

Abstract
Matrix metalloproteinases (MMPs) are zinc-endopeptidases that play roles in numerous pathophysiological processes and therefore are promising drug targets. However, the large size of this family and a lack of highly selective compounds that can be used for imaging or inhibition of specific MMPs members has limited efforts to better define their biological function. Here we describe a protein engineering strategy coupled with small molecule probe design to selectively target individual members of the MMP family. Specifically, we introduce a cysteine residue near the active site of a selected protease that does not alter its overall activity or function but allows direct covalent modification by a small molecule probe containing a reactive electrophile. This specific engineered interaction between the probe and the target protease provides a means to both image and inhibit the modified protease with absolute specificity. Here we demonstrate the feasibility of the approach for two distinct MMP proteases, MMP-12 and MT1-MMP (MMP-14).
INTRODUCTION
The matrix metalloproteinases (MMPs) are a large family of enzymes that have been found to regulate many aspects of both normal cellular biology and pathogenesis of human diseases, most notably cancer1,2. However attempts to develop novel anti-cancer agents that target MMPs have proven to be largely unsuccessful. These failures have been attributed, in part, to a generally poor understanding of the physiological functions of individual members of the family3. MMPs, like most proteases, are regulated by multiple post-translational mechanisms, including synthesis as inactive zymogens and inhibition by endogenously expressed protein inhibitors such as the TIMPs4. These post-translational events hinder their functional analysis by conventional genomic and proteomic methods. In addition, the large size of the family coupled with the relatively well-conserved active site region has prevented the design of highly selective inhibitors5,6. Thus, new tools and reagents that enable direct monitoring of specific MMP activity as well as specific inhibition of these proteases would enable studies of the specific roles of particular MMPs in disease as well as in basic biological processes7.
One method for functional studies of proteases that has proven valuable is the use of small molecule activity-based probes (ABPs)8. These reagents are based on covalent irreversible inhibitors that can be appropriately tagged and designed to target specific proteases. However, the development of suitable probes for the MMPs has proven difficult because these proteases use a water nucleophile for initial attack of the substrate9, preventing the design of inhibitors that directly modify a primary catalytic residue. To overcome this limitation, ABPs for metalloproteases have been designed based on a zinc-chelating hydroxamate and a benzophenone photo-crosslinking group. These probes can be used to covalently label the active sites of many MMP proteases10–13. However, while useful for proteomic, biochemical or cell biological studies of multiple MMP proteases, the overall lack of selectivity of the probes as well as the need for photo-crosslinking to covalently link probes into the active site limits their utility for studies of specific MMPs in complex biological systems.
Here we present a strategy for selective targeting of a single MMP protease using a combination of protein engineering methods and small molecule probe design strategies. This method is based on the design of a mutant protease that contains an engineered cysteine residue that does not alter expression, folding or substrate recognition but that is capable of acting as a latent nucleophile that reacts covalently with a suitably designed probe containing a reactive electrophile (Figure 1a). We chose this general strategy because this type of proximity induced labeling of an engineered cysteine has already been validated for the identification of small molecule ligands14 as well as for covalent labeling of a engineered protein target that has been fused to a small molecule binding domain15. It has also been demonstrated as a way to drive selectivity of a small molecule inhibitor towards a specific kinase target 16 and to target a protease with a native cysteine residue near the active site17. Thus, we believed that this approach could be generally applied as a way to target specific proteases within large families that have highly similar active site structures and substrate cleavage profiles. Using two matrix metalloproteinases, MMP12 and MT1-MMP (MMP14), we demonstrate that it is possible to find locations near the active site that can be mutated to cysteine without altering functional properties. We also demonstrate targeting of the engineered cysteine with a chemical probe that contains a hydroxamate group for binding to the active site zinc coupled with an appropriately placed electrophile. This modification results in permanent covalent labeling and inhibition, thus enabling specific inhibition and imaging studies of a single defined protease target.
Figure 1.

Protease engineering and probe design. (a) The target protease is engineered with a non-catalytic cysteine near the substrate binding pocket. The designed probe contains a reversibly binding reactive group (hydroxamate), an electrophile (α-chloro acetamide), a peptide backbone that drives specificity towards MMPs (in orange) and a fluorescent tag (green). When the hydroxamate binds to the active site of engineered protease, the electrophile irreversibly labels the protein through the engineered cysteine resulting in a permanently labeled and inhibited protease. When the probe binds to WT MMPs the binding is reversible. (b) Sequence alignment of mouse MMPs. The two non-conserved residues (184 – site 1 and 243 – site 2 in MMP12) that were chosen for mutation are indicated by arrows. (c) Superposition of the crystal structures of MMP12 (1JIZ) and MT1-MMP (3MA2). The active site Zn2+ is shown as a red ball. The candidate residues are drawn using stick representation (left). The surface representation of the structure of the inhibitor GM6001 bound to the catalytic domain of MMP12 is also shown (right). The GM6001 is shown as orange sticks. The Zn2+ is shown as a red ball. The two mutation sites are highlighted in yellow.
MATERIALS AND METHODS
Synthesis
For full details on the synthesis of TND124 and TND126 see supporting information.
Cell culture and transfection. MCF7 and HEK293T (both from American Type Culture Collection) were maintained in DMEM supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and antibiotics. All cultures were maintained at 37°C in 5% CO2/95% air. Cells were transfected with purified plasmid DNA by Lipofectamine treatment (LifeTechnologies) as recommended by the manufacturer.
Protein alignment
Protein alignment of the members of the human MMP family was performed using CLUSTALW2 (http://www.clustal.org/)
Construction of expression plasmids
The catalytic domains of wild type or engineered MMPs were recombinantly expressed in E.coli as fusions of the Vibrio cholerae MARTX toxin cysteine protease domain (CPD). The pET22b-mMMP12-CPD construct was constructed by PCR amplification of the sequence encoding the catalytic domain of mouse MMP12 (amino acids 29–267) using pCMV-SPORT6-mMMP12 (Open Biosystems) as a template and primers MMP12-5′ and MMP12-3′ (Table S1, supporting information). All the protein mutants were created using PCR fusion methods (primers MMP12-T184C-5′, MMP12-T184C-3′, MMP12-T243C-5′ and MMP12-T243C-3′ - Table S1, supporting information) and the resulting PCR products were cloned into the NdeI and SalI sites of pET22b-CPDSalI. The pET22b-mMT1-MMP-CPD construct was generated by PCR amplification of the sequence encoding the catalytic domain of mouse MT1-MMP (amino acids 28–318) pCDNA3-mMT1-MMP (a kindly gift from Dr. Rozanov) as a template and primers MT1-MMPcat-5′ and MT1-MMPcat-3′ (Table S1, supporting information). All the protein mutants were created using PCR fusion methods (primers MT1-MMPcat-F198C5′, MT1-MMPcat-F198C3′, MT1-MMPcat-F260C5′, MT1- MMPcat-F260C3′, MT1-MMPcat-Q262C5′ and MT1- MMPcat-Q262C3′ - Table S1, supporting information) and the resulting PCR products were cloned into the NdeI and SacI sites of pET22b-CPDSacI. Full length HA tagged mMT1-MMP was created by inserting an HA tag (YPYDVPDYA) between D515 and E516 as described previously 18. The final oligo was inserted into the pCDNA3.1 plasmid between the HindIII and XbaI cut sites.
