

Abstract
We report the crystal structure of the first prokaryotic aspartic proteinase-like domain identified in the genome of Mycobacterium tuberculosis. A search in the genomes of Mycobacterium species showed that the C-terminal domains of some of the PE family proteins contain two classic DT/SG motifs of aspartic proteinases with a low overall sequence similarity to HIV proteinase. The three-dimensional structure of one of them, Rv0977 (PE_PGRS16) of M. tuberculosis revealed the characteristic pepsin-fold and catalytic site architecture. However, the active site was completely blocked by the N-terminal His-tag. Surprisingly, the enzyme was found to be inactive even after the removal of the N-terminal His-tag. A comparison of the structure with pepsins showed significant differences in the critical substrate binding residues and in the flap tyrosine conformation that could contribute to the lack of proteolytic activity of Rv0977.
Keywords: Crystal structure, Aspartic proteinase domain, Mycobacterium tuberculosis
Graphical Abstract
Highlights
-
•
Aspartic proteinase-like domains have been identified in a prokaryote, Mycobacterium.
-
•
Crystal structure of an aspartic proteinase-like domain in Rv0977 of M. tuberculosis.
-
•
The aspartic proteinase-like domain of Rv0977 has the pepsin fold but no activity.
1. Introduction
Aspartic proteinases from several eukaryotes and retroviruses have been extensively studied. They perform a variety of functions and are implicated in a number of diseases. Aspartic proteinases utilize two aspartate residues to cleave the peptide bond with the help of a water molecule. The active site is formed between two domains, each contributing one catalytic aspartate. In the case of HIV proteinase (HIV PR), a homodimer is formed by two polypeptide chains whereas in eukaryotic aspartic proteases such as pepsin, a single polypeptide chain forms the two domains. It has been postulated that the eukaryotic aspartic proteinase has evolved by gene duplication and fusion of an ancestral gene into a single gene that encodes a single polypeptide chain [1]. Recently there have been a few reports on the presence of aspartic proteinases in bacteria [2,3]. However, structural characterization of these enzymes has not been reported. It is presumed that the structure of the prokaryotic aspartic proteinase might be useful in understanding the evolution of the aspartic proteinases from retroviral protease to eukaryotic proteinases.
A search in the genome of Mycobacterium tuberculosis with HIV PR sequence as a query, picked a sequence in the C-terminal domain of a PE_PGRS protein, Rv0977, with low similarity. PE_PGRS (polymorphic GC-rich repetitive sequence) proteins form the largest subfamily of the PE family. The PE family is a large family of proteins identified for the first time in the genome of M. tuberculosis accounting for ∼6% of potentially coding genes of the genome [4]. The name PE is derived from the motif Pro-Glu (PE) found near the N-terminus in most of the PE proteins [4]. These are found exclusively in pathogenic mycobacterial species [5] thus presenting tremendous possibilities to be probed as drug targets. All members of the PE protein family have a highly conserved N-terminal domain of 110 amino acid residues, followed by a C-terminal segment that varies in size. In addition to PE domains, PE_PGRS proteins have multiple tandem repeats of GGAGGX (where X = any amino acid) motif in the C-terminal segment [6]. In the case of Rv0977, this repeat is followed by an additional sequence at the C-terminus, which consists of two signature motifs of pepsins, DTG and DSG.
Rv0977, a member of the PE_PGRS family of proteins, was identified as one of the surface antigens of M. tuberculosis, and was found to be expressed in high levels during infection [7]. Though not many PE or PE_PGRS proteins are characterized, a limited number of reports are available on the levels of expression of Rv0977, host response and its involvement in antigenic variation [5,8]. Rv0977 was shown to have protective immune response which was attributed to the unique sequence present in the C-terminal domain but the exact nature of this domain was unknown. We have identified this domain to be a putative aspartic proteinase and named it Mtb-AP, and determined its structure.
2. Materials and methods
2.1. Cloning and protein purification of Mtb-AP
The region of M. tuberculosis genomic DNA (58536–59354) encoding the predicted aspartic proteinase domain was amplified by PCR using specific forward and reverse primers and was cloned into pET-28a (+) vector. The gene sequence, confirmed by sequencing was 819 base pairs. The clone was transformed into the BL21 (DE3) strain of Escherichia coli. The transformed cells were grown in Luria Bertani (LB) broth at 37 °C, and protein expression was induced with isopropyl-β-d-1-thiogalactopyranoside (IPTG) to a concentration of 100 μM and grown at 12 °C. Cells were harvested following 8–10 h of induction. The pellet was resuspended in lysis buffer (20 mM Tris–HCl (pH 7.4), 500 mM NaCl and 5 mM imidazole). Cells were lysed by sonication and the lysate was centrifuged at 14,000g at 4 °C for 30 min. The supernatant was loaded on to a Ni-NTA-agarose column and the column was washed with buffer including the same components as that of lysis buffer and subsequently with 20 mM Tris–HCl (pH 7.4), 500 mM NaCl, 20 mM imidazole and eluted with elution buffer containing 20 mM Tris–HCl (pH 7.4), 500 mM NaCl, 100 mM imidazole. The eluted protein was dialysed against a buffer containing 20 mM Tris–HCl (pH 7.4) and 300 mM NaCl.
Se-Met incorporated protein was prepared by metabolic inhibition method. Cells were grown at 37 °C in M9 medium supplemented with glucose and Kao and Michayluk vitamin Solution (Sigma). A mixture of amino acids for inhibiting methionine biosynthetic pathway was added to the culture when OD600 reached 0.6. l-Selenomethionine (Sigma) 25 mg/L and 100 μM IPTG were added an hour later and cells were grown for further 10 h at 12 °C. Purification of Se-Met incorporated enzyme was carried out following the protocol used for the native enzyme. The selenomethionine incorporation was confirmed by mass determination using Electro Spray Ion-Mass Spectrometry (ESI-MS).
