

Abstract
Transforming growth factor-β (TGF-β) is a potent pleiotropic cytokine that regulates mammalian development, differentiation, and homeostasis in essentially all cell types and tissues. TGF-β normally exerts anticancer activities by prohibiting cell proliferation, and by creating cell microenvironments that inhibit cell motility, invasion and metastasis. However, accumulating evidence indicates the process of tumorigenesis, particularly that associated with metastatic progression, confers TGF-β with oncogenic activities, a functional switch known as the “TGF-β Paradox.” The molecular determinants governing the “TGF-β Paradox” are complex and represent an intense area of investigation by researchers in academic and industrial settings. Recent findings link genetic and epigenetic events in mediating the acquisition of oncogenic activity by TGF-β, as does aberrant alterations within tumor microenvironments. These events coalesce to enable TGF-β to direct metastatic progression via the stimulation of epithelial-mesenchymal transition (EMT), which permits carcinoma cells to abandon polarized epithelial phenotypes in favor of apolar mesenchymal-like phenotypes. Attempts to deconstruct the EMT process induced by TGF-β have identified numerous signaling molecules, transcription factors, and microRNAs operant in mediating the initiation and resolution of this complex transdifferentiation event. Besides its ability to enhance carcinoma cell invasion and metastasis, EMT also engenders transitioned cells with stem-like properties, including the acquisition of self-renewal and tumor-initiating capabilities coupled to chemoresistance. Here we review recent findings that delineate the pathophysiological mechanisms whereby EMT stimulated by TGF-β promotes metastatic progression and disease recurrence in human carcinomas.
Keywords: Cancer stem cells, Chemoresistance, Epithelial-mesenchymal transition, Integrins, Metastasis, Transforming growth factor-β, Tumor microenvironment
Introduction
Transforming growth factor-β (TGF-β) is a ubiquitously expressed cytokine that regulates an assortment of biological activities in essentially all cell types and tissues. Besides its role in regulating cell development, differentiation, and survival, TGF-β also inhibits the proliferation of epithelial, endothelial, and hematopoietic cell lineages (Blobe, et al., 2000, Massague, 2008, Tian, et al., 2011). Interestingly, resistance to TGF-β-mediated cytostasis is a hallmark of neoplastic transformation, which ultimately transforms the signals produced by this cytokine into oncogenic activities, particularly enhanced cancer cell invasion and metastasis. This conversion in TGF-β function is known as the “TGF-β Paradox” (Rahimi and Leof, 2007, Schiemann, 2007, Tian and Schiemann, 2009), which underlies the adverse prognosis associated with elevated TGF-β levels in developing carcinomas, including their acquisition of EMT, metastatic, and chemoresistant phenotypes (Taylor, et al., 2010, Tian and Schiemann, 2009, Wendt, et al., 2009a). The molecular mechanisms whereby TGF-β suppresses tumor formation remain to be fully elucidated, as does the manner in which this cytokine promotes the progression of late-stage carcinomas. Historically, investigations into TGF-β action have applied “cell-centric” approaches to interrogate the “TGF-β Paradox,” doing so by monitoring the differential expression of gene transcripts, proteins, and microRNAs that exist between normal and malignant cells as a potential means to explain the dichotomous functions of TGF-β. Although enormous strides have been made through these analyses (Kang, et al., 2003, Minn, et al., 2005, Zavadil, et al., 2001), these findings have collectively failed to explain the contradictory nature of TGF-β, nor have they accurately recapitulated the “TGF-β Paradox” in experimental settings. Due to these limitations, recent studies have begun to investigate the impact that dynamic alterations in carcinoma microenvironments play in dictating the behaviors of TGF-β. Indeed, numerous findings point to a prominent role of EMT in manifesting the “TGF-β Paradox” and its ability to couple TGF-β to metastatic progression in human carcinomas. Here we review recent findings that directly impact our understanding of the role of TGF-β in mediating the initiation and resolution of EMT, and how these events underlie the pathophysiology of TGF-β in human malignancies.
TGF-β signaling
Mammals express three genetically distinct TGF-β ligands (e.g., TGF-βs 1–3) whose mature and biologically active forms are ~97% identical and exhibit virtually indistinguishable actions in vitro (Blobe, Schiemann and Lodish, 2000, Massague, 1998). Individual TGF-β ligands are expressed spatiotemporally during embryogenesis and tissue morphogenesis, events contributing to the vast array of diverse and nonredundant phenotypes displayed by mice lacking distinct TGF-β isoforms (Chang, et al., 2002). TGF-β signaling commences by its binding to three high-affinity receptors, TGF-β type I (TβR-I), type II (TβR-II), and type III (TβR-III or betaglycan) receptors. When expressed, TβR-III is the most abundant TGF-β receptor present on the cell surface where it (i) functions as an accessory receptor that binds and modulates TGF-β function in responsive cells, and (ii) mediates the tumor suppressing activities of TGF-β in tissues of the breast, ovary, prostate, lung, pancreas, kidney, and endometrium (Gatza, et al., 2010). Although TβR-III lacks intrinsic enzymatic activity, TβR-I and TβR-II both harbor Ser/Thr protein kinases in their cytoplasmic domains that initiate intracellular signaling (Feng and Derynck, 2005, Massague and Gomis, 2006, Shi and Massague, 2003). The binding of TGF-β to TβR-II enables this polypeptide to transphosphorylate and activate TβR-I, which subsequently binds, phosphorylates, and stimulates the latent transcription factors, Smad2 and Smad3 (Feng and Derynck, 2005, Massague and Gomis, 2006, Shi and Massague, 2003). Phosphorylated Smad2/3 rapidly form high order complexes with the common Smad, Smad4, which enables the resulting heterotrimeric complexes to take up residence in the nucleus where they regulate transcription in a gene- and cell-specific manner (Feng and Derynck, 2005, Massague and Gomis, 2006, Shi and Massague, 2003). Changes in cell behavior regulated by the activation of Smad2/3 are referred to as “canonical TGF-β signaling” and these events are modulated in all subcellular compartments by numerous effector molecules.
In addition to its activation of canonical Smad2/3 signaling, TGF-β also couples to a variety of noncanonical effector molecules (i.e., Smad2/3-independent), including the (i) MAP kinases, ERK1/2, p38MAPK, and JNK; (ii) cell survival mediators, PI3K, AKT1/2, and mTOR; (iii) inflammation mediators, NF-κB, Cox-2, and prostaglandins; (iv) small GTP-binding proteins, Ras, RhoA, Rac1, and Cdc42; and (v) nonreceptor protein tyrosine kinases, Src, FAK, and Abl (Kang, et al., 2009, Moustakas and Heldin, 2005, Parvani, et al., 2011). Interestingly, recent findings indicate that the coupling of TGF-β to its noncanonical effectors appears inappropriately amplified in metastatic cancer cells, thereby generating a signaling imbalance that overrides and/or dampens the tumor suppressing messages transduced by Smad2/3 in human tumors (Wendt, et al., 2009b). Figure 1 depicts the interactome of noncanonical effector molecules targeted by TGF-β during EMT and metastatic progression. Unfortunately, precisely how these noncanonical effectors are activated by TGF-β in normal and malignant cells remains to be determined definitively, as does the manner in which these pathways become dysregulated and magnified during the acquisition of metastatic phenotypes by cancer cells. These relationships and their regulation by tumor microenvironments during the initiation of EMT and metastatic progression driven by TGF-β are discussed below.
Fig. 1.

The EMT signaling network and transcriptome regulated by TGF-β. Red spheres represent genes whose expression is increased, while Green spheres signify genes whose expression is decreased. Orange spheres depict increased enzymatic activity stimulated by TGF-β during EMT. Lines between nodes indicate a described interaction or transactivation between those molecules during EMT.
Epithelial-mesenchymal transitions (EMTs)
Definition and classical EMT phenotypes
EMT is a normal physiological process that is essential for embryogenesis and tissue morphogenesis, and for tissue remodeling and repair during wound healing (Wendt, Allington and Schiemann, 2009a). However, pathological EMT is increasingly recognized to play an important role during the development of human diseases, including chronic inflammation, fibrosis, rheumatoid arthritis, and cancer invasion and metastasis (Micalizzi, et al., 2010a, Taylor, Parvani and Schiemann, 2010, Thiery, et al., 2009). EMT was first described almost a century ago with the observation that epithelial cells in chick embryos acquire mesenchymal-like features (Thiery, 2002), an event officially recognized as being a discrete physiological process by Greenberg and Hay in 1982 (Greenburg and Hay, 1982). The ability of TGF-β to induce EMT was initially described by Derynck and colleagues in 1994 (Miettinen, et al., 1994). During the intervening years since this important discovery, the findings of numerous studies have coalesced in establishing TGF-β as a master regulator of the initiation and resolution of EMT under a variety of pathophysiological contexts (Taylor, et al., 2010, Wendt, et al., 2009a). Typically, differentiated epithelium is comprised of a single layer of polarized epithelial cells that possess an apical and basolateral orientation, and an actin cytoskeleton organized into defined “cobblestones” due to the concentration of polymerized actin fibers at cell-cell junctions. The EMT process occurs when fully differentiated epithelial cells undergo a transdifferentiation process that gives rise to their bearing fibroblastoid-like phenotypes (Micalizzi, et al, 2010a, Taylor, et al., 2010, Thiery, et al, 2009). Additionally, transdifferentiating epithelial cells exhibit a variety of common molecular features, including (i) downregulated expression of polarized epithelial cell markers (e.g., E-cadherin, ZO-1, β4 integrin, and cytokeratin 19); (ii) rearrangements of the actin, intermediate filaments, and myosin cytoskeletal systems; (iii) redistribution of intracellular organelles; and (iv) upregulated expression of mesenchymal cell markers (e.g., N-cadherin, vimentin, and α-smooth muscle actin) and invasive factors (e.g., MMPs and extracellular matrix molecules) (Micalizzi, et al., 2010a, Taylor et al., 2010, Thiery, et al., 2009). Figure 2 depicts these events and highlights many of the most established and best characterized markers of EMT, including (i) E- and N-cadherins (Bhowmick, et al., 2001a, Hazan, et al., 2000, Miettinen, et al., 1994); (ii) vimentin (Grunert, et al., 2003, Mendez, et al., 2010); (iii) fibronectin (Ignotz and Massague, 1986); (iv) α-smooth muscle actin (Masszi, et al., 2003, Yazhou, et al., 2004); (v) matrix metalloproteinases (Duivenvoorden, et al., 1999, Kim, et al., 2007a, Lee, et al., 2008); and (vi) β3 integrin (Galliher and Schiemann, 2006, Wendt and Schiemann, 2009).
Fig. 2.