Protein expression and purification
For purification of His6-tagged CPD fusion proteins, overnight cultures of the appropriate strain were diluted 1:500 into 1 L 2YT media and grown with shaking at 37°C. When an OD600 of 0.6 was reached, IPTG was added to 250 μM, and cultures were grown for 8 hrs at 16°C. Cultures were pelleted, resuspended in 25 mL lysis buffer [500 mM NaCl, 50 mM Tris-HCl, 1 mM tris(2-carboxyethyl)phosphine (TCEP), pH 7.5, 15 mM imidazole, 10% glycerol] and flash frozen in liquid nitrogen. Lysates were thawed, then lysed by sonication and cleared by centrifugation at 15,000xg for 30 minutes. His6-tagged CPD fusion proteins were affinity purified by incubating the lysates in batch with 0.5–1.0mL Ni-NTA agarose beads (Qiagen) with shaking for 2–4 hrs at 4°C. The binding reaction was pelleted at 1,500 x g, the supernatant was set aside, and the pelleted Ni2+-NTA agarose beads were washed three times with lysis buffer. In some cases, 10% of the Ni2+-NTA beads containing immobilized CPD-His6 fusion proteins were removed, pelleted and then His6-tagged fusion protein eluted using high imidazole buffer [500 mM NaCl, 50 mM Tris-HCl, pH 7.5, 175 mM imidazole, 10% glycerol].
To liberate untagged target proteins into the supernatant fractions, 300–500 μL lysis buffer was added to the Ni2+-NTA beads containing CPD-His6 fusion proteins and the indicated amount of inositol hexakisphosphate (InsP6, Calbiochem) was added. In general, on-bead cleavage was performed by incubating the beads in the presence of 50– 100 μM InsP6 for 1–2 hr at either room temperature or 4°C. The beads were pelleted at 1,500xg, and the supernatant fraction was removed. The beads were then washed 3–4 times with 300–500 μL lysis buffer, and supernatant fractions retained. His6-tagged proteins remaining on the beads (i.e. cleaved CPD-His6) were eluted using high imidazole buffer [500 mM NaCl, 50 mM Tris-HCl, pH 7.5, 175 mM imidazole, 10% glycerol] in 300–500 μL volumes. The elution was repeated 3–4 times, and eluate fractions were collected.
Enzymatic activity
The recombinant enzyme was diluted to a final concentration of 100 nM in the reaction buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10 mM CaCl2, 0.02% NaN3), and 10 μl of 100 μM of the substrate (Mca-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2, Anaspec) was added. The substrate turnover was immediately measured by fluorescent emission over a 1 hour period at 37°C using a Spectramax M5 plate reader (Molecular Devices).
The hydrolysis of the fluorogenic substrate (0–10 μM) by WT or F260C MT1-MMP was measured at 37°C in a continuous plate reader-based protocol. Fluorescence associated with the turnover of the substrate was measured every 30 s for 1 h (λex 325 nm, λem 393 nm) in a Spectramax M5 plate reader (Molecular Devices). The rate of product formation was determined using linear regression of the fluorescence-time data with the plate reader software (SOFTmax Pro, v 4.8; Molecular Devices Inc., Sunnyvale, CA, USA). Linear reaction rates (R2>0.98 for all data) were fitted to the Michaelis-Menten equation using the nonlinear curve fitting facility of Kaleida Graph in order to obtain Vmax and Km values.
Peptide substrate profiling
The peptide substrate profiling was performed using a mixture-based oriented peptide library19. Degenerate positions X were prepared using isokinetic mixtures of the 19 naturally occurring L-amino acids excluding cysteine. The exact proportions of each amino acid at each degenerate position was determined by Edman sequencing of an unacetylated portion of the library. Individual peptides were purified by high-performance liquid chromatography, and characterized by mass spectrometry. To determine the primed-side cleavage specificity, a 1 mM solution of Ac-X-X-X-X-X-XX- X-X-X-[D-Lys]-[D-Lys] in MMP reaction buffer (50 mM HEPES, pH 7.4, 200 mM NaCl, 10 mM CaCl2) was digested at 37°C to 5–10% cleavage and heated to 100°C for 2 min. A 10 μl aliquot was subjected to Edman sequencing on an Applied Biosystems (Foster City, CA) Procise 494 Automated Protein Sequencer at the Tufts University Core Facility (Boston, MA). Amino acid preference in a given cycle was calculated by dividing the amount of a particular residue by the average amount per amino acid residue in that cycle. The data were then corrected for bias present in the library by dividing each value by the relative amount of that particular amino acid in the starting mixture.
In silico modeling
All modeling was performed using the Molecular Operating Environment (MOE) software with the default energy minimization settings. The N238Cys and I179Cys mutations were modeled into the crystal structure of human MMP8 bound to the peptidic hydroxamate inhibitor HONH-iBM-Asn-NHBn(m-NH2) (PDB# 1A85). For each model, mutated residues and all side-chains within 4.5 Å were energy minimized. ABPs were docked into the models by sequentially modifying the side-chains of the HONH-iBM-Asn-NHBn(m-NH2) inhibitor into those of potential ABPs starting from the P1′ position and ending with the addition of the fluorophore. Energy minimization was performed as follows: each side chain in the ABP was built manually followed by three energy minimizations: i) the modified side-chain, ii) the modified side-chain plus all MMP8 residues within 4.5 Å and finally iii) the whole ligand plus all MMP8 residues within 4.5 Å This operation was repeated for each side chain until we obtained models of ABPs bound to MMP8 mutants before covalent modification of the engineered Cys by the α-chloro acetamide electrophile. Multiple residues at the P3′ position were tested to identify the optimal side-chain length to achieve close contacts between the α-chloro acetamide group and the engineered Cys. The P3′ side-chain was build to point either towards Cys179 or Cys238. After obtaining a model of the ABP non-covalently bound to MMP8, we modeled the end-product of the reaction between the engineered Cys and the α-chloro acetamide group, followed by energy minimization of the ABPs and all residues within 4.5 Å For TND124, we found that the hydroxamate moiety remained within binding distance of the active site Zn2+ (Figure S2, supporting information).