2.2. Cloning and protein purification of Mtb-AP without tag
To obtain a construct devoid of the His-tag, the reverse primer containing a stop codon was used along with the previously used forward primer to amplify the gene. The amplified product was cloned into pET-22b (+) vector. Due to the presence of stop codon in the reverse primer the expressed protein did not have the C-terminal His-tag present in the pET-22b (+) vector. The clone devoid of His-tag was confirmed by sequencing. It was transformed into the BL21 (DE3) strain of E. coli cells. The cells were grown and induced similar to His-tagged protein. The pelleted cells were resuspended in lysis buffer containing 25 mM Bis–Tris buffer (pH 6.0). Cells were lysed by sonication and the lysate was centrifuged at 14,000g at 4 °C for 30 min. The supernatant was loaded onto a Q-Sepharose anion exchange column and washed with buffer containing 25 mM Bis–Tris buffer (pH 6.0). The protein was eluted with NaCl gradient from 0 to 1 M by using a gradient mixer. The eluted fractions were loaded on to a SDS–PAGE. The fractions containing Mtb-AP were pooled, concentrated and loaded onto a Sephacryl S-200 HR gel filtration column. The pure fractions obtained from gel-filtration were used for assay and crystallization.
2.3. Crystallization and data collection of Mtb-AP His-tagged construct
Crystals of Mtb-AP were obtained in microbatch method by mixing 2 μl of 6 mg/ml of protein with 2 μl of solution consisting of 0.1 M MES buffer pH 6.5 and 12% PEG 20K (Hampton Research Crystal Screen2, condition # 26). This condition was optimized to get diffraction quality crystals. The native crystals diffracted to a resolution of 2.7 Å at home source. Selenomethionine incorporated protein also crystallized in the native condition. SAD data were collected for these crystals on BM-14 situated at the European Synchrotron Radiation Facility (ESRF), Grenoble, France.
2.4. Structure solution and refinement
The diffraction data were processed using iMosflm [9] and the structure was solved using the SAS protocol of Auto-Rickshaw [10]. The input diffraction data were prepared and converted for use in Auto-Rickshaw using programs of the CCP4 suite [11]. FA values were calculated using the program SHELXC [12]. All of the six heavy atoms were found using the program SHELXD [13]. About 96% of the model was built using the program ARP/wARP [14]. This model was further refined using REFMAC5 [15] from the CCP4 suite, followed by iterative cycles of manual rebuilding using Coot [16] and refinement. Solvent molecules were identified by the automatic water-picking algorithm of Coot. The positions of these automatically picked solvent molecules were manually checked, and a few more were identified on the basis of electron density contoured at 1.0σ in the 2Fo − Fc map and 3.0σ in the Fo − Fc map. Data collection and refinement statistics are shown in Table 1. The geometry of the final model was checked with MolProbity [17]. There were 96% of residues in the favored region of the Ramachandran plot, with the remaining 4% being in the allowed region. All structural figures were prepared using PyMOL (Schrödinger). Structural superpositions were carried out using ssm algorithm of the program Coot [16].
Table 1.
Data collection and refinement statistics.
| Data collection | SAD data set |
|---|---|
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 61.11, 66.16, 69.62 |
| Wavelength (Å) | 0.978 |
| Resolution (Å) | 37.73–1.98 |
| Rsym | 0.132 (0.415)a |
| I/σI | 16.3 (6.9) |
| Completeness (%) | 100 (100) |
| Redundancy | 14.6 (14.3) |
| Refinement | |
| Resolution (Å) | 37.73–1.98 |
| No. reflections in working set | 19,397 |
| No. reflections in test set | 1044 |
| Rwork/Rfree | 0.16/0.21 |
| No. atoms | |
| Protein | 1970 |
| Zn2+ | 1 |
| Ethylene glycol | 124 |
| Water | 209 |
| B-factors (Å2) | |
| Protein | 16.30 |
| Zn2+ | 7.05 |
| Ethylene glycol | 38.47 |
| Water | 28.39 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.017 |
| Bond angles (˚) | 1.62 |
Values in parentheses are for the highest resolution shell.
2.5. Activity assays
The activity of Mtb-AP was tested using denatured haemoglobin, bovine serum albumin (BSA), casein fluorescein isothiocyanate (FITC-Casein), oxidized insulin B-chain and an HIV PR substrate in the pH range 3–7, by adjusting the pH of the substrate solution using 0.1 M citrate buffer and 0.1 M Tris buffer. For denatured haemoglobin, the assay was carried out at pH 3 and 4 only as it precipitates beyond pH 4. The protocol with each of these substrates is briefly explained below. All the assays were performed at 37 °C.
2.5.1. Haemoglobin
40 μl of Mtb-AP (100 μg ml−1) was added to 400 μl of 0.5% denatured haemoglobin and incubated for various time periods (2, 4, 6, 12 and 16 h). The reaction was stopped at respective time intervals by adding 800 μl of 5% trichloroacetic acid (TCA). The precipitate obtained after addition of TCA was removed by centrifugation at 13,000 rpm for 10 min. Pepsin at a concentration of 10 μg ml−1 was used as a positive control. For each reaction, blank was set up in which TCA was added prior to enzyme addition. The absorbance of TCA soluble peptides in the supernatant in each test reaction was recorded at 280 nm against the respective blanks [18,19].
2.5.2. BSA
40 μl of 100 μg ml−1 Mtb-AP was added to 400 μl of 0.5% BSA solution and incubated for different time periods (2, 4 and 8 h) [20]. The rest of the steps were essentially similar to that of the Haemoglobin assay.
2.5.3. FITC-Casein
10 μl of 100 μg ml−1 Mtb-AP was added to 40 μl of 0.25% FITC-Casein (Sigma) and incubated for 60 min. Then, 150 μl of 10% TCA was added to the mixture and further incubated for 60 min. This mixture was then centrifuged for 10 min at 13,000 rpm. 10 μl of the supernatant was added to 10 ml of 500 mM Tris, pH 8.5. The fluorescence emission was recorded at 525 nm by excitation at 490 nm. Blank for each sample was setup without addition of the enzyme. Trypsin (10 μg ml−1) was used as a positive control for which the reaction was carried out at pH 8.
2.5.4. Oxidized insulin B chain
This assay was carried out following the protocol described earlier [21]. 10 μl of 100 μg ml−1 of Mtb-AP was added to 40 μl of 100 μg ml−1 oxidized insulin B chain and incubated for various time periods (2, 4 and 6 h). The assay was terminated by immersing the tubes in an ice bath followed by addition of 700 μl of borate buffer, pH 9.2 and 100 μl of fluorescamine (Sigma) dissolved in acetone (0.3 mg ml−1). Fluorescence emission was measured at 475 nm by excitation at 395 nm. Blank was setup for all the reactions without addition of the enzyme. As a positive control, 0.5 μg ml−1 of pepsin was used. Aliquots were taken at different time points post incubation and analysed for substrate cleavage by mass spectrometry.