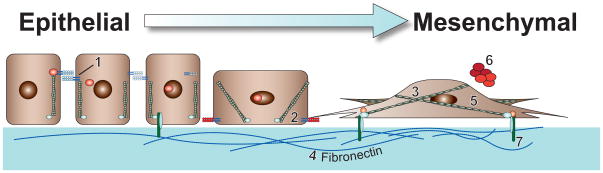
Transdifferentiation from polarized epithelial-like to mesenchymal-like morphologies. Provided numbers depict classical biomarkers of initiated EMT programs, while accompanying boxes describe the EMT-related functions of these highlighted markers. (1) E-cadherin, the primary component of adherens junctions. During EMT, TGF-β represses E-cadherin expression, as well as induces its internalization from the plasma membrane (Miettinen, et al., 1994). (2) N-cadherin, an adhesion molecule that promotes cellular migration (Hazan, et al., 2000). EMT stimulated by TGF-β is associated with increased expression of N-cadherin (Bhowmick, et al., 2001a). (3) Vimentin, an intermediate filament protein that is expressed in all primitive cell types, but not in differentiated epithelial cells. Vimentin may function to drive EMT programs (Mendez, et al., 2010), and also serves as a canonical marker for detecting transdifferentiated mesenchymal cell types (Grunert, et al., 2003). (4) Fibronectin, a critical ECM component whose elevated production by cancer cells is classically associated with EMT programs. TGF-β is a potent inducer of fibronectin production and deposition into the ECM (Ignotz and Massague, 1986). (5) α-Smooth Muscle Actin (α-SMA), a major component of contractile microfilaments and a canonical marker for detecting myofibroblasts. TGF-β stimulation of EMT elicits α-SMA expression (Masszi, et al., 2003), which strongly associates with increased tumor invasion, and with decreased patient survival (Yazhou, et al., 2004). (6) Matrix Metalloproteinases (MMPs), proteolytically degrade basement membranes to allow primary tumor cells to invade the surrounding tissue and intrasvasate into tumor associated vasculature. While several MMPs are induced during EMT, MMP-9 is a well-established target of TGF-β signaling (Duivenvoorden, et al., 1999, Kim, et al., 2007a, Lee, et al., 2008). (7) β3 integrin, a transmembrane protein that physically links the extracellular matrix to intracellular signaling systems and the cytoskeleton via focal adhesion complexes. β3 integrin is rapidly and robustly upregulated by TGF-β and interacts physically with TβR-II via a FAK-dependent mechanism (Galliher and Schiemann, 2006, Wendt and Schiemann, 2009).
An important feature of the EMT process manifests through the disintegration and disassembly of epithelial cell-cell adhesion complexes, including desmosomes and gap, tight, and adherens junctions that function in maintaining epithelial cell polarity and restricting cell motility (Cavallaro and Christofori, 2004, Christofori, 2003). Inactivating the expression and activity of these junctional complexes enables transitioning epithelial cells to abandon their immotile and polarized architectures and assume highly motile and apolar architectures characteristic of mesenchymal-like cells (Fig. 2). For example, tight junctions arise from the collective actions of claudins, occludins, and junctional adhesion molecules (JAMs), which are connected to the actin cytoskeleton via the scaffold proteins zonula occluden (ZO)-1, -2, and -3 (Ebnet, et al., 2004, Schneeberger and Lynch, 2004). Par6 (partitioning-defective 6) also plays an essential role in overseeing the (i) formation of tight junctions; (ii) generation of apical-basolateral polarity; and (iii) initiation of polarized cell migration (Bose and Wrana, 2006). Par6 also interacts physically with TGF-β receptors and can be phosphorylated by TβR-II, leading to the formation of Par6:Smurf1 complexes that promote the ubiquitination and degradation of RhoA (Ozdamar, et al., 2005). These events culminate in the dissolution of tight junctions during EMT stimulated by TGF-β. Moreover, uncoupling the ability of TGF-β to regulate Par6 activity is sufficient to prevent EMT induced by TGF-β (Ozdamar, et al., 2005), as well as its stimulation of breast cancer metastasis in mice (Viloria-Petit, et al., 2009).
In contrast to tight junctions, adherens-based junctional complexes are comprised primarily of the transmembrane protein, epithelial cadherin (E-cad), which connects to the actin cytoskeleton via α- and β-catenins (Niessen, 2007). TGF-β stimulation of EMT inactivates E-cad function by repressing the synthesis of E-cad transcripts, or by delocalizing and internalizing Ecad proteins from the cell membrane, an event coupled to a loss of Rac1 activity (Takaishi, et al., 1997). The dissolution of cell-cell contacts accompanies the formation of prominent actin filaments and the appearance of fibroblastoid-like phenotypes in transitioning epithelial cells. The ability of TGF-β to remodel the actin cytoskeleton transpires through the activation of RhoA (Bhowmick, et al., 2001a, Ridley and Hall, 1992), which together with diminished cell adhesion elicits cell migration and invasion. TGF-β also modulates the adhesion of transitioning epithelial cells by stimulating their expression of matrix metalloproteinases (MMPs), while simultaneously inhibiting that of their inhibitors, the tissue inhibitors of metalloproteinases (TIMPs), which collectively leads to the degradation of various basement membrane constituents such as collagen IV and laminin (Duivenvoorden, et al., 1999). Collectively, these phenotypic and morphologic changes conspire to transform immobile multicellular epithelial sheets into highly motile, independent units that are poised for invasion and metastasis.
Classifying EMT subtypes
The process of EMT has recently been categorized into three distinct subtypes based the biological settings and contexts in which the EMT event is activated. Importantly, the generic features attributed to EMT are readily observed in all of the three major EMT subtypes. For instance, type 1 EMT is activated during embryogenesis and tissue morphogenesis, while type 2 EMT is initiated during tissue regeneration, wound healing, and inflammation-driven fibrosis. Finally, type 3 EMT is employed by carcinoma cells during their acquisition of invasive and metastatic phenotypes (Kalluri and Weinberg, 2009). In the following sections, we highlight evidence linking TGF-β to the regulation of individual EMT subtypes, particularly that of type 3 EMT during metastatic progression stimulated by TGF-β.
Type 1 EMT
Type 1 EMT is synonymous with developmental EMT, and as such, this EMT subtype transpires during embryogenesis to facilitate the faithful synthesis of organs and tissues. Initiation of type 1 EMT promotes gastrulation and the subsequent formation of ectodermal, mesodermal, and endodermal tissues from the invaginating primitive streak (Micalizzi and Ford, 2009, Yang and Weinberg, 2008). In conjunction with the Wnt signaling pathway, primitive streak cells readily undergo EMT when stimulated by the TGF-β superfamily members, Nodal, Vg1, and BMP-2 (Chea, et al., 2005, Conlon, et al., 1994, Raible, 2006, Skromne and Stern, 2001, Zhou, et al., 1993). Type 1 EMT is also initiated during neurulation to facilitate neural tube formation during spinal and cerebral development, and again by neural crest cells to generate the adrenal medulla and the skeletal and peripheral nervous systems (Sauka-Spengler and Bronner-Fraser, 2008). Additional insights associating TGF-β signaling to developmental EMT programs have been gleaned from genetically engineering mice to lack expression of individual TGF-β isoforms and their receptors. For example, homozygous deletion of TGF-β2 elicits perinatal lethality due to multiple defects in the initiation of type 1 EMT during organogenesis and tissue morphogenesis, particularly that linked to atrioventricular valve formation by transitioned endocardial cells (Sanford, et al., 1997). Likewise, disruption of the TGF-β3 locus also produces perinatal lethality resulting from defective type 1 EMT that occurrs during lung and palate development (Kaartinen, et al., 1995). Finally, the expression and activity of TβR-III are essential for TGF-β stimulation of type 1 EMT necessary for normal heart and secondary palate formation (Brown, et al., 1999, Nakajima, et al., 2007).
Type 2 EMT
Type 2 EMT plays an essential role in governing tissue homeostasis via its ability to induce the healing and remodeling of wounded tissues. Unlike type I EMT, transdifferentiation initiated by type 2 EMT is driven by inflammatory reactions, whose termination is sufficient to resolve the EMT event following wound repair (Kalluri and Weinberg, 2009). The series of events that underlie type 2 EMT during wound healing and tissue remodeling are highly complex and require the coordinated interactions between multiple cell types, such as fibroblasts, endothelial, and immune cells, to facilitate the re-epithelialization of damaged epithelium. TGF-β plays a key role in mediating these events, doing so in part by promoting myofibroblast activation and differentiation coupled to extracellular matrix (ECM) remodeling necessary for tissue repair (Taylor, et al., 2010). Interestingly, chronic inflammatory reactions can trigger a dysregulated type 2 EMT response that engenders the formation of a variety of fibroproliferative disorders, particularly in the lung, liver, kidney, and retina (Wynn, 2007, Wynn, 2008). Along these lines, inhibiting collagen cross-linking during mammary fibrosis significantly reduces breast cancer metastasis in mice (Levental, et al., 2009), findings consistent with the notion that desmoplastic and fibrotic reactions enhance the incidence of mammary tumorigenesis (Boyd, et al., 2007). Thus, chemotherapeutic targeting of the TGF-β signaling system may abrogate neoplastic progression by simultaneously inactivating the deleterious activities associated with pathologic type 2 and type 3 EMTs (see below).
Type 3 EMT
Type 3 EMT is synonymous with oncogenic EMT and is essential in facilitating the acquisition of invasive and metastatic phenotypes by developing neoplasms (Micalizzi, et al., 2010a, Taylor, et al., 2010, Thiery, et al., 2009). Oncogenic EMTs are readily distinguished from their normal counterparts by the fact that these events transpire in malignant cells that harbor a host of genetic and epigenetic abnormalities that coalesce in hijacking the EMT process during metastatic dissemination. Importantly, EMT stimulated by TGF-β has been linked to the selection and expansion of cancer stem cells (CSCs) (Ben-Porath, et al., 2008, Mani, et al., 2008, Morel, et al., 2008, Shipitsin, et al., 2007), and by extension, to the development of chemoresistance and disease recurrence (Frank, et al., 2010, Singh and Settleman, 2010). Even more remarkably, recent findings have implicated EMT as the molecular determinant that manifests the “TGF-β Paradox” and the conversion of TGF-β function from that of a tumor suppressor to an inducer of metastatic progression. In the succeeding sections, we present evidence linking EMT to the acquisition of oncogenic TGF-β signaling, as well as provide an overview of how TGF-β stimulates type 3 EMT in developing neoplasms.
The “TGF-β Paradox”
The initial framework underlying the “TGF-β Paradox” began nearly 30 years ago when TGF-β was originally defined by its ability to promote the morphological transformation and anchorage-independent growth of normal rat kidney fibroblasts (NRK-49 cells; (Moses, et al., 1981, Roberts, et al., 1981). Shortly thereafter, contradicting findings obtained in epithelial cells established TGF-β as a potent and powerful inhibitor of cell proliferation (Carr, et al., 1986). Thus, TGF-β stimulates the growth of mesenchymal cell lineages, but inhibits that of epithelial cell lineages. Interestingly, studies from this same era demonstrated that transformed cells secrete 40-fold more TGF-β as compared to their nontransformed counterparts (Anzano, et al., 1985). This important observation strongly suggested that cellular transformation accompanies a loss in the tumor suppressing functions of TGF-β. The merits of this supposition were quickly translated and verified in mouse models of tumorigenesis. For instance, implanting slow-release TGF-β pellets in female mice reversibly inhibits mammary gland growth and morphogenesis (Silberstein and Daniel, 1987). In contrast, inoculating neutralizing TGF-β antibodies enhances the ability of natural killer cells to eradicate breast cancer development and progression in mice (Arteaga, et al., 1993). A specific role for TGF-β in promoting metastasis was established by conditional expression of constitutively-active TGF-β1, which selectively enhances the pulmonary metastasis of mammary tumors without altering their growth and proliferation (Muraoka-Cook, et al., 2004). Likewise, transgenic expression of constitutively-active TGF-β1 in mouse keratinocytes promotes their proliferation and eventual progression to spindle cell carcinoma in a chemical carcinogenesis model of skin cancer (Cui, et al., 1996, Fowlis, et al., 1996). More recently, conditionally deleting TβR-I expression in the oral cavity of mice was observed to facilitate carcinogen-induced tumor formation in part via constitutive PI3K/AKT activity (dBian, et al., 2009), while combining TβR-I haploinsufficiency onto a an Apc(Min/+) background produces twice as many intestinal tumors as compared to wild-type mice (Zeng, et al., 2009). Collectively, these and other findings establish a paradigm whereby TGF-β potently inhibits tumor initiation and the development of early-stage carcinomas, but enthusiastically drives the metastatic progression of late-stage carcinomas.
