

Abstract
A variant allele, ADH1B*48His, also known as ADH1B*2, at the human Alcohol Dehydrogenase 1B gene (ADH1B) is strongly associated with alcoholism in some populations and has an unusual geographic distribution. Strong evidence implies selection has increased the frequency of this allele in some East Asian populations but does not fully explain its geographic pattern. We have studied haplotypes of ten single nucleotide polymorphisms (SNPs) and two short tandem repeat polymorphisms (STRPs) in the ADH1B region in 2,206 individuals from a worldwide set of populations. These SNPs and STRPs define nine common haplogroups most of which have distinct geographic patterns. The haplogroups H5 and H6, both with the derived ADH1B*48His allele, appear restricted to the Middle East and East Asia, respectively. The positively selected H7 is derived from H6 by a new regulatory region variant defining SNP rs3811801 restricted to East Asia. Age estimates of the haplogroups based on the STRPs also agree with the time of the migration events estimated by other studies. H7 is estimated to have expanded recently, around 2,800 years ago, and ancient DNA samples from North China confirm its presence about that time. The dating of the H7 expansion may help understand the selective force on the ADH1B gene.
Keywords: ADH1B, Haplotype evolution, Recent expansion, Geographic distribution
Introduction
The alcohol dehydrogenase (ADH) gene family is one of the most studied gene families in the human genome. In studies over the past 30 years variants in this gene family have been found to be associated not only with alcoholism but also with a large number of disorders including different cancers (Chen et al., 1999; Osier et al., 1999; Eriksson et al., 2001; Edenberg et al., 2006; Matsuo et al., 2007; Triano et al., 2003; Jia et al., 2007). There are several evolutionarily related genes in this family, among which ADH1B (previously ADH2) has been the most frequently studied from the very beginning of molecular studies of ADH (Li et al., 1977). Some protein electrophoretic polymorphisms of the enzymes encoded by ADH1B and the adjacent gene (ADH1C) were found to correspond to differences in the kinetics of the enzymes (e.g. Smith, 1986). Subsequently, the DNA differences were identified. However, as the linkage disequilibrium (LD) in these regions is relatively high, the functional association for some of the polymorphisms might be attributable to the LD with other polymorphisms (Osier et al., 1999). One of the function-associated polymorphisms, ADH1B Arg48His (rs1229984), is believed to be the most important in ADH studies. However, the high LD between the ADH1B*48His allele and the flanking regions may be the reason for the functional associations of all variants in the Class I ADH region (1A, 1B, and 1C), at least in some populations (Osier et al., 1999; Han et al., 2007). The geographic distribution of the ADH1B*48His allele is also dramatic. The frequency of the derived allele of this variant is common to high (>25%) in West Asia and East Asia, but infrequent to absent in the rest of the world, including the regions between West Asia and East Asia (Li et al., 2007c; Borinskaya et al., 2009; Li & Kidd, 2009). The functional association (Chen et al., 1999; Osier et al., 1999; Eriksson et al., 2001; Edenberg et al., 2006; Matsuo et al., 2007) of this variant is found in both areas with differing strength, but not in every population. The high frequency of the ADH1B*48His variant in many East Asian populations is most likely the result of natural selection that favored the chromosomes with that allele (Smith, 1986; Li et al., 2008). A variant in the regulatory region of ADH1B, the derived allele at rs3811801, is possibly responsible for the selection and the particular functional association in East Asia (Li et al., 2008). Because of the high LD and specific geographic distribution, a more globally comprehensive joint analysis of the variants in the region of ADH1B is warranted to more clearly understand the evolution of this gene as humans have diversified.
Our initial phylogenetic analysis of the ADH1B haplogroups included several of the ten single nucleotide polymorphisms (SNPs) in Figure 1 (Li et al., 2007c). However, an understanding of the diversification and migration history also needs to include short tandem repeat polymorphisms (STRPs) because of their higher mutation rates. Here we demonstrate a detailed phylogenetic analysis of ADH1B including the ten SNPs and two STRPs also shown in Figure 1. Detailed classification and age estimates for haplogroups concur with the “Out of Africa” pattern: temporally ordering the distinct haplotypes of West Asia and East Asia and demonstrating a recent expansion of the derived allele of the regulatory variant. Ancient DNA analyses we present here also support the recent expansion. This study provides a more detailed picture of the history of natural selection on ADH1B variants among different East Asian populations.
Figure 1.
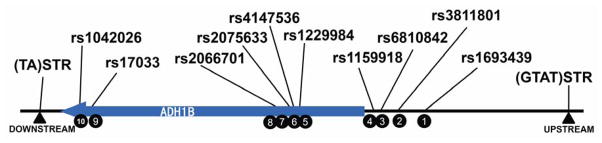
Markers typed in this paper. The centromere is to the left (downstream) and the telomere is to the right. The SNPs are numbered as represented in haplotypes going from the 5′ to 3′ of the gene, cf. Tables 1 and 2.