2.5.5. HIV-1 PR substrate
The substrate of HIV-1 PR (His-Lys-Ala-Arg-Val-Leu*NPhe-Glu-Ala-Nle-Ser) was a kind gift from Dr M.V. Hosur, BARC, Mumbai, India. The assay was carried out following the protocol described earlier [22]. 1 μg of Mtb-AP was added to 200 μM of HIV PR substrate solution at different pH and incubated. UV absorption spectra in the wavelength range 250–350 nm were recorded for the reaction mixture after 2 h of incubation. Decrease in absorbance at 310 nm indicates the hydrolysis of the scissile peptide bond. As a control, absorbance at 310 nm was measured for the substrate alone.
2.6. Pepstatin binding assay
Mtb-AP without His-tag in 20 mM Tris pH 7.4, 300 mM NaCl was adjusted to different pH (3, 4, 5, 6 and 7) using 0.05 M citrate buffer and 0.05 M Tris buffer. The enzyme at a concentration of 0.5 mg ml−1 in different pH buffers was applied to columns containing pepstatin agarose equilibrated with the corresponding pH buffers. The column was washed with 10 bed volumes of buffer of respective pH and the protein was eluted with 0.1 M Tris–HCl buffer pH 8.6, containing 1 M NaCl. The eluted fractions and the agarose beads were loaded on to a SDS PAGE and the protein was visualized by silver staining [19]. Pepsin was used as a positive control. The His-tagged Mtb-AP served as a negative control.
3. Results and discussion
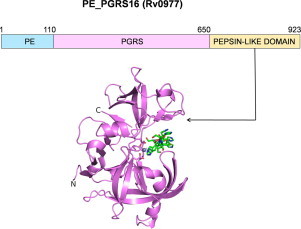
A BLAST search carried out using pepsin sequences as query did not give any positive hits in the Mycobacterium genomes. However, a search with the sequence of HIV PR indicated a weak similarity to the protein Rv0977 of M. tuberculosis with only 15 residues being identical between the two sequences (Fig. 1A). In addition to the PE and PGRS domains, Rv0977 has a C-terminal domain with two aspartic proteinase motifs, one DTG and one DSG, one of them being picked by the sequence search. The length of the polypeptide chain of this domain with 273 residues is intermediate to that of pepsins (∼325 residues) and HIV PR (2×99 residues). The two aspartates are separated by 154 residues. A search with the sequence of this domain revealed the presence of similar domains in two other proteins of M. tuberculosis, Rv1983 (PE_PGRS35) and Rv2519 (PE26) (Fig. 1B), and also in other species of Mycobacterium (Table S1). All the proteins that were identified to contain this domain belong to the PE family. The occurrence of this domain consistently in a single family of proteins in various mycobacterial species encouraged us to take up further analysis of this putative aspartic proteinase domain.
Fig. 1.
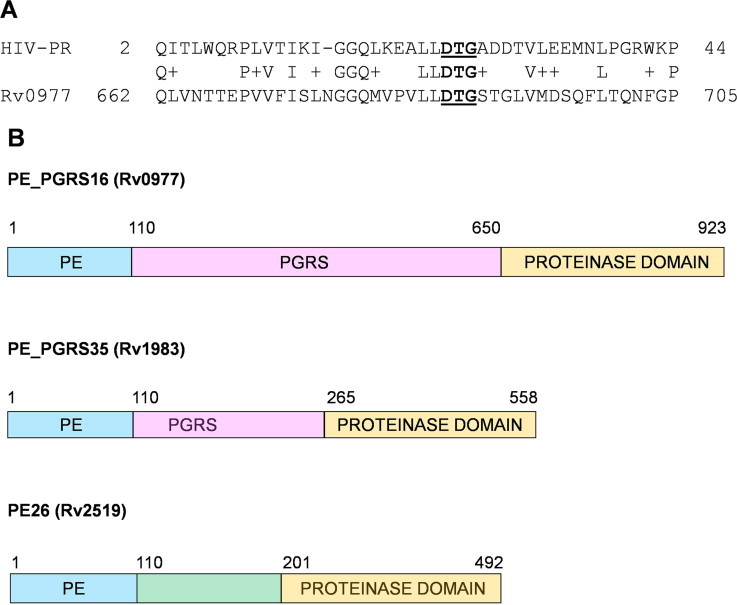
Identification of aspartic proteinase domain in M. tuberculosis. (A) BLAST search results showing the sequence alignment of HIV proteinase with M. tuberculosis PE-PGRS protein, Rv0977. The signature motif DTG is shown in bold and underlined. (B) Domain organization of the three PE-family proteins of M. tuberculosis having the aspartic proteinase domain.
The genes corresponding to the aspartic proteinase domains of the three M. tuberculosis proteins have been cloned and expressed. Only the domain from Rv0977 could be obtained in soluble form. The purified N-terminal His-tagged protein, however, showed no activity with many aspartic proteinase substrates such as haemoglobin, BSA, insulin B chain, FITC-Casein or with the HIV PR substrate [22], His-Lys-Ala-Arg-Val-Leu*NPhe-Glu-Ala-Nle-Ser. Structural studies were simultaneously pursued by setting up crystallization trials. Diffraction data to a resolution of 2.7 Å were collected from a crystal grown in one of the Hampton screen conditions (0.1 M MES buffer pH 6.5 and 12% PEG 20K). Molecular replacement attempts using the structures of pepsins or retroviral proteinases as search models were unsuccessful. Since there are six methionines in the sequence, a Se-Met derivative was prepared and data were collected to 1.98 Å resolution (Table 1) at BM14 beamline of ESRF, Grenoble.