In addition to its ability to induce cytostasis in epithelial cells, TGF-β also governs the behaviors of adjacent fibroblasts and their synthesis and secretion of paracrine factors and ECM molecules that collectively suppress carcinoma development. For instance, targeted deletion of TβR-II in mammary carcinoma cells facilitated the inappropriate activation of two distinct paracrine signaling axes – i.e., SDF-1:CXCR4 and CXCL5:CXCR2 – which led to the recruitment of immature GR1+CD11b+ myeloid cells that drive breast cancer metastasis by inhibiting host tumor immunosurveillance, and by inducing MMP expression (Yang, et al., 2008). Likewise, rendering fibroblasts deficient in TβR-II expression elicited prostate intraepithelial neoplasia and invasive carcinoma of the forestomach resulting from disruptions in tumor suppressing paracrine signaling networks (Bhowmick, et al., 2004). Moreover, similar inactivation of TβR-II in mammary fibroblasts greatly exaggerates the growth and invasion of breast cancer cells simultaneously engrafted under the renal capsule. The proliferation promoting activities of TβR-II-deficient fibroblasts derive from their upregulated expression of TGF-α, MSP (macrophage-stimulating protein), and HGF (hepatocyte growth factor) (Cheng, et al., 2005). Finally, genetic inactivation of Smad4 in T cells elicits carcinoma formation within the gastrointestinal track (e.g., colon, rectum, intestine, and stomach) due to aberrant stromal expansion and signaling (Kim, et al., 2006). Collectively, these findings highlight the paradoxical activities of TGF-β in normal and malignant cells, and in doing so, point to the potential difficulties in developing effective therapies capable of predictably circumventing the context- and stage-specific behaviors of this cytokine in neoplastic tissues.
Mechanisms underlying type 3 EMT driven by TGF-β
TGF-β and transcriptional regulators of EMT
Attempts to deconstruct the EMT process have identified a variety of transcription factors that mediate essential functions in activating the “EMT transcriptome” necessary to bring about epithelial transdifferentiation process (Table 1). Indeed, heterologous expression of the transcription factor Twist, which fulfills a critical role during mesoderm development (Leptin, 1991), results in the initiation of the EMT program. Consistent with a role for EMT in driving carcinoma metastasis, rendering metastatic breast cancer cells deficient in Twist expression abrogates their ability to metastasize (Yang, et al., 2004). Snail-1 is similarly required during mesoderm development and its expression readily represses that of E-cad (Cano, et al., 2000). More recently, Snail-2 (also know as Slug) was identified as a downstream effector of Twist that plays an essential role in facilitating Twist-mediated downregulation of E-cad (Casas, et al., 2011, Mani, et al., 2008). In addition to inducing the expression of Twist (Allington, et al., 2009, Lee, et al., 2008, Mani, et al., 2008), TGF-β also couples to the expression of high mobility group A2 (HMGA2), leading to robust downregulation of E-cad expression concomitant with the induction of Snail-1, Snail-2, and Twist (Thuault, et al., 2006). Along these lines, the findings of several studies support the notion that E-cad plays an active role in maintaining differentiated and polarized epithelial phenotypes, and in fact, E-cad-deficiency is sufficient to elicit a robust EMT in mammary epithelial cells (Battula, et al., 2010, Mani, et al., Weinberg, 2008, Onder, et al., 2008, Taube, et al., 2010). Interestingly, recent evidence suggests that the expression and activity of these EMT transcription factors elicits a “feed forward” response coupled to increased autocrine TGF-β production and signaling (Taube, et al., 2010). Elucidating the molecular mechanisms operant in mediating autocrine TGF-β signaling and its stabilization of the EMT process may shed new light in better understanding the differences between physiological (i.e. Types 1 and 2) and pathological (type 3) EMT. Indeed, prolonged induction of EMT results in the recruitment of DNA methyltransferases and chromatin remodeling enzymes to the promoters of EMT-regulated genes targeted by TGF-β, including estrogen receptor-α and E-cad (Dumont, et al., 2008). Moreover, inhibiting canonical Smad2 signaling either by inactivating its expression or by overexpressing the inhibitory Smad, Smad7, was sufficient to induce a mesenchymal-epithelial transition (MET) initiated by the loss of epigenetic silencing of epithelial-associated genes (Papageorgis, et al., 2010). Collectively, these findings provide an initial framework to begin delineating the sequential nature of events associated with EMT induced by TGF-β, and as well as its ability to dictate epigenetic silencing as a means to stabilize and maintain oncogenic EMT programs.
Table 1.
Master Regulators of TGF-β-induced EMT
| Transcriptional Regulators | Gene target |
|---|---|
| Twist | Snail-2, TGF-β (Taube et al. 2010) |
| Snail | E-cad (Cano et al. 2000), ER-α (Dhasarathy, et al., 2007) |
| HMG2a | Snail-1, Snail-2, Twist (Thuault et al. 2006) |
| Snail-2 | E-cad (Hajra, et al., 2002) |
| Zeb1/2 | E-cad (Comijn, et al., 2001) |
| Dnmt1 | E-cad (Papageorgis et al. 2010) |
| Six1 | TβR-I (Micalizzi et al., 2010b) |
| FOXC2 | Vim*, Fibro*, α-SMA*, N-Cad (Mani, et al., 2007) |
|
| |
| MicroRNA | Transcript target |
|
| |
| miR-200c | ZEB1, ZEB2 (Gregory et al. 2008) |
| miR-9 | E-cad (Ma et al. 2010) |
| miR-155 | RhoA (Kong et al. 2008) |
| miR-21 | Tiam1 (Cottonham et al. 2010, Davis et al. 2008 Zavadil et al. 2007) |
α-Smooth muscle actin; Fibronectin; Vimentin
TGF-β and microRNAs
Recent evidence has associated the expression of microRNAs (miRs) to the initiation of the “TGF-β Paradox” and its promotion of oncogenic TGF-β signaling (Table 1). For instance, EMT induced by TGF-β downregulates expression of the miR-200 family of microRNAs, which facilitates the expression of ZEB1/2 and their repression of E-cad expression in transitioning cells (Gregory, et al., 2008, Korpal, et al., 2008). Moreover, the absence of miR-200 expression has predictive value in identifying highly invasive and metastatic carcinomas (Burk, et al., 2008, Singh, et al., 2008). Interestingly, loss of miR-220c expression has been linked to inactivation of p53 function, an event coupled to the acquisition of EMT and stem cell-like phenotypes (Chang, et al., 2011). Along these lines, we recently identified c-Abl as a potent suppressor of EMT stimulated by TGF-β (Allington, et al., 2009), an event that transpires via c-Abl-mediated reactivation of p53 expression and its induction of p21 (T.M.A. and W.P.S., unpublished observation). Likewise, elevated ZEB1/2 expression and accompanying EMT are sufficient to overcome senescence induced by either p53 activation or EGFR overexpression (Ohashi, et al., 2010). In addition to its regulation of the miR-220 family of microRNAs, TGF-β also stimulates the processing of primary miR-21 transcripts into their pre-miR-21 counterparts, an event requiring the formation of Smad2/3:DROSHA complexes. Interestingly, these miR maturation events transpire normally in Smad4-deficient cells (Davis, et al., 2008), thereby identifying a novel bifurcation in the canonical TGF-β signaling system that couples specifically to miR processing events. TGF-β stimulation of miR-21 and miR-31 expression serve in promoting EMT programs coupled to cell migration and invasion, presumably via downregulated expression of the guanine exchange factor, Tiam1 (Cottonham, et al., 2010, Zavadil, et al., 2007). The induction of miR-155 expression by TGF-β proceeds through a Smad4-dependent pathway and is necessary in mediating downregulated RhoA expression during EMT reactions (Kong, et al., 2008). Finally, Myc amplification has recently been shown to induce miR-9 expression, which enhances breast cancer metastasis by downregulating E-cad expression (Ma, et al., 2010). Although a direct role for TGF-β in regulating these events was not examined, these findings nonetheless may provide new insights into how aberrant Myc expression overrides the cytostatic actions of TGF-β (Alexandrow, et al., 1995, Chen, et al., 2001). Figure 1 depicts the relationship between various microRNAs and TGF-β effector molecules operant in eliciting EMT programs.
Impact of integrins during EMT induced by TGF-β
As discussed above, variations in the composition of the ECM can elicit profound alterations in how normal and malignant cells respond to TGF-β. Mechanistically, these disparate functions of TGF-β have been linked to the expression and activity originating from integrins. For example, we observed TβR-II to interact physically with β3 integrin following its activation by vitronectin, or its elevated expression that transpires during EMT induced by TGF-β (Galliher and Schiemann, 2006, Galliher and Schiemann, 2007, Galliher-Beckley and Schiemann, 2008). Additionally, the formation of β3 integrin:TβR-II complexes is critically dependent upon the expression and activity of FAK, such that depleting FAK expression disrupts the interaction between these receptors and markedly reduces the ability of TGF-β to stimulate breast cancer invasion and metastasis (Wendt and Schiemann, 2009). Interestingly, the FAK effector, p130Cas, was found to bind and sequester Smad3, thereby preventing its phosphorylation by TβR-I and subsequent translocation into the nucleus (Kim, et al., 2008). Along these lines, we observed elevated p130Cas expression to mark metastatic progression, as well as to distort the delicate balance between canonical and noncanonical TGF-β signaling systems in metastatic breast cancers (Wendt, et al., 2009b). Indeed, rendering metastatic breast cancer cells deficient in p130Cas expression drastically reduces breast cancer development and progression stimulated by TGF-β (Wendt, et al., 2009b), presumably by enhancing the sensitivity of p130Cas-deficient cells to apoptotic stimuli (Cabodi, et al., 2006). Interestingly, TβR-II also interacts physically with β1 integrin (Galliher and Schiemann, 2006), which is required for TGF-β to induce EMT and activate p38 MAPK (Bhowmick, et al., 2001b). Taken together, these findings highlight the direct influence integrins and focal adhesion complexes play in promoting EMT and metastatic progression stimulated by TGF-β.
Additional insights into the role of integrins in regulating normal and malignant cell responses to TGF-β have been gleaned through the use of 3D-organotypic culture systems. For instance, inactivating β4 integrin function in normal mammary epithelial cells elicits their formation of dysmorphic acinar structures reminiscent of those produced by malignant mammary epithelial cells. (Weaver, et al., 2002). Conversely, inactivating β1 integrin function in breast cancer cells elicits their formation of spherical acinar structures reminiscent of normal mammary epithelial cells (Weaver, et al., 1997). More recently, collagen has been incorporated within these 3D-organotypic cultures as a means to better recapitulate the microenvironmental compliance experienced by palpable primary tumors, and to initiate mechanotransduction pathways coupled to integrin activation (Paszek, et al., 2005). In undertaking similar analyses, we observed compliant 3D-organotypic cultures to be sufficient in restoring the cytostatic activities of TGF-β in metastatic breast cancer cells that are normally completely resistant to the anti-proliferative activities of TGF-β in traditional 2D-culture systems (Allington, et al., 2009, Taylor, et al., 2011). Importantly, inclusion of type I collagen in these 3D-organotypic cultures to increase their rigidity (i.e., decreased compliance) and initiate mechanotransduction was observed to dose-dependently uncouple TGF-β from the regulation of cell cycle progression (Allington, et al., 2009). Conversely, treating developing organoids with the small molecule TβR-I inhibitor (TβR-I Inhibitor II) stimulated the growth of 4T1 cells in compliant 3D-organotypic cultures wherein TGF-β functions as a tumor suppressor. However, administering this same pharmacologic regimen to rigid 3D-organotypic cultures wherein TGF-β functions as a tumor promoter greatly inhibited the growth of the resultant 4T1 organoids (Taylor, et al., 2011), thereby validating the first in vitro system that recapitulates the “TGF-β Paradox” solely by modulating the tension sensed by malignant MECs. Importantly, the expression and activity of lysyl oxidase stimulated by TGF-β contributes to its acquisition of oncogenic behavior and induction of EMT in normal and malignant mammary epithelial cells (Taylor, et al., 2011). Oncogenic TGF-β signaling brought about by mechanotransduction is partially neutralized by rendering breast cancer cell deficient in lysyl oxidase expression, which enhances E-cad expression and activity in developing organoids (Taylor, et al., 2011). Along these lines, increasing ECM rigidity not only induces dramatic differences in cell proliferation, but also suppresses E-cad expression by promoting EMT programs (Tilghman, et al., 2010), presumably via rigidity-induced TGF-β synthesis and autocrine signaling coupled to integrin-mediated activation of latent TGF-β complexes (Taylor, et al., 2011, Wipff, et al., 2007). Recently, initiation of MET programs and their elevated expression of E-cad was observed to promote the metastatic outgrowth of disseminated breast cancer cells in the lungs of mice (Chao, et al., 2010, Dykxhoorn, et al., 2009). Somewhat paradoxically, E-cad expression was also shown to normalize acinar morphology and growth (Wang, et al., 2002), presumably via heterotypic binding to and regulation of β1 integrin expression (Whittard, et al., 2002, Wu, et al., 2006, Zhang, et al., 2006). As a means to explain these contrasting findings, we recently found that the heterotypic binding of E-cad to β1 integrin suppresses its expression and inhibits the initiation of metastatic outgrowth breast micrometastases. Moreover, the conversion from dormancy to proliferative programs capable of supporting metastatic outgrowth required a transient EMT event, which downregulated E-cad expression and upregulated that of β1 integrin necessary for micrometastatic outgrowth (M.K.W. and W.P.S., unpublished observation). Collectively, these studies provide novel insights into the role of mechanotransduction in mediating oncogenic TGF-β signaling and its coupling to type 3 EMT during distinct stages of the metastatic cascade (Drake, et al., 2009, Wendt, et al., 2010).
