

Background: EB1 is a microtubule (MT) plus-end-binding protein known to influence MT stability.
Results: EB1 is up-regulated in osteoblasts and is required for bone cell differentiation.
Conclusion: EB1 affects β-catenin stability and cooperates at cell-cell adhesion sites to influence gene expression.
Significance: Learning how peripherally targeted proteins interact at cell-cell contact sites is crucial for understanding developmental processes.
Keywords: beta-Catenin, Cell Biology, E-cadherin, Microtubules, Osteoblasts, EB1, Cell Contacts
Abstract
Osteoblasts are differentiated mesenchymal cells that function as the major bone-producing cells of the body. Differentiation cues including ascorbic acid (AA) stimulation provoke intracellular changes in osteoblasts leading to the synthesis of the organic portion of the bone, which includes collagen type I α1, proteoglycans, and matrix proteins, such as osteocalcin. During our microarray analysis of AA-stimulated osteoblasts, we observed a significant up-regulation of the microtubule (MT) plus-end binding protein, EB1, compared with undifferentiated osteoblasts. EB1 knockdown significantly impaired AA-induced osteoblast differentiation, as detected by reduced expression of osteoblast differentiation marker genes. Intracellular examination of AA-stimulated osteoblasts treated with EB1 siRNA revealed a reduction in MT stability with a concomitant loss of β-catenin distribution at the cell cortex and within the nucleus. Diminished β-catenin levels in EB1 siRNA-treated osteoblasts paralleled an increase in phospho-β-catenin and active glycogen synthase kinase 3β, a kinase known to target β-catenin to the proteasome. EB1 siRNA treatment also reduced the expression of the β-catenin gene targets, cyclin D1 and Runx2. Live immunofluorescent imaging of differentiated osteoblasts revealed a cortical association of EB1-mcherry with β-catenin-GFP. Immunoprecipitation analysis confirmed an interaction between EB1 and β-catenin. We also determined that cell-cell contacts and cortically associated EB1/β-catenin interactions are necessary for osteoblast differentiation. Finally, using functional blocking antibodies, we identified E-cadherin as a major contributor to the cell-cell contact-induced osteoblast differentiation.
Introduction
The dynamic nature of bone involves an interplay between bone formation (osteogenesis) and born resorption (osteolysis). It is through constant bone remodeling that vertebrates are able to maintain constant bone mass in disease-free states. The tightly regulated balance of remodeling of bone involves osteoblasts that form new bone and osteoclasts that remove bone. Osteoclast ontogeny and the mechanisms that regulate bone resorption have been studied intensely (1, 2), with much of the therapeutic treatments for bone-wasting disorders targeted toward osteoclasts (3). In light of the capability to target bone resorption mechanisms, there has been a more recent movement toward understanding how the osteoblastic differentiation mechanism can be targeted to enhance bone formation (4, 5). Thus, understanding the molecular mechanisms that control osteoblast differentiation is paramount to identify therapeutic targets in bone wasting disorders.
Mature osteoblasts originate from multipotent mesenchymal stem cells that are induced to differentiate toward an osteoblastic lineage by various factors. The canonical Wnt signaling plays a major role in osteoblast differentiation (6), with a pool of soluble and highly unstable β-catenin transducing the Wnt signal in this pathway (7). The signaling pathway is initiated by the ligation of the Wnt ligand to the Frizzled and LDL receptor-related protein 5/6 (LRP5/6) coreceptors, which initiates a signaling cascade that leads to an intracellular accumulation and nuclear recruitment of β-catenin (8). In the absence of Wnt ligands, cytoplasmic β-catenin is recruited into a destruction complex, which induces β-catenin phosphorylation, ubiquitination, and subsequent proteasomal degradation. Within the canonical pathway, Wnt stimulation promotes dissociation of the destruction complex, inhibits β-catenin degradation, and promotes the translocation of β-catenin to the nucleus and subsequent interaction with T-cell factor/lymphoid enhancer factor (TCF/LEF)4 to initiate transcription of target genes (6, 7). The necessity of β-catenin in osteoblast differentiation and bone development is well established (9–11), wherein osteoblast precursors that lack β-catenin do not differentiate (10).
The differentiation of osteoblasts involves the sequential activation of at least two osteoblast-specific transcription factors, RUNX2 and osterix, which acts further downstream from RUNX2 (12). RUNX2, also known as CBFA1, is considered a master regulator of transcription for early osteoblast genes (6). The presence of a TCF regulatory element on the Runx2 promoter indicates that the canonical Wnt signaling pathway directly regulates Runx2 gene expression in pluripotent mesenchymal and osteoprogenitor cells via the recruitment of β-catenin to the Runx2 gene and thus contributes to osteoblast maturation (13). Runx2 knock-out mice have a severe defect in intramembranous and endochondral ossification (14, 15). RUNX2 is expressed in early stages and throughout osteoblast differentiation and has been shown to bind to and regulate the expression of many osteoblast genes, with RUNX2 binding regions present in the promoter regions of osteocalcin, collagen, and bone sialoprotein genes (16). Interestingly, the ectopic expression of RUNX2 in fibroblasts that are not committed to the osteoblast lineage induces the gene expression of the osteoblast-specific markers, including collagen, bone sialoprotein, and osteocalcin (16).
Aside from the role of β-catenin in the Wnt signaling pathway, β-catenin also has a secondary function at sites of cell-cell contacts at adherens junctions. The transmembrane cell adhesion molecule, E-cadherin, is a major component of adherens junctions in epithelial and other cell types (17–19) that recruits β-catenin and results in the coupling of E-cadherin to the Wnt pathway. The binding of β-catenin to type I cadherins renders a stable pool of membrane-bound β-catenin that regulates and stabilizes these cell-cell contacts (20, 21). High resolution analysis has allowed understanding of the elaborate cell adhesion complex that includes cadherins, catenins, and the F-actin network (22). Adherens junctions also have a microtubule (MT) component, wherein dynamic MTs recruit and control the regional distribution of cadherins at cell-cell contacts (23). MT plus-end binding proteins have been observed to target these adherens junctions (23–26). The end-binding protein, EB1, is one of the best studied MT plus-end binding proteins that stabilizes MTs (27, 28) by forming comet-like structures at the tips of growing microtubules (29, 30). In conjunction with the EB3 family member, EB1 promotes continuous MT growth in cells by inhibiting MT catastrophes (31). Dynamic MT ends are required for the lateral movement and clustering of E-cadherin but are not necessary for E-cadherin surface display (23). EB1 has been shown to target to β-catenin puncta at the cell surface (24, 26) and co-localize with cadherins (23–25). The adenomatous polyposis coli (APC) tumor suppressor protein, which is also an MT plus-end protein, stabilizes complexes with the axin scaffolding protein and the two kinases, glycogen synthase kinase 3β (GSK-3β) and casein kinase 1α, to form the destruction complex and regulate β-catenin protein levels (32). EB1 has been identified in a binding screen for APC (33), and thus EB1 may target APC to MT plus-ends and thus enable the interactions of APC with cortical targets (29). In addition, overexpression of EB1 has been found to promote cellular growth in cancer models via the β-catenin/TCF pathway (34–37).
Given the importance of the Wnt signaling cascade in osteoblast differentiation, in the present study, we identify how osteoblast differentiation is influenced by cytoskeletal elements, namely EB1, the MT plus-end-binding protein. We utilized the MC3T3-E1 mouse preosteoblast cell line to allow molecular manipulation of EB1 protein levels. We show that EB1 is significantly up-regulated in ascorbic acid (AA)-stimulated osteoblasts and that EB1 knockdown significantly impairs the osteoblast differentiation program. Through cell biology analysis, we determine that EB1 interacts with and influences the stability of β-catenin and identify EB1 as an important regulator of cell-cell adhesion-induced osteoblast differentiation.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
Fetal bovine serum was purchased from Wisent Inc. (St-Bruno, Canada). α-Minimal essential medium, Alexa Fluor 488, Oligofectamine, and Lipofectamine 2000 were purchased from Invitrogen. Rat and mouse monoclonal antibodies against EB1 were purchased from Abcam (Cambridge, UK) and Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), respectively. β-Catenin mouse monoclonal antibody was purchased from BD Transduction Laboratories (Mississauga, Canada). Mouse monoclonal active β-catenin antibody was purchased from Millipore (Billerica, MA). Phospho-β-catenin (Ser-33/37/Thr-41) rabbit polyclonal, GSK-3β, phospho-GSK-3α/β rabbit polyclonal (Ser-21/9), and GAPDH HRP rabbit polyclonal antibodies were purchased from Cell Signaling Inc. (Danvers, MA). Anti-E-cadherin rat monoclonal blocking antibody (ECCD-1) was purchased from Calbiochem. Phalloidin was purchased from Invitrogen. Anti-acetylated mouse monoclonal tubulin and mouse monoclonal actin antibodies were purchased from Sigma-Aldrich. Collagen type I α1 rabbit polyclonal antibody was purchased from Cedarlane (Burlington, Canada). All fluorescently labeled and horseradish peroxidase-conjugated secondary antibodies were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). The Pierce chemiluminescence kit was obtained from Thermo Fisher Scientific (Rockford, IL). l-Ascorbic acid powder and control isotype IgG from rat serum were purchased from Sigma-Aldrich.
Cell Culture, Osteoblast Treatments, and Transfections
MC3T3-E1 subclone 4 was acquired from ATCC (American Type Culture Collection, Manassas, VA). The MC3T3-E1 preosteoblast cell line was routinely cultured in α-minimal essential medium without AA, supplemented with heat-inactivated 10% fetal bovine serum, and incubated in a humidified atmosphere at 37 °C with 5% CO2. For all experiments, osteoblasts grown to 80% confluence were passed using 0.05% trypsin and then plated onto 6-well plates (Starstedt Inc.) with 25-mm glass coverslips for immunofluorescent studies. For Western blotting and RNA extraction, cells were grown in 6-well plates without coverslips. For differentiation, osteoblasts were stimulated with AA (50 μg/ml) or 100 ng/ml rhBMP-2 (PeproTech, Rocky Hill, NJ), over a 5-day period, with medium and AA being replenished every second day. For primary cell analysis, cells were isolated from postnatal day 0 C57BL/6J mouse calvaria using a modification of a method described previously (38, 39). For plasmid transfection, osteoblasts were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. For comparative experiments between low and high density growth, cells were plated at 10,000 and 100,000 cells/ml, respectively, and treated with AA for 5 days. For experiments involving E-cadherin-blocking antibody, cells were treated with isotype control IgG or a blocking antibody against E-cadherin (20 μg/ml) at days 2 and 4 during a 5-day stimulation with AA.