Gel labeling
Recombinant enzyme (100 ng) was spiked in rat liver lysates (50 μg) and incubated with different concentrations of the probe for 1h at 37°C. The probes were diluted to the desired concentration from a 1000X stock solution in DMSO. Pre-incubation with NEM (1 mM), EDTA (10 mM), GM6001 (10 μM), TND126 (without Cy5 −10 μM) was performed for 30min at 37°C before addition of TND124 (1 μM). These compounds were also diluted to the final concentration from a 1000X stock solution. After the incubation time, the samples were resolved by 15% SDS-PAGE gel and visualized on a Typhoon flatbed fluorescent laser scanner (GE Healthcare). In the case of the labeling of HEK293T cells lysates transiently expressing MT1-MMP WT or mutant, cells were lysed using hypotonic buffer (10 mM PIPES, 10 mM KCl, 5 mM MgCl2 and 4 mM DTT). Then the lysates were incubated with 0.5 μM of TND124 for 1 hour at 37°C. After the incubation time, the samples were then resolved by 15% SDS-PAGE gel and visualized on a Typhoon flatbed fluorescent laser scanner (GE Healthcare).
Probe washout studies
Recombinant enzyme (1000 ng; WT or T184C MMP12) was incubated with 3 μM of TND124 for 1h at 37°C. Afterwards, samples were dialyzed using Slide-A-Lyzer MINI Dialysis Devices, 10K MWCO (Thermo) for 24h and 48 hours at 4°C. For each time point (before addition of TND124 and 24h and 48h after dialysis), 50 μl of each sample was used to detect the activity of the enzyme using a fluorogenic substrate following the protocol mentioned above.
In vitro TND124 inhibition kinetics
Recombinant MMP12 WT and T184C (10 nM) were incubated with increasing concentrations of TND124 in activity buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10 mM CaCl2, 0.02% NaN3) at 37°C in a 96-well plate. After 1h incubation, 10 μM of the fluorogenic substrate was added and the initial rates (V0) of substrate hydrolysis were determined by fluorescent reading at 37°C for 30 min. V0 values were converted to percentages of residual activity relative to untreated controls. The IC50 values were determined by KaleidaGraph using sigmoidal fit. The final presented IC50 values represent the average of three independent assays.
Inhibition studies of MMP12 activity against natural substrates
Gelatin FITC conjugated and collagen IV FAM conjugated were purchased at Anaspec. Recombinant enzymes (3,000 ng; WT or T184C MMP12) were incubated with TND124 (3 μM) for 1h at 37°C. Afterwards, the samples were diluted 1/10 with the activity buffer containing the gelatin or collagen substrate (at a final concentration of 0.1mg/ml or 0.05 mg/ml respectively) incubated for 48h at 37°C. The samples are centrifuged at 10,000rpm for 10min and the fluorescence of the supernatant is measured at 515 nm using a Spectramax M5 plate reader (Molecular Devices).
Casein zymography
Casein zymography was performed based on a previously described technique with slight modifications20. Recombinant protein (500 ng; WT or T184C MMP12) were incubated with 3 μM of TND124 for 1h at 37°C. The samples were diluted 1/10 with the activity buffer and analyzed by electrophoresis. Electrophoresis was carried out on a sodium dodecyl sulfate (SDS)-10% polyacrylamide gel containing α-casein (Sigma) at a concentration of 1 mg/ml. Gels were then incubated in 2.5% (v/v) Triton X-100 for 30 minutes and soaked for 16 hours in a buffer containing 10 mM CaCl2 and 100 mM NaCl at 37°C. The gels were then stained with Coomassie brillant blue G250. Caseinolytic activity was detected as white lysis zones against the blue background.
Mass spectrometry analysis
Intact mass spectrometry
Recombinant enzymes (50 μg; WT or T184C MMP12) were incubated with TND124 and then analyzed using a Thermo LTQ-FT mass spectrometer (Thermo Fisher Scientific) using FTMS + p NSI Full MS scanning mode, mass range 400.00–2000.00, FT resolution 100,000. The MW of eluted peptides was determined by deconvolution using Isopro 3.0 (MS/MS Software).
Tryptic digestion of labeled protein
Recombinant enzymes (50 μg; WT or T184C MMP12) were incubated with TND124 and digested with 1 μg of trypsin at room temperature for 18 hours. The tryptic reactions were separated on a PLRPS 150 mm ×0.1 mm column (Varian, 5 μM particle size, 300 Å pore size) at a flow rate of 700 nL/min in 0.1% trifluoroacetic acid/water (A):0.1% trifluoroacetic acid/acetonitrile (B). The column was run on a gradient of 10% B to 60% B for 25 min, 60% B to 90 % B for 2 min, held at 90% B for 2 min, then rapidly decreased to 10% B over 0.1 min, then run for 11 min at 10% B. Eluted samples were analyzed using a Thermo LTQ-FT mass spectrometer (Thermo Fisher Scientific) using FTMS + p NSI Full MS scanning mode, mass range 400.00–2000.00, FT resolution 100,000. The MW of eluted peptides was determined by deconvolution using Isopro 3.0 (MS/MS Software). The measured masses were compared to the predicted MW of possible peptide cleavage products determined from the primary sequence of recombinant polypeptides derived from MMP12 (modified or unmodified by TND124) using PROTPARAM at http://ca.expasy.org/tools/protparam.html.
Collagen/Gelatin degradation and invasion assays
Acid-extracted rat tail type I collagen was prepared as previously described and stored in acetic acid (0.035 M)21. Collagen solutions were neutralized using equimolar concentrations of NaOH in a 1x MEM +HEPES (25 mM) buffered solution and allowed to gel at 37°C for 1 h in a 95% air/5% CO2 atmosphere21. For degradation assays, collagen gels (2 mg/ml) were prepared as a thin coating in 4-well chambered coverglass slides. The film was then fluorescently labeled using an AlexaFluor-(488 or 594) Protein Labeling Kit (Invitrogen). Cells were plated atop the collagen film at a density of 2×103 cells/well in MEM supplemented with 10% FBS. In the experiments using the inhibitor TND124, prior to plating atop the collagen film, the cells were incubated in serum free media with 3 μM TND124 for 1.5h at 37°C. Afterwards, cells were washed with media 3 times for 10 min period and plated atop the collagen film as described above except serum-free media was used. Where indicated, gelatin degradation was assessed using heat-denatured type I collagen (60 °C for 1 hr) in place of the native substrate21. All assays were allowed to proceed for 72 h prior to imaging. For invasion assays, collagen gels (2 mg/mL) were prepared at a thickness of 0.8–1.0 mm in 6-well transwell inserts (3 μm pore size, polystyrene; Corning). Cells were plated atop the upper surface of the collagen gels at a density of 5×104 cell per well. Media containing 10% FBS was placed in the lower chamber and serum-free media was added to the upper chamber. Invasion was allowed to proceed for 5 days and quantified in randomly selected fields at 20X on a phase-contrast microscope as described previously21.
Zymography for proMMP2 activation
MT1-MMP activity was confirmed by zymographic analysis of MMP-2 activation when the transfected cells were incubated with human proMMP-2 expressed from deficient Timp2 cells22 as has been described before23.