Material and Methods
Population samples
We typed 2,206 individuals from a global sample of 42 populations. According to population ancestry and geography locations, these 42 populations are categorized into eight groups. The populations and sample sizes are as follows:
Africa: Biaka 69, Mbuti 39, Yoruba 78, Ibo 48, Hausa 39, Chagga 45, Masai 20, and African Americans 91; Middle Eastern: Ethiopian Jews 32, Yemenite Jews 43, Ashkenazi Jews 81, Samaritans 41, and Druze 103; Europe: Adygei 54, Chuvash 42, Russians from Archangelsk 33, Russians from Vologda 48, Finns 36, Danes 51, Irish 115, and European Americans 90; East Asia: Chinese from San Francisco 58, Chinese from Taiwan 50, Hakka 41, Koreans 54, Japanese 48, Ami 40, Atayal 42, and Cambodians 25; Pacific: Nasioi 23 and Micronesians 37; Siberia: Komi Zyriane 47, Khanty 50, and Yakut 51; North America: Cheyenne 56, Pima-Arizona 51, Pima-Mexico 99, and Maya 49; South America: Quechua 23, Ticuna 65, Rondonian Surui 45, and Karitiana 54.
Sample descriptions can be found in the Allele Frequency Database (ALFRED) by searching on the population names or clicking on the sample ID link from the individual SNP frequencies. All volunteers were apparently normal and healthy, with no diagnosis of alcoholism or related disorders except in a subset of the Taiwanese samples. All samples were collected with informed consent under protocols approved by the relevant institutional review boards.
Genetic makers
The two STRPs flanking ADH1B have not previously been reported on or studied; they are described in more detail in supplemental material. Of the 16 SNPs we typed between the two STRPs we chose ten SNPs and two STRPs to define the core haplogroups of ADH1B (Figure 1). The polymorphisms and locations of the STRPs and the ten core SNP markers and their haplotypes are given in Tables 1 and 2. For consistency all SNP alleles are presented on the genomic reference positive strand as ancestral or derived even though various databases give reference alleles on a mixture of positive and negative strands. The six SNPs typed but not included in this analysis are rs1789891, rs9307239, rs3811802, rs1229982, rs28626993, and rs2066702. Additional information on the STRPs and all 16 SNPs is available in ALFRED. We used TaqMan assays from Applied Biosystems to type the SNPs (Livak et al., 1995). The designations of ancestral alleles are based on our typing of 15 non-human apes of five species and are consistent with UCSC designations. The two STRPs in the flanking regions of ADH1B are described in more detail in supplemental material.
Table 1.
The two STRPs and 10 SNPs analyzed at ADH1B, their positions, and ancestral alleles.
| rs number or “name” | position | Positive (+)strand polymorphism | Ancestral allele on + strand |
|---|---|---|---|
| upstream (GTAT)n | 100252794 | ||
| rs1693439 | 100245489 | A/G | G |
| rs3811801 | 100244319 | A/G | G |
| rs6810842 | 100243445 | G/T | G |
| rs1159918 | 100243009 | A/C | A |
| rs1229984 | 100239319 | C/T | C |
| rs4147536 | 100239112 | A/C | C |
| rs2075633 | 100238998 | C/T | T |
| rs2066701 | 100238413 | A/G | G |
| rs17033 | 100228945 | C/T | T |
| rs1042026 | 100228466 | C/T | T |
| downstream (TA)n | 100226379 |
Table 2.
Haplotype labels corresponding to Figure 2 with alleles ordered as in Table 1, top to bottom. The ancestral alleles are in black; the derived alleles are in red.
| Haplotype number | Haplotype with all alleles on + strand, but telomeric to centromeric |
|---|---|
| H1 ancestral |
|
| H1b |
|
| H1c |
|
| H2 |
|
| H2b |
|
| H4 |
|
| H4b |
|
| H3 |
|
| H3b |
|
| H3c |
|
| H5 |
|
| H5b |
|
| H6 |
|
| H7 |
|
Statistical Analyses
Estimates of haplotype frequencies for the ten core SNPs were calculated with the program PHASE2.1 (Stephens et al., 2001; Stephens & Donnelly, 2003). The network (shortest tree) of the haplotypes deemed present was calculated and drawn by the program NETWORK4.201 (Bandelt et al., 1999). Haplogroups were defined based on the clusters in the network. Allele and haplotype frequencies were estimated by gene counting in each population using the phased data for the haplotypes. Haplotypes of just the ten core SNPs corresponded closely to the major NETWORK haplogroups. Because NETWORK cannot accommodate crossovers, the phylogenetic relationships were drawn using a simple parsimony assumption weighting recombination more probable than multiple recurrent mutations.
The distributions of the ADH1B downstream STRP alleles in different geographic regions were displayed as histograms. Variances of STRP allele sizes in different geographic regions and for different haplogroups were calculated using repeat number. The coalescence time of modern human (200,000 years, 10,000 generations) given by other previous dating studies (Cann et al., 1987; Ayala & Escalante,1996; Thomson et al., 2000; Hammer et al., 1998; Mellars, 2006) and the global allele size variance based on our whole world sample were adopted to estimate the mutation rate of the downstream STRP. The ages of the haplogroups were calculated by the age estimate formula for a single STRP (Su et al., 1999; Wilson et al., 2003) and the NETWORK program. The formula used is t=−Neln(1 − V/Neμ), where Ne is the effective population size, V is the STRP variance observed, μ is the mutation rate of the STRP, and t is the resulting number of generations. The inferred migration routes were based upon the variances in different geographic regions and the evolutionary relationships among the haplogroups.