Microarray Analysis
Microarray data were obtained from Ref. 40. MC3T3-E1 osteoblasts were grown in 6-well plates in triplicates and differentiated with AA (50 μg/ml) for 5 days. Total RNA from control and AA-treated osteoblasts was purified from cultured cells using the RNeasy minikit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Samples were hybridized to MOE430.20 GeneChips (Affymetrix, Santa Clara, CA) at the Center for Applied Genomics at SickKids (Toronto, Canada) and analyzed as described previously (40). -Fold changes in gene expression levels were analyzed using Student's unpaired t tests. A critical p value of 0.01 was considered as the criterion to select a significant -fold change in gene expression. Of the total 45,103 genes identified by (40), EB1 and components of Wnt signaling were selected for further investigation in the present study.
siRNA Treatments
MC3T3-E1 cells treated with AA for 2 days were transfected with Oligofectamine according to the manufacturer's instructions using MISSION siRNA (SASI_Mm02_00312684, Sigma-Aldrich) directed against EB1 at a concentration of 20 μm for 72 h in addition to treatment with AA. Control cells were transfected with a MISSION® siRNA universal negative (scrambled) control (Sigma-Aldrich). Protein levels of siRNA-treated cells were analyzed by immunoblotting, immunofluorescence, and quantitative PCR 3 days after transfection.
Cloning and Quantitative PCR
All expression constructs were created with standard PCR-based cloning strategies. Total RNA was isolated from cultured MC3T3-E1 cells using the RNeasy minikit from Qiagen (Hilden, Germany). The cDNA sequences of full-length EB1 and β-catenin were amplified with Superscript One-Step RT-PCR with a platinum Taq kit using the following primer pairs: EB1-F, 5′-aatgctagcaccatggcagtgaat-3′; EB1-R, 5′-attgaattcgatactcttcttgttcctc-3′; β-catenin-F, 5′-aagctagcaccatggctactcaagctgacc-3′; β-catenin-R, 5′-aagctagcaccatggctactcaagctgacc-3′.
The resulting PCR products were cloned in frame to pRetroQ-mcherry-N1 and peGFP-N1 vectors, respectively, using NheI and EcoRI restriction sites for EB1 and NheI and BamHI for β-catenin. Quantitative RT-PCR was performed following a time course of ascorbic acid stimulation, in addition to measuring differences in mRNA levels of cells grown in low or high density environments. For quantitative RT-PCR, total mRNA from MC3T3-E1 cells grown in 6-well plates was extracted using the RNeasy kit (Qiagen, Hilden, Germany). The purity and quantity of RNA were confirmed by a NanoDrop® ND-100 spectrophotometer (Thermo Fisher Scientific). cDNA was synthesized from 1 μg of total RNA in 50-μl reaction mixtures, using the SuperScript III First-Strand Synthesis SuperMix for quantitative RT-PCR from Invitrogen. A control cDNA serial dilution series of known concentration was constructed for each gene to establish a standard curve. Identical volumes of cDNA were loaded for all samples, and samples were run in triplicate. GAPDH was chosen as the reference gene for normalization of the results. The 25-μl real-time quantitative reverse transcriptase polymerase chain reaction (RT-PCR) mixtures contained 1 μl of cDNA, a 10 μm concentration of each primer, and 2× SYBR Green (Bio-Rad). The PCR was carried out at an initial denaturing temperature of 95 °C for 3 min, followed by a protocol consisting of 40 cycles of 30 s at 95 °C, 30 s at 55 °C, and 30 s at 72 °C using a DNA Engine Opticon System (Bio-Rad). After the last cycle, a dissociation curve was generated by first collecting fluorescent signals at 55 °C with subsequent measurements taken at 2-s intervals with increasing temperature up to 95 °C. We confirmed a linear range of amplification for all primers and products, and melting curves were used to verify the absence of nonspecific PCR products. The CT (cycle number at which fluorescence is detected above threshold) was determined by the Opticon Monitor software algorithm. The ΔCT for gene-specific mRNA expression was calculated relative to the CT of GAPDH, a housekeeping gene to control for input cDNA. Relative mRNA expression levels were calculated using the formula 2−ΔCT, by which ΔΔCT = ΔCT sample − ΔCT reference. Three or more independent experiments were performed for each sample and in triplicate, and the corresponding results were normalized to GAPDH and expressed as mean ± S.E. or S.D. if otherwise mentioned. -Fold change was calculated relative to the sample for the same gene with the lowest expression level, which was set to 1. Statistical analyses were conducted using Student's unpaired t tests (p values of <0.05). The specific primers used were as follows: EB1(MAPRE1)-F, 5′-gaatctgacaaagatagaacagttgtg-3′; EB1(MAPRE1)-R, 5′-cactttcttcaaggcaatggat-3′; alkaline phosphatase-F, 5′-cggatcctgaccaaaaacc-3′; alkaline phosphatase-R, 5′-tcatgatgtccgtggtcaat-3′; bone sialoprotein-F, 5′-gaaaatggagacggcgatag-3′; bone sialoprotein-R, 5′-cattgttttcctcttcgtttga-3′; collagen type I, α 1-F, 5′-catgttcagctttgtggacct-3′; collagen type I, α 1-R, 5′-gcagctgacttcagggatgt-3′; osteocalcin-F, 5′-caccatgaggaccctctctc-3′; osteocalcin-R, 5′-tggacatgaaggctttgtca-3′; cyclin D1-F, 5′-gcgtaccctgacaccaatctc-3′; cyclin D1-R, 5′-acttgaagtaagatacggagggc-3′; Runx2-F, 5′-gactgtggttaccgtcatggc-3′; Runx2-R, 5′-acttggtttttcataacagcgga-3′; E-cadherin-F, 5′-caggtctcctcatggctttgc-3′; E-cadherin-R, 5′-cttccgaaaagaaggctgtcc-3′; β-catenin-F, 5′-gatttcaaggtggacgagga-3′; β-catenin-R, 5′-cactgtgcttggcaagttgt-3′; osterix-F, 5′-tctccatctgcctgactcct-3′; osterix-R, 5′-agcgtatggcttctttgtgc-3′; GAPDH-F, 5′-tcaacgaccccttcattgac-3′; GAPDH-R, 5′-atgcagggatgatgttctgg-3′.
Western Blot Analysis
Cells were washed with ice-cold phosphate-buffered saline (PBS), and lysates were collected using radioimmunoprecipitation assay buffer containing protease and phosphatase inhibitor mixtures (1:1000 dilution) from Sigma-Aldrich. Insoluble and soluble fractions were separated by centrifugation at 12,000 × g for 10 min at 4 °C. Protein samples were then boiled in 2× Laemmli buffer for 5 min. Cell lysates were loaded on a 10% SDS-polyacrylamide gel, separated in Tris-glycine buffer, and transferred to nitrocellulose membranes (Bio-Rad). The membranes were blocked for 1 h in 5% nonfat milk diluted in Tris-buffered saline with Tween (TBST; 0.1% Tween 20 in 20 mm Tris-HCl, pH 7.6, 137 mm NaCl) and incubated with primary antibodies against EB1 (1:1000), β-catenin (1:1000), phospho-β-catenin (1:500), active β-catenin (1:800), GSK-3β (1:1000), phospho-GSK-3α/β (1:1000), actin (1:5000), tubulin (1:5000), GAPDH (1:1000), and collagen (1:1500) overnight at 4 °C and then subsequently incubated for 1 h with corresponding horseradish peroxidase-conjugated secondary antibodies. Protein signals were visualized using a Supersignal® West Pico chemiluminescent substrate kit (Fisher) according to the manufacturer's instructions. Relative differences in protein levels were measured with ImageJ software (National Institutes of Health, Bethesda, MD) by determining -fold changes of protein band pixels of treated cells relative to control conditions.
Immunoprecipitations
Cultures of MC3T3-E1 cells were grown to subconfluence on 10-cm tissue culture plates. After the experimental treatment, cells were rinsed twice in PBS, and protein lysates were obtained using radioimmunoprecipitation assay lysis buffer containing a mixture of protease inhibitors (Sigma-Aldrich). Total proteins (450 μg) were precleared overnight with rotation at 4 °C using 50 μl of Protein A/G Plus-agarose (sc-2003, Santa Cruz Biotechnology, Inc.). Lysates were then pooled and normalized for total protein concentration. EB1 and β-catenin antibodies were incubated overnight with the ExactaCruz IP matrix (sc-45042, Santa Cruz) to form IP complexes according to the manufacturer's instructions. An equivalent amount of isotypic IgG (Sigma-Aldrich) was bound to the ExactaCruz IP matrix as a negative control. Then IP matrix-antibody complexes were washed twice with phosphate-buffered saline and then incubated with cell lysates at 4 °C. The IP matrices were pelleted, washed three times with radioimmunoprecipitation assay buffer containing a mixture of protease inhibitors (Sigma-Aldrich), and finally resuspended in 2× Laemmli sample buffer. After separation by SDS-PAGE, the proteins were transferred to a nitrocellulose membrane for Western blotting analysis using EB1 and β-catenin antibody, respectively. Relative differences in protein levels were measured with ImageJ software by determining -fold changes of protein band pixels of cells treated with ascorbic acid during a time course relative to control conditions.
Immunostaining and Fluorescent Imaging
For immunostaining of EB1, α-tubulin, and β-catenin, cells were fixed and permeabilized in ice-cold 100% methanol for 10 min at −20 °C. For actin staining, cells were fixed in 4% PFA for 20 min and permeabilized with 0.1% Triton X-100 in PBS containing 100 mm glycine for 20 min. Cells were blocked in 5% fetal bovine serum in PBS for 1 h and incubated with the indicated primary antibodies in 1% fetal bovine serum in PBS at a dilution of 1:1000 for EB1, 1:10,000 for β-catenin, and 1:10,000 for α-tubulin. Cells were then incubated with appropriate fluorescent secondary antibodies (1:1000) and/or phalloidin (1:500) for 1 h. For nuclear staining, cells were washed twice with H2O and incubated for 10 min with DAPI (1:10,000) before mounting. Slides were mounted using Dako fluorescent mounting medium (DakoCytomation, Carpinteria, CA). Epifluorescent images were acquired with an EC Plan apochromat ×63/1.4 oil objective and AxioCam MRm camera on an Axio Observer Z1 microscope (Carl Zeiss). For live cell imaging, EB1-mCherry and β-catenin-GFP constructs were transfected overnight in osteoblasts that had been exposed to AA for 2 days. Time lapse imaging was carried out using an XL-S1 incubator with TempModule S1, CO2 module S1, heating insert P S1, and heating device humidity S1 connected to an Axio Observer Z1 microscope (Carl Zeiss). All live cell images were captured with an AxioCam MRm camera. Fluorescent and differential interference contrast images were acquired every 10 s for 10 min. Approximately 10 EB1 comets/cell were measured from 15 immunofluorescent images of undifferentiated versus 1-, 3-, and 5-day AA-stimulated osteoblasts using ImageJ software.
