharomyces cerevisiae), nematodes (Caenorhabditis elegans) and flies (Drosophila melanogaster) and significantly reduces age-related oxidative protein damage in mice.16 Furthermore, in chronological aging experiments, the spe1Δ yeast strain that is deficient in ornithine decarboxylase activity exhibits increased mortality, which could be restored to normal levels by supplementation with low doses of Spd or its precursor Put.17 The fact that the anti-aging effect of Spd was phenocopied by the knockout of histone acetylases suggested that Spd can modulate the activity of these enzymes favoring the expression of key longevity proteins including the autophagic machinery reviewed below.17
Autophagy is a pathway that plays an essential role in the conservation of cellular homeostasis by removal of old, supernumerary, damaged or ectopic organelles and/or portions of cytoplasm.18 At least three different types of autophagy have been described; macroautophagy, the best characterized of them, and referred to as autophagy in the rest of this report, involves the sequestration of cytoplasmic materials such as vesicles and old organelles and their degradation by lysosomal enzymes when delivered to lysosomes.19 Two main steps are activated during autophagy induction: autophagosome formation and autophagosome maturation. Initially, from specific sites of cellular cytoplasm, a curved membrane, the phagophore, starts to form and elongates around the cargo materials and closes to form a double-membrane vesicle called the autophagosome. Autophagosomes interact with endocytic or phagocytic compartments and finally fuse with lysosomes to form the mature autolysosome where the enclosed materials are degraded.20
Several genes required for autophagy have been described. Their products, the ATG proteins, form complexes that comprise the core molecular machinery responsible for sequential activation of this pathway.21 These core ATG proteins are composed of four subgroups; two of them are responsible for phagophore elongation by catalyzing specific ubiquitination-like conjugation reactions. The first reaction renders the ATG12–ATG5-ATG16L1 complex that is found in the phagophore membrane. In turn, the LC3 protein is cleaved and conjugated with phosphatidylethanolamine (PE) to form LC3-II that is inserted into the membranes of autophagic vesicles.22 Multiple stress situations (nutrient deprivation, cellular hypoxia, mitochondrial or DNA damage, accumulation of unfolded proteins, etc.) switch on cellular autophagy. As mentioned above, Spd has been recently described as a new modulator of this response. Although the molecular aspects of this regulation are poorly understood, recent studies showed that Spd inhibits histone acetyl transferase activity, leading to upregulation of several ATG genes including ATG7, ATG11 and ATG15.17 Spd is also able to directly induce autophagy in a transcription-independent way when added to culture media. The mechanism is not well elucidated yet but could be due to enhanced deacetylation of essential autophagy-related proteins such as ATG5 and ATG7.23 Furthermore the same concentrations of Spd that exert proautophagic effects also have a marked life span-extending effect on yeast, nematodes and flies. Conversely, genetic inhibition of essential ATG genes abrogates longevity extension induced by Spd, indicating that this polyamine can prolong life span by the induction of autophagy.17
Autophagy has been related to the capacity for infection by Trypanosoma cruzi, the etiologic agent of Chagas disease. Previously we have reported that trypomastigotes, the infective forms of this parasite, localize in LC3-positive compartments during invasion. Increased autophagy in the host cell mediated by a variety of stimuli benefits T. cruzi colonization whereas deficit or absence of ATG genes significantly decreases it.24 Since autophagy is a process involved in the innate immune response, the T. cruzi-autophagy relationship is an interesting aspect to be considered in the development of Chagas disease.25 This neglected illness is endemic in Latin America, with almost 9 million infected people and 28 million at risk of contracting it. Current therapy is only partially effective and elicits serious side effects.26 For these reasons, new pathways and molecules that target them are being studied.27,28 In this sense, polyamine metabolism seems to be a promising target since polyamines are essential for T. cruzi survival. Because the parasite is naturally auxotrophic for putrescine synthesis,14,29 PAs must be obtained from the host environment through TcPAT12 and maybe other transport systems.30,31
In this work we show that the reduction of cellular levels of PAs as a result of DFMO treatment resulted in a strong inhibition of the autophagic response. DFMO was able to inhibit the induction of autophagy in response to starvation or rapamycin treatment in two different mammalian cell lines. DFMO reduced protein levels of LC3 and ATG5, two key proteins required at the initial steps of autophagosome formation. Importantly, pretreatment of cells with DFMO abrogated the increase in T. cruzi cellular infection produced during autophagy induction or in a cell line with a high basal level of autophagy. Considering that DFMO is already approved by the US. Food and Drug Administration and that autophagy inhibitors like wortmannin or 3-methyl adenine are nonspecific and highly toxic drugs,32-34 we propose DFMO as a candidate agent that may be useful to control host autophagy in the setting of particular parasitic infections such as Chagas disease, and potentially in other settings of excessive autophagy such as cancer or heart failure.
Results
DFMO prevents the autophagic response in epithelial and cardiac cells
Stably transfected CHO cells overexpressing GFP-LC3 were pre-incubated with 1 mM DFMO for 48 h to deplete intracellular PAs (see below). After that, treated and control cells were starved (2 h) or incubated with rapamycin (2 h) to induce autophagy. As shown in the Figure 1A, starved cells (Stv) displayed a punctate pattern of GFP-LC3 compared with the diffuse cytosolic distribution observed in control conditions (Ctr). Addition of bafilomycin A1 (BAFA1) to prevent lysosomal degradation further increased autophagosome abundance (Stv+BAFA1). In contrast, DFMO pretreatment suppressed autophagy in cells subjected to starvation for 2 h (DFMO+Stv). The level of autophagic response by scoring the percentage of cells with more than 10 puncta per cell was then analyzed (Fig. 1B). Consistent with previous reports, Stv and rapamycin (Rap) increased autophagy reaching 40% and 50% of cells with GFP-LC3 puncta, respectively, while wortmannin (WM) suppressed starvation-induced autophagy. As expected, BAFA1 treatment increased the percentage of cells with more than 10 puncta to 98%, due to its inhibition of lysosomal clearance of autophagosomes. DFMO pretreatment abrogated the induction of autophagy by starvation (10%, p < 0.001) or rapamycin (13%, p < 0.001) (Fig. 1B), indicating that DFMO suppresses both mechanisms of autophagic induction. Basal autophagy was also slightly reduced in DFMO-treated cells maintained in control medium (Ctr). To discard the possibility that these effects might be unique to the stably transfected CHO cell line, the study was repeated in nontransfected CHO cells using monodansylcadaverine (MDC) (Fig. 1C) and endogenous LC3 (Fig. S1A) to label autophagic compartments. In both cases the percentage of cells with > 10 puncta was quantified and similar results were obtained (Fig. 1D; Fig. S1B). We also evaluated the effect of DFMO pretreatment in HL-1 cardiomyocytes that were transiently transfected with GFP-LC3. The threshold that defines autophagic activation in these cells is up to 20 or 30 LC3 puncta per cell as a cutoff.35,36 Starvation increased the percentage of cells with > 20 puncta per cell from 2% at baseline to 40% after starvation. BAFA1 treatment increased the percentage of cells with > 20 puncta to 75% whereas DFMO pretreatment abrogated the starvation response (14%, p < 0.01) (Fig. 1E). These results indicate that DFMO can abrogate autophagy induced by different stimuli and in different cell lines.