Ancient DNA validation
The expansion age of the derived allele of rs3811801 was investigated using samples of 38 remains from four sites in North China dated from 2500BC to 220AD (Table 3) examined for the East Asian high frequency derived alleles of rs1229984 and rs3811801. The authenticity for the ancient DNA was insured by strict adherence to the criteria suggested by Pääbo et al. (2004). The sampled remains were excavated from the dry loess stratum in the slopes at those sites in which the biochemical preservation is relatively good. Each skeleton was sampled from several different parts: mostly teeth, astragalus, calcaneus, vertebrae, etc. The teeth we chose were all intact and still fixed to the jaw. Repeated amplifications from different samples of each skeleton were always performed. Samples were handled with gloves by a limited number of anthropologists wearing face masks and caps during excavation. During the transportation, the samples were packaged in hermetically sealed plastic bags. The ancient DNA labs in Fudan University (Li et al., 2007b) are strictly controlled following all the criteria for ancient DNA studies (Pääbo et al., 2004), such as routine sterilization by different treatments (DNAse away, positive air pressure, bleach and ultraviolet light irradiation), air filtration, and isolated rooms for different experimental steps (three rooms for sample cleaning, DNA extraction, and PCR cocktail preparation, respectively). No more than three researchers can work in the lab, wearing full body protective clothing, and using dedicated equipment and reagents. Pre-PCR work was performed in the hood in the designated rooms of the lab. PCR cocktails were prepared and sealed carefully before they were removed from the pre-PCR rooms. The PCR room is far from the other rooms and airflow between the PCR room and the other rooms is strictly avoided. The post-PCR facilities and analyses are physically separated from the pre-PCR location and procedures. The extraction protocol was the same as that we have published (Li et al., 2007b). Initial typings of these samples were done at Fudan University. The two SNPs were examined by TaqMan as we do for the modern DNA. Extraction controls of unknown animal bones from the same sites were included. PCRs of each SNP were repeated six times. PCR controls were also included each time. The DNA templates were quantified to 10ng for each PCR. Replications were performed in the lab at Yale University.
Table 3.
Numbers of chromosomes seen for each haplogroups in each different geographic region (N) and the ranges (R) of the repeat lengths of the ADH1B downstream STRP. For these summaries H1b, H2b, and H4b are included with H1, H2, and H3, respectively.
| H1 | H2 | H3 | H4 | H5 | H6 | H7 | Total | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | R | N | R | N | R | N | R | N | R | N | R | N | R | N | R | |
| Sub-Sahara | 305 | 11~35 | 48 | 11~13 | 55 | 16~22 | 449 | 11~35 | 0 | 1 | 18 | 858 | 11~35 | |||
| Middle East | 120 | 11~35 | 41 | 11~13 | 114 | 16~25 | 103 | 11~26 | 207 | 13~32 | 11 | 16~21 | 4 | 17~18 | 600 | 11~35 |
| Europe | 296 | 11~27 | 93 | 11~13 | 244 | 16~26 | 259 | 11~29 | 25 | 11~26 | 4 | 17~20 | 17 | 17~18 | 938 | 11~29 |
| Siberia | 76 | 11~25 | 92 | 11~12 | 46 | 16~21 | 63 | 11~25 | 2 | 24 | 0 | 17 | 17~20 | 296 | 11~25 | |
| E.&S.E. Asia | 19 | 11~25 | 30 | 11~12 | 53 | 15~19 | 81 | 11~25 | 1 | 11 | 202 | 16~26 | 330 | 16~26 | 716 | 11~26 |
| Oceania | 2 | 19~20 | 20 | 11~12 | 18 | 16~21 | 54 | 15~27 | 1 | 21 | 24 | 17~18 | 1 | 17 | 120 | 11~27 |
| N.America | 11 | 11~23 | 230 | 11~13 | 6 | 16~29 | 255 | 11~25 | 8 | 11~22 | 0 | 510 | 11~25 | |||
| S.America | 6 | 11~21 | 269 | 11~13 | 1 | 19 | 98 | 11~26 | 0 | 0 | 374 | 11~26 | ||||
| Total | 835 | 11~35 | 823 | 11~13 | 537 | 15~26 | 1362 | 11~35 | 244 | 11~32 | 242 | 16~26 | 369 | 16~26 | 4412 | 11~35 |
RESULTS
Allele and haplotype data
The allele frequencies for the SNPs and the STRPs are available on the ALFRED website for all of the populations studied. For the ADH1B upstream STRP, six alleles were observed, from 10 repeats (77bp) to 15 repeats (97bp). For the ADH1B downstream STRP, 25 alleles were observed, from 11 repeats (195bp) to 35 repeats (243bp). The frequency of the shortest 11-repeat allele of the downstream STRP is unusually high (10.3%), suggesting a minimum limit or “absorbing boundary” for repeat number of this STRP, i.e., a size at which the mutation rate becomes very low. The ten SNPs define nine common haplotypes across the 17.023kb extent from rs1693439 to rs1042026. Table 1 gives the positions and alleles of these ten SNPs using the allele on the genome reference strand for all SNPs. Thus, the haplotypes were defined from telomeric (5′ of the gene) to centromeric (3′ of the gene) with alleles on the positive strand. Nine common haplotypes were observed (Table 2). Each of those nine haplotypes defines a haplogroup involving the STRP alleles.