Mineralization Assay
MC3T3-E1 cells were plated at an initial density of 100,000 cells/well. 50 μg/ml AA in fresh medium was exchanged every 2 days until confluence was reached. Upon confluence, 10 mm β-glycerophosphate (Sigma) was added at each medium change. Cultures were transfected with siRNA as indicated previously, beginning with day 9 of a 23-day culture period. Every 4 days, until day 21, cells were treated with siRNA to maintain EB1 knockdown. At day 21, cultures were washed, and a recovery period of 3 days was allowed before ALP/von Kossa staining. Cells were fixed in 4% paraformaldehyde for 15 min, washed, and incubated with AS MX phosphate/Fast Red Violet LB in 0.2 m Tris-HCl (pH 8.3), followed by incubation with 2.5% silver nitrate solution. All reagents were purchased from Sigma-Aldrich. Images were taken using a Zeiss Axioplan 2 epifluorescent microscope with a color AxioCam HRc camera.
RESULTS
EB1 Is Up-regulated in AA-stimulated Osteoblasts
We recently performed a microarray analysis to identify the up-regulated Rab GTPases that are important for procollagen trafficking during osteoblast differentiation (40). Our microarray analysis revealed an expected up-regulation of the components of the Wnt signaling pathway (Fig. 1A), in particular Wnt10a and Wnt11, which are known to signal through β-catenin to influence osteoblast differentiation (41, 42). Interestingly, the microarray analysis also revealed that the MT plus-end-binding protein, EB1, was elevated over 2-fold in MC3T3-E1 cells stimulated with AA (50 μg/ml) for 5 days, compared with control cells (Fig. 1A). We validated the microarray analysis using quantitative RT-PCR and observed a nearly 5-fold increase in EB1 transcript in 5-day stimulated osteoblasts, compared with undifferentiated cells (Fig. 1B). Enhanced EB1 protein was likewise detected using immunoblotting of cell lysates during the course of AA stimulation (Fig. 1C). Osteoblast differentiation is typified by dramatic collagen up-regulation (43–45), and EB1 protein increased in parallel with procollagen production in AA-stimulated osteoblasts (Fig. 1C). Epifluorescent inspection of endogenous EB1 in control cells or cells fixed at day 1, 3, or 5 post-AA stimulation revealed characteristic comets of EB1 at the ends of MTs (Fig. 1D). The EB1 comets were longer and more prominent in osteoblasts at later stages of AA-induced differentiation (Fig. 1, D and E). Thus, the osteoblast differentiation profile includes up-regulation of the MT plus-end-binding protein, EB1. Brighter EB1 comets were observed in AA-treated primary mouse calvaria osteoblasts, and these cells showed a significant increase in EB1 mRNA and a modest increase in total EB1 protein during AA induction of osteoblast marker genes, compared with control unstimulated cells (supplemental Fig. 1).
FIGURE 1.

EB1 is up-regulated in AA-treated osteoblasts. A, microarray analysis revealed components of the Wnt signaling pathway and EB1 mRNA levels that were up-regulated in 5-day AA-treated MC3T3-E1 osteoblast cells, compared with control cells. B, quantitative RT-PCR validation revealed a significant increase in EB1 mRNA expression levels in AA-treated osteoblasts over a 5-day time course, when compared with undifferentiated control cells (ctl). All of the signal intensities were normalized against that of the GAPDH gene. The time course was done in triplicate, and data are plotted as the mean ± S.E. from three independent experiments (*, p < 0.05, Student's t test). C, Western blot analysis showing the levels of EB1 protein during 5 days of differentiation with AA. Top, endogenous EB1 expression was detected using an anti-EB1 antibody. Middle, total collagen levels during AA stimulation. Bottom, loading of equal amounts of protein was confirmed using the GAPDH antibody. -Fold increase in EB1 levels normalized to GAPDH is shown numerically below the blot. *, significant -fold increase compared with control cells (p < 0.05). D, representative immunofluorescent images showing the changes in EB1 protein expression and distribution in 1-, 3-, and 5-day AA-treated osteoblasts, compared with untreated control cells. EB1 is shown in red and MTs in green. Immunostaining with an anti-EB1 antibody shows the typical comet-like pattern of growing MT plus-ends, from the center of the cells outward. The arrows highlight increasing comets extending toward the cell periphery. E, quantification of EB1 comet length showed a significant increase following 5 days of AA treatment, compared with unstimulated control cells. Average comet length was determined from 10 comets/cell from a total of 15 images/treatment day, and data are shown as mean ± S.D. (error bars) (*, p < 0.01, Student's t test). Scale bars, 10 μm.
EB1 Is Required for Osteoblast Differentiation
Due to the progressive and significant increase in EB1 levels following AA stimulation, we next asked whether EB1 is essential for osteoblast differentiation. After 2 days of AA stimulation, MC3T3-E1 cells were then exposed to either scrambled or EB1 siRNA for the final 72 h of AA stimulation. Immunostaining and immunoblotting with an EB1 antibody were performed to confirm the successful protein knockdown of EB1 (Fig. 2, A and B). EB1 knockdown induced MT curvature and reduced levels of stabilized MTs, as detected by immunoblotting with an acetylated α-tubulin antibody (Fig. 2, A and B) (46, 47), in agreement with previous findings. RNA was collected from cells exposed to scrambled or EB1 siRNA, and total mRNA was probed for the levels of specific bone cell markers, including alkaline phosphatase, bone sialoprotein, collagen type I α1, and osteocalcin using quantitative RT-PCR (Fig. 2C). In the 5-day AA-treated cultures, EB1 siRNA treatment caused a significant reduction in EB1 mRNA as expected (Fig. 2C). Interestingly, EB1 siRNA treatment also caused significant decreases in alkaline phosphatase, bone sialoprotein, collagen type I α1, and osteocalcin mRNA levels, compared with scrambled siRNA-treated cells (Fig. 2C). Thus, EB1 is essential for early osteoblast differentiation induced by AA stimulation.
FIGURE 2.

EB1 knockdown impairs AA-induced osteoblast differentiation. A, immunoblot analysis revealed a decrease of EB1 protein expression following EB1 knockdown by siRNA for 72 h in 5-day AA-stimulated MC3T3-E1 cells. Control cells were treated with scrambled (scr) siRNA. The -fold decrease in EB1 level following EB1 siRNA treatment was normalized to β-actin and is shown relative to scrambled siRNA-treated cells. Immunoblot analysis of 5-day AA-simulated osteoblast cell lysates revealed that, concomitant with EB1 reduction, there is a significant decrease in acetylated α-tubulin levels, compared with scrambled siRNA-treated cells. *, significant -fold decrease compared with scrambled siRNA-treated cells (p < 0.05). B, immunostaining for EB1 (red) and MTs (green) confirmed the protein knockdown of EB1 in siRNA-treated cells and waviness and alteration of the MT network (arrows). Scale bars, 10 μm. C, effect of EB1 knockdown on osteoblast differentiation. Cells were differentiated with AA for 5 days and transfected with scrambled or EB1 siRNAs for the final 72 h. RNA was prepared from scrambled and EB1 siRNA-treated cells, and total RNA was probed for the levels of specific bone cell markers, including alkaline phosphatase, bone sialoprotein, collagen type I α1, and osteocalcin using quantitative RT-PCR analysis. Signal intensity was normalized against that of the GAPDH gene. Each sample was done in triplicate, and data are plotted as the mean ± S.E. (error bars) of results from four independent experiments (*, p < 0.05, Student's t test showing the significant –fold change compared with control scrambled siRNA-treated cells).
After establishing the importance of EB1 in early osteoblast differentiation events, we next examined whether EB1 was essential for bone mineralization. Bone mineralization, assessed by ALP and von Kossa staining, was strongly reduced in cultures treated with EB1 siRNA, compared with scrambled siRNA (supplemental Fig. 2). These results are in agreement with the notion that mineralization is a carefully orchestrated process, strongly influenced by increasing inorganic phosphate availability with ALP activity (48). Appropriate trafficking and delivery of matrix vesicles, rich in calcium, phosphate, and phosphoproteins, could also be at a disadvantage in cells with reduced EB1 levels (49). Thus, EB1 is also necessary for late stage osteoblast-mediated matrix mineralization.
EB1 Knockdown Impacts β-Catenin Levels and Distribution in Osteoblasts
We next examined the role of EB1 in the canonical Wnt signaling pathway, which has been implicated in BMP-2-induced osteoblast differentiation (50, 51). We first performed a detailed time course analysis of β-catenin mRNA expression in cells treated with AA for 5 days. β-Catenin transcript levels responded strongly to AA stimulation (Fig. 3A). A protein expression time course in cells treated with AA was performed with immunoblotting, and a marked up-regulation of β-catenin protein was also observed, compared with untreated cells (Fig. 3B). We then examined the temporal regulation of GSK-3β, the kinase that phosphorylates β-catenin and marks it for destruction by the proteasome (52). The total levels of GSK-3β did not change throughout osteoblast differentiation (Fig. 3B), but an increase in phospho-GSK-3β (Ser-9) was observed 3 and 5 days after AA simulation (Fig. 3B), indicating a steady inactivation of this kinase (53) following AA exposure. Therefore, OB differentiation includes a steady enrichment of β-catenin protein levels and, in parallel, a down-regulation of GSK-3β activity.
FIGURE 3.

EB1 is important for β-catenin levels and distribution during osteoblast differentiation. A, quantitative RT-PCR confirmed a significant increase in β-catenin mRNA expression levels in 5-day AA-treated osteoblasts, when compared with undifferentiated control cells (ctl). Signal intensities were normalized against GAPDH values. The time course was done in triplicate, and data are plotted as the mean ± S.D. (error bars) from three independent experiments (*, p < 0.05, Student's t test). B, time course immunoblotting analysis throughout a 5-day AA stimulation regime showed a significant increase in total β-catenin as well as a significant increase in phospho-GSK-3β (both normalized to GAPDH levels); total GSK-3β was unaltered throughout the course of AA stimulation. Numbers indicate -fold changes from control cells from three independent trials (p < 0.05, Student's t test). C, following EB1 knockdown by siRNA for 72 h, immunoblot analysis of 5-day AA-simulated osteoblast cell lysates revealed that, concomitant with EB1 reduction, there is a significant decrease in β-catenin levels and a significant increase in phospho-β-catenin levels, compared with scrambled (scr) siRNA-treated cells. Numbers indicate -fold changes from scrambled siRNA-treated cells (p < 0.05, Student's t test). D, quantitative RT-PCR, after 72-h EB1 siRNA knockdown, displayed an accompanying significant decrease in β-catenin mRNA expression levels, compared with scrambled siRNA-treated cells. Values are presented as mean ± S.D. from three independent experiments (*, p < 0.01). E, immunofluorescence analysis of 5-day AA-treated osteoblasts exposed to control, scrambled siRNA showed robust recruitment of β-catenin at the cell cortex (arrows) and within the nucleus (asterisks); however, these β-catenin localizations were visibly diminished in cells treated with EB1 siRNA. Scale bars, 10 μm.