Figure 1. DFMO inhibits the autophagic response of epithelial and cardiac cells. CHO cells or stably transfected CHO cells overexpressing GFP-LC3 were grown in full nutrient medium in the presence (+DFMO) or absence of 1 mM DFMO during 48 h. Subsequently cells were incubated for 2 h in control (Ctr) or starvation medium (Stv) or in control medium supplemented with 50 ng/μl rapamycin (Rap) alone (None) or in the presence of 100 nM wortmannin (+WM); 100 nM bafilomycin A1 (+BAFA1) or 1 mM DFMO (+DFMO) as indicated under Materials and Methods. (A) Confocal images depict GFP-LC3 distribution under the indicated conditions. (B) Graph shows the percentage of cells with more than 10 puncta per cell in each condition. Data represent the mean ± SEM of at least three independent experiments (number of counted cells ≈100). Significantly different from control: ***p < 0.001. (C) CHO cells were treated as before and then were incubated for 30 min with 100 μM MDC and analyzed by in vivo confocal microscopy. Images depict differential interference contrast (DIC) and MDC labeling of cells under the indicated conditions. (D) Percentage of cells with more than 10 MDC-labeled dots per cell in each condition. Data represent the mean ± SEM of at least three independent experiments (number of counted cells ≈100). Significantly different from control: ***p < 0.001. (E) HL-1 cells transiently overexpressing GFP-LC3 were grown in control medium in the presence or absence of 1 mM DFMO (+DFMO) and then incubated for 4 h in control (Ctr) or starvation (Stv) medium alone (None) or in the presence of 100 nM bafilomycin A1 (+BAFA1) or 1 mM DFMO (+DFMO). The percentage of cells with more than 20 GFP-LC3 dots per cell was quantified in each condition. Data represent the mean ± SEM of at least three independent experiments (number of counted cells ≈100). Significantly different from controls: **p < 0.01. Scale bars: 10 μm.
The inhibitory action of DFMO on autophagy is time and concentration dependent and is related to spermidine levels
In CHO and HL-1 cells, both overexpressing GFP-LC3, we tested different DFMO concentrations from 0.1 to 10 mM for different periods of time. CHO cells treated with 1 or 10 mM DFMO for 2 h in starvation media showed a 40% reduction of autophagy (Fig. 2A, 1 mM–2 h and 10 mM–2 h, see details in Materials and Methods). The maximal inhibitory effect was achieved when 1 mM DFMO was applied 48 h before the starvation stimulus (Fig. 2A, 1 mM–48 h). However, in HL-1 cells the addition of 1 mM DFMO incubated for 4 h in starvation media did not significantly reduce the response (Fig. 2B, 1 mM–4 h) whereas the addition of 10 mM DFMO under the same conditions was able to reduce autophagy by ≈40% (Fig. 2B, 10 mM–4 h). Similar to CHO cells, 48 h pretreatment with 1 mM DFMO significantly attenuated autophagy by 65% (Fig. 2B). In contrast, pretreatment for 48 h with 0.1 or 0.5 mM DFMO did not interfere with the autophagic response in either cell line (data not shown). Based on these results, we used a 48 h pretreatment with 1 mM DFMO in our subsequent studies.
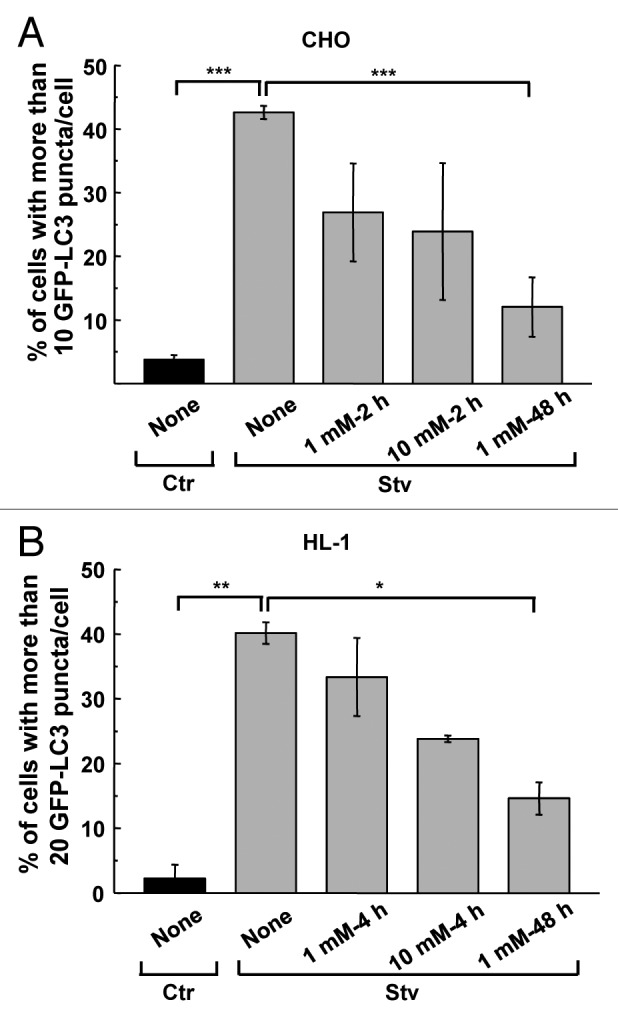
Figure 2. DFMO suppresses autophagy after prolonged incubation time. CHO or HL-1 cells, both overexpressing GFP-LC3, were incubated in control (Ctr) or starvation medium (Stv) in the presence or absence of different concentrations of DFMO (from 0.1 to 10 mM) for different times (2, 4 or 48 h). (A) Percentage of cells with more than 10 puncta per cell at the indicated conditions. Data represent the mean ± SEM of at least three independent experiments (number of counted cells ≈100). Significantly different from control: ***p < 0.001. (B) Percentage of cells with more than 20 dots of LC3 per cell at the indicated conditions. Data represent the mean ± SEM of at least three independent experiments (number of counted cells ≈100). Significantly different from control: **p < 0.01, *p < 0.05.
In eukaryotic cells, DFMO treatment depletes intracellular PAs through specific inhibition of ODC1 enzymatic activity.37 As explained in the introduction, previous work has shown that Spd promotes autophagy.17 To determine if the inhibition of the autophagic response observed in DFMO treated cells was the result of PAs depletion, we measured the intracellular levels of the main PAs (Spd and Spm) in CHO cells subjected to the same conditions as described above. Intracellular concentration of Spd (ng of Spd in 5 × 106 cells, see details in Materials and Methods) were not significantly modified in Ctr, Stv or WM conditions; however Rap treatment elicited a significant increase of this PA compared with the control. Interestingly, DFMO-treated cells displayed undetectable levels of Spd after Stv or Rap treatments (Fig. 3A). In contrast, no variations were observed in Spm levels in all the conditions analyzed (Fig. 3B). We confirmed the previous result by analyzing the autophagic activity of cells by microscopy after adding Spd to media (see details in Materials and Methods). As shown in Figure 3C the addition of Spd under starvation conditions did not increase the percentage of cells with > 10 MDC dots/cell. Conversely, addition of both DFMO and Spd 48 h before starvation significantly recovered the level of autophagosomes to the values obtained in Stv media, indicating that DFMO’s inhibitory action is directly attributed to depletion of Spd. A similar conclusion was obtained by quantification of endogenous LC3 dots in each condition (Fig. 3D). Taken together, these results indicate that the reduction in the level of autophagic response caused by DFMO is likely a consequence of Spd depletion.

Figure 3. DFMO treatment decreases intracellular levels of spermidine. CHO cells were grown in full nutrient medium in the presence (+DFMO) or absence of 1 mM DFMO during 48 h and then were incubated for 2 h in control (Ctr) or starvation medium (Stv) or in control medium supplemented with 50 ng/μl rapamycin (Rap) alone (None) or in the presence of 100 nM wortmannin (+WM) or 1 mM DFMO (+DFMO). Intracellular pools of polyamines were determined by HPLC after benzoylation as indicated in Materials and Methods. Concentrations (ng /5 × 10 6 cells) of spermidine (A) and spermine (B) are depicted for the indicated conditions. All values are the mean ± SD of two determinations. Significantly different from control: *p < 0.05, **p < 0.01. (C and D) CHO cells were grown in full nutrient media alone (None) or in the presence of 1 mM DFMO (+DFMO), 100 μM Spd (+Spd) or 1 mM DFMO plus 100 μM Spd (+DFMO +Spd) during 48 h and then were incubated for 2 h in control (Ctr) or starvation medium (Stv) followed by MDC labeling or immunodetection of endogenous LC3 protein. Percentage of cells with more than 10 MDC-labeled dots per cell (C) or 10 endogenous LC3 puncta per cell (D) was calculated for each condition. Data represent the mean ± SEM of at least three independent experiments (number of counted cells ≈100). Significantly different from control: *p < 0.05, **p < 0.01.