Network and taxonomy
The inferred phylogenetic relationships of the nine common haplotypes are displayed in Figure 2. By parsimony, H1b, H2, H2b, H3, H3b, H3c, H4, H4a, and H5 can all be explained by accumulation of mutations from the ancestral H1. H3 was derived from H1b by three SNP mutations. H2 and H4 are derived by accumulation of successive mutations from H1. H5 was derived from H1b by the non-synonymous mutation to ADH1B*48His. H6 was formed by the recombination between H5 and H3, and also contains the derived ADH1B*48His allele from H5. In addition to the data shown in Table 2 and Figure 2, this recombination event is supported by multiple flanking SNPs not part of the current analysis (Han et al., 2007). H7 was derived from H6 by the mutation of the regulatory region SNP (rs3811801).
Figure 2.

Evolutionary schema for the thirteen globally most common haplotypes. While the circle sizes are not strictly proportional to overall frequency, the shadings within each circle represent the proportion of that haplotype occurring in each geographic region by the colors corresponding to regions in the map. H1b, the 14th haplotype, is very rare but does complete the evolutionary connections. H1c and H5b are not rare but cannot be explained by a single event (crossover or mutation) but have a more complex evolutionary background; the fine dotted lines indicate some of the allelic relationships.
The geographic distribution of the haplogroups is also dramatic (Figure 2; Table 3). Almost all of the Native Americans belong to H2 and H4. The ancestral haplotype H1 is nearly absent out of Africa. H5 occurs mainly in the Middle East, while the evolutionarily derived H6 and H7 are mainly in East Asia. Only a very few individuals among the Chuvash, Adygei, and Ashkenazi Jews belong to H7, which might have been introduced from the East very recently, as these populations lie along historic trade routes, e.g., the Silk Road. The low STRP diversity of these few H7 individuals in the West also supports the recent arrival of the haplogroup from East Asia. The ADH1B haplotypes containing the derived ADH1B*48His allele associated with altered enzyme function are distinctly different in West Asia (H5) from those in East Asia (H6 & H7) (Table 3). The six SNPs not included in the core demonstrate a more complex pattern of haplotype evolution by generating additional side branches, crossovers, etc., all of which are rare globally and, at most, uncommon in any single population. They do not alter the pattern of these core haplotypes presented and analyzed in this paper.
STRP Diversity
As the diversity of the downstream STRP is much higher than that of the upstream STRP, the more informative downstream STRP is the focus of analyses. The total diversity of the upstream STRP varies by geographic region (Figure 3). With the exception of Oceania, where the two major peaks are consistent with the known considerable differences between Melanesians and Micronesians, there is a general decrease in variance with distance from Africa if the peak at 11 repeats is ignored. The most common non-11 repeat allele also varies by geographic region. Excepting the alleles at or adjacent to the assumed absorbing lower boundary of allele sizes, the variances of the allele size distributions show a clinal reduction from Africa through Eurasia to the New World. This reduction is consistent with the African origin of modern human and a relatively recent expansion into the rest of the world relative to the mutation rate(s) at this STRP.
Figure 3.

The overall distribution of repeat sizes for the downstream STRP by geographic region. Variances are given for each of the peaks in each region except for the apparent absorbing boundary at 11 repeats.
More significant than the geographic pattern per se is that the distributions of the downstream STRP alleles differ by geography because the STRP alleles differ by core haplotype of the ten SNPs (Figure 4). The STRP variances by core haplotype (i.e., within each haplogroup) are also quite different. In H1 (including H1b) and H4, there are single major peaks around 21 repeats. In H2 (primarily H2b), the peak is around the shortest 11-repeat allele. There are two peaks in H3 around 16 and 19 repeats, and a sharp gap at 18 repeats, suggesting multiple lineages of this haplotype not identified by these 10 SNPs. The peak in H5 is also around 19 repeats, while the peak in H6 moves to 18 repeats, and that in H7 to 17 repeats. H7 contains the lowest STRP diversity, also indicating this haplotype is the youngest.
Figure 4.

The STRP size distributions associated with each of the seven major SNP-based haplotypes. Compared to Figure 2, H1 here also includes H1b; H2 here also includes H2b; H4 here also includes H4b. H3 and H5 are not pooled with the minor haplotypes shown in Figure 2. Shadings of each bar in the histograms represent the proportion of that haplotype occurring in each geographic region by the coloring of regions in the map.
The supposed migration routes of ADH1B haplogroups are illustrated in Figure 5. H1, H1b, H2, H3, and H4 formed a background for the Old World. Only H2, H2b, and H4 were introduced into the New World, where H2b and H4 became more common. H5 originated in the Middle East and has hardly expanded beyond that region. Because both contributing haplotypes exist in Southwest Asia, H6 may have originated in that region and then expanded into East Asia and Oceania. Wherever it originated, H6 became the major haplogroup in East Asia and more recently gave birth to H7 in East Asia.
Figure 5.
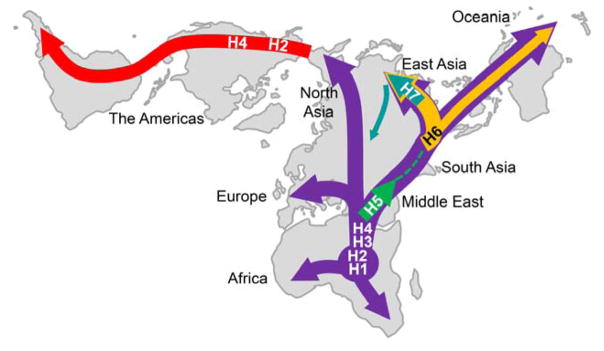
Migration routes of the ADH1B haplogroups
Age estimates of the haplogroups
The ages of the haplogroups were estimated using the downstream STRP variances. The mutation rate was estimated to be 2.474×10−3 per generation based on the well accepted coalescence time of the modern human (10,000 generations) and the variance of our whole world sample (23.26). With this mutation rate, the ages of the haplogroups were estimated in Table 4 by two methods. H1 is as old as the whole human population. The age of H2 is not reliable because the lengths of the STRP alleles of this haplogroup are about at the minimum limit and not appropriate to the formula. The age of H5 is very close to the most accepted time of the “Out of Africa” event and it appears to have been born in the Middle East, the geographic location for the first modern human emergence “out of Africa”. The youngest haplogroup is H7 with an expansion age of only around 1,600 to 4,000 years. As we assumed only 20 years for a generation and the coalescence time for all modern humans may be longer, these ages were most probably underestimated.