Live cell labeling and immunofluorescence
Cells were transfected with lipofectamine and seeded in 8 well chamber slides (Lab-tek Nunc) at a density of 5×104 cells/well. After 24h, they were incubated with 1.5 μM TND124 or TND126 for 1h at 37°C followed by multiple washes with media. The monolayer was washed with PBS and fixed with 4% paraformaldehide for 20 min at room temperature. The cells were then permeabilized with PBS, 0.1% triton X-100 and 1% heat-inactivated goat serum for an hour. The cells were then washed with PBS and incubated with anti-HA tag (1:100; Covance clone 16B12) primary antibody in blocking buffer (PBS with 1% heat-inactivated goat serum) for 1 hour at room temperature. The cells were then washed with PBS and incubated with AlexaFluor 488 conjugated secondary antibody (1:1000; Invitrogen) for 30 min. The cells were treated and mounted using Vectashield with DAPI (Vector laboratories) for imaging using a Zeiss LSM700 confocal microscope. In the case of pre-incubation with a general MMP inhibitor, cells were treated with 10 μM GM6001 for 30min, washed and incubated with the probe TND124 (1.5 μM) as described.
Zebrafish
Fish strain and maintenance
Wild-type zebrafish (Danio rerio) were maintained under standard laboratory conditions. Embryos were collected from natural matings, reared at 28.5 °C, and staged according to age and morphology as described24
Morpholino injections, rescue and Alcian Blue staining
The mt1-mmpa MO and wild-type mt1-mmpa pCS2 clone were previously described25. The F260C mutant mt1-mmpa pCS2 construct was generated by PCR mutagenesis by overlap extension. Capped synthetic mRNA was generated by Sp6 RNA polymerase in vitro transcription (Ambion). For rescue experiments, one cell-stage embryos were injected with 8 ng of mmp14a MO followed immediately by injection with 200 pg of mRNA encoding either wildtype or mutant mt1-mmpa. Embyros were incubated at 28.5°C until 5 days post-fertilization, fixed using 4% paraformaldehyde in PBS, and craniofacial cartilage elements were visualized using Alcian blue as described 25.
Imaging of embryos
Using a pressure-based microinjection apparatus (WPI), one-cell stage embryos were coinjected with 2 nl of 50 μM TND124 and 300 pg of either wild type or modified synthetic mmp14a mRNA. At either the 8-somite or 18-somite stage, embryos were fixed in 4% PFA/PBS overnight at 4 °C followed by PBS washing and manual dechorionation. Embryos were washed 3X for 5 min each in PBS/0.1% Triton X-100. DIC and Cy5 images of the developing posterior trunk regions were taken at 200x magnification using a Leica DMI6000 B inverted microscope equipped with a Hamamatsu ORCA ER digital camera and Simple PCI software.
RESULTS
Engineering the protease
We first set out to identify residues that would be sufficiently proximal to an active site bound probe to allow modification but that would not cause changes in the overall substrate selectivity of the protease when mutated to cysteine. Therefore, we looked for non-conserved residues that are unlikely to contribute to folding or substrate binding but that are near the substrate-binding pocket of the enzyme. The MMPs have a high degree of structural conservation in the active site region and high-resolution X-ray structures of several members of this family are available. A preliminary search of the primary sequences of the human MMPs revealed two possible candidates (corresponding to residues 198 and 260 in the protein alignment, Figure 1b). These sites mapped to regions near the S2′ substrate binding pocket and were in close proximity to a bound inhibitor (Figure 1c). We initially chose MMP12 as a target because this protease has been expressed in recombinant form and very little is known about its primary biological function other than it is mainly secreted by macrophages26 and is involved in several pathologies such as emphysema27.
In order to assess the possible sites for introduction of the cysteine residue, we first had to express the mutants in recombinant form. We initially expressed the catalytic domains of three mutant forms of MMP12 (T184C, T243C and the double mutant T184C/T243C) in E.coli using a protein expression platform recently developed in our lab. This method makes use of the cysteine protease domain (CPD) of the bacterial MARTX toxin that autocatallytically processes itself upon addition of inistol hexakis phosphate (IP6)28. One of the main benefits of this approach is that it avoided refolding of the protein from inclusion bodies as had been published for the purification of active MMP1229. We found that while all three of the cysteine mutants were proteolytically active towards a fluorogenic substrate, both T243C and T184C/T243C mutant enzymes had a reduced activity (Figure 2a).
Figure 2.

Engineering MMP12 and the corresponding selective probe TND124. (a) Progress curves for processing of a quenched fluorescent peptide substrate (Mca-PLGL-Dpa-AR-NH2, where MCA (7-methoxycoumarin-4-yl)acetate) is the fluorophore and Dnp (dinitrophenyl) is the quencher) by WT, T184C, T243C and T184CT243C MMP12 catalytic domains. (b) Structure of the general MMP inhibitor GM6001, the designed probe TND124 and the control probe TND126. The electrophile (α-chloro acetamide) is shown in blue; the electrophile coordinating the zinc (hydroxamate) in TND124 or the control methyl amide in TND126 is shown in red and the fluorescent tag (Cy5) is shown in green. (c) WT or T184C MMP12 catalytic domains were added to rat liver lysates. The general MMP inhibitor GM6001 (10 μM) was added prior to labeling of the samples by addition of TND124 at the indicated concentrations. Samples were analyzed by SDS-PAGE followed by scanning of the gel for Cy5 fluorescence using a flatbed laser scanner. The location of the labeled MMP12 catalytic domain is indicated with an arrowhead. (d) Recombinant WT or T184C MMP12 catalytic domains were incubated with TND124 for 30 minutes followed by dialysis for 48 hours. The residual activity of both proteases was measured after 24 and 48 hours. Data are presented as average of activity from an experiment performed in triplicate (error bars ± SD; bottom panel). (e) Recombinant WT MMP12 and T184C MMP12 catalytic domains were incubated with DMSO (−) or 3 μM TND124 (+) and activity was detected by casein zymography. The white signals are produced by degradation of the substrate by active MMP12. Note the specific inhibition of this activity only in the T184C sample treated with TND124.
Therefore, we chose to focus our efforts on the T184C variant that had activity that was essentially identical to the wild type enzyme.
Because mutation of residues near the active site of the protease could result in a change in overall substrate binding, we next decided to assess the specificity of the T184C mutant protease using a peptide library method19. This method involves incubation of the protease with a diverse pool of peptides and then measuring the most optimal substrates by sequencing of the cleaved peptides (Figure S1, supporting information). These results indicated that the T184C mutant had nearly identical substrate selectivity as the WT enzyme with only very minor changes in P2′ position where we observed a modest preference for phenylalanine, leucine, methionine and tyrosine residues. Since the majority of substrate specificity of the MMP family is derived from the P1′ position as well as from binding of the substrate to the caboxy-terminal hemopexin domain, the T184C mutant is likely to have a substrate specificity that is virtually identical to the WT enzyme
Engineering of the probe
In order to design a suitable probe that contains an electrophile in close proximity to the engineered cysteine residue, we started with the broad spectrum MMP inhibitor GM6001 that has been used to make photocrosslinkable probes10 (Figure 2b). Using the structure of a hydroxamate- based inhibitor bound to the active site of MMP8 (Figure S2, supporting information), we designed a probe, TND124, that contains the hydroxamic acid group and primary P1′ recognition group of GM6001 to drive tight binding in the active site. Furthermore, we incorporated an α-chloro acetamide electrophile positioned at the P2′ position and a Cy5 fluorescent tag in the P3′ position (Figure 2b). Based on docking of this probe into the active site of MMP8, the electrophile was predicted to lie within 1.8 Å of the engineered cysteine (Figure S2, supporting information). The electrophile was selected due to its previously reported restricted reactivity profile towards cysteines30. We designed a synthetic route that comprised standard peptide chemistry to build the main scaffold and click chemistry to introduce the electrophile at the end of the synthesis (Supporting information).