To examine the involvement of EB1 in regulating β-catenin, we performed knockdown experiments. Using immunoblotting, we examined total cellular levels of β-catenin and observed a significant reduction in β-catenin protein levels in cells treated with EB1 siRNA versus control cells, which showed the expected high levels of β-catenin after 5 days of AA stimulation (Fig. 3C). To determine the possible mechanism behind the reduced β-catenin levels in EB1 siRNA-treated cells, we next examined the phosphorylation status of this protein. Immunoblotting of EB1 siRNA-treated lysates showed increased levels of phospho-β-catenin at sites that are phosphorylated by GSK-3β (Ser-33/37) (54), compared with scrambled siRNA-treated cells (Fig. 3C). Quantitative RT-PCR analysis confirmed that a significant decrease in β-catenin transcript levels also occurred in EB1-depleted cells, compared with scrambled siRNA-treated cells (Fig. 3D). To determine which cellular pool of β-catenin was impacted by EB1 knockdown, we next examined the distribution of β-catenin using immunofluorescence. In 5-day AA-stimulated control cells treated with scrambled siRNA, there was robust recruitment of β-catenin to sites of cell-cell contact (24) (Fig. 3E). β-Catenin was also localized to the nuclei of differentiating osteoblasts (Fig. 3E), as described previously (55). β-Catenin showed a marked difference in localization in cells treated with EB1 siRNA, with reduced accumulation at the cell cortex as well as marginal staining within the nuclei (Fig. 3E). Altogether, these results indicated that EB1 is important for the protection of β-catenin from GSK-3β-mediated proteolytic destruction during osteoblast differentiation.
Growing evidence suggests that cross-talk exists between the Wnt pathway and other established osteoblast differentiation pathways (6, 20, 56). BMP-2 is a well studied inducer of osteoblast differentiation. Recent papers (51, 57) have demonstrated that BMP-2 cooperates with β-catenin during differentiation and has the potential to up-regulate Wnt-specific genes. It was of interest to test whether EB1 also impacted BMP-2-mediated differentiation. Gene markers of osteoblast differentiation were first assessed through quantitative RT-PCR. Apart from collagen type I α1, bone sialoprotein, alkaline phosphatase, and osteocalcin transcript levels increased in cells upon the addition of BMP-2, relative to unstimulated control cells (Fig. 4A). Interestingly, EB1 mRNA expression was not enhanced by BMP-2 stimulation (Fig. 4B). Only a modest, but not significant, increase in EB1 protein levels (Fig. 4C) and a similar EB1 cellular distribution (Fig. 4D) were observed in BMP-2-treated cells, compared with unstimulated cells. However, when cells were treated with EB1 siRNA (Fig. 4E), expression of all major osteoblast differentiation marker genes was significantly decreased, compared with scrambled siRNA-treated cells (Fig. 4F). Therefore, the presence of EB1 is also required for BMP-2-induced osteoblast differentiation.
FIGURE 4.

EB1 is important for BMP-2 induced osteoblast differentiation. A, quantitative RT-PCR analysis revealed a significant increase in the specific osteoblast markers, bone sialoprotein (BSP), ALP, and osteocalcin (OCN), in BMP-2-treated cells, compared with control cells. Signal intensities were normalized against that of the GAPDH gene. The time course was performed in triplicate, and data are plotted as the mean ± S.D. (error bars) from three independent experiments (*, p < 0.01, Student's t test). Interestingly, collagen type I α1 and EB1 (B) mRNA levels were not elevated during the 5-day BMP-2 treatment. C, Western blot analysis of EB1 protein levels showed a modest, but not significant, increase after 5 days of differentiation with BMP-2, compared with control cells. Equal loading of lysate was confirmed with β-actin protein levels. -Fold increase in EB1 levels, normalized to β-actin, is shown numerically below the blot. D, similar EB1 distribution patterns between control and 5-day BMP-2-treated MC3T3-E1 cells are shown in representative epifluorescent images. EB1 is shown in red, and MTs are shown in green. Scale bar, 20 μm. A representative immunoblot of EB1 siRNA knockdown efficiency is shown in E. F, effect of EB1 siRNA treatment on osteoblast differentiation. Cells were treated with BMP-2 for 5 days and were subjected to scrambled or EB1 siRNA for the final 72 h. The osteoblast bone-specific markers, bone sialoprotein, ALP, osteocalcin, collagen type I α1, and β-catenin, were significantly decreased in EB1 siRNA-treated cells, compared with scrambled siRNA control cells. RNA samples from scrambled siRNA- and EB1 siRNA-treated cells were taken in triplicate, and data are presented as mean ± S.D. of three independent experiments (*, p < 0.01).
Requirement of EB1 for β-Catenin-mediated Transcriptional Events
EB1 siRNA caused a visible reduction in nuclear β-catenin in AA-stimulated OBs. Studies in numerous cell types have shown that β-catenin translocates to the nucleus to activate various TCF/LEF genes (6, 7). We used quantitative RT-PCR to assay the gene products of two common targets of β-catenin in 5-day AA-stimulated osteoblasts that had been exposed to either EB1 siRNA or scrambled siRNA for 72 h. We first examined the transcript levels of cyclin D1, a cell cycle protein that is transcriptionally regulated by β-catenin (58), that was significantly up-regulated in our microarray analysis (Fig. 1A). Quantitative RT-PCR analysis confirmed that cyclin D1 levels were significantly increased over 5 days of exposure to AA (Fig. 5A). EB1 siRNA treatment significantly reduced the levels of cyclin D1 in 5-day AA-stimulated OBs, compared with scrambled siRNA-treated cells (Fig. 5B). We next performed similar analyses and probed for Runx2 levels in these cells because Runx2 has been shown to be regulated by β-catenin (13). We first validated that AA stimulation caused a significant up-regulation of this major transcription regulator (Fig. 5C). There was a significant decrease in Runx2 levels in EB1 siRNA-treated cells, compared with control cells (Fig. 5D). Influence of EB1 on a second master regulator of osteoblast differentiation and maturation, osterix (59–61), was also assessed. Quantitative RT-PCR results displayed a striking increase in osterix mRNA levels over a 5-day AA stimulation period. osterix, which is largely accepted to act downstream of RUNX2 (12, 59), also showed a pronounced reduction in transcript levels when cells were exposed to EB1 siRNA (Fig. 5, E and F). Similar results were also observed with BMP-2-treated cultures (supplemental Fig. 3). Runx2 and osterix mRNA levels were significantly up-regulated over a 5-day stimulation period with BMP-2. Whereas cyclin D1 mRNA levels increased, albeit non-significantly over the 5-day period, cells that were treated with EB1 siRNA showed a marked decrease in Runx2, osterix, and cyclin D1 transcript levels. Thus, EB1 knockdown has pronounced effects on β-catenin target genes in osteoblasts and affects regulators of both early osteogenic cell commitment and later differentiation stages.
FIGURE 5.

Cyclin D1, Runx2, and osterix mRNA levels significantly increase during osteoblast differentiation and are impacted by EB1 knockdown. A, C, and E, total RNA was collected and analyzed for cyclin D1 expression (A), Runx2 (C), or osterix (E) by quantitative RT-PCR analysis. Cyclin D1, Runx2, and osterix mRNA levels increased in MC3T3-E1 cells differentiated with AA over a 5-day time period. Each sample was done in triplicate, and data were normalized to GAPDH and are plotted as the mean ± S.E. (error bars). (*, p < 0.05, Student's t test). B, D, and F, EB1 siRNA affects cyclin D1, Runx2, and osterix mRNA expression levels. Cells were treated with AA for 5 days and transfected with EB1 siRNA or scrambled (scr) siRNA for the final 72 h. Total RNA was collected and analyzed for cyclin D1 expression (B), Runx2 (D), or osterix (F) by quantitative RT-PCR analysis. EB1 siRNA treatment significantly reduced the transcript levels of all three genes in AA-treated cells, compared with scrambled siRNA-treated cells (*, p < 0.05, Student's t test). Each sample was done in triplicate, and data are plotted as the mean ± S.E.
EB1 and β-Catenin Interact at Cell-Cell Adhesion Sites in Osteoblasts
To gain more insight into potential interactions of EB1 with β-catenin, we next turned to live cell epifluorescent imaging. We cloned and subcloned EB1 and β-catenin into fluorescent mammalian expression vectors. MC3T3-E1 osteoblasts were transiently co-transfected with EB1-mCherry and β-catenin-GFP, followed by stimulation with AA. EB1-mCherry showed characteristic comet-like movement from the putative centrosome toward the cell periphery (Fig. 6A and supplemental Movie 1) (29). β-Catenin-GFP distribution was cytosolic with notable accumulations in the nucleus and at the cell edge (Fig. 6A). Epifluorescent microscopy provided high temporal imaging, and EB1-mCherry comets were frequently observed penetrating the cortex at cell-cell interaction regions that were rich in β-catenin-GFP (Fig. 6A and supplemental Movie 1). EB1 has been reported to physically complex with β-catenin in differentiating muscle cells (28). To definitively determine if EB1 and β-catenin were interacting and whether these interactions changed during the course of osteoblast differentiation, we performed reciprocal immunoprecipitations of these proteins during a 5-day AA time course (Fig. 6B). The reverse immunoprecipitations revealed an interaction between EB1 and β-catenin that increased as osteoblast differentiation proceeded (Fig. 6B). This finding, together with our live cell imaging results, indicates that enhanced EB1 protein and cortical targeting probably drive the elevated associations with β-catenin during osteoblast differentiation.
FIGURE 6.