Impact of growth factor receptors during EMT induced by TGF-β
As mentioned above, recent advancements in the use of 3D-organotypic cultures has shed new light into the importance of cell and tumor microenvironments in regulating TGF-β stimulation of EMT. In a similar avenue of research, recent studies are now interrogating the specific impact played by other tumor-derived growth factors and chemokines in coupling TGF-β to EMT programs. For instance, the expression and activity of Cox-2 are essential in facilitating TGF-β stimulation of EMT in normal and malignant mammary epithelial cells (Neil, et al., 2008, Tian and Schiemann, 2010). Importantly, the coupling of TGF-β to Cox-2 expression is increased synergistically by co-stimulation with EGF and its activation of MAP kinases (Saha, et al., 1999). Interestingly, TGF-β can transactivate EGFR via a Src-dependent pathway coupled to the activation of TACE/ADAM17 (TNF-α-converting enzyme) and its cleavage of EGF-like ligands (Murillo, et al., 2005, Wang, et al., 2009a). Along these lines, two-photon intravital imaging analyses assisted in identifying a paracrine signaling axis comprised of carcinoma cell-derived colony stimulating factor-1 (CSF-1) and macrophage-derived epidermal growth factor (EGF) that facilitates tumor cell migration and invasion (Patsialou, et al., 2009, Wang, et al., 2007, Wyckoff, et al., 2004). Importantly, TGF-β plays an essential role in establishing this paracrine signaling network through its ability to upregulate CSF-1 receptor expression (Patsialou, et al., 2009). EGFR also activates transcription factor STAT3 (signal transducer and activator of transcription-3), which induces Twist expression necessary to induce autocrine TGF-β signaling (Lo, et al., 2007). We recently demonstrated that TGF-β stimulation of EMT confers invasive activity to EGFR in part by (i) stabilizing its expression at the cell surface, and (ii) facilitating its coupling to Src and p38 MAPK (Wendt, et al., 2010). Interestingly, chronic activation of EMT programs by TGF-β results in the acquisition of resistance to EGFR inhibitors (Barr, et al., 2008). Thus, these findings support the notion that transient EMT programs support oncogenic EGFR signaling, while those encompassing prolonged EMT programs support “kinase switching” away from EGFR to instead depend upon the actions of FGFR or PDGFR (Thomson, et al., 2008). Finally, insulin-like growth factor-1 (IGF-1) and its receptor (IGF1R) also induce cell transformation, invasion, and metastasis (Zha and Lackner, 2010), presumably via an EMT event involving the actions of Snail-2, ZEB1/2, and Akt2 (Graham, et al., 2008, Irie, et al., 2005, Kim, et al., 2007b, Sivakumar, et al., 2009). Along these lines, we find IGF1R expression to render transitioning mammary epithelial cells resistant to apoptotic stimuli when undergoing EMT induced by TGF-β, and to play an essential role in mediating cell migration and invasion stimulated by TGF-β (M.T. and W.P.S., unpublished observation). Future studies need to determine the molecular mechanisms that elicit “kinase switching,” as well as the spatiotemporal events that influence their initiation and resolution.
EMT, cancer stem cells, and chemoresistance
EMT and cancer stem cells
Solid tumors are notoriously heterogeneous with respect to their collective morphologies, proliferative indices, genetic lesions, and responsiveness to chemotherapeutic regimens (Visvader, 2011). Housed within this heterogeneous environment lies a subpopulation of cancer stem cells (CSCs) that possess self-renewal and tumor-initiating properties that give rise to metastatic progression and disease recurrence (Pardal, et al., 2003). Although considerable debate remains regarding the “true” identity of CSCs, several recent studies have identified a variety of proteins that serve in marking tumor-initiating cells (Table 2). For example, CD44+/CD24−/low, CD29hi/CD24med, or aldehyde dehydrogenase 1 (ALDH1) can differentiate breast CSCs from those lacking tumor-initiating properties (Al-Hajj, et al., 2003, Ginestier, et al., 2007), while detecting expression of CD133 has been proposed as a marker for CSCs derived from cancers of the brain, colon and pancreatic cancers (Hermann, et al., 2007, O’Brien, et al., 2007, Ricci-Vitiani, et al., 2007). Moreover, cellular efflux of Hoechst stains can visualize a heterogeneous “side-population” of cells enriched in stem cell-like properties (Challen and Little, 2006), as can detecting the expression of EpCAM (epithelial cell adhesion molecule) (Munz, et al., 2009). Importantly, the initiation of EMT programs elicits the selection and expansion of CSCs (Ben-Porath, et al., 2008, Morel, et al., 2008, Shipitsin, et al., 2007), leading to the development of chemoresistant phenotypes and disease recurrence (Creighton, et al., 2010, Singh and Settleman, 2010, Turley, et al., 2008). Consistent with its function as a master regulator of EMT, TGF-β stimulation of carcinoma cells facilitates the appearance of CSCs in an EMT-dependent manner, suggesting that targeting the TGF-β signaling system can suppress tumor development and progression by inhibiting CSC function. Accordingly, inactivation of TGF-β signaling in metastatic breast cancers results in a MET that reestablished a more epithelial-like phenotype in these aggressive cells, while elevated TGF-β signatures associate with metastatic progression and poorer clinical outcomes (Shipitsin, et al., 2007). Future studies need to determine the impact on tumor development, progression, and recurrence that may be achieved by combining targeted TGF-β chemotherapies with agents capable of selectively targeting CSCs, including the delivery of (i) chemotherapeutic agents coupled to monoclonal antibodies directed at CSC cell surface markers (Deonarain, et al., 2009) or (ii) pharmaceuticals capable of inactivating CSCs drug transporters (Baguley, 2010).
Table 2.
CSC markers and their relationship to chemoresistant phenotypes
| CSC markers | Cancer type | Conferred drug resistance |
|---|---|---|
| CD133+/CD117+ | Ovarian | Carboplatin; Paclitaxel; Cisplatin (Hu, et al., 2010, Shi, et al., 2010) |
| Stro-1+ | Osteosarcoma | Doxorubicin (Adhikari, et al., 2010) |
| CD117+/ABCG2+ | Prostate | Arsenite; Cisplatin; Paclitaxel; Adriamycin; Methotrexate (Liu, et al., 2010, Tokar, et al., 2010) |
| CD133+/ABCG2+ | SCLC* | 5-FU, Cisplatin; Etoposide (Gutova, et al., 2007, Wang, et al., 2010) |
| CD34+CD38− | AML* | Ara-C (Ishikawa, et al., 2007, Saito, et al., 2010) |
| CD34− | CML* | Imatinib (Lemoli, et al., 2009) |
| uPAR+ | SCLC* | 5-FU*, Cisplatin; Etoposide (Gutova, et al. 2007, Wang et al 2010) |
| Hoechst+ | Liver | Doxorubicin; Methotrexate (Zhang, et al., 2010) |
| CD29hi/CD24med | Breast | Paclitaxel; Cisplatin (Shafee, et al., 2008, To, et al., 2010) |
| CD44+/CD24−/low | Breast & Pancreatic | Paclitaxel; Cisplatin; Gemcitabine (Hermann et al. 2007, Hong et al. 2009, Shafee et al. 2008, To et al. 2010) |
| CD44+ | Pancreatic | Gemcitabine (Hermann et al. 2007, Hong et al. 2009) |
| CD133+ | Brain & Colon | Carboplatin; Paclitaxel; Irinotecan; Temozolomide; Etoposide; Oxaliplatin; 5-FU;Cyclophosphamide (Dylla, et al., 2008, Fang, et al., 2010, Liu, et al., 2006, Ong, et al., 2010, Todaro, et al., 2007) |
5-FU, 5-fluorouracil; AML, Acute myeloid leukemia; CML, chronic myelogenous leukemia; SCLC, Small cell lung cancer
EMT and chemoresistance
The most common failure in successfully treating cancer patients is their development of chemoresistance, which typically falls into two major categories: (i) de novo drug resistance, which manifests in the absence of drug selection due to a pre-existing phenotype; and (ii) acquired drug resistance, which manifests as an adaptive response to drug selection. Mechanistically, acquired drug resistance typically reflects the ability of cells to upregulate their expression of drug efflux transporters or metabolizing enzymes, or to downregulate their drug-induced apoptotic machinery (Gottesman, 2002). As mentioned above, the initiation of EMT programs underlies disease progression and the emergence of chemoresistance, which has led to the notion that inactivating EMT or inducing MET may serve to resensitize carcinoma cells to previously effective treatment regimens (Table 2). For instance, anti-EGFR inhibitors such as Erlotinib, Gefitinib, or Cetuximab are widely used clinically to treat a variety of human cancers; however, the effectiveness of these drugs is inversely correlated to the completeness of the EMT program and acquisition of mesenchymal-like phenotypes attained in cancers of the lung, head and neck (Chin, et al., 2008, Frederick, et al., 2007, Thomson, et al., 2005, Thomson, et al., 2008, Yauch, et al., 2005). Likewise, ZEB1-driven EMT in pancreatic cancers elicits their resistance to Gemcitabine, 5-fluorouricil (5-FU), and Cisplatin resistance (Arumugam, et al., 2009). Importantly, rendering pancreatic cancer cells deficient in the expression of either ZEB1 or Notch was sufficient to elicit a MET and restore sensitivity to these cytotoxic agents (Arumugam, et al., 2009, Wang, et al., 2009b). Along these lines, the appearance of acquired resistance reflected the completion of EMT programs in (i) ovarian cancer cells that developed resistance to paclitaxel (Kajiyama, et al., 2007); (ii) bladder cancer cells that developed resistance to radiotherapy and Cisplatin, an event mediated by ZEB2 (Sayan, et al., 2009); and (iii) luminal breast cancer cells that developed resistance to either Paclitaxel, Docetaxel, or Doxorubicin, events mediated by downregulated expression of ER-α and upregulated expression of TβR-II (Iseri, et al., 2011). Interestingly, Doxorubicin treatment of breast cancer cells elicits an EMT program sufficient to enhance their metastasis to the lungs and bone in mice. Importantly, administration of the TβR-I antagonist neutralized the metastasis promoting activities of Doxorubicin (Bandyopadhyay, et al., 2010), suggesting that the ability of cytotoxic agents to induce EMT depends upon the initiation of autocrine TGF-β signaling. Future studies need to determine the overall reliance of EMT induced by TGF-β in mediating acquired resistance to chemotherapeutics, as well as to develop reliable biomarkers capable to typing the function of TGF-β within these developing and progressing neoplasms.