To determine if the probe was able to covalently label the engineered MMP12, we performed mass spectrometry analysis of the WT and T184C MMP12 after treatment with the probe. Analysis of intact mass confirmed that TND124 formed a single covalent modification of the engineered cysteine (Figure S3, supporting information). Tryptic digestion of the labeled protein further confirmed that the peptide containing the engineered cysteine shifted by a mass consistent with the addition of a single TND124 molecule (an increase of 1193 amu), thus demonstrating specific modification of the engineered cysteine. We next needed to determine if this confirmed modification was specific for the target protein by performing a labeling study in complex protein extracts. Therefore, we added either mutant or wild-type MMP12 to crude tissue extracts, incubated with TND124 and monitored probe labeled proteins by SDS-PAGE followed by fluorescence scanning (Figure 2c). Under these conditions, TND124 showed highly selective labeling of the cysteine engineered catalytic domain with negligible background labeling when used at low probe concentrations (< 1 μM). Importantly, we did not observe labeling of the WT target and pretreatment of the samples with the general MMP inhibitor, GM6001, completely blocked labeling indicating that probe binding to the engineered protease was dependent on enzymatic activity as well as the cysteine residue.
Finally, we sought to determine if the covalent labeling of the engineered protease resulted in irreversible inhibition of enzyme activity. We therefore performed a kinetic inhibition study using a fluorogenic substrate (Figure S4a, supporting information). This data confirmed that TND124 inhibited the T184C mutant with increased potency relative to the WT enzyme (IC50=0.12 ± 0.01 μM for T184C versus IC50=0.75 ± 0.05 μM for WT). More importantly, the T184C protease, unlike the WT form, was unable to regain activity after removal of the probe by dialysis (Figure 2d). We also obtained similar results for natural protein substrate processing where only the activity of the engineered enzyme against collagen or gelatin was inhibited (Figure 4b, c supporting information). Finally irreversible inhibition of only the engineered enzyme was confirmed using casein zymography (Figure 2e). Overall these results demonstrate that while the probe is a weak, reversible inhibitor of the WT enzyme, it acts as a potent irreversible inhibitor of the engineered cysteine variant of the enzyme.
Engineering MT1-MMP
After demonstrating the feasibility of our approach using MMP-12, we wanted to see if this method was generalizable across the MMP family. We therefore chose to apply our strategy to one of the most intensely studied members of the MMP family, MT1-MMP (MMP-14). This protease is a trans-membrane protease that has been shown to be involved in the regulation of cell migration in processes such as tumor growth and metastasis31,32. Because of the similarity in the catalytic domain of all of the MMP family members, we were able to focus on residues corresponding to the locations of the mutants tested in MMP12. Therefore, we generated catalytically active wild-type MT1-MMP as well as the F198C and F260C mutants. We also tried a third mutation site (Q262). As observed for MMP12, both the F198C and F260C mutants were proteolytically active, with the F260C mutant being closer in activity level to the wild type enzyme (Figure 3a). Interestingly, the Q262C mutant showed a complete loss of protease activity highlighting the fact that some residues near the active site cannot be mutated without affecting activity. Detailed enzyme kinetic analyses revealed that F260C had a 2 fold higher Km than the wild type protease but a similar Vmax (Figure 3b), indicating that the F260C mutant can reach the same catalytic rate as the wild type enzyme. The specificity of F260C MT1-MMP was further tested using the peptide substrate library and, like MMP12, we found that it had nearly identical primary substrate selectivity compared to the WT enzyme with only very minor changes in P1′ position (Figure S5, supporting information).
Figure 3.

Engineering MT1-MMP. (a) Progress curves for processing of a quenched fluorescent peptide substrate (Mca-PLGL-Dpa-AR-NH2) by WT, F198C, F260C and Q262C MT1-MMP catalytic domains. (b) Kinetic analysis of the enzyme activity of WT and F260C MT1-MMP using the fluorogenic substrate in (a). Data are presented as the average of three independent experiments (error bars ±standard deviation [SD]). (c) Recombinant WT or F260C MT1-MMP catalytic domains were added to rat liver lysate followed by preincubation with DMSO, GM6001 (10 μM), EDTA (10 mM), or NEM (1 mM). Samples were then labeled with TND124 (1 μM) and analyzed by SDS-PAGE followed by in-gel fluorescent scanning for Cy5. The location of the MT1-MMP catalytic domain is indicated. (d) Recombinant WT or F260C MT1-MMP catalytic domains were incubated with TND124 over a range of concentrations and the activity of the enzyme against the MCA-PLGL-Dpa-AR-NH2 substrate was measured. Data are plotted as percent activity relative to enzyme incubated with DMSO (IC50 = 1 ±0.1 μM for the mutant protease versus an IC50 = 2.8 ±0.8 μM for the WT enzyme). Data are presented as average of three independent experiments (error bars ±SD).
Finally, we checked the selectivity of the probe for the engineered mutant by labeling the recombinant protein in tissue extracts (Figure 3c). As observed for the MMP12 mutants, TND124 showed robust labeling of the F260C mutant protein with no detectable labeling of the WT enzyme. In addition, pre-incubation of the samples with a cysteine alkylating agent (N-ethyl maleimide, NEM) or a chelating agent (EDTA) confirmed that the probe required the engineered cysteine as well as the zinc in the active site for effective labeling. We also generated a control probe (TND126) lacking the hydroxamate group but containing the α-chloro acetamide group (Figure S6a, supporting information). This probe did not label the F260C MT1-MMP (Figure S6b, supporting information) nor did it block labeling of the enzyme by TND124 (Figure 3c), confirming that the zinc coordination by the hydroxamate anchors the probe in the active site to facilitate irreversible modification of the enzyme. Consistent with this result, kinetic inhibition studies using the peptide substrate confirmed that TND124 inhibited the activity of F260C MT1-MMP (IC50 = 1 ± 0.1 μM for the mutant protease versus IC50 = 2.8 ±0.8 μM for the WT enzyme; Figure 3d).
