

Abstract
Ethidium bromide (EB) is known to inhibit cleavage of bacterial rRNA precursors by Escherichia coli ribonuclease III, a dsRNA-specific nuclease. The mechanism of EB inhibition of RNase III is not known nor is there information on EB-binding sites in RNase III substrates. We show here that EB is a reversible, apparently competitive inhibitor of RNase III cleavage of small model substrates in vitro. Inhibition is due to intercalation, since (i) the inhibitory concentrations of EB are similar to measured EB intercalation affinities; (ii) substrate cleavage is not affected by actinomycin D, an intercalating agent that does not bind dsRNA; (iii) the EB concentration dependence of inhibition is a function of substrate structure. In contrast, EB does not strongly inhibit the ability of RNase III to bind substrate. EB also does not block substrate binding by the C-terminal dsRNA-binding domain (dsRBD) of RNase III, indicating that EB perturbs substrate recognition by the N-terminal catalytic domain. Laser photocleavage experiments revealed two ethidium-binding sites in the substrate R1.1 RNA. One site is in the internal loop, adjacent to the scissile bond, while the second site is in the lower stem. Both sites consist of an A-A pair stacked on a CG pair, a motif which apparently provides a particularly favorable environment for intercalation. These results indicate an inhibitory mechanism in which EB site-specifically binds substrate, creating a cleavage-resistant complex that can compete with free substrate for RNase III. This study also shows that RNase III recognition and cleavage of substrate can be uncoupled and supports an enzymatic mechanism of dsRNA cleavage involving cooperative but not obligatorily linked actions of the dsRBD and the catalytic domain.
INTRODUCTION
Double-helical RNA is a target for recognition by diverse cellular and viral proteins involved in the processing, modification, transport, translation and degradation of RNA (1–4). For example, the Drosophila Staufen protein binds double-stranded (ds)RNA structures and participates in mRNA localization (5,6), while the ADAR family of RNA-editing enzymes modulate gene expression by catalyzing site-specific deamination of adenosines within dsRNA elements (7). The protein kinase PKR and members of the 2-5A synthetase family are activated by dsRNA during the mammalian antiviral response. As a countermeasure, specific viral proteins can bind and sequester dsRNA (2,3). The recently characterized phenomena of RNA interference (RNAi) and post-transcriptional gene silencing (PTGS) also involve the synthesis, recognition and processing of dsRNA (8–12).
Members of the ribonuclease III (RNase III) family specifically recognize and cleave dsRNA and are involved in a variety of post-transcriptional regulatory mechanisms. RNase III is highly conserved in the bacteria and orthologs occur in fungi, plants, animals and even a virus (13,14). The most studied member of the family is RNase III of Escherichia coli (EC 3.1.24) (15–18). Escherichia coli RNase III cleaves the primary transcript of the rRNA operons, creating the immediate precursors of 16S, 23S and 5S rRNAs (19). Recent studies indicate that rRNA processing is a conserved role for RNase III family members (20–22). Escherichia coli RNase III also converts cellular and viral mRNA precursors to their translationally most active forms (23–25) and can cleave within mRNA coding sequences, causing translation inhibition (26). RNase III participates in antisense (AS) RNA action by cleaving duplex structures produced by AS RNA binding to target sequences (27). Escherichia coli RNase III also autoregulates its expression through cleavage of a double-stranded element within the 5′-leader of its message, which promotes rapid subsequent decay (28–30).
Escherichia coli RNase III is active as a homodimer and requires a divalent metal ion (preferably Mg2+) to hydrolyze phosphodiesters, providing 5′-phosphate and 3′-hydroxyl product termini (16). RNase III processing reactions can be faithfully reconstructed in vitro using small model substrates containing the requisite reactivity epitopes and using physiologically relevant salt concentrations (31–33). In low salt and/or in the presence of Mn2+ additional cleavages occur at secondary sites, which are not normally recognized in vivo (34,35). The C-terminal portion of the RNase III polypeptide contains a dsRNA-binding domain (dsRBD) (36), a motif present in many other dsRNA-binding proteins (4,5), which is important for substrate recognition in vitro and in vivo (A.K.Amarasinghe, S.Su, W.Sun, R.W.Simons and A.W.Nicholson, manuscript in preparation). The N-terminal portion of the RNase III polypeptide contains the catalytic (nuclease) domain, which exhibits an array of conserved residues, at least one of which (Glu117) has been shown to be important for cleavage but not for substrate binding (37,38). Although it is now generally accepted that the RNase III mechanism of action requires the participation of both the dsRBD and the catalytic domain, the fundamental steps in dsRNA recognition and cleavage by RNase III have not been defined.