Considering that PAs are important regulators of DNA replication, we have also studied the effect of DFMO treatment on cell proliferation and cytotoxicity by performing the alamarBlue assay (see details in Materials and Methods). Our results obtained from different cell lines show that in the conditions used, DFMO did not significantly impair cell proliferation or viability (Fig. S2).
DFMO hampers autophagosome formation
To examine the specific mode of action of DFMO on the autophagic response, we evaluated the protein levels of LC3 in CHO cells subjected to various treatments. The specific anti-LC3 antibody detects two bands with a relative electrophoretic mobility of 18 and 16 kDa corresponding to LC3-I and the lipidated form, LC3-II, respectively (Fig. 4A, upper panel). To compare the abundance of LC3-I and -II in different conditions, we quantified the bands by densitometry and compared with TUBULIN level (Fig. 4A, bottom panel). To overcome variation between experiments we normalized the values of LC3-II/TUB ratio to the value of the control for each experiment. Figure 4B shows the values of normalized LC3-II/TUB ratio obtained from four independent experiments. Cells subjected to starvation exhibited an ≈2.0- to 4.0-fold increase in the normalized LC3-II/TUBULIN ratio, which was prevented by wortmannin administration (+WM) (Fig. 4B). Similar to the scoring of autophagy by microscopy, inhibition of lysosomal degradation with BAFA1 significantly increased the abundance of LC3-II. Pretreatment of cells with DFMO prevented the expected increase in LC3-II after starvation or rapamycin treatment (Fig. 4B). These results indicate that DFMO pretreatment prevented the induction of autophagy by starvation or rapamycin and confirmed the findings previously obtained by microscopic analysis.

Figure 4. DFMO affects autophagosome formation. CHO cells were grown in full nutrient medium in the presence or absence of 1 mM DFMO (+DFMO) during 48 h and then were incubated for 2 h in control (Ctr) or starvation medium (Stv) or in control medium supplemented with 50 ng/ μl rapamycin (Rap) alone (None) or in the presence of 100 nM wortmannin (+ WM); 100 nM bafilomycin A1 (+BAFA1) or 1 mM DFMO (+DFMO) as indicated in Materials and Methods. Afterwards, cells were lysed with sample buffer and the samples were subjected to western blot analysis to detect endogenous LC3 and TUBULIN proteins as indicated in Materials and Methods. (A) Representative immunoblots are depicted. (B) Densitometry was performed using NIH ImageJ. The LC3-II/TUBULIN normalized to control and (C) the LC3-II/LC3-I ratios were calculated. Data represent the mean ± SEM of four independent experiments. Significantly different from control: ***p < 0.001, *p < 0.05. (D) To evaluate autophagic flux, similar experiments were performed in the presence or absence of 100 nM BAFA1, which blocks lysosomal degradation. (E) LC3-II/TUBULIN ratio was calculated. Data are representative of three independent experiments. Significantly different from control: **p < 0.01, ns: non-significant.
Autophagy inhibitors have different mechanisms of action that can be assessed by western blot.38,39 Some inhibitors like wortmannin abolish the early steps of autophagosome formation and prevent LC3 lipidation whereas others like BAFA1 prevent lysosomal degradation, resulting in autophagosome (and LC3-II) accumulation in cells. Analysis of LC3 in cells subjected to starvation or rapamycin treatment revealed that the LC3-II/LC3-I ratio was not altered by DFMO pretreatment. In contrast, starved cells incubated with wortmannin showed a decreased LC3-II/LC3-I ratio and an increase in LC3-I (Fig. 4C). To exclude the possibility of accelerated flux as an explanation for the low level of LC3-II in DFMO-treated cells, we added BAFA1 to prevent lysosomal degradation in starved cells that were pretreated with DFMO. We found that BAFA1 did not result in accumulation of LC3-II in DFMO-treated cells in both, control or starvation conditions, indicating that the flux was not accelerated (Fig. 4D and E).
We next calculated the absolute amount of LC3 (LC3 I + II) relative to TUBULIN from the same immunoblots. Total levels of LC3 were not generally modified in almost all treatments with exception of BAFA1, which increased LC3 levels due to reduced protein degradation. Of note, DFMO caused a significant reduction (p < 0.01 and p < 0.001 under Stv and Rap treatments respectively) in the total LC3 expression levels (Fig. 5A). In addition to the reduction in LC3 abundance in DFMO-treated cells, ATG5, a protein required for the first steps of autophagosome formation, was similarly decreased (Fig. 5B). Given the transcriptional effects of Spd on autophagy gene expression,17 it seems likely that Spd depletion by DFMO treatment (Fig. 3A) suppresses autophagy by downregulating the expression of rate-limiting factors such as LC3 and ATG5.
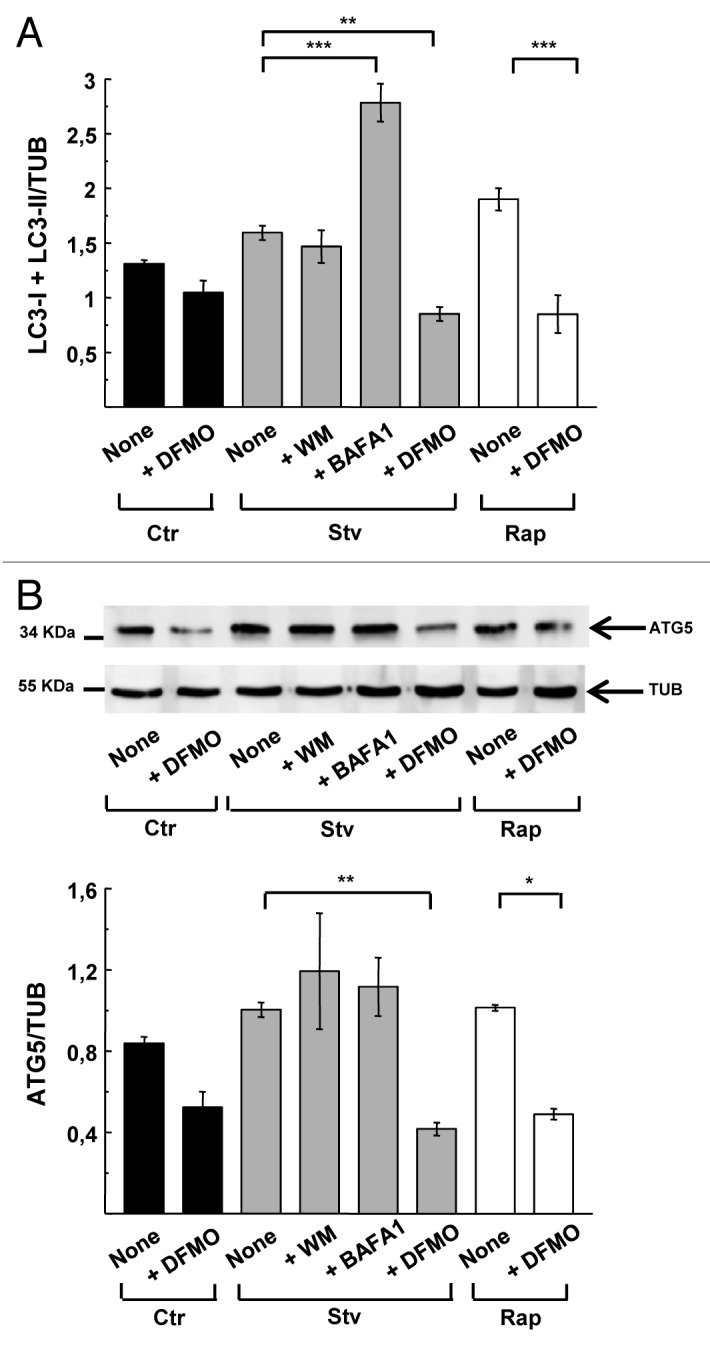
Figure 5. DFMO treatment diminishes LC3 and ATG5 protein levels. CHO cells were grown in full nutrient medium and treated as indicated in Figure 4. After cell lysis, detection of endogenous LC3, ATG5 and TUBULIN proteins by western blot was performed. (A) LC3-I+LC3-II/TUBULIN ratio obtained from experiments depicted in Figure 4. Band intensity was quantified with the ImageJ program. Significantly different from control: **p < 0.01, ***p < 0.001. (B) Upper panel: representative immunoblot of three experiments corresponding to ATG5 detection is depicted. Lower panel: quantification of the ATG5/TUBULIN ratio. Data are representative of three independent experiments. Significantly different from control: **p < 0.01, *p < 0.05.