Functional validation of the cysteine engineered MT1-MMP in cells
To determine if the newly identified cysteine mutant form of MT1-MMP could be synthesized, folded and correctly trafficked to the cell surface, we transfected the full-length WT and F260C mutant MT1-MMP in MCF-7 cells. These cells do not display proteolytic activity against collagen substrates and are devoid of tissue-invasive behavior 33. However, upon expression of MT1-MMP, the cells have been shown to hydrolyze subjacent substrates (e.g. native type I collagen as well as heat-denatured type I collagen/gelatin) and to display collagen-invasive activity 33. Therefore, we performed gelatin degradation and collagen invasion assays with MCF7 cells expressing either WT or F260C MT1-MMP. Using either construct, the MCF-7 cells degraded the underlying substrate while also showing comparable invasive activity, thereby demonstrating that the F260C mutant is expressed in an active form that is capable of degrading native substrates (Figure S7, supporting information and Figure 4a).
Figure 4.
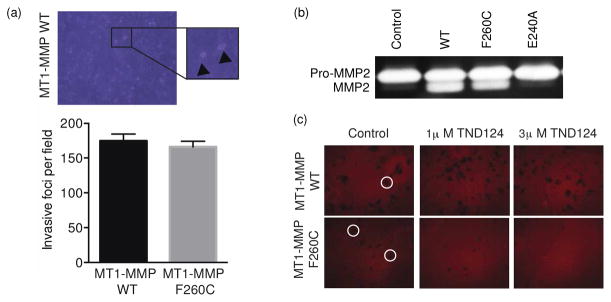
The engineered MT1-MMP is expressed and is functional in cells. (a) MCF7 cells were transfected with full length WT or F260C mutant MT1-MMP and then plated on a type-I collagen matrix. A typical image is shown to indicate the foci that are formed when the cells invade through the matrix (marked by an arrowhead). These foci were counted and the average number of invasive foci per field was plotted for WT and F260C expressing cells (error bars ±SD). (b) Zymography showing the activation of proMMP2 to MMP2 by media from cells expressing WT or F260C MT1-MMP. (c) Cells that were transfected with WT or F260C MT1-MMP were incubated with TND124 at the indicated concentrations, washed and plated onto a layer of fluorescently labeled gelatin. Typical sites of degradation (highlighted by circles) are observed as dark patches in the surface.
To further test the function of the F260C variant, we measured the ability of cells expressing the mutant to process the native MT1-MMP substrate, pro-MMP2 to its active form. To this end, we collected conditioned media from cells that secreted pro-MMP2, incubated it with HEK293T cells expressing either the WT or the F260C variant MT1-MMP and then measured the extent of pro- MMP2 processing to the mature MMP2 using gel zymography (Figure 4b). These results confirmed that both the WT and the F260C variant cleaved pro-MMP2 while the non-transfected MCF-7 cells nor MCF-7 cells expressing the proteolytically-inactive, catalytic site mutant, E240A MT1-MMP were unable to process the zymogen.
Finally, to determine if the engineered cysteine mutant MT1-MMP could be specifically inhibited, we treated cells expressing the different MT1-MMP variants with the probe and after various washouts, plated the cells atop a gelatin-coated matrix. Under these conditions, the probe only inhibited gelatin degradation in cells expressing the mutant enzyme, thus demonstrating that TND124 is selective for the engineered form of MT1-MMP (Figure 4c).
Specific imaging of cysteine engineered MT1-MMP activity in cells
To further validate our protein engineering strategy, we tested the ability of the probe to specifically label the active pools of the F260C variant MT1-MMP in an intact cell system. Biochemical analysis confirmed labeling of the mature, active form of MT1-MMP (i.e, the 55 kDa mature form of the protease generated after prodomain processing 1) only in lysates from cells expressing the F260C variant (Figure 5a). Labeling was also specifically competed away by pre-incubation with GM6001, suggesting that the labeling was specific for the active cysteine engineered variant.
Figure 5.

Specific labeling and imaging of the engineered MT1-MMP in cells. (a) Cell lysates of untransfected HEK293T cells (control) or cells expressing the full length WT or F260C MT1-MMP were pre-treated with GM6001 (+) or DMSO (−) followed by labeling with TND-124 (0.5 μM). Samples were analyzed by SDS-PAGE followed by in-gel fluorescence scanning for Cy5. The location of the mature 55 kDa MT1-MMP is indicated. (b) HEK293T cells were transfected with a full-length WT, F260C or catalytically inactive mutant E240A MT1-MMP (all tagged with HA tag) and incubated with TND124 (red, 1.5 μM) at 37°C or at 4°C to block re-uptake of the expressed MT1-MMP (bottom row). Cells were washed, fixed and stained with an anti-HA antibody (green) for MT1-MMP and DAPI nuclear stain (blue). Scale bars are 20 μm.
To assess the selectivity of labeling, we treated HEK293T cells that were transiently transfected with HA-tagged WT or F260C MT1-MMP with TND124 for an hour. Following extensive washing, the cells were fixed, stained with anti-HA tag antibody and visualized by microscopy. Importantly, the probe showed highly specific labeling of cells that were positive for expression of the F260C variant of MT1-MMP (Figure 5b) with no signal detected in untransfected cells or cells expressing WT or the proteolytically- inactive E240A mutant. In addition, preincubating the cells with GM6001 (Figure S8, supporting information) or using a negative control probe TND126 (Figure S9, supporting information) confirmed that the labeling was specific to active pools of the engineered MT1-MMP variant. The probe signal was mainly localized to intracellular, endosomal-like compartments, as well as to cellular protrusions. This could be the result of internalization of the MT1-MMP protein after surface labeling, similar to what has been reported when the WT protease is expressed in intact cells34. In support of this, when we lowered the incubation temperature to 4 °C to slow endocytosis, we found that MT1-MMP labeling was confined primarily to the cell surface (Figure 5b).
Functional expression and imaging of cysteine engineered MT1-MMP in zebrafish
As a final confirmation of the MT1-MMP mutant to retain normal functional activity in vivo, we expressed the mutant gene in zebrafish. The zebrafish genome encodes two maternally expressed isoforms homologous to human MT1-MMP: MT1-MMPa and MT1-MMPb25. Knockdown of MT1-MMPa/b using anti-sense morpholino oligonucleotides (MO)25 was shown to induce a craniofacial morphogenesis phenotype. Importantly, this phenotype could be rescued by co-injection of the MO with synthetic mt1- mmp mRNA. Therefore, we could determine if the F260C variant MT1-MMP was functional in the context of a whole organism by assessing its ability to rescue the knockdown phenotype. We found that when synthetic F260C mt1-mmpa mRNA was co-injected with a MO directed against the zebrafish MT1-MMP, we observed a dramatic reduction in the number of fish with the morphant phenotype (Figure 6a). Importantly, the F260C mutant gene rescued the phenotype to the same extent as the wt mt1-mmpa suggesting that the cysteine-engineered variant is functionally competent even when expressed in a whole organism (Figure 6b).
Figure 6.