Table 4.
The expansion age estimates of the haplogroups
| Estimates by formula of Su et al. | Estimates by NETWORK | |||
|---|---|---|---|---|
| Variance | Generations | Thousand Years | Thousand Years | |
| H1 | 23.32 | 10029 | 200.6 | 185.4±25.6 |
| H2 | 0.19 | 77 | 1.5 | 4.1±2.5 |
| H3 | 3.86 | 1576 | 31.5 | 32.7±9.1 |
| H4 | 13.24 | 5539 | 110.8 | 135.8±28.5 |
| H5 | 6.44 | 2692 | 53.8 | 66.7±16.9 |
| H6 | 1.90 | 783 | 15.7 | 21.4±8.6 |
| H7 | 0.31 | 126 | 2.5 | 2.8±1.2 |
Average generation time (although variable with life expectancy over time) assumed to be 20 years over the long time span considered.
Allele frequencies in the ancient DNA from North China
As haplogroup H7 with the derived alleles of rs1229984 and rs3811801 probably expanded very recently, we examined these two SNPs in the ancient samples from North China dated from 2,500BC to 220AD. If the hypothesis is correct, the derived allele frequency of rs3811801 in the ancient Chinese would not be as high as it is in the modern Chinese. Half of our ancient samples were successfully amplified (Table 5). The derived allele of rs3811801 is definitely present but the frequency in the 4,000 years old population sample is 10% (2/20). The highest frequency of 33% (2/6) occurs in the 2,000 years old Dongxiaoying population sample. Pooling the three more recent samples, including the two in which the derived allele was not seen gives an even lower frequency of 11% (2/18). Even with the small sample sizes the 10% and 11% estimates are significantly lower (p=0.0297 and p=0.0466, respectively, by Fisher’s exact test) than the frequency in the modern population (>50%) in the same region. These data support a recent increase in the frequency of this derived allele.
Table 5.
Haplotype frequencies of rs3811801-rs1229984 (sites 2 and 5 in Figure 1) among the ancient Chinese samples
| Site | N | Success N | Number of each haplotype | Age | Province | County | Period | ||
|---|---|---|---|---|---|---|---|---|---|
| CG | CA | TA | |||||||
| Taoshi | 16 | 10 | 18 | 0 | 2 | ~2500BC-2000BC | Shanxi | Xiangfen | Neolithic age |
| Shangma | 6 | 2 | 4 | 0 | 0 | 770BC-476BC | Shanxi | Houma | Eastern Zhou Dynasty |
| Lingzhi | 7 | 4 | 7 | 1 | 0 | 475BC-221BC | Shandong | Zibo | Warring States Period |
| Dongxiaoying | 9 | 3 | 3 | 1 | 2 | 206BC-220AD | Shandong | Tengzhou | Han Dynasty |
Discussion
Redisplay of the Out of Africa pattern
ADH1B diversification and inferred migration and evolutionary histories display the well supported “Out of Africa” pattern of modern human origins and diversification. The SNP-based haplotypes and the variances of the ADH1B downstream STRP both indicate that the populations in Africa are the oldest and those in Oceania and the Americas are much younger. The ancestral haplotype exists primarily in Africa; the most derived haplotypes exist primarily in East Asia. The pattern also reflects a successive founder model (Liu et al., 2006) with the expansion across Eurasia and into the Pacific and the Americas.
We assume that the coalescence time of the ADH1B gene is no older than that of the modern human population, around 200,000 years (Cann et al., 1987; Ayala & Escalante,1996; Thomson et al., 2000; Hammer et al., 1998; Mellars, 2006). The coalescence time of the H5 haplogroup in the Middle East is then estimated to be around 50,000 to 70,000 years, which is within the well accepted time frame for modern humans expanding out of Africa. If the real coalescence time of the ADH1B gene is shorter than our assumption and the diversification of the gene started some time after the origin of modern humans, the ages of the haplogroups may be even younger than our estimates. Conversely, if we assume a somewhat longer generation time, the age estimate for H5 would be older but still within a generally agreed upon interval. It is also possible that STRP variation persisted through speciation and the true coalescent of modern STRP alleles predates speciation. However, substantial overestimates for the ages are unlikely, as our results agree with those from most other genetic studies (Cann et al., 1987; Ayala & Escalante,1996; Thomson et al., 2000; Hammer et al., 1998; Mellars, 2006).
A bottleneck into the New World
The haplogroup pattern of the New World is quite different from that of the Old World. H2b and H4 are predominant in the New World, while other haplogroups are almost absent, essentially lost along the way as modern humans entered the New World. It is believed that all of the Native Americans we studied derived from a small population that arrived in the northwest tip of the New World (Schurr & Sherry, 2004; Zegura et al., 2004), a bottleneck sufficient to explain the absence of H1 and H3 in Native Americans. This bottleneck effect is also supported by the much lower variance of the STRP in America than in Siberia.