The structure of Mtb-AP solved by SAD phasing revealed the fold and the catalytic site architecture typical of pepsins (Fig. 2). There are 23 extra residues from the vector at the N-terminus of the cloned fragment of 273 residues of the protein domain. Electron density for the region −21 to −10 which includes the six histidine residues from the tag and from residues 3 to 271, except Ala266 was seen clearly. No density was observed for the side chains of Asn83, Leu85, Asn122, Pro125, Thr265, Ile267 and Thr271. Unexpectedly, the hexahistidine tag was found to occupy the entire substrate binding pocket with two of the histidines and the catalytic aspartates coordinating a Zn2+ ion (Fig. 3), located at the position of the catalytic water molecule found in the crystal structures of other aspartic proteinases. The active site of the protein was completely blocked as evident from the crystal structure thus explaining the lack of activity of the protein. The presence of Zn2+ ion in the active site was unexpected, as the crystallization condition does not contain the ion. Neither the culturing medium nor the purification buffers had Zn2+ ion, hence it is likely that the Zn2+ ion was taken up from the cell. The presence of zinc ion was confirmed by atomic absorption spectroscopy. Similar coordination between the catalytic residues and the Zn2+ ion is observed in histo-aspartic proteinase of Plasmodium falciparum [23]. In this structure, apart from the catalytic His32 and Asp215, a water molecule and the side chain of Glu278 from a neighbouring molecule interact with the Zn2+ ion.
Fig. 2.
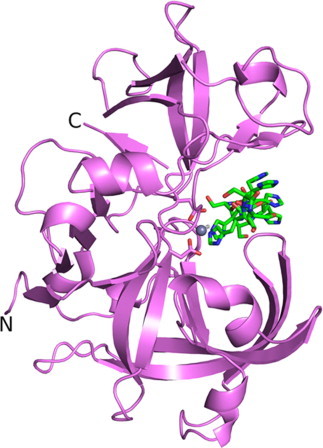
Crystal structure of Mtb-AP shown in cartoon representation. The catalytic aspartates, the bound His-tag residues (green) are shown in stick representation and the bound zinc ion as a grey sphere. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 3.
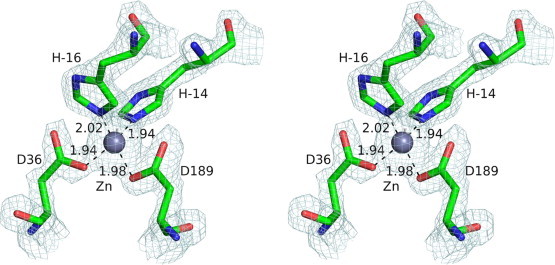
Stereo image of the coordination of the zinc atom with His-tag residues and catalytic aspartates. 2Fo − Fc electron density map contoured at 2.0σ level is shown.
3.1. The overall fold
The fold of the domain is closer to pepsins than to retroviral proteases with one exception in topology (Fig. 4). A six stranded β-sheet is formed between the N- and the C-terminal of Mtb-AP by three strands from each domain. The predominantly β-sheet structure including the unique ψ-loops which harbour the catalytic aspartates and the hydrophobic-hydrophobic-glycine (HHG) motif [24] is retained in the present structure except that the HHG motif of the C-terminal domain is replaced by Asn-Thr-Gly residues. There are three 310-helices and three α-helices in Mtb-AP. One of the α-helices, His250-Gln256 of the C-terminal domain, connected to the central β-strand of the interdomain β-sheet (Fig. 4) is conserved in pepsins as well as in HIV PR. The other two α-helices are in similar positions to those in pepsins. The location of two of the three 310-helices, Ser129-Met134 and Pro135-Asn139, corresponds to a single α-helix in pepsins, the break being caused by the proline residue at 135 (Fig. 5). The third 310-helix aligns with the long loop in pepsins which has an α-helix and a 310-helix (Fig. 5). The loop lengths of Mtb-AP seem to lie between those of the retroviral proteins (Fig. 6A) and pepsins (Fig. 6B).
Fig. 4.
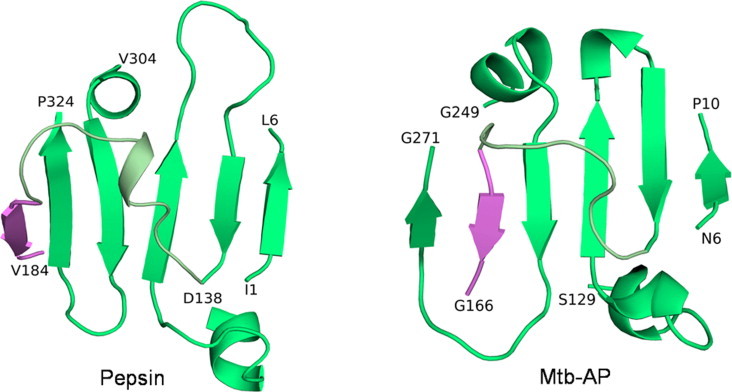
The interdomain β-sheet topology of pepsin and Mtb-AP. The first β-strand of the C-terminal domain is shown in pink.
Fig. 5.
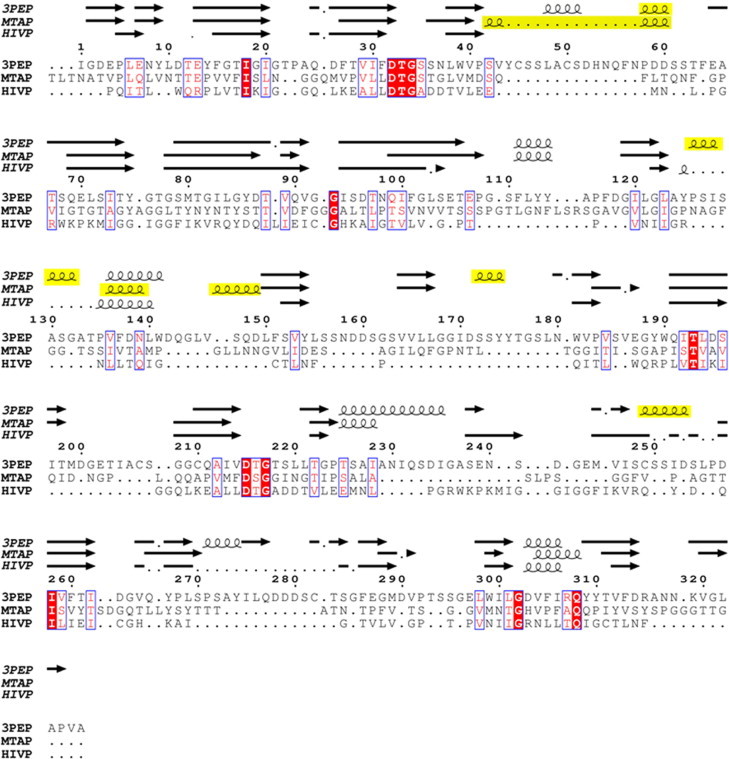
Structure-based sequence alignment of pepsin, Mtb-AP and HIV proteinase. 310 helices are highlighted in yellow. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 6.