Reduction of host cell spermidine impairs T. cruzi infection
Previously our laboratory reported that the protozoan parasite Trypanosoma cruzi interacts with autophagosomes during infection.24 Furthermore, induction of host cell autophagy before the infection significantly increases the percentage of infected cells.25 These observations were extended to different host cell types and T. cruzi strains, showing the relevance of this phenomenon.25 In searching for a safe autophagy inhibitor that might prevent T. cruzi entry into the cell, we tested the effect of DFMO on T. cruzi infection in different conditions and host cell lines. CHO cells overexpressing GFP-LC3 were pretreated for 48 h with 1 mM DFMO with or without the addition of Spd. After that, cells were starved for 2 h and then infected with T. cruzi of the CL Brener strain for 6 h (MOI = 10) in the same conditions. After fixation, parasites were detected by indirect immunofluorescence using a specific antibody against T. cruzi (see the scheme in Fig. 6A and details in Materials and Methods). In agreement with our previous results, induction of autophagy increased the level of T. cruzi infection (30%, p < 0.001) whereas DFMO significantly reduced the percentage of infected cells under starvation conditions (12%, p < 0.001) (Fig. 6B). The addition of Spd abolished the effect of DFMO (28%, p < 0.001), indicating that similar to the autophagic response, the reduction in the level of T. cruzi infection caused by DFMO is a consequence of Spd depletion.

Figure 6. DFMO treatment impairs T. cruzi infection. CHO cells overexpressing GFP-LC3 were grown in the presence (+DFMO) or absence of DFMO or DFMO plus spermidine (+DFMO+Spd) during 48 h and then were incubated for 2 h in control (Ctr) or starvation medium (Stv) alone (None) or with the indicated compounds. Cells were then infected with T. cruzi of the CL Brener strain (MOI = 10) for 6 h in the same conditions. After fixation parasites were detected by indirect immunofluorescence using a specific antibody. (A) Outline of the experiment. (B) Quantification of percentage of infected cells under the indicated conditions. Data represent the mean ± SEM of at least three independent experiments (number of counted cells ≈100). Significantly different from control: ***p < 0.001. (C) Confocal images depicting GFP-LC3 localization around T. cruzi vacuoles under the indicated conditions. Scale bars: 10 μm. (D) HL-1 cells transiently overexpressing GFP-LC3 were grown in control medium in the presence (+DFMO) or absence of 1 mM DFMO during 48 h and then incubated for 3 h in control medium alone (Ctr) or supplemented with 50 ng/ μl rapamycin (Rap) or in starvation medium alone (Stv) or supplemented with 100 nM bafilomycin A1 (+ BAFA1) or 1 mM DFMO (+ DFMO) or TAT- ATG5K130R protein (+TAT-ATG5K130R). After that, cells were infected with T. cruzi of the Brazil heart strain (MOI = 10) for 1 h in the same conditions. After fixation parasites were detected by indirect immunofluorescence using a specific antibody. The percentage of infected cells at the indicated conditions was quantified. Data represent the mean ± SEM of at least three independent experiments (number of counted cells ≈100). Significantly different from controls: *p < 0.05. Scale bars: 10 μm. (E) CHO and Vero cells were grown in the presence or absence of DFMO during 48 h and then were infected with T. cruzi of the CL Brener strain (MOI = 10) for 24 h in the same conditions. After detection of the parasites, the percentage of infected cells at the indicated conditions was calculated. Data represent the mean ± SEM of at least three independent experiments (number of counted cells ≈100). Significantly different from control: *p < 0.05.
In our previous work we have shown that GFP-LC3 protein decorates the membrane of the T. cruzi parasitophorous vacuole (TcPV) formed during trypomastigote invasion.24,25 Despite the significant reduction of cellular infection in DFMO-treated cells, host GFP-LC3 was still recruited to the TcPV in all conditions studied (Fig. 6C). This result indicates that DFMO does not modify the capacity of T. cruzi to interact with autophagic compartments or with LC3.
We also studied the effect of DFMO on T. cruzi infection in HL-1 cells expressing GFP-LC3. Inducing autophagy by Stv or Rap increased the percentage of infected cells (Fig. 6D), while inhibition of autophagy with the cell-permeable protein TAT- ATG5K130R,40 significantly reduced T. cruzi infection. Similarly, DFMO pretreatment reversed the starvation-enhanced infection rate in HL-1 cells (Fig. 6D). Interestingly, although BAFA1 blocks flux but not autophagy initiation, it attenuated starvation-enhanced infection, suggesting that the autophagic flux is important for successful infection. This is consistent with our previous observation that lysosomal and autolysosomal compartments participate in T. cruzi invasion. When autophagy is activated by starvation or other stimulus, autophagosomes rapidly fuse with preformed lysosomes to form autolysosomes41 that may then be exploited by T. cruzi to augment its access to the host cell.
Since basal autophagy was also reduced in DFMO-treated cells (Fig. 1B; Fig. 4E) we analyzed the effect of DFMO pretreatment on T. cruzi entry into host cells under basal conditions. Inclusion of DFMO during 24 h infection with T. cruzi failed to modify infection in CHO cells but significantly decreased infection in Vero cells in similar conditions (Fig. 6E). By confocal microscopy, using MDC to label autophagosomes, we have observed that basal autophagy is low in CHO cells, but higher in Vero cells and that DFMO treatment was able to decrease this autophagy under control conditions (Fig. S3). This observation may explain the higher rate of T. cruzi infection and the benefit of DFMO treatment in Vero cells compared with CHO cells. Taken together these results indicate that depletion of Spd abrogates T. cruzi infection with an efficacy that is roughly proportional to the level of autophagy.
Reduction of T. cruzi infection by DFMO is impaired in MEF atg5 knockout cells
In previous work we have shown that T. cruzi infection was significantly reduced in MEF cells null for atg5.24 Considering that autophagy is important for the beneficial effects of DFMO on T. cruzi infection, we next studied the action of this compound on parasite infection in these cells and compared with their corresponding controls. MEF WT and atg5−/− (MEF atg5 KO) cells were pretreated for 48 h with 1 mM DFMO before infection with T. cruzi for 24 h (MOI = 10). After fixation cells were processed to detect the parasites by indirect immunofluorescence using a specific antibody against T. cruzi followed by staining with FITC-phalloidin to visualize cell perimeters (Fig. 7A). As expected, a significant reduction in the percentage of infected cells was quantified in MEF KO cells at 24 h after infection (10%, p < 0.01) (Fig. 7B). DFMO produced a significant reduction of T. cruzi infection in MEF WT cells (5%, p < 0.001). In atg5 KO MEFs, infection was quite low, and addition of DFMO provided little additional benefit, corroborating the previous results and suggesting that DFMO inhibits T. cruzi infection by suppressing autophagy. We conclude that downregulation of autophagic machinery by DFMO suppresses autophagy and impairs T. cruzi colonization of cells.