New haplogroups along the way to East Asia
The haplogroups with the derived ADH1B*48His allele, H5, H6, and H7, are of most interest in ADH studies. These haplogroups occur only outside of sub-Saharan Africa, but in different geographic regions. H5 occurs mostly in the Middle East, while H6 and H7 occur almost exclusively in eastern Eurasia. These two regions are separated by South Asia and Central Asia. In South Asia, the ADH1B*48His allele is almost absent; in Central Asia, the frequency of this allele is also very low (Li et al., 2007c; Borinskaya et al., 2009; Li & Kidd, 2009). However, most of the examined samples from Central Asia with this allele belong to H7, the derived haplogroup from H6 (Li et al., 2008), and the STRP diversity of H7 in Central Asia is low. Therefore, most probably these chromosomes were introduced from East Asia recently. H6 occurs mostly in East Asia, Southeast Asia, and Oceania. H6 clearly arose as a recombinant between H5 and H3, probably in the Middle East, but its geographic origin is uncertain. Given that it likely increased in frequency in East Asia due to selection, the region of highest frequency cannot be assumed to be the geographic origin.
The STRP distributions
An interesting observation is that the overall STRP distribution in each region of the world is composed of the distributions in each haplogroup. The differences in the distributions are a shift in frequencies of the haplogroups and the consequences of the “bottleneck” as the single chromosome on which a new mutation defined a new haplotype. For example, comparing Figures 3 and 4, one sees that the shift in the most common allele between Europe and the Middle East is attributable to the high frequency of H5 in the Middle East. In East and Southeast Asia, the peak is shifted to 17 repeats because of the high frequencies of H6 and H7. Clearly, during the time for the new haplotype to become common, the STRP must generate new diversity but, barring recombination, that will be centered around the initial STRP allele on the initial chromosome. Thus, age estimates based on only the STRP distribution are subject to error. Our use of the individual haplogroups will have minimized that problem. Moreover, being able to define the ancestral-to-derived relationships of the haplotypes adds a clear constraint on time estimates.
The recent expansion events in East Asia
The estimated ages of H6 and H7 both indicate relatively recent coalescents or expansion times of the haplogroups. H6 expanded around 15 to 21 thousand years ago, which is around the end of the last Ice Age (Shi et al., 1989). Sufficient archaeological discoveries have revealed that modern human activities appeared and increased in East Asia around that time (Chang, 1976). This age estimate suggests that the natural history of East Asians is relatively short, in agreement with Y chromosome and mtDNA data (Zhang et al., 2007; Li et al., 2007a) and many autosomal loci (e.g. Chu et al., 1998). The expansion of H6 might have coincided with its diffusion into East Asia as the Ice Age ended and the climate turned warmer. The age of H7 is estimated at only around 2.8 thousand years. This young age is unexpected with respect to the high frequency of H7 in present East Asia, but can be explained by the strongly supported positive selection on this haplogroup with the derived allele of the regulatory region SNP (rs3811801) (Li et al., 2008). Therefore, this selection must have begun increasing the allele frequency in East Asian populations by at least 2.5 thousand years ago since the ancient DNA studies found the allele in both 4000 and ~2500 year old samples. The estimated age has a standard deviation of 1.2 thousand years, which means the selection/expansion event might have happened or begun as early as 4 thousand years ago. This could be a poor estimate because selection might violate the assumptions of the age estimate by reducing the variance over what it would have been. Also, the mutation rate may not be constant, as indicated by the apparent absorbing boundary at 11 repeats. A slightly lower mean size of the STRPs might indicate a smaller mutation rate (Brinkmann et al., 1998). We note that the STRP allele sizes flanking H7 are at the low ends of the distributions seen around H1, H3, H4, H5, and H6.
Collecting more data on the frequency of haplogroup H7 in ancient Chinese populations is very desirable to help build on or, if necessary, re-evaluate the evidence we have accumulated. The evidence from the ancient DNA samples we have studied adds an important and independent line of evidence to our understanding of the past evolution of the ADH1B haplotypes helping to determine when H7 appeared and began to increase in frequency in East Asia. Studying more ancient population samples at a variety of locations could also help locate more precisely the likely origin and expansion profile of H7. This in turn would give us more context in space and time for understanding the selective force(s) that have affected the evolution of the ADH1B haplogroup. However, even though our ancient DNA samples are small, the results have shown that the frequency of H7 was significantly lower in the past than today, confirming that haplogroup H7 has increased in frequency from what it was 4,500 to 2,000 years ago. This places clear temporal, and hence environmental and cultural, bounds on the “forces” responsible for the selection of this haplotype and presumably the ADH1B*48His allele.
Supplementary Material
Acknowledgments
This work was supported in part by USPHS grants AA009379 and GM057672 and by NSF grant BCS0840570 to KKK, and also in part by the National Natural Science Foundation of China No.39993420 to LJ. We thank all the colleagues who have helped us assemble the population samples, as well as the Coriell Institute for Medical Research (NIGMS Human Genetic Cell Repository) and the National Laboratory for the Genetics of Israeli Populations at Tel-Aviv University. Special thanks are due the many hundreds of individuals from these populations who volunteered to give blood samples for studies such as this.