Expression and imaging of MT1-MMP in zebrafish. (a) Wild-type, day 5 zebrafish embryos in which expression of the mt1-mmpa gene was knocked down with specific antisense morpholinos (mt1-mmpa MO) and day 5 embryos in which the mmp14a MO was injected along with mRNA expressing the Wild Type (mt1-mmpa MO + WT mRNA) or F260C mutant mt1-mmpa (mt1-mmpa MO + F260C mRNA) were stained with alcian blue to visualize cartilage elements. (b) Total number of fish observed with wild type, mild mutant and sever mutant phenotypes were determined and plotted for fish rescued with the WT or F260C mutant mt1-mmpa mRNA. (c) Zebrafish embryo at the single cell stage were either not injected (no probe), injected with the TND124 probe (2 nL of 50 μM DMSO stock) alone (no mRNA), or injected with probe and 300 pg of WT (WT MT1-MMPa) or F260C mutant (F260C MT1-MMPa) mmp14a mRNA. Embryos were fixed, washed and imaged for Cy5 fluorescence at either the 8 or 18 somite stage. Images are from representative embryos selected from a total of ~20 embryos that gave similar results. Scale bars are 100 μM for full images and 25 μM for inset images.
As a final test of the method, we wanted to determine if the probe could be used to label active pools of the engineered cysteine MT1-MMP in the whole zebrafish embryo. Therefore, we injected embryos at the single cell stage with the mRNA for either the WT or F260C variant of MT1-MMP and then fixed, washed and imaged the embryos at the 8-somite stage (Figure 6c). While there was some level of background fluorescence in both embryos that had not been injected with mRNA and in embryos injected with WT MT1-MMP mRNA, we were consistently able to observe specific accumulation and staining in embryos that had been injected with the F260C variant MT1-MMP. These data suggest that the probe is sufficiently selective and stable in vivo for imaging studies of MT1-MMP activity in whole fish.
DISCUSSION
We present here a general method that can be used to specifically target a protease by combining the use of protein engineering and small molecule design. One of the major limitations in collective efforts taken to date to understand protease function is the difficulty in developing small molecules that can be used to specifically image or inhibit a single protease target in the context of a whole cell or organism. In the case of the matrix metalloprotineases, there are a number of hurdles including the lack of a reactive nucleophilic residue in the active site and the high degree of similarity in the substrate and inhibitor binding sites within the catalytic domains of this multigene family. Therefore, methods that permit the use of a small molecule to inhibit or image a single MMP target with absolute specificity should prove transformative in terms of our ability to characterize the functions of these important proteases.
Here we show that it is possible to identify a site on MMPs that can be mutated to cysteine without dramatically altering enzyme activity. Importantly, these cysteine variants can be expressed in cells where they function with similar efficiency as wild type enzymes, supporting the contention that these mutations do not interfere with protein folding or cellular trafficking. In the case of MT1- MMP, the specificity of the mutant protease (as characterized by substrate library hydrolysis) was slightly modified in the P1′ position. However, we find that the mutant protease could process natural substrates and replace the WT protease in a complex organism like zebrafish. This result is likely explained by the fact that MT1-MMP is a multidomain protease and its substrate preference and binding is not only dependent on the interactions of the substrate within the shallow active site pocket, but also within exosites located in other domains such as the hemopexin domain35.
We have designed a first generation probe containing a reactive electrophile that specifically labels the cysteine mutant protease. Furthermore, the probe acts as a potent, irreversible inhibitor of the engineered protease, while only acting as a weak reversible inhibitor of the wild type enzyme. Because the probe directly labels only the active form of the cysteine engineered protease it can be used to both specifically block activity and also image localization of the active protease. Therefore, this new method will allow studies of the relationship between MT1-MMP cellular trafficking and the regulation of its proteolytic activity, something that has been studied only using indirect methods. This strategy could be particularly valuable for studies the role of MT1-MMP in signaling pathways (i.e. Wnt signaling pathway) or in remodeling of the extracellular matrix (ECM) during embryogenesis.
While the TND124 probe has proven to be valuable for imaging and inhibition studies of MMP-12 and MT1-MMP, we believe that it will be possible to further improve the utility of next generation probes by modulating overall reactivity of the electrophile as well as potency of the main probe scaffold for specific targets. In all of our lysate labeling studies, we observed a background signal at higher probe concentrations that is likely due to the reactivity of the α-chloro acetamide. Regardless, we are encouraged by the fact that the probe produces very minimal background signals when used to label intact cells that either do not express the engineered protease or that express the catalytically inactive protease. This discrepancy in background signal in lysates relative to intact cells is likely a result of reduced probe access to intracellular protein targets and the distinct conditions of the labeling reaction when used in cell culture compared to lysates. Therefore, labeling specificity should be assessed in both systems in order to more clearly assess probe selectivity.
CONCLUSION
We have developed a general strategy that allows specific targeting of a protease with small molecule inhibitors and imaging probes. This method makes use of protease engineering methods to generate the selective probe/protease interaction. While we have demonstrated that the engineered protease can be expressed in an active form in cells, we realize that to apply the method to relevant studies of MMP function, we will need to replace the native gene with the engineered mutant under the control of its native promoter. Fortunately, there have been a number of new “genome editing” techniques such as the TALEN effectors36 that have been recently described that enable direct modification of genomic DNA. These methods will allow introduction of the cysteine mutation into the native gene sequence to generate stable cell lines that only express the engineered protease. These cells, and eventually whole animals, expressing the mutant protease could then be used for complex studies of MMP function. We are currently working to establish such cells expressing both of the validated MMP mutants described here. We have also successfully applied this general method to the cysteine protease targets, caspase-1 and caspase-8 (see Xiao et al. manuscript) suggesting that this approach could be applied to virtually any other protease target of interest in the genome including aspartyl proteases, which have yet to be targeted with ABPs.
Supplementary Material
Acknowledgments
We thank the Bogyo lab for useful discussions and specifically Aimee Shen for help in cloning, PCR-based mutagenesis and protein expression and Andrew Guzzetta for help with the mass spectrometry analysis of full length MMPs. We thank Chris Overall and Charlotte Morrison for the TIMP2 deficient cells and the zymography protocols and Dimitri Rozanov for the full length MT1-MMP plasmid. This work was supported by National Institutes of Health grants R01- EB005011 and R21-EB012311 (to MB). M.M. was supported by AGAUR through the Beatriu de Pinos program. Y.D. was supported by a James Hudson Brown-Alexander Brown Coxe postdoctoral fellowship from Yale University School of Medicine.
Footnotes
Notes
The authors declare no competing financial interest
Supporting Information. Supplementary methods, Synthetic Schemes, Table S1 and Figures S1-S9. This material is available free of charge via Internet at http://pubs.acs.org.