Figure 7. DFMO has less effect on T. cruzi infection in MEF atg5 knockout cells. MEF cells (MEF WT) and atg5−/− cells (MEF atg5 KO) were grown in the presence (+DFMO) or absence (Ctr) of 1 mM DFMO during 48 h and then were infected with T. cruzi of the CL Brener strain (MOI = 10) for 24 h in the same conditions. After fixation, parasites were detected by indirect immunofluorescence using a specific antibody followed by incubation with FITC-phalloidin to label cell perimeters. (A) Confocal images depict intracellular parasites (arrows) in each type of cell and condition. (B) Quantification of the percentage of infected cells at the indicated conditions. Data represent the mean ± SEM of at least two independent experiments (number of counted cells ≈100). Significantly different from control: ***p < 0.001, **p < 0.01. Scale bars: 10 μm.
Discussion
In this work, we propose DFMO as an agent that can be useful to limit autophagy in settings where this process may be disadvantageous. Other autophagy inhibitors (e.g., wortmannin or 3-methyl adenine) have off-target effects that contribute to toxicity.32-34 DFMO is a modified ornithine that irreversibly binds and inhibits ODC1, one of the rate-limiting enzymes in polyamine synthesis. We have shown that DFMO treatment caused a significant reduction in intracellular Spd levels (Fig. 3A) whereas Spm levels were not affected (Fig. 3B). As a consequence of Spd depletion, some cellular processes regulated by Spd (e.g., autophagy) are diminished. It is well known that PAs are important regulators of cell proliferation and differentiation;1 despite this, we did not observe cytotoxicity or an effect on cell proliferation under the conditions used (Fig. S2).
Spd was recently recognized as an autophagy inducer and life-span extending compound.17,23 Spd inhibits histone acetyl transferases present in the nucleus and cytosol;17,42 unopposed deacetylases (notably SIRTUINS) lower the acetylation status of the autophagy proteins and their transcriptional regulators, resulting in activation of the pathway. SIRTUINS, which are NAD+-dependent deacetylases, stimulate autophagy through deacetylation of certain targets. However, the regulatory network is complex: Atg3 acetylation by the histone acetyl transferase Esa1 plays a key role in autophagy induction by controlling Atg3 and Atg8 interaction and lipidation of Atg8 in yeast.43 Thus, protein acetylation is emerging as a key regulatory mechanism for autophagy. The results presented in this work show that Spd depletion mediated by DFMO abrogated the autophagic response induced by starvation or rapamycin treatment. This result suggests that in addition to its capacity to induce autophagy through transcriptional regulation, Spd may also participate in the post-translational control of autophagy triggered by other stimuli and may serve as a consumable cofactor during autophagosome formation. The results indicate that Spd depletion suppresses autophagy through a mechanism distinct from wortmannin (which inhibits PtdIns3K) and BAFA1 (which prevents lysosomal degradation). Our analysis of ATG5 expression and LC3 expression and lipidation indicates that Spd depletion reduced ATG5 and LC3 total levels but did not alter the LC3-II/-I ratio (Fig. 4C). Given that ATG5 and LC3 are rate limiting, it is easy to understand how DFMO would constrain autophagy without directly inhibiting the machinery.
Previously we showed that induction of canonical autophagy benefits host cell colonization by tissue culture-derived trypomastigotes (TCT) and that the T. cruzi parasitophorous vacuole (TcPV) has characteristics of an autolysosome.24 In this context we tested DFMO against T. cruzi cellular infection. We have demonstrated that similar to other autophagic inhibitors, DFMO significantly reduced T. cruzi infection of cells during starvation. This effect was reversed by supplementing the culture media with Spd, confirming that DFMO attenuation of autophagy and T. cruzi infection is due to the reduction of intracellular levels of Spd. Interestingly, DFMO pretreatment did not interfere with the capacity of T. cruzi to interact with autophagosomes, LC3 or autophagic derived compartments.
According to the literature there are two main mechanisms that could explain LC3 localization during T. cruzi invasion. One possibility is the fusion of autolysosomes to the plasma membrane and to the TcPV membrane or, alternatively, direct recruitment of LC3 to these structures by a mechanism that could resemble the recently described LC3 acquisition by Salmonella-containing vacuoles.44 In our previously published work, we have shown the presence of autolysosomes moving toward the T. cruzi entry sites.24,25 Additionally, the colocalization between LC3 and TcPV was significantly reduced in the presence of the microtubule toxin vinblastine24 indicating that autolysosome transport is required for LC3 recruitment to the parasite vacuole. However, the fact that after DFMO treatment LC3 is still localized in the TcPV indicates that some direct LC3 recruitment could occur. On the other hand, T. cruzi needs lysosomal and autolysosomal compartments to complete invasion of the host cell.24,45 Independently of LC3 behavior, compounds that abrogate autolysosomal formation will result in a significant reduction in T. cruzi infection. We noted that DFMO efficacy for suppressing infection was proportional to the level of autophagic activity. Cells with high basal autophagy as Vero cells were infected in greater magnitude than CHO cells, while the autophagy deficient MEF atg5−/− cells were less infected than the corresponding wild type MEFs. This observation leads us to speculate that the rate of infection may be proportional to the availability of autolysosomes to participate in formation of the parasitophorous vacuoles. Therefore, it is likely that DFMO, by reducing the ATG machinery, suppresses canonical autophagosome formation and transport, thereby limiting the compartments available for T. cruzi entry. Other authors showed that starvation and rapamycin increase the infection of T. cruzi and T. dionosii metacyclic trypomastigotes (MT) during the infection period but not if cells are treated under these conditions before infection.46,47 In spite of this discrepancy observed between TCT and MT, both cases support a role for autophagy during the first steps of infection. In contrast, induction of autophagy after parasite entry does not have a significant effect over T. cruzi replication and intracellular parasite load.24,48
This work presents a potential new use for DFMO, a drug described a long time ago for treatment of sleeping sickness,13 and which has been widely studied in different classes of cancers for its antiproliferative actions.5,6 Here, we have demonstrated the inhibitory action of DFMO on autophagy and elucidated a possible mechanism of action. Importantly, the reduction of T. cruzi infection by suppressing autophagy with DFMO depletion of polyamines opens a new field of application for this compound that is based on closing the cellular gateway for this parasite. Studies in animal models will be conducted to test the efficacy of this drug on in vivo T. cruzi infection alone or in combination therapy.
Materials and Methods
Reagents
Minimal essential medium (α-MEM, 11900-024) and Dulbecco’s Modified Eagle’s Medium (D-MEM, 12100), fetal bovine serum (FBS, 16000-044), Earle’s Balanced Salt Solution (EBSS, 14155063) and penicillin and streptomycin (15640055) were obtained from Gibco BRL/Life technologies. Monodansylcadaverine (MDC, 30432), wortmannin (W1628), bafilomycin A1 (B1793), DFMO (D-193), TRITC (P1951) and FITC (P5282)-conjugated phalloidin were purchased from Sigma-Aldrich. Rapamycin (R-5000) was purchased from LC Laboratories. The alamarBlue reactive (DAL-1025) was purchased from Biosource-Life Technologies. The polyclonal rabbit anti-LC3 (L7543) and goat anti-human HRP (A8667) antibodies were purchased from Sigma-Aldrich, the polyclonal rabbit anti-ATG5 (AB78073) was purchased from Abcam, and the polyclonal mouse anti-β TUBULIN (E7) was obtained from Developmental Studies Hybridoma Bank. A polyclonal rabbit antibody against T. cruzi was kindly provided by Dr. David Engman (Northwestern University, Chicago, IL) and a polyclonal human antibody against T. cruzi (AbJDO) was obtained from an infected patient. The secondary antibody Cy3-conjugated anti-Goat IgG (305-166-003) was purchased from Jackson Immuno-Research Laboratories. The DNA marker Hoechst 33342 (H3570) was purchased from Life Technologies. All other chemicals were from Sigma-Aldrich.