Footnotes
Web resources
ALFRED: http://alfred.med.yale.edu
Specific information on the ADH1B polymorphisms: http://alfred.med.yale.edu/alfred/recordinfo.asp?condition=loci.locus_uid='LO000002C
UCSC Genome Browser: http://www.genome.ucsc.edu/
None of the authors has any conflicts of interest related to the data presented here.
References
- Ayala FJ, Escalante AA. The evolution of human populations: a molecular perspective. Mol Phylogenet Evol. 1996;5:188–201. doi: 10.1006/mpev.1996.0013. [DOI] [PubMed] [Google Scholar]
- Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- Borinskaya S, Kal’ina N, Marusin A, Faskhutdinova G, Morozova I, Kutuev I, Koshechkin V, Khusnutdinova E, Stepanov V, Puzyrev V, Yankovsky N, Rogaev E. Distribution of the alcohol dehydrogenase ADH1B*47His allele in Eurasia. Am J Hum Genet. 2009;84:89–92. doi: 10.1016/j.ajhg.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann B, Klintschar M, Neuhuber F, Hühne J, Rolf B. Mutation rate in human microsatellites: influence of the structure and length of the tandem repeat. Am J Hum Genet. 1998;62:1408–1415. doi: 10.1086/301869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cann RL, Stoneking M, Wilson AC. Mitochondrial DNA and human evolution. Nature. 1987;325:31–36. doi: 10.1038/325031a0. [DOI] [PubMed] [Google Scholar]
- Chang KC. Early Chinese Civilization. Cambridge: Harvard Yenching Institute; 1976. [Google Scholar]
- Chen CC, Lu RB, Chen YC, Wang MF, Chang YC, Li TK, Yin SJ. Interaction between the functional polymorphisms of the alcohol-metabolism genes in protection against alcoholism. Am J Hum Genet. 1999;65:795–807. doi: 10.1086/302540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu JY, Huang W, Kuang SQ, Wang JM, Xu JJ, Chu ZT, Yang ZQ, Lin KQ, Li P, Wu M, Geng ZC, Tan CC, Du RF, Jin L. Genetic Relationship of populations in China. Proc Natl Acad Sci USA. 1998;95:11763–11768. doi: 10.1073/pnas.95.20.11763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edenberg HJ, Xuei X, Chen HJ, Tian H, Wetherill LF, Dick DM, Almasy L, Bierut L, Bucholz KK, Goate A, Hesselbrock V, Kuperman S, Nurnberger J, Porjesz B, Rice J, Schuckit M, Tischfield J, Begleiter H, Foroud T. Association of alcohol dehydrogenase genes with alcohol dependence: a comprehensive analysis. Hum Mol Genet. 2006;15:1539–1549. doi: 10.1093/hmg/ddl073. [DOI] [PubMed] [Google Scholar]
- Eriksson CJ, Fukunaga T, Sarkola T, Chen WJ, Chen CC, Ju JM, Cheng AT, Yamamoto H, Kohlenberg-Muller K, Kimura M, Murayama M, Matsushita S, Kashima H, Higuchi S, Carr L, Viljoen D, Brooke L, Stewart T, Foroud T, Su J, Li TK, Whitfield JB. Functional relevance of human adh polymorphism. Alcohol Clin Exp Res. 2001;5(Suppl ISBRA):157S–163S. doi: 10.1097/00000374-200105051-00027. [DOI] [PubMed] [Google Scholar]
- Hammer MF, Karafet T, Rasanayagam A, Wood ET, Altheide TK, Jenkins T, Griffiths RC, Templeton AR, Zegura SL. Out of Africa and back again: nested cladistic analysis of human Y chromosome variation. Mol Biol Evol. 1998;15:427–441. doi: 10.1093/oxfordjournals.molbev.a025939. [DOI] [PubMed] [Google Scholar]
- Han Y, Gu S, Oota H, Osier MV, Pakstis AJ, Speed WC, Kidd JR, Kidd KK. Evidence of Positive Selection on a Class I ADH Locus. Am J Hum Genet. 2007;80:441–456. doi: 10.1086/512485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia C, Liu T, Liu Z, Li M, Hu M. Joint effects of eNOS gene T-786C and ADH2 Arg48His polymorphisms on the risk of premature coronary artery disease. Thromb Res. 2007;120:679–684. doi: 10.1016/j.thromres.2006.12.023. [DOI] [PubMed] [Google Scholar]
- Li TK, Bosron WF, Dafeldecker WP, Lange LG, Vallee BL. Isolation of pi-alcohol dehydrogenase of human liver: is it a determinant of alcoholism? Proc Natl Acad Sci USA. 1977;74:4378–4381. doi: 10.1073/pnas.74.10.4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Cai X, Winograd-Cort ER, Wen B, Cheng X, Qin Z, Liu W, Liu Y, Pan S, Qian J, Tan CC, Jin L. Mitochondrial DNA diversity and population differentiation in southern East Asia. Am J Phys Anthropol. 2007a;134:481–488. doi: 10.1002/ajpa.20690. [DOI] [PubMed] [Google Scholar]
- Li H, Huang Y, Mustavich LF, Zhang F, Tan JZ, Wang LE, Qian J, Gao MH, Jin L. Y chromosomes of Prehistoric People along the Yangtze River. Hum Genet. 2007b;122:383–388. doi: 10.1007/s00439-007-0407-2. [DOI] [PubMed] [Google Scholar]