Conclusion
The current EMT paradigm states that oncogenic EMT is a metastable and transient process that engenders carcinoma cells the ability to escape the confines of the primary tumor and disseminate throughout the body via the systemic circulation. Along these lines, EMT is proficient in generating unique cell populations endowed with self-renewal and stem-like qualities, which renders transitioned cells insensitive to apoptosis, hypoxia, and traditional chemotherapies. At present, it remains uncertain where “EMT-competent” carcinoma cells originate and reside within heterogeneous tumor microenvironments, as well as the extent to which these events can be driven by TGF-β at distinct stages of tumor development. Likewise, it remains ambiguous as to whether the EMT phenotype is stable or malleable throughout the evolution of a given tumor and its spatiotemporal susceptibility to the EMT process (Fig. 3). Equally unclear is the precise role played by “EMT-incompetent” cells in (i) supporting the selection, expansion, and dissemination of transitioned carcinoma cells during metastatic progression; (ii) enabling transitioned cells to subvert the effects of neoadjuvant chemotherapies; and (iii) promoting transitioned cells to escape metastatic dormancy and reinitiate proliferative programs coupled to lethal bouts of disease recurrence. Future studies need to address and fill these knowledge gaps, and in doing so, science and medicine may develop the tools necessary to circumvent the “TGF-β Paradox” and its ability to promote EMT, metastatic progression, and disease recurrence in patients harboring metastatic disease.
Fig. 3.
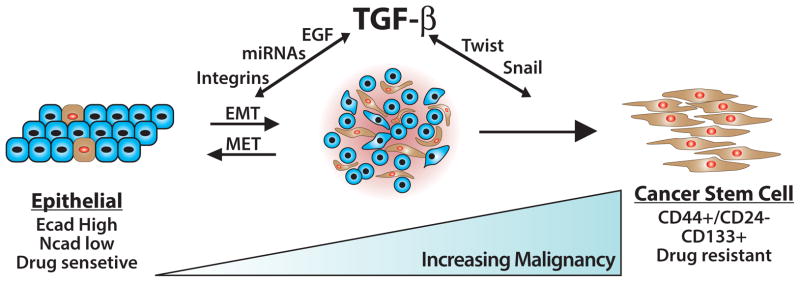
The consequences of EMT programs induced by TGF-β. Administration of TGF-β readily elicits the formation of a metastable EMT state in normal and malignant cells. The restoration of epithelial phenotypes transpires through mesenchymal-epithelial transitions (METs), which occur normally during developmental EMTs and perhaps during metastatic outgrowth associated with oncogenic EMTs. Prolonged exposure of cells to TGF-β or other EMT-initiating factors supports the continued development and expansion of cancer stem cells, which collectively are chemoresistant and underlie disease recurrence. How, when, and where cancer stems cells undergo MET during secondary tumor outgrowth remains to be determined definitively.
Acknowledgments
We thank members of the Schiemann Laboratory for critical comments and reading of the manuscript. W.P.S. was supported by grants from the National Institutes of Health (CA129359), the Komen Foundation (BCTR0706967), the Department of Defense (BC084651); and Case Comprehensive Cancer Center and the University Hospitals Seidman Cancer Center. M.K.W. was supported by the American Cancer Society (PF-09-120-01-CS).
References
- Adhikari AS, Agarwal N, Wood BM, Porretta C, Ruiz B, Pochampally RR, Iwakuma T. CD117 and Stro-1 identify osteosarcoma tumor-initiating cells associated with metastasis and drug resistance. Cancer Res. 2010;70:4602–4612. doi: 10.1158/0008-5472.CAN-09-3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrow MG, Kawabata M, Aakre M, Moses HL. Overexpression of the c-Myc oncoprotein blocks the growth-inhibitory response but is required for the mitogenic effects of transforming growth factor β1. Proc Natl Acad Sci U S A. 1995;92:3239–3243. doi: 10.1073/pnas.92.8.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allington TM, Galliher-Beckley AJ, Schiemann WP. Activated Abl kinase inhibits oncogenic transforming growth factor-β signaling and tumorigenesis in mammary tumors. FASEB J. 2009;23:4231–4243. doi: 10.1096/fj.09-138412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzano MA, Roberts AB, De Larco JE, Wakefield LM, Assoian RK, Roche NS, Smith JM, Lazarus JE, Sporn MB. Increased secretion of type β transforming growth factor accompanies viral transformation of cells. Mol Cell Biol. 1985;5:242–247. doi: 10.1128/mcb.5.1.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arteaga CL, Hurd SD, Winnier AR, Johnson MD, Fendly BM, Forbes JT. Anti-transforming growth factor (TGF)-β antibodies inhibit breast cancer cell tumorigenicity and increase mouse spleen natural killer cell activity. Implications for a possible role of tumor cell/host TGF-β interactions in human breast cancer progression. J Clin Invest. 1993;92:2569–2576. doi: 10.1172/JCI116871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam T, Ramachandran V, Fournier KF, Wang H, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey DJ, Choi W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69:5820–5828. doi: 10.1158/0008-5472.CAN-08-2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baguley BC. Multiple drug resistance mechanisms in cancer. Mol Biotechnol. 2010;46:308–316. doi: 10.1007/s12033-010-9321-2. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay A, Wang L, Agyin J, Tang Y, Lin S, Yeh IT, De K, Sun LZ. Doxorubicin in combination with a small TGFβ inhibitor: a potential novel therapy for metastatic breast cancer in mouse models. PLoS One. 2010;5:e10365. doi: 10.1371/journal.pone.0010365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr S, Thomson S, Buck E, Russo S, Petti F, Sujka-Kwok I, Eyzaguirre A, Rosenfeld-Franklin M, Gibson NW, Miglarese M, Epstein D, Iwata KK, Haley JD. Bypassing cellular EGF receptor dependence through epithelial-to-mesenchymal-like transitions. Clin Exp Metastasis. 2008;25:685–693. doi: 10.1007/s10585-007-9121-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battula VL, Evans KW, Hollier BG, Shi Y, Marini FC, Ayyanan A, Wang RY, Brisken C, Guerra R, Andreeff M, Mani SA. Epithelial-mesenchymal transition-derived cells exhibit multilineage differentiation potential similar to mesenchymal stem cells. Stem Cells. 2010;28:1435–1445. doi: 10.1002/stem.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, Neilson EG, Moses HL. TGF-β signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–851. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- Bhowmick NA, Ghiassi M, Bakin A, Aakre M, Lundquist CA, Engel ME, Arteaga CL, Moses HL. Transforming growth factor-β1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell. 2001a;12:27–36. doi: 10.1091/mbc.12.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick NA, Zent R, Ghiassi M, McDonnell M, Moses HL. Integrin β1 signaling is necessary for transforming growth factor-β activation of p38MAPK and epithelial plasticity. J Biol Chem. 2001b;276:46707–46713. doi: 10.1074/jbc.M106176200. [DOI] [PubMed] [Google Scholar]
- Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor-β in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- Bose R, Wrana JL. Regulation of Par6 by extracellular signals. Curr Opin Cell Biol. 2006;18:206–212. doi: 10.1016/j.ceb.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Boyd NF, Guo H, Martin LJ, Sun L, Stone J, Fishell E, Jong RA, Hislop G, Chiarelli A, Minkin S, Yaffe MJ. Mammographic density and the risk and detection of breast cancer. N Engl J Med. 2007;356:227–236. doi: 10.1056/NEJMoa062790. [DOI] [PubMed] [Google Scholar]
- Brown CB, Boyer AS, Runyan RB, Barnett JV. Requirement of type III TGF-β receptor for endocardial cell transformation in the heart. Science. 1999;283:2080–2082. doi: 10.1126/science.283.5410.2080. [DOI] [PubMed] [Google Scholar]
- Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9:582–589. doi: 10.1038/embor.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabodi S, Tinnirello A, Di Stefano P, Bisaro B, Ambrosino E, Castellano I, Sapino A, Arisio R, Cavallo F, Forni G, Glukhova M, Silengo L, Altruda F, Turco E, Tarone G, Defilippi P. p130Cas as a new regulator of mammary epithelial cell proliferation, survival, and HER2-Neu oncogene-dependent breast tumorigenesis. Cancer Res. 2006;66:4672–4680. doi: 10.1158/0008-5472.CAN-05-2909. [DOI] [PubMed] [Google Scholar]
- Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- Carr BI, Hayashi I, Branum EL, Moses HL. Inhibition of DNA synthesis in rat hepatocytes by platelet-derived type β transforming growth factor. Cancer Res. 1986;46:2330–2334. [PubMed] [Google Scholar]
- Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, Yang J. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res. 2011;71:245–254. doi: 10.1158/0008-5472.CAN-10-2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer. 2004;4:118–132. doi: 10.1038/nrc1276. [DOI] [PubMed] [Google Scholar]
- Challen GA, Little MH. A side order of stem cells: the SP phenotype. Stem cells. 2006;24:3–12. doi: 10.1634/stemcells.2005-0116. [DOI] [PubMed] [Google Scholar]
- Chang CJ, Chao CH, Xia W, Yang JY, Xiong Y, Li CW, Yu WH, Rehman SK, Hsu JL, Lee HH, Liu M, Chen CT, Yu D, Hung MC. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13:317–323. doi: 10.1038/ncb2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H, Brown CW, Matzuk MM. Genetic analysis of the mammalian transforming growth factor-β superfamily. Endocr Rev. 2002;23:787–823. doi: 10.1210/er.2002-0003. [DOI] [PubMed] [Google Scholar]
- Chao YL, Shepard CR, Wells A. Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol Cancer. 2010;9:179. doi: 10.1186/1476-4598-9-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chea HK, Wright CV, Swalla BJ. Nodal signaling and the evolution of deuterostome gastrulation. Dev Dyn. 2005;234:269–278. doi: 10.1002/dvdy.20549. [DOI] [PubMed] [Google Scholar]
- Chen CR, Kang Y, Massague J. Defective repression of c-Myc in breast cancer cells: A loss at the core of the transforming growth factor β growth arrest program. Proc Natl Acad Sci U S A. 2001;98:992–999. doi: 10.1073/pnas.98.3.992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng N, Bhowmick NA, Chytil A, Gorksa AE, Brown KA, Muraoka R, Arteaga CL, Neilson EG, Hayward SW, Moses HL. Loss of TGF-β type II receptor in fibroblasts promotes mammary carcinoma growth and invasion through upregulation of TGF-α-, MSP- and HGF-mediated signaling networks. Oncogene. 2005;24:5053–5068. doi: 10.1038/sj.onc.1208685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin TM, Quinlan MP, Singh A, Sequist LV, Lynch TJ, Haber DA, Sharma SV, Settleman J. Reduced Erlotinib sensitivity of epidermal growth factor receptor-mutant non-small cell lung cancer following cisplatin exposure: a cell culture model of second-line erlotinib treatment. Clin Cancer Res. 2008;14:6867–6876. doi: 10.1158/1078-0432.CCR-08-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofori G. Changing neighbours, changing behaviour: cell adhesion molecule-mediated signalling during tumour progression. EMBO J. 2003;22:2318–2323. doi: 10.1093/emboj/cdg228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7:1267–1278. doi: 10.1016/s1097-2765(01)00260-x. [DOI] [PubMed] [Google Scholar]
- Conlon FL, Lyons KM, Takaesu N, Barth KS, Kispert A, Herrmann B, Robertson EJ. A primary requirement for nodal in the formation and maintenance of the primitive streak in the mouse. Development. 1994;120:1919–1928. doi: 10.1242/dev.120.7.1919. [DOI] [PubMed] [Google Scholar]