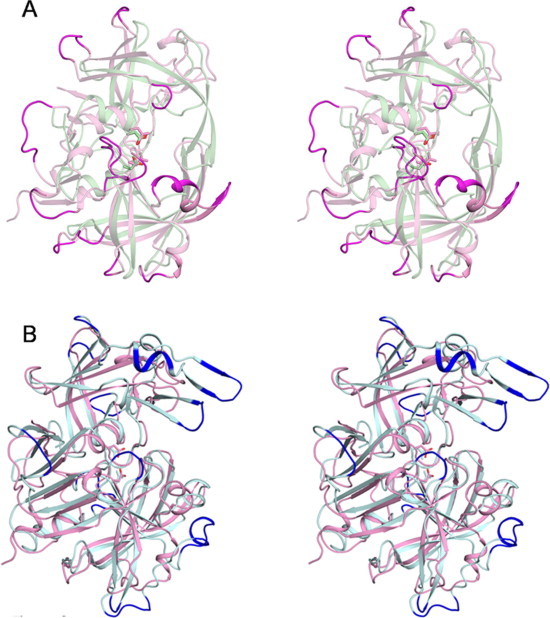
Stereo view of the structural superposition of (A) Mtb-AP (pink) on HIV proteinase dimer (green) and (B) Mtb-AP (pink) on Pepsin (cyan). Longer loops in each superposition are highlighted. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.2. The interdomain β-sheet topology
Six antiparallel β-strands form a large β-sheet between the N and the C-terminal domains in the case of eukaryotic aspartic proteinases. The C-terminal domain of Mtb-AP has a different topology with two β-strands of the sheet arranged parallel to each other (Fig. 4), a consequence of the loop between the N- and the C-terminal domains being shorter. Thus, this structure represents a new subclass of the aspartic proteinase family.
3.3. The histidine-tag
The N-terminal residues −21 to −16 form a short helix which includes three histidine residues (−18 to −16) of the His-tag. The other three histidine residues from −15 to −13 are in an extended conformation. Two of the histidine residues (−16 and −14) coordinate with the Zn2+ ion present at the active site (Fig. 3) In addition, the His-tag peptide is stabilized by a few more interactions with the protein atoms and many interactions with solvent molecules (Fig. S1). Extended peptides of similar size were found to occupy the binding pockets in propepsin [25] and a high pH form of cathepsin D [26] in which a lysine residue directly interacts with the catalytic aspartates.
Subsequently, the protein was expressed without the His-tag and purified using ion exchange and gel filtration. The purified protein appears to be well-folded with a predominantly β-sheet structure similar to the His-tagged construct as observed in the CD spectrum (Fig. S2) and free of Zn2+ ion as confirmed by atomic absorption spectroscopy. However, no activity was detected with the substrates listed above. Also, the protein did not bind to a pepstatin-agarose column indicating that it is pepstatin-insensitive.
3.4. The HHG motif
The residues Val-Leu-Gly present in the N-terminal HHG motif of Mtb-AP are similar in nature to those in the eukaryotic aspartic proteinases. The HHG motif of the C-terminal domain is replaced by Asn-Thr-Gly residues in Mtb-AP. In pepsins, the amide nitrogens of the catalytic aspartates are hydrogen bonded to the backbone carbonyl oxygen of the HHG motif residue (Fig. 7A) but in Mtb-AP the amide nitrogen of catalytic Asp189 is hydrogen bonded to the Oγ atom of Thr248 from the HHG motif (Fig. 7B). This threonine is conserved in all predicted aspartic proteinases of Mycobacterium species (Fig. S3). In pepsins, the side chain of a conserved Thr218 (pepsin numbering) makes a hydrogen bond with the side chain of catalytic Asp215 (pepsin numbering) (Fig. 7A). The proposed function of the hydrogen bond is to make the aspartate negatively charged at low pH [27]. In Mtb-AP, this threonine residue is replaced by a glycine (Gly192). The residue Asn247 of Mtb-AP which replaces the first hydrophobic residue of the conserved C-terminal HHG motif found in all other aspartic proteases, plays the role of Thr218 of pepsins in making a hydrogen bond with the catalytic Asp189 (Fig. 7A). This asparagine is also conserved in all mycobacterial aspartic proteinases (Fig. S3).
Fig. 7.
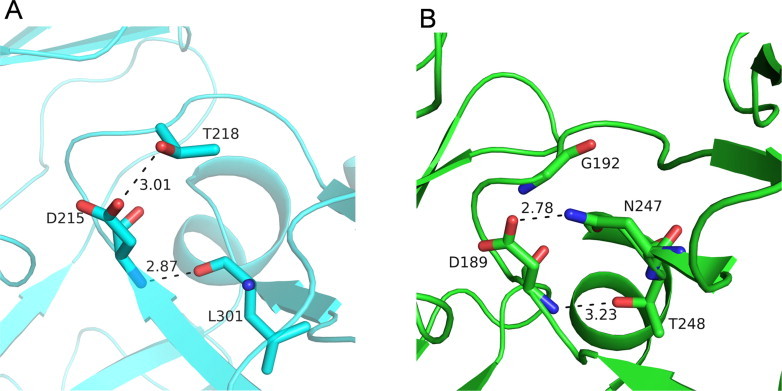
Interaction between the catalytic aspartate with the HHG motif residues in the C-terminal domain (A) Pepsin and (B) Mtb-AP. Residues involved in hydrogen bonding interactions are shown in stick representation. Hydrogen bonds are shown as dashed lines.