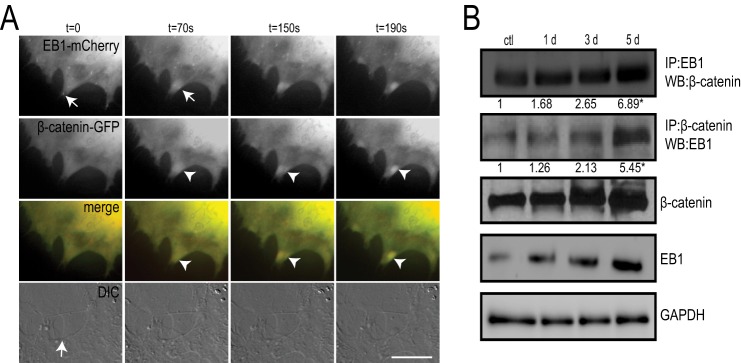
EB1 associates with β-catenin. A, live cell epifluorescent imaging of MC3T3-E1 cells co-transfected with β-catenin-GFP and EB1-mCherry. Panels show targeting of EB1-mCherry comets (arrows) into cortical regions at the site of cell-cell contacts (arrow in differential interference contrast (DIC) still image). Accumulation of β-catenin-GFP (arrowheads) occurs shortly after EB1-mCherry targeting to the cell periphery. Time is indicated in seconds. Scale bar, 10 μm. B, immunoprecipitation with EB1 or β-catenin antibodies reveals an interaction between EB1 and β-catenin that increases with osteoblast differentiation. MC3T3-E1 cells were stimulated with AA for 1, 3, and 5 days, and lysates were prepared and subjected to SDS-PAGE followed by immunoblot analysis for reciprocal antibodies, as indicated. Densitometry analysis was performed on the resulting bands from three independent experiments, and the mean precipitated protein levels are numerically presented as -fold increases relative to control treatments. Western blots are also shown of total cell lysates collected for the immunoprecipitations. GAPDH was used as a loading control. Experiments were repeated three times with similar results.
E-cadherin Mediates Cell-Cell Contact-induced Osteoblast Differentiation
It has been established that the osteoblast differentiation program and bone formation commence when cells have reached confluence (62, 63). We verified this observation in AA-treated MC3T3-E1 cells, and bone sialoprotein, alkaline phosphatase, collagen type I α1, osteocalcin, cyclin D1, and Runx2 were significantly elevated in high confluence versus low confluence cultures (data not shown). Interestingly, EB1 levels also increased in cells grown in high confluence (data not shown). This suggested that cell-cell contacts could be the mediating stimulus behind confluence-driven, AA-induced osteoblast differentiation. As a result, we examined the necessity of cadherins in this process. Although other groups have shown a role for cadherin 11 and N-cadherin for various stages of osteoblast maturation (64–71), we decided to investigate the role of E-cadherin, due to its well established role in recruiting β-catenin to cell adherens structures in other cell types (72–75). Additionally, our microarray analysis revealed that E-cadherin was specifically and significantly up-regulated at the time courses we were interested in, compared with undifferentiated osteoblasts (Fig. 1A), whereas no significant increase in expression of N-cadherin or cadherin 11 were observed following AA stimulation (not shown). We first confirmed that E-cadherin was up-regulated in 5-day AA-stimulated osteoblasts using quantitative RT-PCR. Indeed, compared with control cells, there was a nearly 5-fold increase in E-cadherin expression in AA-stimulated osteoblasts (Fig. 7A). Although we were unable able to find suitable antibodies to detect E-cadherin expression by immunoblotting or immunofluorescence, which has also been experienced by other osteoblast cadherin researchers (64, 65), we did find a suitable E-cadherin antibody for live cell inhibitory assays. To delineate a role for E-cadherin in osteoblast differentiation, cells of equivalent confluence were stimulated with AA and exposed to either E-cadherin-blocking antibody (20 μg/ml) or a rat IgG isotype control at day 2 and day 4. At the end of 5 days, we examined β-catenin distribution in these cells and observed greatly diminished immunostaining for β-catenin in the cell periphery of these cells, despite a robust recruitment of nuclear β-catenin in the absence of functional E-cadherin (Fig. 7B). The specific decrease in cortical β-catenin was confirmed by comparing peripheral β-catenin signal intensity levels at sites of cell-cell contact between the two antibody treatments (Fig. 7C). Cortical β-catenin has been shown to be stabilized by CD82-mediated inactivation of GSK-3β and casein kinase 1α (21). We observed a slight, but not significant, decrease in phospho-GSK-3β (Ser-9) levels in E-cadherin antibody-treated cells, compared with control cells, and the total GSK-3β protein levels were unchanged by these treatments (data not shown). Total RNA was purified, and the effect of E-cadherin blocking on osteoblast differentiation was determined using quantitative RT-PCR to the relevant osteoblast genes described earlier. Quantitative RT-PCR analysis revealed that alkaline phosphatase and bone sialoprotein gene expression were significantly attenuated, compared with control isotype IgG-treated cells (Fig. 7E). Collagen type I α1 levels were reduced, but not significantly, following E-cadherin-blocking experiments. There was also no significant difference in osteocalcin levels in cells treated with E-cadherin antibody, compared with isotype IgG-treated control cells (Fig. 7E). E-cadherin inhibition also had no effects on the message level of EB1 in AA-stimulated osteoblasts (Fig. 7E). Interestingly, E-cadherin blocking affected Runx2 expression but had no effect on cyclin D1 expression (Fig. 7D). Thus, part of the cell contact-induced osteoblast differentiation is mediated through E-cadherin, which has a selective impact on the cortical localization of β-catenin.
FIGURE 7.

E-cadherin is up-regulated in differentiated osteoblasts and partially contributes to osteoblast differentiation. A, quantitative RT-PCR analysis demonstrated that E-cadherin mRNA expression levels significantly increase in 5-day AA-treated MC3T3-E1 cells, compared with undifferentiated control cells (*, p < 0.05, Student's t test). The time course of E-cadherin expression was done in triplicate, and data are plotted as the mean ± S.E. (error bars). B, the addition of an E-cadherin-blocking antibody during AA treatment caused a visible reduction in β-catenin at the cortex region, compared with cells treated with control (ctl) isotype IgG, detected by immunostaining for β-catenin. No difference in nuclear β-catenin was observed following E-cadherin inhibition, compared with control cells. Boxed regions of equivalent size are representative of regions chosen for signal intensity analysis. C, β-catenin intensity at the cell periphery is significantly diminished upon E-cadherin blockade, compared with isotype antibody-treated cells. Six regions of cell-cell contact were chosen per cell, and presented values are the mean of 120 positions ± S.D. (*, p < 0.001). Scale bars, 10 μm. D, Runx2 mRNA levels significantly decreased in osteoblasts differentiated with AA for 5 days and treated with E-cadherin-blocking antibody, compared with control cells exposed to isotype IgG (*, p < 0.05, Student's t test). There was no significant effect of the E-cadherin antibody on the relative mRNA levels of cyclin D1 in differentiated osteoblasts. Data are plotted as the mean ± S.E. from three independent experiments. E, EB1 and osteoblast differentiation markers are differentially influenced by the blocking of E-cadherin. A significant -fold change reduction in alkaline phosphatase and bone sialoprotein mRNA levels was observed when cells were treated with the E-cadherin-blocking antibody throughout the AA stimulation time course (*, p < 0.05, Student's t test) relative to control antibody-treated cells. EB1 mRNA levels were unaffected by E-cadherin inhibition, as was osteocalcin, compared with isotype IgG control cells. Collagen type I α1 expression was reduced in E-cadherin-blocking antibody-treated cells but not significantly when compared with control cells (**, p = 0.05, Student's t test).
DISCUSSION
Here we show that cell confluence is a major determining factor for AA-induced osteoblast differentiation, with key osteoblast genes and EB1 expression levels dependent on highly confluent monolayers of cells. Osteoblast differentiation is dependent on EB1, with major Wnt-dependent gene events being affected in its absence. The differentiation of muscle cells also relies on EB1 expression (28), and it will be of interest to determine if there are other developmental processes that require this MT plus-end-binding protein. Another EB1 family member, EB3, is also involved in later stages of muscle cell elongation (28). Although we cannot rule out an EB3 contribution to osteoblast differentiation, EB3 levels were not up-regulated in our microarray studies, so we did not pursue this further.
In AA-stimulated OBs, EB1 levels and MT comet lengths increased throughout differentiation, similar to what we observed with CLIP-170 patterning during macrophage activation (76). Similar to CLIP-170 down-regulation in macrophages (76), EB1 knockdown diminished the levels of acetylated α-tubulin, further validating a role for EB1 in MT stabilization (31) in osteoblasts. BMP-2 stimulation or AA exposure to primary calvaria osteoblasts did not cause an increase in EB1 comet lengths, suggesting that comet length enhancement in AA-stimulated cells may be more relevant for MT stabilization and not necessarily osteoblast differentiation. EB1 knockdown markedly reduced β-catenin levels in both the nucleus and cell periphery in osteoblasts in AA-stimulated MC3T3-E1 cells. Because EB1 is largely a cytosolic protein in these cells (determined through confocal analysis and immunoblotting analysis of nuclear fractions; data not shown), its association with β-catenin is most likely functionally relevant at the cell cortex. However, β-catenin also localizes to centrosomes (77, 78), and we cannot rule out that functional interactions between EB1 and β-catenin occur at this key MT-nucleating center. Diminished surface E-cadherin and β-catenin was observed in muscle cells with reduced EB1 (28), implicating an MT trafficking defect of these adherens proteins. Interestingly, the neuronal protein Semaphorin 3A (Sema3A) causes β-catenin enrichment at growth cones (79). Sema3A promotes GSK-β-induced MT changes and facilitates axonal transport (80–83). However, EB1 knockdown in fibroblasts had no effect on cadherin and β-catenin density at cell-cell contacts (26). Although EB1 could be important for the delivery of β-catenin to adherens sites in osteoblasts, our most pronounced findings implicate a requirement for EB1 for the global stability of β-catenin (see the model in Fig. 8). Indeed, our results from EB1 knockdown of BMP-2-treated cells corroborate this notion. EB1 and β-catenin transcript levels were not altered during normal BMP-2 induction. However, once treated with EB1 siRNA, a strong reduction in β-catenin levels correlated with a severely diminished differentiation program. With the loss of β-catenin and presumably Wnt-enhanced pathway activation, BMP-2-mediated differentiation was critically affected.
FIGURE 8.