- Cottonham CL, Kaneko S, Xu L. miR-21 and miR-31 converge on TIAM1 to regulate migration and invasion of colon carcinoma cells. J Biol Chem. 2010;285:35293–35302. doi: 10.1074/jbc.M110.160069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creighton CJ, Chang JC, Rosen JM. Epithelial-mesenchymal transition (EMT) in tumor-initiating cells and its clinical implications in breast cancer. J Mammary Gland Biol Neoplasia. 2010;15:253–260. doi: 10.1007/s10911-010-9173-1. [DOI] [PubMed] [Google Scholar]
- Cui W, Fowlis DJ, Bryson S, Duffie E, Ireland H, Balmain A, Akhurst RJ. TGFβ1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell. 1996;86:531–542. doi: 10.1016/s0092-8674(00)80127-0. [DOI] [PubMed] [Google Scholar]
- Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454:56–61. doi: 10.1038/nature07086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dBian Y, Terse A, Du J, Hall B, Molinolo A, Zhang P, Chen W, Flanders KC, Gutkind JS, Wakefield LM, Kulkarni AB. Progressive tumor formation in mice with conditional deletion of TGF-β signaling in head and neck epithelia is associated with activation of the PI3K/Akt pathway. Cancer Res. 2009;69:5918–5926. doi: 10.1158/0008-5472.CAN-08-4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deonarain MP, Kousparou CA, Epenetos AA. Antibodies targeting cancer stem cells: a new paradigm in immunotherapy? MAbs. 2009;1:12–25. doi: 10.4161/mabs.1.1.7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhasarathy A, Kajita M, Wade PA. The transcription factor Snail mediates epithelial to mesenchymal transitions by repression of estrogen receptor α. Mol Endocrinol. 2007;21:2907–2918. doi: 10.1210/me.2007-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JM, Strohbehn G, Bair TB, Moreland JG, Henry MD. ZEB1 enhances transendothelial migration and represses the epithelial phenotype of prostate cancer cells. Mol Biol Cell. 2009;20:2207–2217. doi: 10.1091/mbc.E08-10-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duivenvoorden WC, Hirte HW, Singh G. Transforming growth factor β1 acts as an inducer of matrix metalloproteinase expression and activity in human bone-metastasizing cancer cells. Clin Exp Metastasis. 1999;17:27–34. doi: 10.1023/a:1026404227624. [DOI] [PubMed] [Google Scholar]
- Dumont N, Wilson MB, Crawford YG, Reynolds PA, Sigaroudinia M, Tlsty TD. Sustained induction of epithelial to mesenchymal transition activates DNA methylation of genes silenced in basal-like breast cancers. Proc Natl Acad Sci. 2008;105:14867–14872. doi: 10.1073/pnas.0807146105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykxhoorn DM, Wu Y, Xie H, Yu F, Lal A, Petrocca F, Martinvalet D, Song E, Lim B, Lieberman J. miR-200 enhances mouse breast cancer cell colonization to form distant metastases. PLoS One. 2009;4:e7181. doi: 10.1371/journal.pone.0007181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dylla SJ, Beviglia L, Park IK, Chartier C, Raval J, Ngan L, Pickell K, Aguilar J, Lazetic S, Smith-Berdan S, Clarke MF, Hoey T, Lewicki J, Gurney AL. Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS One. 2008;3:e2428. doi: 10.1371/journal.pone.0002428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebnet K, Suzuki A, Ohno S, Vestweber D. Junctional adhesion molecules (JAMs): more molecules with dual functions? J Cell Sci. 2004;117:19–29. doi: 10.1242/jcs.00930. [DOI] [PubMed] [Google Scholar]
- Fang DD, Kim YJ, Lee CN, Aggarwal S, McKinnon K, Mesmer D, Norton J, Birse CE, He T, Ruben SM, Moore PA. Expansion of CD133(+) colon cancer cultures retaining stem cell properties to enable cancer stem cell target discovery. Br J Cancer. 2010;102:1265–1275. doi: 10.1038/sj.bjc.6605610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng XH, Derynck R. Specificity and versatility in TGF-β signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- Fowlis DJ, Cui W, Johnson SA, Balmain A, Akhurst RJ. Altered epidermal cell growth control in vivo by inducible expression of transforming growth factor β1 in the skin of transgenic mice. Cell Growth Differ. 1996;7:679–687. [PubMed] [Google Scholar]
- Frank NY, Schatton T, Frank MH. The therapeutic promise of the cancer stem cell concept. J Clin Invest. 2010;120:41–50. doi: 10.1172/JCI41004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick BA, Helfrich BA, Coldren CD, Zheng D, Chan D, Bunn PA, Jr, Raben D. Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non-small cell lung carcinoma. Mol Cancer Ther. 2007;6:1683–1691. doi: 10.1158/1535-7163.MCT-07-0138. [DOI] [PubMed] [Google Scholar]
- Galliher AJ, Schiemann WP. Beta3 integrin and Src facilitate transforming growth factor-β mediated induction of epithelial-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 2006;8:R42. doi: 10.1186/bcr1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galliher AJ, Schiemann WP. Src phosphorylates Tyr284 in TGF-β type II receptor and regulates TGF-β stimulation of p38 MAPK during breast cancer cell proliferation and invasion. Cancer Res. 2007;67:3752–3758. doi: 10.1158/0008-5472.CAN-06-3851. [DOI] [PubMed] [Google Scholar]
- Galliher-Beckley AJ, Schiemann WP. Grb2 binding to Tyr284 in TβR-II is essential for mammary tumor growth and metastasis stimulated by TGF-β. Carcinogenesis. 2008;29:244–251. doi: 10.1093/carcin/bgm245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatza CE, Oh SY, Blobe GC. Roles for the type III TGF-β receptor in human cancer. Cell Signal. 2010;22:1163–1174. doi: 10.1016/j.cellsig.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- Graham TR, Zhau HE, Odero-Marah VA, Osunkoya AO, Kimbro KS, Tighiouart M, Liu T, Simons JW, O’Regan RM. Insulin-like growth factor-I-dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 2008;68:2479–2488. doi: 10.1158/0008-5472.CAN-07-2559. [DOI] [PubMed] [Google Scholar]
- Greenburg G, Hay ED. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J Cell Biol. 1982;95:333–339. doi: 10.1083/jcb.95.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- Grunert S, Jechlinger M, Beug H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol. 2003;4:657–665. doi: 10.1038/nrm1175. [DOI] [PubMed] [Google Scholar]
- Gutova M, Najbauer J, Gevorgyan A, Metz MZ, Weng Y, Shih CC, Aboody KS. Identification of uPAR-positive chemoresistant cells in small cell lung cancer. PLoS One. 2007;2:e243. doi: 10.1371/journal.pone.0000243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajra KM, Chen DY, Fearon ER. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer research. 2002;62:1613–1618. [PubMed] [Google Scholar]
- Hazan RB, Phillips GR, Qiao RF, Norton L, Aaronson SA. Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J Cell Biol. 2000;148:779–790. doi: 10.1083/jcb.148.4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–323. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Hong SP, Wen J, Bang S, Park S, Song SY. CD44-positive cells are responsible for gemcitabine resistance in pancreatic cancer cells. Int J Cancer. 2009;125:2323–2331. doi: 10.1002/ijc.24573. [DOI] [PubMed] [Google Scholar]
- Hu Z, Zhang Z, Guise T, Seth P. Systemic delivery of an oncolytic adenovirus expressing soluble transforming growth factor-β receptor II-Fc fusion protein can inhibit breast cancer bone metastasis in a mouse model. Hum Gene Ther. 2010;21:1623–1629. doi: 10.1089/hum.2010.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignotz RA, Massague J. Transforming growth factor-β stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem. 1986;261:4337–4345. [PubMed] [Google Scholar]
- Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, Natesan S, Brugge JS. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J Cell Biol. 2005;171:1023–1034. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iseri OD, Kars MD, Arpaci F, Atalay C, Pak I, Gunduz U. Drug resistant MCF-7 cells exhibit epithelial-mesenchymal transition gene expression pattern. Biomed Pharmacother. 2011;65:40–45. doi: 10.1016/j.biopha.2010.10.004. [DOI] [PubMed] [Google Scholar]
- Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, Nakamura R, Tanaka T, Tomiyama H, Saito N, Fukata M, Miyamoto T, Lyons B, Ohshima K, Uchida N, Taniguchi S, Ohara O, Akashi K, Harada M, Shultz LD. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25:1315–1321. doi: 10.1038/nbt1350. [DOI] [PubMed] [Google Scholar]
- Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J. Abnormal lung development and cleft palate in mice lacking TGF-β3 indicates defects of epithelial-mesenchymal interaction. Nat Genet. 1995;11:415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- Kajiyama H, Shibata K, Terauchi M, Yamashita M, Ino K, Nawa A, Kikkawa F. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int J Oncol. 2007;31:277–283. [PubMed] [Google Scholar]
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JS, Liu C, Derynck R. New regulatory mechanisms of TGF-β receptor function. Trends Cell Biol. 2009;19:385–394. doi: 10.1016/j.tcb.2009.05.008. [DOI] [PubMed] [Google Scholar]
- Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, Guise TA, Massague J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- Kim BG, Li C, Qiao W, Mamura M, Kasprzak B, Anver M, Wolfraim L, Hong S, Mushinski E, Potter M, Kim SJ, Fu XY, Deng C, Letterio JJ. Smad4 signalling in T cells is required for suppression of gastrointestinal cancer. Nature. 2006;441:1015–1019. doi: 10.1038/nature04846. [DOI] [PubMed] [Google Scholar]
- Kim ES, Sohn YW, Moon A. TGF-β-induced transcriptional activation of MMP-2 is mediated by activating transcription factor (ATF)2 in human breast epithelial cells. Cancer Lett. 2007a;252:147–156. doi: 10.1016/j.canlet.2006.12.016. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Litzenburger BC, Cui X, Delgado DA, Grabiner BC, Lin X, Lewis MT, Gottardis MM, Wong TW, Attar RM, Carboni JM, Lee AV. Constitutively active type I insulin-like growth factor receptor causes transformation and xenograft growth of immortalized mammary epithelial cells and is accompanied by an epithelial-to-mesenchymal transition mediated by NF-κB and snail. Mol Cell Biol. 2007b;27:3165–3175. doi: 10.1128/MCB.01315-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Seok Kang Y, Soo Kim J, Shin NY, Hanks SK, Song WK. The integrin-coupled signaling adaptor p130Cas suppresses Smad3 function in transforming growth factor-β signaling. Mol Biol Cell. 2008;19:2135–2146. doi: 10.1091/mbc.E07-10-0991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong W, Yang H, He L, Zhao JJ, Coppola D, Dalton WS, Cheng JQ. MicroRNA-155 is regulated by the transforming growth factor β/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol Cell Biol. 2008;28:6773–6784. doi: 10.1128/MCB.00941-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910–14914. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, Albig AR, Regner M, Schiemann BJ, Schiemann WP. Fibulin-5 initiates epithelial-mesenchymal transition (EMT) and enhances EMT induced by TGF-β in mammary epithelial cells via a MMP-dependent mechanism. Carcinogenesis. 2008;29:2243–2251. doi: 10.1093/carcin/bgn199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoli RM, Salvestrini V, Bianchi E, Bertolini F, Fogli M, Amabile M, Tafuri A, Salati S, Zini R, Testoni N, Rabascio C, Rossi L, Martin-Padura I, Castagnetti F, Marighetti P, Martinelli G, Baccarani M, Ferrari S, Manfredini R. Molecular and functional analysis of the stem cell compartment of chronic myelogenous leukemia reveals the presence of a CD34- cell population with intrinsic resistance to imatinib. Blood. 2009;114:5191–5200. doi: 10.1182/blood-2008-08-176016. [DOI] [PubMed] [Google Scholar]