One approach to determine the RNase III mechanism of action is to examine the effect of small molecule inhibitors. Such compounds include substrate mimics and transition state analogs and have been informative probes of other RNases (see for example 39,40). It was reported that the nucleic acid intercalating agent ethidium bromide (EB) could inhibit E.coli RNase III cleavage of the bacterial rRNA precursor (30S RNA) in vitro (41). Although inhibition was assumed to reflect EB intercalation into 30S RNA, the mechanism of inhibition was not determined nor was the drug binding site(s) identified. The Schizosaccharomyces pombe RNase III ortholog Pac1p is also inhibited by low (micromolar) concentrations of EB (13). We show here that EB is a reversible, apparently competitive inhibitor of E.coli RNase III. We demonstrate that an ethidium-dependent change in RNA structure can uncouple RNase III binding and cleavage of substrate, indicating a RNase III mechanism of action involving cooperative but not obligatorily linked actions of the dsRBD and the catalytic domain.
MATERIALS AND METHODS
Materials
Water was deionized and distilled. Chemicals were reagent grade or molecular biology grade and were purchased from Fisher Scientific (Chicago, IL) or Sigma (St Louis, MO). Escherichia coli bulk stripped tRNA was purchased from Sigma and further purified by repeated phenol extraction and ethanol precipitation. The radiolabeled nucleotides [γ-32P]ATP (3000 Ci/mmol) and [α-32P]CTP (3000 Ci/mmol) were from Dupont-NEN (Boston, MA). Calf intestinal alkaline phosphatase was purchased from Roche Molecular Biochemicals (Indianapolis, IN). Genetically modified M-MLV reverse transcriptase (Superscript II) was obtained from Life Technologies (Gaithersburg, MD). Restriction enzymes and T4 polynucleotide kinase were from New England Biolabs (Beverly, MA). T7 RNA polymerase was purified from an overexpressing bacterial strain as described (42,43). EB and actinomycin D (AD) were purchased from Sigma and used without further purification. EB solutions were prepared in water and stored at 4°C, protected from light. A molar extinction coefficient of 5450 M–1cm–1 (480 nm, H2O) (44) was used to determine concentrations. AD solutions were prepared in water and a molar extinction coefficient of 33 600 M–1cm–1 (240 nm, methanol) (45) was used to determine concentrations. RNase III was purified from an overexpressing bacterial strain as an N-terminal (His)6-tagged protein as described (46). The (His)6 sequence has no significant effect on RNase III processing efficiency or cleavage specificity (46). For the purposes of this report (His)6–RNase III will be referred to as RNase III. The purification and properties of the catalytically inactive (His)6–RNase III[Glu117Gln] mutant are described elsewhere (47). The purification and biochemical properties of (His)6–dsRBD will be described elsewhere (A.K.Amarasinghe, S.Su, W.Sun, R.W.Simons and A.W.Nicholson, manuscript in preparation). Briefly, the segment of the RNase III gene encoding amino acids 148–226 was cloned into plasmid pET-15b and the dsRBD purified as an N-terminal, (His)6-tagged species.
RNase III processing substrates were synthesized as described using oligodeoxynucleotide templates and T7 RNA polymerase (46). The oligodeoxynucleotides were synthesized by the Wayne State Macromolecular Core Facility or by Life Technologies and were further purified by denaturing gel electrophoresis as described (46). The sequences of the templates for R1.1 RNA and R1.1[WC] RNA, as well as the 18 nt promoter oligonucleotide, are provided elsewhere (48,49). Briefly, RNA was synthesized in internally 32P-labeled form by including [α-32P]CTP (final specific activity 43 Ci/mol) in the transcription reactions. Alternatively, RNA was 5′-32P-labeled by treating dephosphorylated, unlabeled transcript with T4 polynucleotide kinase and [γ-32P]ATP (3000 Ci/mmol). The radiolabeled RNAs were purified by electrophoresis in polyacrylamide gels containing 7 M urea (46) and stored at –20°C in Tris–EDTA buffer (pH 7).