References
- 1.Rowe RG, Weiss SJ. Annu Rev Cell Dev Biol. 2009;25:567–595. doi: 10.1146/annurev.cellbio.24.110707.175315. [DOI] [PubMed] [Google Scholar]
- 2.Kessenbrock K, Plaks V, Werb Z. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Decock J, Thirkettle S, Wagstaff L, Edwards DR. J Cell Mol Med. 2011;15:1254–1265. doi: 10.1111/j.1582-4934.2011.01302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hadler-Olsen E, Fadnes B, Sylte I, Uhlin-Hansen L, Winberg JO. Febs J. 2011;278:28–45. doi: 10.1111/j.1742-4658.2010.07920.x. [DOI] [PubMed] [Google Scholar]
- 5.Fingleton B. Semin Cell Dev Biol. 2008;19:61–68. doi: 10.1016/j.semcdb.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lia NG, Shib ZH, Tang YP, Duan JA. Curr Med Chem. 2009;16:3805–3827. doi: 10.2174/092986709789178037. [DOI] [PubMed] [Google Scholar]
- 7.Knapinska A, Fields GB. Chembiochem. 2012;13:2002–2020. doi: 10.1002/cbic.201200298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paulick MG, Bogyo M. Curr Opin Genet Dev. 2008;18:97–106. doi: 10.1016/j.gde.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coleman JE. Curr Opin Chem Biol. 1998;2:222–234. doi: 10.1016/s1367-5931(98)80064-1. [DOI] [PubMed] [Google Scholar]
- 10.Saghatelian A, Jessani N, Joseph A, Humphrey M, Cravatt BF. Proc Natl Acad Sci USA. 2004;101:10000–10005. doi: 10.1073/pnas.0402784101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan EWS, Chattopadhaya S, Panicker RC, Huang X, Yao SQ. J Am Chem Soc. 2004;126:14435–14446. doi: 10.1021/ja047044i. [DOI] [PubMed] [Google Scholar]
- 12.Wang J, Uttamchandani M, Li J, Hu M, Yao S. Chemical Communications. 2006.
- 13.Sieber SA, Niessen S, Hoover HS, Cravatt BF. Nat Chem Biol. 2006;2:274–281. doi: 10.1038/nchembio781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erlanson DA, Braisted AC, Raphael DR, Randal M, Stroud RM, Gordon EM, Wells JA. Proc Natl Acad Sci USA. 2000;97:9367–9372. doi: 10.1073/pnas.97.17.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Z, Jing C, Gallagher SS, Sheetz MP, Cornish VW. J Am Chem Soc. 2012;134:13692–13699. doi: 10.1021/ja303374p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blair JA, Rauh D, Kung C, Yun CH, Fan QW, Rode H, Zhang C, Eck MJ, Weiss WA, Shokat KM. Nat Chem Biol. 2007;3:229–238. doi: 10.1038/nchembio866. [DOI] [PubMed] [Google Scholar]
- 17.Hagel M, Niu D, St Martin T, Sheets MP, Qiao L, Bernard H, Karp RM, Zhu Z, Labenski MT, Chaturvedi P, Nacht M, Westlin WF, Petter RC, Singh J. Nat Chem Biol. 2011;7:22–24. doi: 10.1038/nchembio.492. [DOI] [PubMed] [Google Scholar]
- 18.Chun TH, Sabeh F, Ota I, Murphy H, McDonagh KT, Holmbeck K, Birkedal-Hansen H, Allen ED, Weiss SJ. J Cell Biol. 2004;167:757–767. doi: 10.1083/jcb.200405001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turk BE, Huang LL, Piro ET, Cantley LC. Nat Biotechnol. 2001;19:661–667. doi: 10.1038/90273. [DOI] [PubMed] [Google Scholar]
- 20.Gibbs DF, Warner RL, Weiss SJ, Johnson KJ, Varani J. Am J Respir Cell Mol Biol. 1999;20:1136–1144. doi: 10.1165/ajrcmb.20.6.3483. [DOI] [PubMed] [Google Scholar]
- 21.Sabeh F, Ota I, Holmbeck K, Birkedal-Hansen H, Soloway P, Balbin M, Lopez-Otin C, Shapiro S, Inada M, Krane S, Allen E, Chung D, Weiss SJ. J Cell Biol. 2004;167:769–781. doi: 10.1083/jcb.200408028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morrison CJ, Butler GS, Bigg HF, Roberts CR, Soloway PD, Overall CM. J Biol Chem. 2001;276:47402– 47410. doi: 10.1074/jbc.M108643200. [DOI] [PubMed] [Google Scholar]
- 23.Tam EM, Morrison CJ, Wu YI, Stack MS, Overall CM. Proc Natl Acad Sci USA. 2004;101:6917–6922. doi: 10.1073/pnas.0305862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 25.Coyle RC, Latimer A, Jessen JR. Exp Cell Res. 2008;314:2150–2162. doi: 10.1016/j.yexcr.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 26.Shapiro SD, Kobayashi DK, Ley TJ. J Biol Chem. 1993;268:23824–23829. [PubMed] [Google Scholar]
- 27.Churg A, Wang RD, Tai H, Wang X, Xie C, Dai J, Shapiro SD, Wright JL. Am J Respir Crit Care Med. 2003;167:1083–1089. doi: 10.1164/rccm.200212-1396OC. [DOI] [PubMed] [Google Scholar]
- 28.Shen A, Lupardus PJ, Morell M, Ponder EL, Sadaghiani AM, Garcia KC, Bogyo M. PLoS ONE. 2009;4:e8119. doi: 10.1371/journal.pone.0008119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parkar AA, Stow MD, Smith K, Panicker AK, Guilloteau JP, Jupp R, Crowe SJ. Protein Expr Purif. 2000;20:152–161. doi: 10.1006/prep.2000.1280. [DOI] [PubMed] [Google Scholar]
- 30.Weerapana E, Simon GM, Cravatt BF. Nat Chem Biol. 2008;4:405–407. doi: 10.1038/nchembio.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strongin AY. Biochim Biophys Acta. 2010;1803:133– 141. doi: 10.1016/j.bbamcr.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Radisky ES, Radisky DC. J Mammary Gland Biol Neoplasia. 2010;15:201–212. doi: 10.1007/s10911-010-9177-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ota I, Li XY, Hu Y, Weiss SJ. Proc Natl Acad Sci U S A. 2009;106:20318–20323. doi: 10.1073/pnas.0910962106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Remacle A, Murphy G, Roghi C. J Cell Sci. 2003;116:3905–3916. doi: 10.1242/jcs.00710. [DOI] [PubMed] [Google Scholar]
- 35.Overall CM. Mol Biotechnol. 2002;22:51–86. doi: 10.1385/MB:22:1:051. [DOI] [PubMed] [Google Scholar]
- 36.Hockemeyer D, Wang H, Kiani S, Lai CS, Gao Q, Cassady JP, Cost GJ, Zhang L, Santiago Y, Miller JC, Zeitler B, Cherone JM, Meng X, Hinkley SJ, Rebar EJ, Gregory PD, Urnov FD, Jaenisch R. Nat Biotechnol. 2011;29:731–734. doi: 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.