- Leptin M. Twist and Snail as positive and negative regulators during Drosophila mesoderm development. Genes Dev. 1991;5:1568–1576. doi: 10.1101/gad.5.9.1568. [DOI] [PubMed] [Google Scholar]
- Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL, Yu JS. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Xu F, Du X, Lai D, Zhao Y, Huang Q, Jiang L, Huang W, Cheng W, Liu Z. Establishment and characterization of multi-drug resistant, prostate carcinoma-initiating stem-like cells from human prostate cancer cell lines 22RV1. Mol Cell Biochem. 2010;340:265–273. doi: 10.1007/s11010-010-0426-5. [DOI] [PubMed] [Google Scholar]
- Lo HW, Hsu SC, Xia W, Cao X, Shih JY, Wei Y, Abbruzzese JL, Hortobagyi GN, Hung MC. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007;67:9066–9076. doi: 10.1158/0008-5472.CAN-07-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, Teruya-Feldstein J, Reinhardt F, Onder TT, Valastyan S, Westermann F, Speleman F, Vandesompele J, Weinberg RA. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010;12:247–256. doi: 10.1038/ncb2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani SA, Yang J, Brooks M, Schwaninger G, Zhou A, Miura N, Kutok JL, Hartwell K, Richardson AL, Weinberg RA. Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc Natl Acad Sci U S A. 2007;104:10069–10074. doi: 10.1073/pnas.0703900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J. TGF-β signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- Massague J. TGF β in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J, Gomis RR. The logic of TGFβ signaling. FEBS Lett. 2006;580:2811–2820. doi: 10.1016/j.febslet.2006.04.033. [DOI] [PubMed] [Google Scholar]
- Masszi A, Di Ciano C, Sirokmany G, Arthur WT, Rotstein OD, Wang J, McCulloch CA, Rosivall L, Mucsi I, Kapus A. Central role for Rho in TGF-β1-induced α-smooth muscle actin expression during epithelial-mesenchymal transition. Am J Physiol Renal Physiol. 2003;284:F911–924. doi: 10.1152/ajprenal.00183.2002. [DOI] [PubMed] [Google Scholar]
- Mendez MG, Kojima S, Goldman RD. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010;24:1838–1851. doi: 10.1096/fj.09-151639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. Journal of mammary gland biology and neoplasia. 2010a;15:117–134. doi: 10.1007/s10911-010-9178-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micalizzi DS, Ford HL. Epithelial-mesenchymal transition in development and cancer. Future Oncol. 2009;5:1129–1143. doi: 10.2217/fon.09.94. [DOI] [PubMed] [Google Scholar]
- Micalizzi DS, Wang CA, Farabaugh SM, Schiemann WP, Ford H. Homeoprotein Six1 increases TGF-β type I receptor and converts TGF-β signaling from suppressive to supportive for tumor growth. Cancer Res. 2010b;70:10371–10380. doi: 10.1158/0008-5472.CAN-10-1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF-β induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol. 1994;127:2021–2036. doi: 10.1083/jcb.127.6.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE. 2008;3:e2888. doi: 10.1371/journal.pone.0002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses HL, Branum EL, Proper JA, Robinson RA. Transforming growth factor production by chemically transformed cells. Cancer Res. 1981;41:2842–2848. [PubMed] [Google Scholar]
- Moustakas A, Heldin CH. Non-Smad TGF-β signals. J Cell Sci. 2005;118:3573–3584. doi: 10.1242/jcs.02554. [DOI] [PubMed] [Google Scholar]
- Munz M, Baeuerle PA, Gires O. The emerging role of EpCAM in cancer and stem cell signaling. Cancer Res. 2009;69:5627–5629. doi: 10.1158/0008-5472.CAN-09-0654. [DOI] [PubMed] [Google Scholar]
- Muraoka-Cook RS, Kurokawa H, Koh Y, Forbes JT, Roebuck LR, Barcellos-Hoff MH, Moody SE, Chodosh LA, Arteaga CL. Conditional overexpression of active transforming growth factor β1 In vivo accelerates metastases of transgenic mammary tumors. Cancer Res. 2004;64:9002–9011. doi: 10.1158/0008-5472.CAN-04-2111. [DOI] [PubMed] [Google Scholar]
- Murillo MM, del Castillo G, Sanchez A, Fernandez M, Fabregat I. Involvement of EGF receptor and c-Src in the survival signals induced by TGF-β1 in hepatocytes. Oncogene. 2005;24:4580–4587. doi: 10.1038/sj.onc.1208664. [DOI] [PubMed] [Google Scholar]
- Nakajima A, Ito Y, Asano M, Maeno M, Iwata K, Mitsui N, Shimizu N, Cui XM, Shuler CF. Functional role of transforming growth factor-β type III receptor during palatal fusion. Dev Dyn. 2007;236:791–801. doi: 10.1002/dvdy.21090. [DOI] [PubMed] [Google Scholar]
- Neil JR, Johnson KM, Nemenoff RA, Schiemann WP. Cox-2 inactivates Smad signaling and enhances EMT stimulated by TGF-beta through a PGE2-dependent mechanisms. Carcinogenesis. 2008;29:2227–2235. doi: 10.1093/carcin/bgn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessen CM. Tight junctions/adherens junctions: basic structure and function. J Invest Derm. 2007;127:2525–2532. doi: 10.1038/sj.jid.5700865. [DOI] [PubMed] [Google Scholar]
- O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- Ohashi S, Natsuizaka M, Wong GS, Michaylira CZ, Grugan KD, Stairs DB, Kalabis J, Vega ME, Kalman RA, Nakagawa M, Klein-Szanto AJ, Herlyn M, Diehl JA, Rustgi AK, Nakagawa H. Epidermal growth factor receptor and mutant p53 expand an esophageal cellular subpopulation capable of epithelial-to-mesenchymal transition through ZEB transcription factors. Cancer Res. 2010;70:4174–4184. doi: 10.1158/0008-5472.CAN-09-4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68:3645–3654. doi: 10.1158/0008-5472.CAN-07-2938. [DOI] [PubMed] [Google Scholar]
- Ong CW, Kim LG, Kong HH, Low LY, Iacopetta B, Soong R, Salto-Tellez M. CD133 expression predicts for non-response to chemotherapy in colorectal cancer. Mod Pathol. 2010;23:450–457. doi: 10.1038/modpathol.2009.181. [DOI] [PubMed] [Google Scholar]
- Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL. Regulation of the polarity protein Par6 by TGFβ receptors controls epithelial cell plasticity. Science. 2005;307:1603–1609. doi: 10.1126/science.1105718. [DOI] [PubMed] [Google Scholar]
- Papageorgis P, Lambert AW, Ozturk S, Gao F, Pan H, Manne U, Alekseyev YO, Thiagalingam A, Abdolmaleky HM, Lenburg M, Thiagalingam S. Smad signaling is required to maintain epigenetic silencing during breast cancer progression. Cancer Res. 2010;70:968–978. doi: 10.1158/0008-5472.CAN-09-1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003;3:895–902. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- Parvani JG, Taylor MA, Schiemann WP. Noncanonical TGF-β signaling during mammary tumorigenesis. J Mammary Gland Biol Neoplasia. 2011 doi: 10.1007/s10911-011-9207-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Patsialou A, Wyckoff JB, Wang Y, Goswami S, Stanley ER, Condeelis JS. Invasion of human breast cancer cells in vivo requires both paracrine and autocrine loops involving the colony-stimulating factor-1 receptor. Cancer Res. 2009;69:9498–9506. doi: 10.1158/0008-5472.CAN-09-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimi RA, Leof EB. TGF-β signaling: a tale of two responses. J Cell Biochem. 2007;102:593–608. doi: 10.1002/jcb.21501. [DOI] [PubMed] [Google Scholar]
- Raible DW. Development of the neural crest: achieving specificity in regulatory pathways. Curr Opin Cell Biol. 2006;18:698–703. doi: 10.1016/j.ceb.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- Roberts AB, Anzano MA, Lamb LC, Smith JM, Sporn MB. New class of transforming growth factors potentiated by epidermal growth factor: isolation from non-neoplastic tissues. Proc Natl Acad Sci U S A. 1981;78:5339–5343. doi: 10.1073/pnas.78.9.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha D, Datta PK, Sheng H, Morrow JD, Wada M, Moses HL, Beauchamp RD. Synergistic induction of cyclooxygenase-2 by transforming growth factor-β1 and epidermal growth factor inhibits apoptosis in epithelial cells. Neoplasia. 1999;1:508–517. doi: 10.1038/sj.neo.7900051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Uchida N, Tanaka S, Suzuki N, Tomizawa-Murasawa M, Sone A, Najima Y, Takagi S, Aoki Y, Wake A, Taniguchi S, Shultz LD, Ishikawa F. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat Biotechnol. 2010;28:275–280. doi: 10.1038/nbt.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFβ knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauka-Spengler T, Bronner-Fraser M. A gene regulatory network orchestrates neural crest formation. Nat Rev Mol Cell Biol. 2008;9:557–568. doi: 10.1038/nrm2428. [DOI] [PubMed] [Google Scholar]
- Sayan AE, Griffiths TR, Pal R, Browne GJ, Ruddick A, Yagci T, Edwards R, Mayer NJ, Qazi H, Goyal S, Fernandez S, Straatman K, Jones GD, Bowman KJ, Colquhoun A, Mellon JK, Kriajevska M, Tulchinsky E. SIP1 protein protects cells from DNA damage-induced apoptosis and has independent prognostic value in bladder cancer. Proc Natl Acad Sci U S A. 2009;106:14884–14889. doi: 10.1073/pnas.0902042106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiemann WP. Targeted TGF-β chemotherapies: friend or foe in treating human malignancies? Expert Rev Anticancer Ther. 2007;7:609–611. doi: 10.1586/14737140.7.5.609. [DOI] [PubMed] [Google Scholar]
- Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol. 2004;286:C1213–1228. doi: 10.1152/ajpcell.00558.2003. [DOI] [PubMed] [Google Scholar]
- Shafee N, Smith CR, Wei S, Kim Y, Mills GB, Hortobagyi GN, Stanbridge EJ, Lee EY. Cancer stem cells contribute to cisplatin resistance in BRCA1/p53-mediated mouse mammary tumors. Cancer Res. 2008;68:3243–3250. doi: 10.1158/0008-5472.CAN-07-5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi MF, Jiao J, Lu WG, Ye F, Ma D, Dong QG, Xie X. Identification of cancer stem cell-like cells from human epithelial ovarian carcinoma cell line. Cell Mol Life Sci. 2010;67:3915–3925. doi: 10.1007/s00018-010-0420-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, Nikolskaya T, Serebryiskaya T, Beroukhim R, Hu M, Halushka MK, Sukumar S, Parker LM, Anderson KS, Harris LN, Garber JE, Richardson AL, Schnitt SJ, Nikolsky Y, Gelman RS, Polyak K. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11:259–273. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Silberstein GB, Daniel CW. Reversible inhibition of mammary gland growth by transforming growth factor-β. Science. 1987;237:291–293. doi: 10.1126/science.3474783. [DOI] [PubMed] [Google Scholar]
- Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Spoelstra NS, Jean A, Howe E, Torkko KC, Clark HR, Darling DS, Shroyer KR, Horwitz KB, Broaddus RR, Richer JK. ZEB1 expression in type I vs type II endometrial cancers: a marker of aggressive disease. Mod Pathol. 2008;21:912–923. doi: 10.1038/modpathol.2008.82. [DOI] [PubMed] [Google Scholar]
- Sivakumar R, Koga H, Selvendiran K, Maeyama M, Ueno T, Sata M. Autocrine loop for IGF-I receptor signaling in SLUG-mediated epithelial-mesenchymal transition. Int J Oncol. 2009;34:329–338. [PubMed] [Google Scholar]