Cell culture
CHO cells were maintained in flasks in α-MEM supplemented with 10% FBS and antibiotics (control medium) at 37°C in an atmosphere of 95% air and 5% CO2 and were plated on glass coverslips in 12- or 24-well plates to 80% confluence before experiments. Stably transfected CHO cells overexpressing EGFP-LC3 were grown in flasks in the same media supplemented with 0.2 mg/ml geneticin (Sigma, A1720) and 10% FBS and antibiotics at 37°C in an atmosphere of 95% air and 5% CO2 and plated as before. Vero and MEF cells were grown in D-MEM supplemented with 10% FBS and antibiotics at 37°C in an atmosphere of 95% air and 5% CO2. HL-1 cells were maintained in flasks coated with gelatin-fibronectin (Sigma, G1890 and F1141, respectively), at 37°C in an atmosphere of 5% CO2 in Claycomb medium (Sigma, 51800C) supplemented with 2 mM L-glutamine (Invitrogen, 25030-164), 10% fetal bovine serum (Thermo Fisher Scientific, SH30071.03), 0.1 mM norepinephrine (Sigma, A0937), and 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B (Sigma, A2942). For experiments HL-1 cells were plated onto 35-mm glass-bottomed culture dishes (MatTek Co.) and cultured in the same medium.
GFP-LC3 adenovirus infection
Formation of autophagosomes in HL-1 cells was quantified via fluorescence imaging of GFP-LC3 puncta. Cells were infected for 2 h with adenovirus encoding GFP-LC3 at a concentration of 10 plaque-forming units/cell, and 18 h later cells were subjected to the indicated experimental conditions. Cells were fixed for 15 min with 4% formaldehyde in phosphate-buffered saline, pH 7.4.
Quantification of autophagy
Autophagy was induced by amino acid starvation. CHO or HL-1 cells overexpressing GFP-LC3 grown in 24-well plates or 35-mm glass-bottomed culture dishes were washed three times with PBS and incubated with control medium or with 0.5 ml Earle’s balanced salt solution (EBSS, Sigma; Starvation medium) at 37°C for 2 h (for CHO cells) or 4 h (for HL-1 cells) in the presence or the absence of the drugs (100 nM wortmannin or 100 nM BAFA1). Alternatively, autophagy was induced by treatment with rapamycin (50 ng/μl) for 2 h in full-nutrient medium without antibiotics. In some experiments cells were pretreated with 1 or 10 mM DFMO for 2 h or with 0.1, 0.5 or 1 mM DFMO for 48 h before induction of autophagy. Cells were fixed with 3% paraformaldehyde in PBS for 15 min at room temperature, washed with PBS, and blocked with 50 mM NH4Cl in PBS, and then were mounted with Mowiol 4-88 Reagent (Calbiochem, 475904) and examined by confocal microscopy, using an Olympus Confocal FV1000 microscope and processed with the program FV10-ASW 1.7. Alternatively CHO cells were subjected to indirect immunofluorescence to detect endogenous LC3 using a specific antibody. The percentage of cells with more than 10 or 20 LC3 dots/cell was determined. Confocal images of 10 random fields were quantified, representing around 100 cells per experiment. Data are presented as mean values and error bars indicate the SEM from at least two independent experiments. Statistical calculations (Student and Tukey’s tests, significant differences were ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001) were made using Kyplot statistical software and graphs were plotted with Microsoft Power Point.
Monodansylcadaverine staining
CHO cells grown in 6-well plates were washed three times with PBS and incubated in control or starvation medium at 37°C for 2 h in the presence or the absence of drugs. Cells under DFMO treatment were pretreated with 1 mM DFMO for 48 h before induction of autophagy. Autophagic vacuoles were labeled with MDC by incubating cells grown on coverslips with 100 μM MDC in PBS at 37°C for 30 min. After incubation, cells were washed four times with PBS and immediately analyzed by confocal microscopy, using an Olympus Confocal FV1000 microscope and processed with the program FV10-ASW 1.7. The percentage of cells with more than 10 MDC dots/cell was determined. Confocal images of 10 random fields were quantified, representing around 100 cells per experiment. Data are presented as mean values and error bars indicate the SEM from at least three independent experiments. Statistical calculations (Student and Tukey’s tests, significant differences were ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001) were made using Kyplot statistical software and graphs were plotted with Microsoft Power Point.
AlamarBlue assay
The AlamarBlue assay was conducted according to the manufacturer instructions. CHO, HL-1, Vero and MEF cells grown in 96-well plates were washed three times to PBS and incubated in control medium in the presence or the absence of 1 mM DFMO at 37°C for 48 h. After that, cells were washed and alamarBlue reactive was added with the medium and incubated for 6 h at 37°C before the measurement. The level of cell growth based on detection of cell metabolic activity was proportional to the absorbance at 540 nm. Figure S2 shows the results of one experiment conducted with triplicate samples, representative of two independent experiments.
Western blot analysis
CHO cells grown in 6-well plates were washed three times with PBS and incubated with control or starvation medium at 37°C for 2 h in the presence or the absence of the drugs. Cells under DFMO treatment were pretreated with 1 mM DFMO for 48 h before experiment. Cells were lysed with sample buffer and protein samples were run on a 12.5% polyacrylamide gel and transferred to Hybond-ECL (Amersham) nitrocellulose membranes. The membranes were blocked ON in Blotto at 4°C (5% non-fat milk, 0.1% Tween 20, and PBS), washed twice with PBS and incubated with a primary antibody anti-LC3 (1:800 dilution) or anti-ATG5 (1:500 dilution) followed by a peroxidase-conjugated secondary antibody (1:10,000 dilution). Anti-TUBULIN (1:300 dilution) was used to detect TUBULIN as a loading control. The corresponding bands were detected using an enhanced chemiluminescence detection kit (Amersham, RPN2109) and the band was detected using Fujifilm LAS-4000.
Determination of polyamine intracellular concentrations by HPLC
CHO cells were grown in 12-well plates, washed three times with PBS and incubated with control medium or with 0.5 ml Earle’s balanced salt solution (EBSS, Sigma; starvation medium) at 37°C for 2 h in the presence or the absence of the drugs (0.2 µM mM wortmannin or 0.1 µM mM BAFA1). Alternatively, autophagy was induced by treatment with rapamycin (50 ng/μl) for 2 h in full nutrient medium without antibiotics. In some experiments cells were pretreated with 1 mM DFMO for 48 h before induction of autophagy. Afterwards the cells were collected and resuspended in 0.5 ml of PBS (5.106 cells/ml); 25 μl of 100% trichloroacetic acid were added, and after mixing and centrifugation (5 min, 12,000 g) the supernatant was diluted with 2 ml of 2 M NaOH. After adding 5 μl of benzoyl chloride and incubating for 30 min at 37°C, the mixture was extracted twice with 1 ml of chloroform. The combined organic phase was washed with 1 ml of water and then evaporated until dry. The residue was resuspended in a mixture of water–methanol (45/55 v/v). The resulting samples were injected into a Beckman ODS (C18) column for HPLC analysis, and the elution was performed with a methanol gradient (from 55% to 87%). The detection of benzoyl derivatives of polyamines was done with a UV spectrophotometer operated at 254 nm.5,6,49 CHO cells were resuspended in 0.2 M perchloric acid. After centrifugation, NaOH was added to the supernatant fraction and different aliquots treated with benzoyl chloride. The benzoyl derivatives were separated by HPLC on a Beckman system equipped with a C/s reverse-phase column and analyzed spectrophotometrically.
Propagation of Trypanosoma cruzi
CL Brener strain of T. cruzi was provided by Dr. Juan Jose Cazzulo (Inst. de Investigaciones Biotecnológicas. IIB-UNSAM, Bs. As., Argentina) and T. cruzi Brazil heart strain was kindly provided by Dr. David Engman (Northwestern University, Chicago, IL, USA). Infective tissue cell derived trypomastigotes (TCT) from CL Brener were prepared as described in Romano et al.24 Briefly, Vero cells plated in T25 flasks at 37°C in D-MEM supplemented with 3% FBS and antibiotics (infection medium) were infected with TCT suspensions of each T. cruzi strains for 1 d in infection medium at 37°C in an atmosphere of 95% air and 5% CO2 followed for 4 to 6 d more in axenic infection medium. Free TCT elicited by cellular lysis were harvested from the medium and centrifuged at 600 × g for 15 min at 20°C. The supernatants were discarded and pellets containing T. cruzi (trypomastigotes and amastigotes) were covered with 2 or 3 ml of infection medium and incubated for 3 h at 37°C in an atmosphere of 95% air and 5% CO2 to allow the swim-up of trypomastigotes. TCT were counted in the Neubauer camera and used in the experiments.