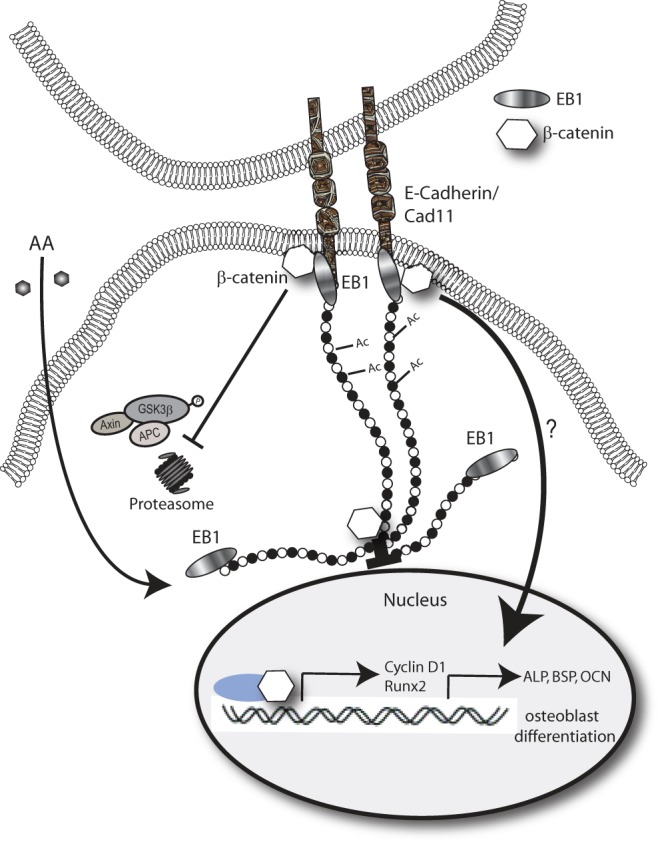
Proposed model of EB1 involvement in AA-induced osteoblast differentiation. AA stimulation of osteoblasts induces EB1 expression to promote MT nucleation and stabilization (Ac MTs). MT growth into cadherin-mediated cell-cell contacts promotes β-catenin stabilization at the cell cortex, protecting it from GSK3β-directed proteolytic degradation. β-Catenin can either be localized within the nucleus, at the centrosome, or at the cell periphery. Peripherally associated and stabilized β-catenin is important for early osteoblast gene expression mediated by RUNX2, in particular for alkaline phosphatase and bone sialoprotein gene expression.
The confluence-mediated stimulation of osteoblast differentiation is at least partially mediated by E-cadherin. In other systems, in vivo experimental up-regulation of cadherins results in decreased β-catenin signaling (84–87), and conversely, reduced expression of cadherins amplifies β-catenin nuclear signaling (88). These studies promoted the notion that cadherins sequester cytosolic pools of β-catenin away from the nucleus. In AA-stimulated osteoblasts, we observed a significant increase in E-cadherin levels during differentiation that was not inhibitory toward β-catenin and, in fact, paralleled up-regulated β-catenin levels and cortical and nuclear accumulation and activity. E-cadherin functional blocking antibodies did not impair β-catenin-dependent cyclin D1 expression, further suggesting that this cadherin does not have a negative, β-catenin-sequestering function.
The coordination and cross-talk of the adhesive or nuclear pools of β-catenin are not well understood. In our study, both pools were negatively impacted by reduced EB1, which could be directly linked to an overall destabilization of β-catenin in the absence of MT targeting to the cortex. Interestingly, E-cadherin-blocking antibodies preferentially reduced the cortically associated pool of β-catenin, allowing us to functionally discriminate between these two populations. Although we observed normal β-catenin nuclear recruitment and cyclin D1 expression in the absence of E-cadherin signaling, expression of Runx2 and the downstream osteoblast genes alkaline phosphatase and bone sialoprotein was significantly attenuated. This supports the idea that the osteogenic role of β-catenin is probably not solely based on canonical Wnt signals (89), based on scarce evidence establishing that Runx2 is activated by TCF/LEF-dependent mechanisms (90, 91). A related β-catenin protein, p120 catenin, can influence gene expression by regulating the cytoplasmic and nuclear distribution of the transcription factor Kaiso. The levels and cellular distributions of both p120ct and Kaiso are modulated in tumorigenic cells (92).
Osteocalcin gene expression was also not impacted by treating osteoblasts with E-cadherin-blocking antibodies. Interestingly, the most pronounced reduction in osteocalcin mRNA levels in BMP-2-stimulated human neonatal cells occurred when both N- and E-cadherin-neutralizing antibodies had disrupted cellular adhesions (93). Coordination of multiple cadherins may be necessary for full induction of certain osteoblast genes. However, collagen type I α1 mRNA levels were modestly affected by E-cadherin inhibition, and it has been established that collagen production is required for AA-induced osteocalcin expression (94, 95). Full collagen transcription activation may require other potential compensatory cadherins, such as N-cadherin and cadherin11. These cadherins have been shown to strongly impact osteoblast differentiation at various stages of development (64–71). Cadherin 11 knockdown affects β-catenin abundance (66), and double knockdown of both N-cadherin and cadherin 11 caused reduced cyclin D1 expression in calvaria cells (66). N-cadherin is thought to promote early commitment to osteogenesis, whereas cadherin 11 is involved in later maintenance and differentiation of the osteoblast lineage (66). Because MC3T3-E1 preosteoblasts are considered to be committed to the osteoblast lineage (96), cadherin 11 is most likely to have overlapping functions with E-cadherin. However, we cannot rule out the possibility that other cell confluent-dependent structures, such as gap junctions (97), are necessary for full activation of osteoblast differentiation genes.
In conclusion, we have identified a critical role for the MT-binding protein EB1 in maintaining the global stability of β-catenin in osteoblasts. EB1 protein levels presumably increase to match and sustain the growing β-catenin protein intracellular content for the rapid proliferative stages of osteoblast differentiation. Our live epifluorescent imaging revealed dynamic clusters of β-catenin that grow and persist at cell-cell contact regions targeted by EB1. It will be of great interest to determine the role of the dynamic population of peripherally targeted β-catenin in osteoblast cell-cell interactions and to determine how cadherins cooperate with EB1 to regulate the cytoplasmic pools of β-catenin. Preservation of the cortical pools of β-catenin seems to be dispensable for the expression of early osteoblast genes, which is instead highly dependent on cell confluence and EB1 and cadherin signaling. Cell surface proteins are ideal candidates for therapeutic intervention in diseases of the bone, and our study warrants continued focus on the interplay of these proteins and the impact of these peripheral events on osteoblast gene expression.
Supplementary Material
Acknowledgment
We thank Tanya Zappitelli for critical reading of the manuscript.
This work was supported in part by Natural Science and Engineering Research Council (NSERC) Grant RGPIN 298538-09 (to R. E. H.)
All of the microarray data used in this study are available from the Gene Expression Omnibus (GEO) database (www.ncbi.nlm.nih.gov/projects/geo/) under accession number GSE37676.

This article contains supplemental Figs. 1–3 and Movie 1.
- TCF
- T-cell factor
- LEF
- lymphoid enhancer factor
- MT
- microtubule
- APC
- adenomatous polyposis coli
- GSK-3β
- glycogen synthase kinase 3β
- AA
- ascorbic acid
- IP
- immunoprecipitation
- ALP
- alkaline phosphatase.
REFERENCES
- 1. Asagiri M., Takayanagi H. (2007) The molecular understanding of osteoclast differentiation. Bone 40, 251–264 [DOI] [PubMed] [Google Scholar]
- 2. Boyle W. J., Simonet W. S., Lacey D. L. (2003) Osteoclast differentiation and activation. Nature 423, 337–342 [DOI] [PubMed] [Google Scholar]
- 3. Edwards J. R., Weivoda M. M. (2012) Osteoclasts. Malefactors of disease and targets for treatment. Discov. Med. 13, 201–210 [PubMed] [Google Scholar]
- 4. Canalis E., Giustina A., Bilezikian J. P. (2007) Mechanisms of anabolic therapies for osteoporosis. N. Engl. J. Med. 357, 905–916 [DOI] [PubMed] [Google Scholar]
- 5. Hoeppner L. H., Secreto F. J., Westendorf J. J. (2009) Wnt signaling as a therapeutic target for bone diseases. Expert Opin. Ther. Targets 13, 485–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lin G. L., Hankenson K. D. (2011) Integration of BMP, Wnt, and notch signaling pathways in osteoblast differentiation. J. Cell. Biochem. 112, 3491–3501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Davis E. K., Zou Y., Ghosh A. (2008) Wnts acting through canonical and noncanonical signaling pathways exert opposite effects on hippocampal synapse formation. Neural Dev. 3, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. MacDonald B. T., Tamai K., He X. (2009) Wnt/beta-catenin signaling. Components, mechanisms, and diseases. Dev. Cell 17, 9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Day T. F., Guo X., Garrett-Beal L., Yang Y. (2005) Wnt/β-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 8, 739–750 [DOI] [PubMed] [Google Scholar]
- 10. Hill T. P., Später D., Taketo M. M., Birchmeier W., Hartmann C. (2005) Canonical Wnt/β-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev. Cell 8, 727–738 [DOI] [PubMed] [Google Scholar]
- 11. Hu H., Hilton M. J., Tu X., Yu K., Ornitz D. M., Long F. (2005) Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development 132, 49–60 [DOI] [PubMed] [Google Scholar]
- 12. Nakashima K., de Crombrugghe B. (2003) Transcriptional mechanisms in osteoblast differentiation and bone formation. Trends Genet. 19, 458–466 [DOI] [PubMed] [Google Scholar]
- 13. Gaur T., Lengner C. J., Hovhannisyan H., Bhat R. A., Bodine P. V., Komm B. S., Javed A., van Wijnen A. J., Stein J. L., Stein G. S., Lian J. B. (2005) Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J. Biol. Chem. 280, 33132–33140 [DOI] [PubMed] [Google Scholar]
- 14. Komori T., Yagi H., Nomura S., Yamaguchi A., Sasaki K., Deguchi K., Shimizu Y., Bronson R. T., Gao Y. H., Inada M., Sato M., Okamoto R., Kitamura Y., Yoshiki S., Kishimoto T. (1997) Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89, 755–764 [DOI] [PubMed] [Google Scholar]
- 15. Otto F., Thornell A. P., Crompton T., Denzel A., Gilmour K. C., Rosewell I. R., Stamp G. W., Beddington R. S., Mundlos S., Olsen B. R., Selby P. B., Owen M. J. (1997) Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 89, 765–771 [DOI] [PubMed] [Google Scholar]
- 16. Ducy P., Zhang R., Geoffroy V., Ridall A. L., Karsenty G. (1997) Osf2/Cbfa1. A transcriptional activator of osteoblast differentiation. Cell 89, 747–754 [DOI] [PubMed] [Google Scholar]