Substrate cleavage assay
Cleavage assays were performed as described (46). Short reaction times were employed, as well as low enzyme and substrate concentrations (relative to the Km), so that initial cleavage velocities would be maximally responsive to any inhibitory effect of EB (50). To remove intermolecular aggregates formed during storage at –20°C 5′-32P-labeled RNA was heated at 100°C for 30 s in TE buffer, then snap cooled on ice. The RNA was added to a reaction mix containing buffer consisting of 160 mM NaCl, 30 mM Tris–HCl (pH 8), 0.01 mg/ml tRNA, 0.1 mM EDTA, 0.1 mM DTT and 5% glycerol. EB was added, as appropriate, at the specified concentrations, followed by RNase III. Samples were incubated at 37°C for 5 min and cleavage initiated by adding MgCl2 (pre-warmed to 37°C, 10 mM final concentration). Reactions were stopped by adding a bromophenol blue/xylene cyanol dye mix containing 20 mM EDTA, 20% sucrose and 7 M urea in TBE buffer and aliquots (∼8000 d.p.m.) analyzed by electrophoresis (350 V) in a 15% polyacrylamide gel containing TBE buffer and 7 M urea. Reactions were visualized by autoradiography at –70°C using Fuji Rx film and intensifying screens and were quantitated either by radioanalytical imaging (Ambis) or by phosphorimaging (Molecular Dynamics Storm 860 system).
Substrate binding assay
Gel shift assays were carried out essentially as described (46). Briefly, 5′-32P-labeled RNA was heated and snap cooled, then added to a reaction containing binding buffer consisting of 160 mM NaCl, 30 mM Tris–HCl (pH 8), 10 mM CaCl2, 0.1 mM EDTA, 0.1 mM DTT, 5% glycerol and 0.01 µg/µl tRNA. EB was added, as appropriate, at the specified concentrations, followed by RNase III. The samples were incubated at 37°C for 10 min, then placed on ice for ∼20 min. Aliquots were loaded onto a non-denaturing 6% polyacrylamide gel (80:1 acrylamide:bisacrylamide) containing TBE buffer and CaCl2 (10 mM) and electrophoresed at 120 V for ∼3 h (4°C) using TBE buffer containing 10 mM CaCl2. Experiments were visualized by autoradiography at –70°C using Fuji Rx film and intensifying screens and were quantitated by radioanalytical imaging or phosphorimaging. The (His)6–RNase III[Glu117Gln] mutant (47) was used to determine apparent dissociation constants (Kd). We also found that low (≤2 µM) concentrations of EB gave more variable results in the cleavage and binding assays. This behavior of EB at low concentrations has been noted elsewhere (51) and is attributed to non-specific interactions of EB in the reaction environment. The kinetic studies therefore generally employed EB concentrations ≥5 µM.
Photochemical mapping of ethidium-binding sites
The photochemical mapping of EB intercalation sites in RNA is based on previous protocols (52,53). We employed reverse transcription–primer extension (RT–PE) analysis to detect sites of RNA chain breakage resulting from laser irradiation of ethidium–RNA complexes. To map EB-binding sites in tRNA an unmodified tRNAVal transcript was prepared by transcribing SalI-linearized plasmid pFVAL119 (54) with T7 RNA polymerase and gel purifying the 107 nt RNA as described (46). To map EB-binding sites in R1.1 RNA a synthetic oligodeoxynucleotide was prepared which encoded an extended version of R1.1 RNA (see Fig. 7B). The additional 30 nt at the 3′-end of the R1.1 RNA provided a binding site for the RT primer, which had the sequence 5′-TAAACCTTAAGGTTCTCCTATCTCGAGTCG-3′. The sequence of the oligodeoxynucleotide template encoding the extended version of R1.1 RNA was 5′-TAAACCTTAAGGTTCTCCTATCTCGAGTCGTATTAACCGGAAGAAGGTCAATCATAAAGGCCACT-CTTGCGAATGACCTTGAGTTTGTCCCTCTATAGTGAGCTCTCCCTATAGTGAGTCGTATTA-3′. The oligodeoxynucleotide was transcribed in vitro using T7 RNA polymerase and the 108 nt RNA gel purified as described.
Figure 7.


Photocleavage mapping of the ethidium-binding sites in R1.1 RNA. Laser irradiation and RT–PE analysis were performed as described in Materials and Methods. Irradiation was for 25 min. (A) RT–PE analysis of the irradiated R1.1 RNA–EB complex, as revealed by phosphorimaging. The complete experiments are shown in lanes 3 and 4 (40 and 5 min reaction times, respectively). The asterisks indicate the main positions of EB-dependent photocleavage [A62 and A58; see (B)]. The control experiments are shown in lanes 1, 2, 5 and 6. Lane 1, analysis of R1.1 RNA irradiated in the absence of EB; lane 2, analysis of R1.1 RNA and EB with no irradiation; lanes 5 and 6, analysis of R1.1 RNA in the absence of EB or irradiation at two reactions times (40 and 5 min, respectively); lanes 7–10, PE reactions carried out in the presence of each of the four dideoxynucleoside 5′-triphosphates; lane 11, position of the RT primer. (B) Diagram of R1.1 RNA showing the positions of the two ethidium-binding sites (indicated by stippled bars). The dotted line connecting C27 and G57 indicates a tentative hydrogen bonding interaction stabilized by ethidium binding (see Discussion). The RT primer is shown bound to its complementary sequence at the 3′-end of R1.1 RNA.