3.5. The substrate binding site and activity
A comparison of the structure of Mtb-AP with its structural equivalents human pepsin (PDB code: 1PSO) and rhizopuspepsin (PDB code: 3APR) in complex with inhibitors showed that the subsites of Mtb-AP appear to be similar to those of reported pepsins [28,29] with some small but significant differences (Fig. 8 and Table S2). S1: The flap residue tyrosine (Tyr64) along with glycine (Gly66) is well conserved in the family of eukaryotic aspartic proteinases. Mtb-AP has an additional alanine (Ala65) between Tyr64 and Gly66 (Fig. S4). A phenylalanine insertion corresponding to Ala65 of Mtb-AP was observed in the case of cockroach allergen Bla g 2 which was found to be inactive [30]. When pepstatin was docked in the binding pocket of Mtb-AP, the Ala65 side chain and the flap Tyr64 were found to make short contacts with it. The side chain of the conserved flap tyrosine has a different conformation (χ1 = 173°) when compared to that (χ1 = −54° in porcine pepsin) of eukaryotic aspartic proteinases (Fig. S5A). This conformation is stabilized by the hydrogen bond between tyrosine hydroxyl group and the side chain of the catalytic Asp36. Similar conformation of tyrosine was also observed in the crystal structures of chymosin [31] and saccharopepsin [32] (Fig. S5B) and was postulated to be responsible for keeping the enzymes in a self-inhibited state. The alanine insertion along with the altered conformer of tyrosine makes the S1 pocket narrower and hence, it is difficult for the substrate to enter the active site. S2: The Oγ atom of Thr77 of porcine pepsin makes a hydrogen bond with the amide nitrogen of P2 residue. This threonine is either an aspartate or a serine in other pepsins, but is replaced by a glycine (Gly67) in Mtb-AP (Fig. 8). S3: The position occupied by Ile193 of Mtb-AP is generally a Thr/Ser/Asn, the side chain of which makes a conserved hydrogen bond with substrate backbone nitrogen atom of P3 residue. This substrate stabilizing interaction is lost in Mtb-AP due to the presence of Ile193. Moreover, Ile193, which forms the mouth of S3 subsite projects out, occluding the S3 subsite (Fig. 9). S1′: This subsite contains the first hydrophobic residue present in the conserved C-terminal HHG motif of eukaryotic aspartic proteinases. In Mtb-AP, the hydrophilic residue Asn247 which replaces the first residue of the C-terminal HHG motif forms the S1′ pocket (Fig. 8). Thus we can speculate that Mtb-AP might require a hydrophilic residue at P1′ position. S2′: Some of the residues of S2′ pocket which can interact with and stabilize the substrate are either not conserved or absent because of the deletion of the loops and thus making this binding pocket wide open (Fig. 9). These differences could contribute to the lack of activity of Mtb-AP.
Fig. 8.
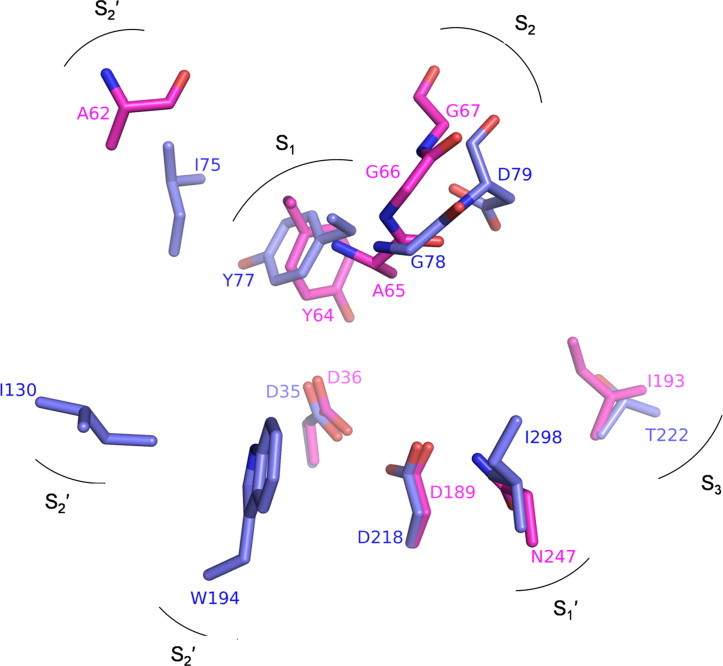
Differences in the subsite residues of Mtb-AP (magenta) and rhizopuspepsin (violet). Residues equivalent to Ile130 and Trp194 of rhizopuspepsin are absent in Mtb-AP because of truncated loops. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 9.
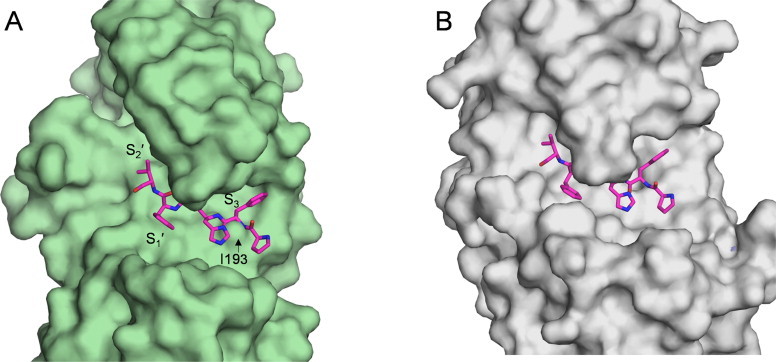
Surface representation of the active site of (A) Mtb-AP and (B) rhizopuspepsin (PDB code: 3APR). A modified peptide inhibitor of rhizopuspepsin is shown at a structural equivalent site in Mtb-AP. Ile193 of Mtb-AP which forms the mouth of S3 pocket is shown with arrow.
Similar to Mtb-AP, proteolytic activity was not detected in most of the members of an extensive family of proteins, the pregnancy-associated glycoproteins (PAGs) even though their sequences are about 50% identical to pepsins [33,34]. Changes in the DTG motif explain the lack of activity in some cases (Asp215 to Gly in ovine PAG-1, Gly34 to Ala in bovine PAG-1 and porcine PAG-1). In a few other PAGs with conserved catalytic site residues, the presence of the sequence EPV instead of QDL for the residues 148–150 of pepsins was suggested to be the cause of inactivity [35]. This region is involved in interactions between the two domains and any changes in this region might affect interdomain orientation altering the function. However, proteolytic activity was reported recently in three PAGs, bovine PAG-2 amp PAG-12 [36] and porcine PAG-2 [37], all of them having the EPV sequence, when a modified peptide was used as the substrate.