- Skromne I, Stern CD. Interactions between Wnt and Vg1 signalling pathways initiate primitive streak formation in the chick embryo. Development. 2001;128:2915–2927. doi: 10.1242/dev.128.15.2915. [DOI] [PubMed] [Google Scholar]
- Takaishi K, Sasaki T, Kotani H, Nishioka H, Takai Y. Regulation of Cell-Cell Adhesion by Rac and Rho Small G Proteins in MDCK Cells. J Cell Biol. 1997;139:1047–1059. doi: 10.1083/jcb.139.4.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW, Hollier BG, Ram PT, Lander ES, Rosen JM, Weinberg RA, Mani SA. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A. 2010;107:15449–15454. doi: 10.1073/pnas.1004900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MA, Amin J, Kirschmann DA, Schiemann WP. Lysyl oxidase contributes to mechanotransduction-mediated regulation of transforming growth factor-β signaling in breast cancer cells. Neoplasia. 2011;13 doi: 10.1593/neo.101086. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MA, Parvani JG, Schiemann WP. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-β in normal and malignant mammary epithelial cells. J Mammary Gland Biol Neoplasia. 2010;15:169–190. doi: 10.1007/s10911-010-9181-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Thomson S, Buck E, Petti F, Griffin G, Brown E, Ramnarine N, Iwata KK, Gibson N, Haley JD. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 2005;65:9455–9462. doi: 10.1158/0008-5472.CAN-05-1058. [DOI] [PubMed] [Google Scholar]
- Thomson S, Petti F, Sujka-Kwok I, Epstein D, Haley JD. Kinase switching in mesenchymal-like non-small cell lung cancer lines contributes to EGFR inhibitor resistance through pathway redundancy. Clin Exp Metastasis. 2008;25:843–854. doi: 10.1007/s10585-008-9200-4. [DOI] [PubMed] [Google Scholar]
- Thuault S, Valcourt U, Petersen M, Manfioletti G, Heldin CH, Moustakas A. Transforming growth factor-β employs HMGA2 to elicit epithelial-mesenchymal transition. J Cell Biol. 2006;174:175–183. doi: 10.1083/jcb.200512110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian M, Neil JR, Schiemann WP. Transforming growth factor-β and the hallmarks of cancer. Cell Signal. 2011;23:951–962. doi: 10.1016/j.cellsig.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian M, Schiemann WP. The TGF-β paradox in human cancer: an update. Future Oncol. 2009;5:259–271. doi: 10.2217/14796694.5.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian M, Schiemann WP. PGE2 receptor EP2 mediates the antagonistic effect of COX-2 on TGF-β signaling during mammary tumorigenesis. FASEB J. 2010;24:1105–1116. doi: 10.1096/fj.09-141341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilghman RW, Cowan CR, Mih JD, Koryakina Y, Gioeli D, Slack-Davis JK, Blackman BR, Tschumperlin DJ, Parsons JT. Matrix rigidity regulates cancer cell growth and cellular phenotype. PLoS One. 2010;5:e12905. doi: 10.1371/journal.pone.0012905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To K, Fotovati A, Reipas KM, Law JH, Hu K, Wang J, Astanehe A, Davies AH, Lee L, Stratford AL, Raouf A, Johnson P, Berquin IM, Royer HD, Eaves CJ, Dunn SE. Y-box binding protein-1 induces the expression of CD44 and CD49f leading to enhanced self-renewal, mammosphere growth, and drug resistance. Cancer Res. 2010;70:2840–2851. doi: 10.1158/0008-5472.CAN-09-3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todaro M, Alea MP, Di Stefano AB, Cammareri P, Vermeulen L, Iovino F, Tripodo C, Russo A, Gulotta G, Medema JP, Stassi G. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell. 2007;1:389–402. doi: 10.1016/j.stem.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Tokar EJ, Qu W, Liu J, Liu W, Webber MM, Phang JM, Waalkes MP. Arsenic-specific stem cell selection during malignant transformation. J Natl Cancer Inst. 2010;102:638–649. doi: 10.1093/jnci/djq093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turley EA, Veiseh M, Radisky DC, Bissell MJ. Mechanisms of disease: epithelial-mesenchymal transition--does cellular plasticity fuel neoplastic progression? Nat Clin Pract Oncol. 2008;5:280–290. doi: 10.1038/ncponc1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viloria-Petit AM, David L, Jia JY, Erdemir T, Bane AL, Pinnaduwage D, Roncari L, Narimatsu M, Bose R, Moffat J, Wong JW, Kerbel RS, O’Malley FP, Andrulis IL, Wrana JL. A role for the TGFβ-Par6 polarity pathway in breast cancer progression. Proc Natl Acad Sci U S A. 2009;106:14028–14033. doi: 10.1073/pnas.0906796106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE. Cells of origin in cancer. Nature. 2011;469:314–322. doi: 10.1038/nature09781. [DOI] [PubMed] [Google Scholar]
- Wang B, Yang H, Huang YZ, Yan RH, Liu FJ, Zhang JN. Biologic characteristics of the side population of human small cell lung cancer cell line H446. Chin J Cancer. 2010;29:254–260. doi: 10.5732/cjc.009.10330. [DOI] [PubMed] [Google Scholar]
- Wang F, Hansen RK, Radisky D, Yoneda T, Barcellos-Hoff MH, Petersen OW, Turley EA, Bissell MJ. Phenotypic reversion or death of cancer cells by altering signaling pathways in three-dimensional contexts. J Natl Cancer Inst. 2002;94:1494–1503. doi: 10.1093/jnci/94.19.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SE, Xiang B, Zent R, Quaranta V, Pozzi A, Arteaga CL. Transforming growth factor β induces clustering of HER2 and integrins by activating Src-focal adhesion kinase and receptor association to the cytoskeleton. Cancer Res. 2009a;69:475–482. doi: 10.1158/0008-5472.CAN-08-2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Wyckoff JB, Goswami S, Wang Y, Sidani M, Segall JE, Condeelis JS. Coordinated regulation of pathways for enhanced cell motility and chemotaxis is conserved in rat and mouse mammary tumors. Cancer Res. 2007;67:3505–3511. doi: 10.1158/0008-5472.CAN-06-3714. [DOI] [PubMed] [Google Scholar]
- Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, Ali S, Abbruzzese JL, Gallick GE, Sarkar FH. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the Notch signaling pathway. Cancer Res. 2009b;69:2400–2407. doi: 10.1158/0008-5472.CAN-08-4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver VM, Peterson OW, Wang F, Larabell CA, Briand P, Damsky C, Bissell MJ. Reversion of the malignant phenotype of human breast cancer cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137:231–245. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver VM, Lelievre S, Lakins JN, Chrenek MA, Jones JC, Giancotti F, Werb Z, Bissell MJ. beta4 integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell. 2002;2:205–216. doi: 10.1016/s1535-6108(02)00125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt MK, Allington TM, Schiemann WP. Mechanisms of the epithelial-mesenchymal transition by TGF-β. Future Oncol. 2009a;5:1145–1168. doi: 10.2217/fon.09.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt MK, Schiemann WP. Therapeutic targeting of the focal adhesion complex prevents oncogenic TGF-β signaling and metastasis. Breast Cancer Res. 2009;11:R68. doi: 10.1186/bcr2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt MK, Smith JA, Schiemann WP. p130Cas is required for mammary tumor growth and transforming growth factor-β-mediated metastasis through regulation of Smad2/3 activity. J Biol Chem. 2009b;284:34145–34156. doi: 10.1074/jbc.M109.023614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt MK, Smith JA, Schiemann WP. Transforming growth factor-β-induced epithelial-mesenchymal transition facilitates epidermal growth factor-dependent breast cancer progression. Oncogene. 2010;29:6485–6498. doi: 10.1038/onc.2010.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittard JD, Craig SE, Mould AP, Koch A, Pertz O, Engel J, Humphries MJ. E-cadherin is a ligand for integrin α2β1. Matrix Biol. 2002;21:525–532. doi: 10.1016/s0945-053x(02)00037-9. [DOI] [PubMed] [Google Scholar]
- Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J Cell Biol. 2007;179:1311–1323. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Liang YL, Li Z, Jin J, Zhang W, Duan L, Zha X. Positive expression of E-cadherin suppresses cell adhesion to fibronectin via reduction of alpha5beta1 integrin in human breast carcinoma cells. J Cancer Res Clin Oncol. 2006;132:795–803. doi: 10.1007/s00432-006-0128-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyckoff JB, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, Graf T, Pollard JW, Segall JE, Condeelis JS. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004;64:7022–7029. doi: 10.1158/0008-5472.CAN-04-1449. [DOI] [PubMed] [Google Scholar]
- Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. 2007;117:524–529. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, Carbone DP, Matrisian LM, Richmond A, Lin PC, Moses HL. Abrogation of TGF β signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13:23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yauch RL, Januario T, Eberhard DA, Cavet G, Zhu W, Fu L, Pham TQ, Soriano R, Stinson J, Seshagiri S, Modrusan Z, Lin CY, O’Neill V, Amler LC. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of Erlotinib in lung cancer patients. Clin Cancer Res. 2005;11:8686–8698. doi: 10.1158/1078-0432.CCR-05-1492. [DOI] [PubMed] [Google Scholar]
- Yazhou C, Wenlv S, Weidong Z, Licun W. Clinicopathological significance of stromal myofibroblasts in invasive ductal carcinoma of the breast. Tumour Biol. 2004;25:290–295. doi: 10.1159/000081394. [DOI] [PubMed] [Google Scholar]
- Zavadil J, Bitzer M, Liang D, Yang YC, Massimi A, Kneitz S, Piek E, Bottinger EP. Genetic programs of epithelial cell plasticity directed by transforming growth factor-β. Proc Natl Acad Sci U S A. 2001;98:6686–6691. doi: 10.1073/pnas.111614398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavadil J, Narasimhan M, Blumenberg M, Schneider RJ. Transforming growth factor-β and microRNA:mRNA regulatory networks in epithelial plasticity. Cells Tissues Organs. 2007;185:157–161. doi: 10.1159/000101316. [DOI] [PubMed] [Google Scholar]
- Zeng Q, Phukan S, Xu Y, Sadim M, Rosman DS, Pennison M, Liao J, Yang GY, Huang CC, Valle L, Di Cristofano A, de la Chapelle A, Pasche B. Tgfbr1 haploinsufficiency is a potent modifier of colorectal cancer development. Cancer Res. 2009;69:678–686. doi: 10.1158/0008-5472.CAN-08-3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha J, Lackner MR. Targeting the insulin-like growth factor receptor-1R pathway for cancer therapy. Clin Cancer Res. 2010;16:2512–2517. doi: 10.1158/1078-0432.CCR-09-2232. [DOI] [PubMed] [Google Scholar]
- Zhang N, Li R, Tao KS, Cao DY, Ti ZY, Ding R, Cai L, Zhang FQ, Dou KF. Characterization of a stem-like population in hepatocellular carcinoma MHCC97 cells. Oncol Rep. 2010;23:827–831. [PubMed] [Google Scholar]
- Zhang W, Alt-Holland A, Margulis A, Shamis Y, Fusenig NE, Rodeck U, Garlick JA. E-cadherin loss promotes the initiation of squamous cell carcinoma invasion through modulation of integrin-mediated adhesion. J Cell Sci. 2006;119:283–291. doi: 10.1242/jcs.02738. [DOI] [PubMed] [Google Scholar]
- Zhou X, Sasaki H, Lowe L, Hogan BL, Kuehn MR. Nodal is a novel TGF-β-like gene expressed in the mouse node during gastrulation. Nature. 1993;361:543–547. doi: 10.1038/361543a0. [DOI] [PubMed] [Google Scholar]