- Li H, Mukherjee N, Soundararajan U, Tárnok Z, Barta C, Khaliq S, Mohyuddin A, Kajuna SLB, Mehdi SQ, Kidd JR, Kidd KK. Geographically separate increases in the frequency of the derived ADH1B*48His allele in eastern and western Asia. Am J Hum Genet. 2007c;81:842–846. doi: 10.1086/521201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Gu S, Cai X, Speed WC, Pakstis AJ, Golub EI, Kidd JR, Kidd KK. Ethnic Related Selection for an ADH Class I Variant within East Asia. PLoS ONE. 2008;3:e1881. doi: 10.1371/journal.pone.0001881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Kidd KK. Low Allele Frequency of ADH1B*47His in West China and Different ADH1B Haplotypes in Western and Eastern Asia. Am J Hum Genet. 2009;84:92–94. [Google Scholar]
- Liu H, Prugnolle F, Manica A, Balloux F. A Geographically Explicit Genetic Model of Worldwide Human-Settlement History. Am J Hum Genet. 2006;79:230–237. doi: 10.1086/505436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Marmaro J, Todd JA. Towards fully automated genome-wide polymorphism screening. Nat Genet. 1995;9:341–342. doi: 10.1038/ng0495-341. [DOI] [PubMed] [Google Scholar]
- Matsuo K, Hiraki A, Hirose K, Ito H, Suzuki T, Wakai K, Tajima K. Impact of the Alcohol-Dehydrogenase (ADH) 1C and ADH1B polymorphisms on drinking behavior in nonalcoholic Japanese. Hum Mutat. 2007;28:506–510. doi: 10.1002/humu.20477. [DOI] [PubMed] [Google Scholar]
- Mellars P. Going East: New Genetic and Archaeological Perspectives on the Modern Human Colonization of Eurasia. Science. 2006;313:796–800. doi: 10.1126/science.1128402. [DOI] [PubMed] [Google Scholar]
- Osier MV, Pakstis AJ, Kidd JR, Lee JF, Yin SJ, Ko HC, Edenberg HJ, Lu RB, Kidd KK. Linkage disequilibrium at the ADH2 and ADH3 loci and risk of alcoholism. Am J Hum Genet. 1999;64:1147–1157. doi: 10.1086/302317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pääbo S, Poinar H, Serre D, Jaenicke-Despres V, Hebler J, Rohland N, Kuch M, Krause J, Vigilant L, Hofreiter M. Genetic analyses from ancient DNA. Annu Rev Genet. 2004;38:645–679. doi: 10.1146/annurev.genet.37.110801.143214. [DOI] [PubMed] [Google Scholar]
- Schurr TG, Sherry ST. Mitochondrial DNA and Y chromosome diversity and the peopling of the Americas: evolutionary and demographic evidence. Am J Hum Biol. 2004;16:420–439. doi: 10.1002/ajhb.20041. [DOI] [PubMed] [Google Scholar]
- Shi YF, Cui ZJ, Li JJ. Quaternary glacier in Eastern China and the climate fluctuation. Beijing: Science Press; 1989. [Google Scholar]
- Smith M. Genetics of Human Alcohol and Aldehyde Dehydrogenases. In: Harris H, Hirschhorn K, editors. Advances in Human Genetics. Vol. 15. New York, NY: Plenum Press; 1986. [DOI] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Donnelly P. A comparison of Bayesian methods for haplotype reconstruction. Am J Hum Genet. 2003;73:1162–1169. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su B, Xiao J, Underhill P, Deka R, Zhang W, Akey J, Huang W, Shen D, Lu D, Luo J, Chu J, Tan J, Shen P, Davis R, Cavalli-Sforza L, Chakraborty R, Xiong M, Du R, Oefner P, Chen Z, Jin L. Y-Chromosome evidence for a northward migration of modern humans into Eastern Asia during the last Ice Age. Am J Hum Genet. 1999;65:1718–1724. doi: 10.1086/302680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson R, Pritchard JK, Shen P, Oefner PJ, Feldman MW. Recent common ancestry of human Y chromosomes: evidence from DNA sequence data. Proc Natl Acad Sci U S A. 2000;97:7360–7365. doi: 10.1073/pnas.97.13.7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triano EA, Slusher LB, Atkins TA, Beneski JT, Gestl SA, Zolfaghari R, Polavarapu R, Frauenhoffer E, Weisz J. Class I Alcohol Dehydorgenase is highly expressed in normal human mammary epithelium but not in invasive breast cancer: Implications for breast carcinogenesis. Cancer Res. 2003;63:3092–3100. [PubMed] [Google Scholar]
- Wilson IJ, Weale ME, Balding DJ. Inferences from DNA data: population histories, evolutionary processes and forensic match probabilities. J R Stat Soc Ser A Stat Soc. 2003;166:155–188. [Google Scholar]
- Zegura SL, Karafet TM, Zhivotovsky LA, Hammer MF. High-resolution SNPs and microsatellite haplotypes point to a single, recent entry of Native American Y chromosomes into the Americas. Mol Biol Evol. 2004;21:164–175. doi: 10.1093/molbev/msh009. [DOI] [PubMed] [Google Scholar]
- Zhang F, Su B, Zhang YP, Jin L. Genetic studies of human diversity in East Asia. Philos Trans R Soc Lond B Biol Sci. 2007;362:987–995. doi: 10.1098/rstb.2007.2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.