Infection experiments
CHO, Vero or HL-1 cells overexpressing GFP-LC3 grown in 24-well plates were incubated in the presence or the absence of 1 mM DFMO or 1 mM DFMO plus 100 μM spermidine for 48 h in control medium before autophagy induction. Cells were then treated in control or starvation media for the indicated times in the presence or the absence of the drugs followed by infection with TCT from T. cruzi CL Brener strain (MOI = 10) for 6 h. The protein TAT ATG5K130R was added to the media at a concentration of ~200 nM every 30 min until the end of the experiment. Cells were then fixed with 3% paraformaldehyde solution in PBS for 15 min at room temperature, washed with PBS, and quenched with 50 mM NH4Cl. Subsequently, cells were permeabilized with 1% saponin in PBS containing 1% bovine serum albumin (BSA), and then incubated with the primary antibody AbJDO against Trypanosoma cruzi (1:50) and detected by incubation with HRP conjugated goat anti human (1:200) and Cy3 conjugated rabbit anti-goat (1:700) secondary antibodies. Cells were mounted with Mowiol containing the DNA marker Hoechst and examined by confocal microscopy.
MEF WT and MEF atg5−/− (MEF atg5 KO) cells grown in 24-well plates were incubated in the presence or the absence of 1 mM DFMO for 48 h in control medium before infection with T. cruzi of the CL Brener strain (MOI = 10) for 24 h in the same conditions. After fixation, parasites were detected by indirect immunofluorescence as indicated above. Cell perimeters were visualized by detection of Actin with FITC-conjugated phalloidin. Cells were mounted with Mowiol containing the DNA marker Hoechst and examined by confocal microscopy.
Supplementary Material
Acknowledgements
We are grateful to Dr. David Engman, who shared with us his knowledge about T. cruzi strains and also provided us the T. cruzi Brazil heart strain. We also thank Milton Osmar Aguilera and Alejandra Medero for technical assistance. Work in this area has been partly supported by grants from Comisión Nacional Salud Investiga (Ministerio de Salud de la Nación), CONICET-Fundación Fulbright, Secretaría de Ciencia, Técnica y Posgrado (Sectyp, Universidad Nacional de Cuyo), Fundación Bunge y Born and Agencia Nacional de Promoción Científica y Tecnológica (PICT# 2008 2235) to P.S.R. and PICT 2008 # 0192 to M.I.C.
Glossary
Abbreviations:
- AMD1
S-adenosylmethionine decarboxylase 1
- ATG5
autophagy-related 5
- BAFA1
bafilomycin A1
- DFMO
α-difluoromethylornithine
- LC3
microtubule-associated protein 1 light chain 3
- MDC
monodansylcadaverine
- ODC1
ornithine descarboxylase 1
- PAs
polyamines
- Put
putrescine
- Rap
rapamycin
- Spd
spermidine
- Spm
spermine
- TUB
tubulin
- TcPV
T. cruzi parasitophorous vacuole
- TCT
tissue culture-derived trypomastigotes
- WM
wortmannin
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/24709
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/24709
References
- 1.Igarashi K, Kashiwagi K. Polyamines: mysterious modulators of cellular functions. Biochem Biophys Res Commun. 2000;271:559–64. doi: 10.1006/bbrc.2000.2601. [DOI] [PubMed] [Google Scholar]
- 2.Birkholtz LM, Williams M, Niemand J, Louw AI, Persson L, Heby O. Polyamine homoeostasis as a drug target in pathogenic protozoa: peculiarities and possibilities. Biochem J. 2011;438:229–44. doi: 10.1042/BJ20110362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Almrud JJ, Oliveira MA, Kern AD, Grishin NV, Phillips MA, Hackert ML. Crystal structure of human ornithine decarboxylase at 2.1 A resolution: structural insights to antizyme binding. J Mol Biol. 2000;295:7–16. doi: 10.1006/jmbi.1999.3331. [DOI] [PubMed] [Google Scholar]
- 4.McCann PP, Pegg AE. Ornithine decarboxylase as an enzyme target for therapy. Pharmacol Ther. 1992;54:195–215. doi: 10.1016/0163-7258(92)90032-U. [DOI] [PubMed] [Google Scholar]
- 5.Casero RA, Jr., Marton LJ. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat Rev Drug Discov. 2007;6:373–90. doi: 10.1038/nrd2243. [DOI] [PubMed] [Google Scholar]
- 6.Gerner EW, Meyskens FL., Jr. Polyamines and cancer: old molecules, new understanding. Nat Rev Cancer. 2004;4:781–92. doi: 10.1038/nrc1454. [DOI] [PubMed] [Google Scholar]
- 7.Pegg AE. Mammalian polyamine metabolism and function. IUBMB Life. 2009;61:880–94. doi: 10.1002/iub.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wallace HM. The polyamines: past, present and future. Essays Biochem. 2009;46:1–9. doi: 10.1042/bse0460001. [DOI] [PubMed] [Google Scholar]
- 9.Elmets CA, Athar M. Targeting ornithine decarboxylase for the prevention of nonmelanoma skin cancer in humans. Cancer Prev Res (Phila) 2010;3:8–11. doi: 10.1158/1940-6207.CAPR-09-0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casero RA, Pegg AE. Polyamine catabolism and disease. Biochem J. 2009;421:323–38. doi: 10.1042/BJ20090598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerner EW, Meyskens FL., Jr. Combination chemoprevention for colon cancer targeting polyamine synthesis and inflammation. Clin Cancer Res. 2009;15:758–61. doi: 10.1158/1078-0432.CCR-08-2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bailey HH, Kim K, Verma AK, Sielaff K, Larson PO, Snow S, et al. A randomized, double-blind, placebo-controlled phase 3 skin cancer prevention study of alpha-difluoromethylornithine in subjects with previous history of skin cancer. Cancer Prev Res (Phila) 2010;3:35–47. doi: 10.1158/1940-6207.CAPR-09-0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heby O, Persson L, Rentala M. Targeting the polyamine biosynthetic enzymes: a promising approach to therapy of African sleeping sickness, Chagas’ disease, and leishmaniasis. Amino Acids. 2007;33:359–66. doi: 10.1007/s00726-007-0537-9. [DOI] [PubMed] [Google Scholar]
- 14.Carrillo C, Cejas S, González NS, Algranati ID. Trypanosoma cruzi epimastigotes lack ornithine decarboxylase but can express a foreign gene encoding this enzyme. FEBS Lett. 1999;454:192–6. doi: 10.1016/S0014-5793(99)00804-2. [DOI] [PubMed] [Google Scholar]
- 15.Carrillo C, Cejas S, Cortés M, Ceriani C, Huber A, González NS, et al. Sensitivity of trypanosomatid protozoa to DFMO and metabolic turnover of ornithine decarboxylase. Biochem Biophys Res Commun. 2000;279:663–8. doi: 10.1006/bbrc.2000.3996. [DOI] [PubMed] [Google Scholar]
- 16.Madeo F, Tavernarakis N, Kroemer G. Can autophagy promote longevity? Nat Cell Biol. 2010;12:842–6. doi: 10.1038/ncb0910-842. [DOI] [PubMed] [Google Scholar]
- 17.Eisenberg T, Knauer H, Schauer A, Büttner S, Ruckenstuhl C, Carmona-Gutierrez D, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009;11:1305–14. doi: 10.1038/ncb1975. [DOI] [PubMed] [Google Scholar]
- 18.Martinou JC, Kroemer G. Autophagy: evolutionary and pathophysiological insights. Biochim Biophys Acta. 2009;1793:1395–6. doi: 10.1016/j.bbamcr.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 19.Cuervo AM. The plasma membrane brings autophagosomes to life. Nat Cell Biol. 2010;12:735–7. doi: 10.1038/ncb0810-735. [DOI] [PubMed] [Google Scholar]