4. Conclusions
The polypeptide chain of Mtb-AP is shorter than the eukaryotic proteases. However, the fold of the enzyme has been preserved by shortening of the loops. The parallel arrangement of two strands in the interdomain β-sheet topology instead of the antiparallel arrangement present in all aspartic proteinases has happened because of shortened loop length but to preserve the same fold. The hydrophilic residues Asn247 and Thr248 substituted in place of hydrophobic residue of the C-terminal HHG motifs have been conserved in all predicted aspartic protease domains of Mycobacterium species. These two residues play a crucial role in preserving the hydrogen bonds made by Asp189, a structural feature essential for catalysis and to maintain the active site geometry.
The alanine insertion in the flap region along with the altered conformation of tyrosine, compared to pepsins, are likely to contribute to the inactivity of the protein by preventing the entry of the substrate into the active site. In addition, the critical substrate binding residues of the S2 and S3 pockets of pepsins are different in Rv0977 and thus the enzyme may not be able to bind the substrate strongly. These residues in Rv2519 and MMAR_1538 are similar in nature to those in pepsins. Hence, it is likely that these proteins could be active.
With the fold and the required geometry of the catalytic site strikingly similar to known aspartic proteinases, it is unlikely that Mtb-AP lacks any protease activity. Though it was found to be insensitive to common pepsin substrates, it is possible that Mtb-AP has a narrow specificity which could be detected with an appropriate substrate. However, it is clear that further extensive investigations are required to establish the substrate specificity of the protein if it turns out to be an aspartic proteinase, or to find whether it has an entirely different function in vivo to understand the exact mechanism of host–pathogen interaction involving Mtb-AP. As in the case of PAGs, smaller peptides might be required instead of protein substrates to detect the activity especially when the activity is weak. Even though the proteolytic cleavage occurs between two specific residues, detection of new specificities is not trivial as it is necessary to generate and screen a large number of longer peptides as the interactions on either side of the scissile bond are also crucial for binding. The wider binding pocket in Mtb-AP makes it even more difficult to explore and establish its exact substrate specificity.
The unique features outlined above makes the M. tuberculosis protein distinctly different from the others in terms of substrate specificity, which needs to be explored. A small number of pepsin-like bacterial aspartic proteinases have been reported, but no structure is available so far. The structure of Mtb-AP supports the hypothesis [3] suggested while reporting the newly identified single chain aspartic prtoteinases in bacteria that the bacterial and eukaryotes might have diverged after gene duplication and fusion event. In spite of the lack of detection of the function, it is important to report the discovery of the aspartic proteinases in the pathogenic mycobacterial species, especially since the three-dimensional structure reveals the potential of the protein to be an aspartic proteinase with two aspartates optimally positioned for catalysis. As Rv0977 has been identified as the surface antigen, it could become a potential target for designing drugs against tuberculosis once the in vivo function is established.
Accession code. The coordinates and structure factors of Mtb-AP have been deposited in the Protein Data Bank with accession code PDB: 4EHC.
Competing financial interests
The authors declare no competing financial interests.
Acknowledgements
Native data were collected at the National X-ray Data Collection Facility at the Molecular Biophysics Unit, Indian Institute of Science. SAD data were collected at beamline station BM14 of the European Synchrotron Radiation Facility (ESRF) and is funded by the Department of Biotechnology (DBT). Financial support from the DBT is acknowledged.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Supplementary material
Supplementary material associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.fob.2013.05.004.
Appendix. Supplementary materials
Supplementary materials for Crystal structure of a putative aspartic proteinase domain of the M. tuberculosis cell surface antigen PE_PGRS16.
References
- 1.Tang J., James M.N., Hsu I.N., Jenkins J.A., Blundell T.L. Structural evidence for gene duplication in the evolution of the acid proteases. Nature. 1978;271:618–621. doi: 10.1038/271618a0. [DOI] [PubMed] [Google Scholar]
- 2.Rawlings N.D., Bateman A. Pepsin homologues in bacteria. BMC Genomics. 2009;10:437. doi: 10.1186/1471-2164-10-437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simoes I., Faro R., Bur D., Kay J., Faro C. Shewasin A, an active pepsin homolog from the bacterium Shewanella amazonensis. FEBS J. 2011;278:3177–3186. doi: 10.1111/j.1742-4658.2011.08243.x. [DOI] [PubMed] [Google Scholar]
- 4.Cole S.T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 5.Singh P.P., Parra M., Cadieux N., Brennan M.J. A comparative study of host response to three Mycobacterium tuberculosis PE_PGRS proteins. Microbiology. 2008;154:3469–3479. doi: 10.1099/mic.0.2008/019968-0. [DOI] [PubMed] [Google Scholar]
- 6.Brennan M.J., Delogu G. The PE multigene family: a ‘molecular mantra’ for mycobacteria. Trends Microbiol. 2002;10:246–249. doi: 10.1016/s0966-842x(02)02335-1. [DOI] [PubMed] [Google Scholar]
- 7.Dheenadhayalan V., Delogu G., Sanguinetti M., Fadda G., Brennan M.J. Variable expression patterns of Mycobacterium tuberculosis PE_PGRS genes: evidence that PE_PGRS16 and PE_PGRS26 are inversely regulated in vivo. J. Bacteriol. 2006;188:3721–3725. doi: 10.1128/JB.188.10.3721-3725.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Talarico S., Zhang L., Marrs C.F., Foxman B., Cave M.D., Brennan M.J., Yang Z. Mycobacterium tuberculosis PE_PGRS16 and PE_PGRS26 genetic polymorphism among clinical isolates. Tuberculosis (Edinb.). 2008;88:283–294. doi: 10.1016/j.tube.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Battye T.G., Kontogiannis L., Johnson O., Powell H.R., Leslie A.G. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 2011;67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Panjikar S., Parthasarathy V., Lamzin V.S., Weiss M.S., Tucker P.A. Auto-rickshaw: an automated crystal structure determination platform as an efficient tool for the validation of an X-ray diffraction experiment. Acta Crystallogr. D Biol. Crystallogr. 2005;61:449–457. doi: 10.1107/S0907444905001307. [DOI] [PubMed] [Google Scholar]