- 17. Boller K., Vestweber D., Kemler R. (1985) Cell-adhesion molecule uvomorulin is localized in the intermediate junctions of adult intestinal epithelial cells. J. Cell Biol. 100, 327–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Collares-Buzato C. B., Jepson M. A., McEwan G. T., Simmons N. L., Hirst B. H. (1994) Junctional uvomorulin/E-cadherin and phosphotyrosine-modified protein content are correlated with paracellular permeability in Madin-Darby canine kidney (MDCK) epithelia. Histochemistry 101, 185–194 [DOI] [PubMed] [Google Scholar]
- 19. Collares-Buzato C. B., McEwan G. T., Jepson M. A., Simmons N. L., Hirst B. H. (1994) Paracellular barrier and junctional protein distribution depend on basolateral extracellular Ca2+ in cultured epithelia. Biochim. Biophys. Acta 1222, 147–158 [DOI] [PubMed] [Google Scholar]
- 20. Nelson W. J., Nusse R. (2004) Convergence of Wnt, β-catenin, and cadherin pathways. Science 303, 1483–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chigita S., Sugiura T., Abe M., Kobayashi Y., Shimoda M., Onoda M., Shirasuna K. (2012) CD82 inhibits canonical Wnt signalling by controlling the cellular distribution of β-catenin in carcinoma cells. Int. J. Oncol. 41, 2021–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ishiyama N., Ikura M. (2012) The three-dimensional structure of the cadherin-catenin complex. Subcell. Biochem. 60, 39–62 [DOI] [PubMed] [Google Scholar]
- 23. Stehbens S. J., Paterson A. D., Crampton M. S., Shewan A. M., Ferguson C., Akhmanova A., Parton R. G., Yap A. S. (2006) Dynamic microtubules regulate the local concentration of E-cadherin at cell:cell contacts. J. Cell Sci. 119, 1801–1811 [DOI] [PubMed] [Google Scholar]
- 24. Bellett G., Carter J. M., Keynton J., Goldspink D., James C., Moss D. K., Mogensen M. M. (2009) Microtubule plus-end and minus-end capture at adherens junctions is involved in the assembly of apico-basal arrays in polarised epithelial cells. Cell Motil. Cytoskeleton 66, 893–908 [DOI] [PubMed] [Google Scholar]
- 25. Ligon L. A., Holzbaur E. L. (2007) Microtubules tethered at epithelial cell junctions by dynein facilitate efficient junction assembly. Traffic 8, 808–819 [DOI] [PubMed] [Google Scholar]
- 26. Shaw R. M., Fay A. J., Puthenveedu M. A., von Zastrow M., Jan Y. N., Jan L. Y. (2007) Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell 128, 547–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wen Y., Eng C. H., Schmoranzer J., Cabrera-Poch N., Morris E. J., Chen M., Wallar B. J., Alberts A. S., Gundersen G. G. (2004) EB1 and APC bind to mDia to stabilize microtubules downstream of Rho and promote cell migration. Nat. Cell Biol. 6, 820–830 [DOI] [PubMed] [Google Scholar]
- 28. Zhang T., Zaal K. J., Sheridan J., Mehta A., Gundersen G. G., Ralston E. (2009) Microtubule plus-end binding protein EB1 is necessary for muscle cell differentiation, elongation and fusion. J. Cell Sci. 122, 1401–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tirnauer J. S., Bierer B. E. (2000) EB1 proteins regulate microtubule dynamics, cell polarity, and chromosome stability. J. Cell Biol. 149, 761–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Morrison E. E., Wardleworth B. N., Askham J. M., Markham A. F., Meredith D. M. (1998) EB1, a protein which interacts with the APC tumour suppressor, is associated with the microtubule cytoskeleton throughout the cell cycle. Oncogene 17, 3471–3477 [DOI] [PubMed] [Google Scholar]
- 31. Komarova Y., De Groot C. O., Grigoriev I., Gouveia S. M., Munteanu E. L., Schober J. M., Honnappa S., Buey R. M., Hoogenraad C. C., Dogterom M., Borisy G. G., Steinmetz M. O., Akhmanova A. (2009) Mammalian end binding proteins control persistent microtubule growth. J. Cell Biol. 184, 691–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brocardo M., Henderson B. R. (2008) APC shuttling to the membrane, nucleus and beyond. Trends Cell Biol. 18, 587–596 [DOI] [PubMed] [Google Scholar]
- 33. Su L. K., Burrell M., Hill D. E., Gyuris J., Brent R., Wiltshire R., Trent J., Vogelstein B., Kinzler K. W. (1995) APC binds to the novel protein EB1. Cancer Res. 55, 2972–2977 [PubMed] [Google Scholar]
- 34. Fujii K., Kondo T., Yokoo H., Yamada T., Iwatsuki K., Hirohashi S. (2005) Proteomic study of human hepatocellular carcinoma using two-dimensional difference gel electrophoresis with saturation cysteine dye. Proteomics 5, 1411–1422 [DOI] [PubMed] [Google Scholar]
- 35. Wang Y., Zhou X., Zhu H., Liu S., Zhou C., Zhang G., Xue L., Lu N., Quan L., Bai J., Zhan Q., Xu N. (2005) Overexpression of EB1 in human esophageal squamous cell carcinoma (ESCC) may promote cellular growth by activating β-catenin/TCF pathway. Oncogene 24, 6637–6645 [DOI] [PubMed] [Google Scholar]
- 36. Nishigaki R., Osaki M., Hiratsuka M., Toda T., Murakami K., Jeang K. T., Ito H., Inoue T., Oshimura M. (2005) Proteomic identification of differentially expressed genes in human gastric carcinomas. Proteomics 5, 3205–3213 [DOI] [PubMed] [Google Scholar]
- 37. El-Rifai W., Frierson H. F., Jr., Harper J. C., Powell S. M., Knuutila S. (2001) Expression profiling of gastric adenocarcinoma using cDNA array. Int. J. Cancer 92, 832–838 [DOI] [PubMed] [Google Scholar]
- 38. Aubin J. E., Heersche J. N., Merrilees M. J., Sodek J. (1982) Isolation of bone cell clones with differences in growth, hormone responses, and extracellular matrix production. J. Cell Biol. 92, 452–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nabavi N., Urukova Y., Cardelli M., Aubin J. E., Harrison R. E. (2008) Lysosome dispersion in osteoblasts accommodates enhanced collagen production during differentiation. J. Biol. Chem. 283, 19678–19690 [DOI] [PubMed] [Google Scholar]
- 40. Nabavi N., Pustylnik S., Harrison R. E. (2012) RabGTPase mediated procollagen trafficking in ascorbic acid stimulated osteoblasts. PLoS One 7, e46265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cawthorn W. P., Bree A. J., Yao Y., Du B., Hemati N., Martinez-Santibañez G., MacDougald O. A. (2012) Wnt6, Wnt10a and Wnt10b inhibit adipogenesis and stimulate osteoblastogenesis through a β-catenin-dependent mechanism. Bone 50, 477–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Friedman M. S., Oyserman S. M., Hankenson K. D. (2009) Wnt11 promotes osteoblast maturation and mineralization through R-spondin 2. J. Biol. Chem. 284, 14117–14125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Scherft J. P., Heersche J. N. (1975) Accumulation of collagen-containing vacuoles in osteoblasts after administration of colchicine. Cell Tissue Res. 157, 353–365 [DOI] [PubMed] [Google Scholar]
- 44. Bellows C. G., Aubin J. E., Heersche J. N., Antosz M. E. (1986) Mineralized bone nodules formed in vitro from enzymatically released rat calvaria cell populations. Calcif. Tissue Int. 38, 143–154 [DOI] [PubMed] [Google Scholar]
- 45. Leblond C. P. (1989) Synthesis and secretion of collagen by cells of connective tissue, bone, and dentin. Anat. Rec. 224, 123–138 [DOI] [PubMed] [Google Scholar]
- 46. Piperno G., LeDizet M., Chang X. J. (1987) Microtubules containing acetylated α-tubulin in mammalian cells in culture. J. Cell Biol. 104, 289–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bulinski J. C., Gundersen G. G. (1991) Stabilization of post-translational modification of microtubules during cellular morphogenesis. BioEssays 13, 285–293 [DOI] [PubMed] [Google Scholar]
- 48. Beck G. R., Jr. (2003) Inorganic phosphate as a signaling molecule in osteoblast differentiation. J. Cell. Biochem. 90, 234–243 [DOI] [PubMed] [Google Scholar]
- 49. Golub E. E. (2009) Role of matrix vesicles in biomineralization. Biochim. Biophys. Acta 1790, 1592–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bain G., Müller T., Wang X., Papkoff J. (2003) Activated β-catenin induces osteoblast differentiation of C3H10T1/2 cells and participates in BMP2 mediated signal transduction. Biochem. Biophys. Res. Commun. 301, 84–91 [DOI] [PubMed] [Google Scholar]
- 51. Rawadi G., Vayssière B., Dunn F., Baron R., Roman-Roman S. (2003) BMP-2 controls alkaline phosphatase expression and osteoblast mineralization by a Wnt autocrine loop. J. Bone Miner. Res. 18, 1842–1853 [DOI] [PubMed] [Google Scholar]
- 52. Sadot E., Conacci-Sorrell M., Zhurinsky J., Shnizer D., Lando Z., Zharhary D., Kam Z., Ben-Ze'ev A., Geiger B. (2002) Regulation of S33/S37 phosphorylated β-catenin in normal and transformed cells. J. Cell Sci. 115, 2771–2780 [DOI] [PubMed] [Google Scholar]
- 53. Sutherland C., Leighton I. A., Cohen P. (1993) Inactivation of glycogen synthase kinase-3 beta by phosphorylation. New kinase connections in insulin and growth-factor signalling. Biochem. J. 296, 15–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu C., Li Y., Semenov M., Han C., Baeg G. H., Tan Y., Zhang Z., Lin X., He X. (2002) Control of β-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108, 837–847 [DOI] [PubMed] [Google Scholar]
- 55. Qiang Y. W., Hu B., Chen Y., Zhong Y., Shi B., Barlogie B., Shaughnessy J. D., Jr. (2009) Bortezomib induces osteoblast differentiation via Wnt-independent activation of β-catenin/TCF signaling. Blood 113, 4319–4330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang R., Oyajobi B. O., Harris S. E., Chen D., Tsao C., Deng H. W., Zhao M. (2013) Wnt/β-catenin signaling activates bone morphogenetic protein 2 expression in osteoblasts. Bone 52, 145–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mbalaviele G., Sheikh S., Stains J. P., Salazar V. S., Cheng S. L., Chen D., Civitelli R. (2005) β-Catenin and BMP-2 synergize to promote osteoblast differentiation and new bone formation. J. Cell. Biochem. 94, 403–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tetsu O., McCormick F. (1999) β-Catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398, 422–426 [DOI] [PubMed] [Google Scholar]