- 20.Yoshimori T. [Autophagy as a bulk protein degradation system: it plays various roles] Tanpakushitsu Kakusan Koso. 2004;49(Suppl):1029–32. [PubMed] [Google Scholar]
- 21.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–32. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 22.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–31. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Madeo F, Eisenberg T, Büttner S, Ruckenstuhl C, Kroemer G. Spermidine: a novel autophagy inducer and longevity elixir. Autophagy. 2010;6:160–2. doi: 10.4161/auto.6.1.10600. [DOI] [PubMed] [Google Scholar]
- 24.Romano PS, Arboit MA, Vázquez CL, Colombo MI. The autophagic pathway is a key component in the lysosomal dependent entry of Trypanosoma cruzi into the host cell. Autophagy. 2009;5:6–18. doi: 10.4161/auto.5.1.7160. [DOI] [PubMed] [Google Scholar]
- 25.Romano PS, Cueto JA, Casassa AF, Vanrell MC, Gottlieb RA, Colombo MI. Molecular and cellular mechanisms involved in the Trypanosoma cruzi/host cell interplay. IUBMB Life. 2012;64:387–96. doi: 10.1002/iub.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guedes PM, Silva GK, Gutierrez FR, Silva JS. Current status of Chagas disease chemotherapy. Expert Rev Anti Infect Ther. 2011;9:609–20. doi: 10.1586/eri.11.31. [DOI] [PubMed] [Google Scholar]
- 27.Astelbauer F, Walochnik J. Antiprotozoal compounds: state of the art and new developments. Int J Antimicrob Agents. 2011;38:118–24. doi: 10.1016/j.ijantimicag.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 28.Romanha AJ, Castro SL, Soeiro MdeN, Lannes-Vieira J, Ribeiro I, Talvani A, et al. In vitro and in vivo experimental models for drug screening and development for Chagas disease. Mem Inst Oswaldo Cruz. 2010;105:233–8. doi: 10.1590/S0074-02762010000200022. [DOI] [PubMed] [Google Scholar]
- 29.Carrillo C, Cejas S, Huber A, González NS, Algranati ID. Lack of arginine decarboxylase in Trypanosoma cruzi epimastigotes. J Eukaryot Microbiol. 2003;50:312–6. doi: 10.1111/j.1550-7408.2003.tb00141.x. [DOI] [PubMed] [Google Scholar]
- 30.Carrillo C, Canepa GE, Algranati ID, Pereira CA. Molecular and functional characterization of a spermidine transporter (TcPAT12) from Trypanosoma cruzi. Biochem Biophys Res Commun. 2006;344:936–40. doi: 10.1016/j.bbrc.2006.03.215. [DOI] [PubMed] [Google Scholar]
- 31.Hasne MP, Coppens I, Soysa R, Ullman B. A high-affinity putrescine-cadaverine transporter from Trypanosoma cruzi. Mol Microbiol. 2010;76:78–91. doi: 10.1111/j.1365-2958.2010.07081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abbas HK, Mirocha CJ. Isolation and purification of a hemorrhagic factor (wortmannin) from Fusarium oxysporum (N17B) Appl Environ Microbiol. 1988;54:1268–74. doi: 10.1128/aem.54.5.1268-1274.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bosch U, Mirocha CJ, Abbas HK, di Menna M. Toxicity and toxin production by Fusarium isolates from New Zealand. Mycopathologia. 1989;108:73–9. doi: 10.1007/BF00436056. [DOI] [PubMed] [Google Scholar]
- 34.Gunther R, Abbas HK, Mirocha CJ. Acute pathological effects on rats of orally administered wortmannin-containing preparations and purified wortmannin from Fusarium oxysporum. Food Chem Toxicol. 1989;27:173–9. doi: 10.1016/0278-6915(89)90066-5. [DOI] [PubMed] [Google Scholar]
- 35.Brady NR, Hamacher-Brady A, Yuan H, Gottlieb RA. The autophagic response to nutrient deprivation in the hl-1 cardiac myocyte is modulated by Bcl-2 and sarco/endoplasmic reticulum calcium stores. FEBS J. 2007;274:3184–97. doi: 10.1111/j.1742-4658.2007.05849.x. [DOI] [PubMed] [Google Scholar]
- 36.Yuan H, Perry CN, Huang C, Iwai-Kanai E, Carreira RS, Glembotski CC, et al. LPS-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. Am J Physiol Heart Circ Physiol. 2009;296:H470–9. doi: 10.1152/ajpheart.01051.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pegg AE, Shantz LM, Coleman CS. Ornithine decarboxylase as a target for chemoprevention. J Cell Biochem Suppl. 1995;22:132–8. doi: 10.1002/jcb.240590817. [DOI] [PubMed] [Google Scholar]
- 38.Rubinsztein DC, Cuervo AM, Ravikumar B, Sarkar S, Korolchuk V, Kaushik S, et al. In search of an “autophagomometer”. Autophagy. 2009;5:585–9. doi: 10.4161/auto.5.5.8823. [DOI] [PubMed] [Google Scholar]
- 39.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–5. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 40.Huang C, Liu W, Perry CN, Yitzhaki S, Lee Y, Yuan H, et al. Autophagy and protein kinase C are required for cardioprotection by sulfaphenazole. Am J Physiol Heart Circ Physiol. 2010;298:H570–9. doi: 10.1152/ajpheart.00716.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lawrence BP, Brown WJ. Autophagic vacuoles rapidly fuse with pre-existing lysosomes in cultured hepatocytes. J Cell Sci. 1992;102:515–26. doi: 10.1242/jcs.102.3.515. [DOI] [PubMed] [Google Scholar]
- 42.Mariño G, Morselli E, Bennetzen MV, Eisenberg T, Megalou E, Schroeder S, et al. Longevity-relevant regulation of autophagy at the level of the acetylproteome. Autophagy. 2011;7:647–9. doi: 10.4161/auto.7.6.15191. [DOI] [PubMed] [Google Scholar]
- 43.Yi C, Ma M, Ran L, Zheng J, Tong J, Zhu J, et al. Function and molecular mechanism of acetylation in autophagy regulation. Science. 2012;336:474–7. doi: 10.1126/science.1216990. [DOI] [PubMed] [Google Scholar]
- 44.Kageyama S, Omori H, Saitoh T, Sone T, Guan JL, Akira S, et al. The LC3 recruitment mechanism is separate from Atg9L1-dependent membrane formation in the autophagic response against Salmonella. Mol Biol Cell. 2011;22:2290–300. doi: 10.1091/mbc.E10-11-0893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andrade LO, Andrews NW. The Trypanosoma cruzi-host-cell interplay: location, invasion, retention. Nat Rev Microbiol. 2005;3:819–23. doi: 10.1038/nrmicro1249. [DOI] [PubMed] [Google Scholar]
- 46.Maeda FY, Alves RM, Cortez C, Lima FM, Yoshida N. Characterization of the infective properties of a new genetic group of Trypanosoma cruzi associated with bats. Acta Trop. 2011;120:231–7. doi: 10.1016/j.actatropica.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 47.Martins RM, Alves RM, Macedo S, Yoshida N. Starvation and rapamycin differentially regulate host cell lysosome exocytosis and invasion by Trypanosoma cruzi metacyclic forms. Cell Microbiol. 2011;13:943–54. doi: 10.1111/j.1462-5822.2011.01590.x. [DOI] [PubMed] [Google Scholar]
- 48.Pinheiro RO, Nunes MP, Pinheiro CS, D’Avila H, Bozza PT, Takiya CM, et al. Induction of autophagy correlates with increased parasite load of Leishmania amazonensis in BALB/c but not C57BL/6 macrophages. Microbes Infect. 2009;11:181–90. doi: 10.1016/j.micinf.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 49.Morgan DM. Determination of polyamines as their benzoylated derivatives by HPLC. Methods Mol Biol. 1998;79:111–8. doi: 10.1385/0-89603-448-8:111. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.