- 11.Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sheldrick G.M., Hauptman H.A., Weeks C.M., Miller R., Usón I. Ab initio phasing. In: Rossmann M.G., Arnold E., editors. International Tables for Crystallography. IUCr and Kluwer Academic Publishers; Dordrecht: 2001. pp. 333–351. Vol. F. [Google Scholar]
- 13.Schneider T.R., Sheldrick G.M. Substructure solution with SHELXD. Acta Crystallogr. D Biol. Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 14.Perrakis A., Morris R., Lamzin V.S. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 15.Murshudov G.N., Vagin A.A., Dodson E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 16.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 17.Davis I.W., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nielsen P.K., Foltmann B. Purification and characterization of porcine pepsinogen B and pepsin B. Arch. Biochem. Biophys. 1995;322:417–422. doi: 10.1006/abbi.1995.1483. [DOI] [PubMed] [Google Scholar]
- 19.Wunschmann S., Gustchina A., Chapman M.D., Pomes A. Cockroach allergen Bla g 2: an unusual aspartic proteinase. J. Allergy Clin. Immunol. 2005;116:140–145. doi: 10.1016/j.jaci.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 20.ten Have T., Dekkers E., Kay J., Phylip L.H., van Kan J.A. An aspartic proteinase gene family in the filamentous fungus Botrytis cinerea contains members with novel features. Microbiology. 2004;150:2475–2489. doi: 10.1099/mic.0.27058-0. [DOI] [PubMed] [Google Scholar]
- 21.Kumar A., Bhosale M., Reddy S., Srinivasan N., Nandi D. Importance of non-conserved distal carboxyl terminal amino acids in two peptidases belonging to the M1 family: thermoplasma acidophilum Tricorn interacting factor F2 and Escherichia coli Peptidase N. Biochimie. 2009;91:1145–1155. doi: 10.1016/j.biochi.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 22.Kumar M., Prashar V., Mahale S., Hosur M.V. Observation of a tetrahedral reaction intermediate in the HIV-1 protease–substrate complex. Biochem. J. 2005;389:365–371. doi: 10.1042/BJ20041804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhaumik P., Xiao H., Parr C.L., Kiso Y., Gustchina A., Yada R.Y., Wlodawer A. Crystal Structures of the Histo-Aspartic Protease (HAP) from Plasmodium falciparum. J. Mol. Biol. 2009;388:520–540. doi: 10.1016/j.jmb.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pearl L.H., Taylor W.R. A structural model for the retroviral proteases. Nature. 1987;329:351–354. doi: 10.1038/329351a0. [DOI] [PubMed] [Google Scholar]
- 25.Hartsuck J.A., Koelsch G., Remington S.J. The high-resolution crystal structure of porcine pepsinogen. Proteins. 1992;13:1–25. doi: 10.1002/prot.340130102. [DOI] [PubMed] [Google Scholar]
- 26.Lee A.Y., Gulnik S.V., Erickson J.W. Conformational switching in an aspartic proteinase. Nat. Struct. Biol. 1998;5:866–871. doi: 10.1038/2306. [DOI] [PubMed] [Google Scholar]
- 27.Andreeva N.S., Rumsh L.D. Analysis of crystal structures of aspartic proteinases: on the role of amino acid residues adjacent to the catalytic site of pepsin-like enzymes. Protein Sci. 2001;10:2439–2450. doi: 10.1110/ps.25801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujinaga M., Chernaia M.M., Tarasova N.I., Mosimann S.C., James M.N.G. Crystal structure of human pepsin and its complex with pepstatin. Protein Sci. 1995;4:960–972. doi: 10.1002/pro.5560040516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suguna K., Padlan E.A., Smith C.W., Carlson W.D., Davies D.R. Binding of a reduced peptide inhibitor to the aspartic proteinase from Rhizopus chinensis: implications for a mechanism of action. Proc. Natl. Acad. Sci. U.S.A. 1987;84:7009–7013. doi: 10.1073/pnas.84.20.7009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gustchina A., Li M., Wünschmann S., Chapman M.D., Pomés A., Wlodawer A. Crystal structure of cockroach allergen Bla g 2, an unusual zinc binding aspartic protease with a novel mode of self-inhibition. J. Mol. Biol. 2005;348:433–444. doi: 10.1016/j.jmb.2005.02.062. [DOI] [PubMed] [Google Scholar]
- 31.Andreeva N., Dill J., Gilliland G.L. Can enzymes adopt a self-inhibited form? Results of X-ray crystallographic studies of chymosin. Biochem. Biophys. Res.Commun. 1992;184:1074–1081. doi: 10.1016/0006-291x(92)90701-l. [DOI] [PubMed] [Google Scholar]
- 32.Gustchina A., Li M., Phylip L.H., Lees W.E., Kay J., Wlodawer A. An unusual orientation for Tyr75 in the active site of the aspartic proteinase from Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 2002;295:1020–1026. doi: 10.1016/s0006-291x(02)00742-8. [DOI] [PubMed] [Google Scholar]
- 33.Xie S.C., Low B.G., Nagel R.J., Kramer K.K., Anthony R.V., Zoli A.P., Beckers J.F., Roberts R.M. Identification of the major pregnancy-specific antigens of cattle and sheep as inactive members of the aspartic proteinase family. Proc. Natl. Acad. Sci U.S.A. 1991;88:10247–10251. doi: 10.1073/pnas.88.22.10247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xie S., Green J., Bixby J.B., Szafranska B., DeMartini J.C., Hecht S., Roberts R.M. The diversity and evolutionary relationships of the pregnancy-associated glycoproteins, an aspartic proteinase subfamily consisting of many trophoblast-expressed genes. Proc. Natl. Acad. Sci. U.S.A. 1997;94:12809–12816. doi: 10.1073/pnas.94.24.12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hughes A.L., J A. Green, Piontkivska H., Roberts R.M. Aspartic proteinase phylogeny and the origin of pregnancy-associated glycoproteins. Mol. Biol. Evol. 2003;20:1940–1945. doi: 10.1093/molbev/msg217. [DOI] [PubMed] [Google Scholar]
- 36.Telugu B.P.V.L., Palmier M.O., van Doren S.R., Green J.A. An examination of the proteolytic activity for bovine pregnancy-associated glycoprotein 2 and 12. Biol. Chem. 2010;391:259–270. doi: 10.1515/BC.2010.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Telugu B.P.V.L., Green J.A. Characterization of the peptidase activity of recombinant porcine pregnancy-associated glycoprotein-2. J. Biochem. 2008;144:725–732. doi: 10.1093/jb/mvn127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials for Crystal structure of a putative aspartic proteinase domain of the M. tuberculosis cell surface antigen PE_PGRS16.