- 59. Javed A., Chen H., Ghori F. Y. (2010) Genetic and transcriptional control of bone formation. Oral Maxillofac. Surg. Clin. North Am. 22, 283–293, v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Soltanoff C. S., Yang S., Chen W., Li Y. P. (2009) Signaling networks that control the lineage commitment and differentiation of bone cells. Crit. Rev. Eukaryot. Gene Expr. 19, 1–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sinha K. M., Zhou X. Genetic and molecular control of osterix in skeletal formation. J. Cell. Biochem. 114, 975–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Whitson S. W., Harrison W., Dunlap M. K., Bowers D. E., Jr., Fisher L. W., Robey P. G., Termine J. D. (1984) Fetal bovine bone cells synthesize bone-specific matrix proteins. J. Cell Biol. 99, 607–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Whitson S. W., Whitson M. A., Bowers D. E., Jr., Falk M. C. (1992) Factors influencing synthesis and mineralization of bone matrix from fetal bovine bone cells grown in vitro. J. Bone Miner. Res. 7, 727–741 [DOI] [PubMed] [Google Scholar]
- 64. Cheng S. L., Lecanda F., Davidson M. K., Warlow P. M., Zhang S. F., Zhang L., Suzuki S., St John T., Civitelli R. (1998) Human osteoblasts express a repertoire of cadherins, which are critical for BMP-2-induced osteogenic differentiation. J. Bone Miner. Res. 13, 633–644 [DOI] [PubMed] [Google Scholar]
- 65. Cheng S. L., Shin C. S., Towler D. A., Civitelli R. (2000) A dominant negative cadherin inhibits osteoblast differentiation. J. Bone Miner. Res. 15, 2362–2370 [DOI] [PubMed] [Google Scholar]
- 66. Di Benedetto A., Watkins M., Grimston S., Salazar V., Donsante C., Mbalaviele G., Radice G. L., Civitelli R. (2010) N-cadherin and cadherin 11 modulate postnatal bone growth and osteoblast differentiation by distinct mechanisms. J. Cell Sci. 123, 2640–2648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Guntur A. R., Rosen C. J., Naski M. C. (2012) N-cadherin adherens junctions mediate osteogenesis through PI3K signaling. Bone 50, 54–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Haÿ E., Laplantine E., Geoffroy V., Frain M., Kohler T., Müller R., Marie P. J. (2009) N-cadherin interacts with axin and LRP5 to negatively regulate Wnt/β-catenin signaling, osteoblast function, and bone formation. Mol. Cell. Biol. 29, 953–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mbalaviele G., Shin C. S., Civitelli R. (2006) Cell:cell adhesion and signaling through cadherins. Connecting bone cells in their microenvironment. J. Bone Miner. Res. 21, 1821–1827 [DOI] [PubMed] [Google Scholar]
- 70. Lemonnier J., Haÿ E., Delannoy P., Lomri A., Modrowski D., Caverzasio J., Marie P. J. (2001) Role of N-cadherin and protein kinase C in osteoblast gene activation induced by the S252W fibroblast growth factor receptor 2 mutation in Apert craniosynostosis. J. Bone Miner. Res. 16, 832–845 [DOI] [PubMed] [Google Scholar]
- 71. Ferrari S. L., Traianedes K., Thorne M., Lafage-Proust M. H., Genever P., Cecchini M. G., Behar V., Bisello A., Chorev M., Rosenblatt M., Suva L. J. (2000) A role for N-cadherin in the development of the differentiated osteoblastic phenotype. J. Bone Miner. Res. 15, 198–208 [DOI] [PubMed] [Google Scholar]
- 72. Goichberg P., Shtutman M., Ben-Ze'ev A., Geiger B. (2001) Recruitment of β-catenin to cadherin-mediated intercellular adhesions is involved in myogenic induction. J. Cell Sci. 114, 1309–1319 [DOI] [PubMed] [Google Scholar]
- 73. Aberle H., Schwartz H., Kemler R. (1996) Cadherin-catenin complex. Protein interactions and their implications for cadherin function. J. Cell. Biochem. 61, 514–523 [DOI] [PubMed] [Google Scholar]
- 74. Jou T. S., Stewart D. B., Stappert J., Nelson W. J., Marrs J. A. (1995) Genetic and biochemical dissection of protein linkages in the cadherin-catenin complex. Proc. Natl. Acad. Sci. U.S.A. 92, 5067–5071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chen Y. T., Stewart D. B., Nelson W. J. (1999) Coupling assembly of the E-cadherin/β-catenin complex to efficient endoplasmic reticulum exit and basal-lateral membrane targeting of E-cadherin in polarized MDCK cells. J. Cell Biol. 144, 687–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Binker M. G., Zhao D. Y., Pang S. J., Harrison R. E. (2007) Cytoplasmic linker protein-170 enhances spreading and phagocytosis in activated macrophages by stabilizing microtubules. J. Immunol. 179, 3780–3791 [DOI] [PubMed] [Google Scholar]
- 77. Bahmanyar S., Guiney E. L., Hatch E. M., Nelson W. J., Barth A. I. (2010) Formation of extra centrosomal structures is dependent on β-catenin. J. Cell Sci. 123, 3125–3135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Huang P., Senga T., Hamaguchi M. (2007) A novel role of phospho-β-catenin in microtubule regrowth at centrosome. Oncogene 26, 4357–4371 [DOI] [PubMed] [Google Scholar]
- 79. Hida T., Yamashita N., Usui H., Nakamura F., Sasaki Y., Kikuchi A., Goshima Y. (2012) GSK3β/axin-1/β-catenin complex is involved in semaphorin3A signaling. J. Neurosci. 32, 11905–11918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Fan J., Raper J. A. (1995) Localized collapsing cues can steer growth cones without inducing their full collapse. Neuron 14, 263–274 [DOI] [PubMed] [Google Scholar]
- 81. Goshima Y., Ito T., Sasaki Y., Nakamura F. (2002) Semaphorins as signals for cell repulsion and invasion. J. Clin. Invest. 109, 993–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Goshima Y., Kawakami T., Hori H., Sugiyama Y., Takasawa S., Hashimoto Y., Kagoshima-Maezono M., Takenaka T., Misu Y., Strittmatter S. M. (1997) A novel action of collapsin. Collapsin-1 increases antero- and retrograde axoplasmic transport independently of growth cone collapse. J. Neurobiol. 33, 316–328 [DOI] [PubMed] [Google Scholar]
- 83. Tran T. S., Kolodkin A. L., Bharadwaj R. (2007) Semaphorin regulation of cellular morphology. Annu. Rev. Cell Dev. Biol. 23, 263–292 [DOI] [PubMed] [Google Scholar]
- 84. Fagotto F., Funayama N., Gluck U., Gumbiner B. M. (1996) Binding to cadherins antagonizes the signaling activity of β-catenin during axis formation in Xenopus. J. Cell Biol. 132, 1105–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Orsulic S., Huber O., Aberle H., Arnold S., Kemler R. (1999) E-cadherin binding prevents β-catenin nuclear localization and β-catenin/LEF-1-mediated transactivation. J. Cell Sci. 112, 1237–1245 [DOI] [PubMed] [Google Scholar]
- 86. Heasman J., Crawford A., Goldstone K., Garner-Hamrick P., Gumbiner B., McCrea P., Kintner C., Noro C. Y., Wylie C. (1994) Overexpression of cadherins and underexpression of β-catenin inhibit dorsal mesoderm induction in early Xenopus embryos. Cell 79, 791–803 [DOI] [PubMed] [Google Scholar]
- 87. Sanson B., White P., Vincent J. P. (1996) Uncoupling cadherin-based adhesion from wingless signalling in Drosophila. Nature 383, 627–630 [DOI] [PubMed] [Google Scholar]
- 88. Cox R. T., Kirkpatrick C., Peifer M. (1996) Armadillo is required for adherens junction assembly, cell polarity, and morphogenesis during Drosophila embryogenesis. J. Cell Biol. 134, 133–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Salazar V. S., Mbalaviele G., Civitelli R. (2008) The pro-osteogenic action of β-catenin requires interaction with BMP signaling, but not Tcf/Lef transcriptional activity. J. Cell. Biochem. 104, 942–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Glass D. A., 2nd, Bialek P., Ahn J. D., Starbuck M., Patel M. S., Clevers H., Taketo M. M., Long F., McMahon A. P., Lang R. A., Karsenty G. (2005) Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell 8, 751–764 [DOI] [PubMed] [Google Scholar]
- 91. Kato M., Patel M. S., Levasseur R., Lobov I., Chang B. H., Glass D. A., 2nd, Hartmann C., Li L., Hwang T. H., Brayton C. F., Lang R. A., Karsenty G., Chan L. (2002) Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J. Cell Biol. 157, 303–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wang Y., Li L., Li Q., Xie C., Wang E. (2012) Expression of P120 catenin, Kaiso, and metastasis tumor antigen-2 in thymomas. Tumour Biol. 33, 1871–1879 [DOI] [PubMed] [Google Scholar]
- 93. Haÿ E., Lemonnier J., Modrowski D., Lomri A., Lasmoles F., Marie P. J. (2000) N- and E-cadherin mediate early human calvaria osteoblast differentiation promoted by bone morphogenetic protein-2. J. Cell. Physiol. 183, 117–128 [DOI] [PubMed] [Google Scholar]
- 94. Xiao G., Cui Y., Ducy P., Karsenty G., Franceschi R. T. (1997) Ascorbic acid-dependent activation of the osteocalcin promoter in MC3T3-E1 preosteoblasts. Requirement for collagen matrix synthesis and the presence of an intact OSE2 sequence. Mol. Endocrinol. 11, 1103–1113 [DOI] [PubMed] [Google Scholar]
- 95. Franceschi R. T. (1999) The developmental control of osteoblast-specific gene expression. Role of specific transcription factors and the extracellular matrix environment. Crit. Rev. Oral Biol. Med. 10, 40–57 [DOI] [PubMed] [Google Scholar]
- 96. Sudo H., Kodama H. A., Amagai Y., Yamamoto S., Kasai S. (1983) In vitro differentiation and calcification in a new clonal osteogenic cell line derived from newborn mouse calvaria. J. Cell Biol. 96, 191–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lecanda F., Warlow P. M., Sheikh S., Furlan F., Steinberg T. H., Civitelli R. (2000) Connexin43 deficiency causes delayed ossification, craniofacial abnormalities, and osteoblast dysfunction. J. Cell Biol. 151, 931–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.