To carry out photoirradiation, purified RNA (∼2 pmol) was combined with the specified concentration of EB in 20 µl buffer (see Fig. 7 legend) in an ultraclear 0.5 ml polypropylene tube. Irradiation was performed at ambient temperature for the indicated time with the 532 nm line (∼2 W) from a Millenia Xs diode pumped CW visible laser, having a beam width of >2 mm. Following irradiation the samples were stored at –20°C prior to further analysis. For RT–PE analysis 32P-labeled primer (∼4 × 104 d.p.m.) was annealed to an aliquot of the RNA (∼0.1 pmol) by heating at 90°C for 2 min, after which it was cooled to room temperature and placed on ice. M-MLV reverse transcriptase (Superscript II) (200 U) was added, along with the supplied buffer and four dNTPs, and the reaction incubated at 42°C for the specified time. Formamide-containing dye mix (2/3 vol) was added and the reaction products electrophoresed (31 V/cm) at room temperature in an 8% polyacrylamide sequencing gel (0.2 mm thickness) containing TBE buffer and 7 M urea. Sequence ladders were generated by carrying out a separate RT–PE reaction on unmodified RNA and in the presence of each of the four dideoxy NTPs. The 32P-labeled cDNA products were visualized by phosphorimaging and the sites of EB cleavage mapped using the sequencing ladders as reference lanes and by the fact that EB-dependent RNA photocleavage provides an intact 5′ nucleotide at the breakage site (53), which directly corresponds to a reverse transcriptase stop site.
RESULTS
EB inhibits RNase III cleavage of small model substrates in vitro
EB (Fig. 1A) is a phenanthridine derivative which binds double-helical RNA and DNA by intercalation. The binding event causes localized partial unwinding and lengthening of the double helix, without disrupting base pairing, and increases helix stability (reviewed in 55,56). It was reported that EB at 1.5 mM blocks E.coli RNase III cleavage of the bacterial 30S rRNA precursor in vitro (41). Inhibition was assumed to be due to drug binding to double-helical structures recognized by RNase III. However, the mechanism of EB action was not described, nor were the EB-binding sites identified. The ∼5500 nt 30S RNA most likely contains many EB-binding sites which, along with the large size of the RNA, would complicate a biochemical analysis of the mechanism of inhibition. We instead examined the ability of EB to inhibit cleavage of a small substrate encoded by bacteriophage T7. The R1.1 processing signal (Fig. 1B) is positioned between genes 1.0 and 1.1 in the T7 genetic early region (23) and is cleaved by RNase III at a single site within the internal loop to provide the mature 5′- and 3′-ends of the flanking mRNAs (23). An imino proton NMR study confirmed the overall secondary structure of R1.1 RNA (57) and the 60 nt transcript can be efficiently cleaved in vitro at the canonical site by purified RNase III (33,48). R1.1[WC] RNA (Fig. 1B) is a smaller, fully base paired variant of R1.1 RNA and is cleaved by RNase III at two sites across the helical stem (49). In this regard the pattern of cleavage of R1.1[WC] RNA is representative of other fully double-stranded substrates for RNase III (48,58).
Figure 1.

(A) Structure of EB. (B) Sequences and secondary structures of R1.1 RNA and R1.1[WC] RNA. R1.1 RNA is based on the phage T7 R1.1 processing signal (23), while R1.1[WC] RNA is a smaller, fully base paired variant of R1.1 RNA (49). The arrows indicate the RNase III cleavage sites.
To assess the inhibitory effect of EB 5′-32P-labeled R1.1 RNA was combined with the specified amounts of EB, followed by addition of RNase III and cleavage initiated by adding Mg2+. The reaction time was chosen to allow only limited cleavage of substrate, so as to provide maximum sensitivity to any inhibitory effect of EB. Figure 2A shows that EB blocks cleavage of R1.1 RNA, with a half-maximal inhibition observed at ∼8 µM EB and no cleavage detected at 40 µM EB. Essentially the same inhibitory profile is obtained if EB is added after RNase III but prior to Mg2+ addition (data not shown). The ethidium cation is the inhibitory agent, since cleavage is unaffected by adding equivalent amounts of sodium bromide (data not shown). We also examined the effect of EB on RNase III cleavage of R1.1[WC] RNA. The results (Fig. 2B) show that EB also inhibits cleavage of R1.1[WC] RNA, with a half-maximal inhibitory concentration of ∼4 µM. The different shapes of the EB inhibition curves for R1.1 RNA and R1.1[WC] RNA (see Fig. 2D) suggest that RNA, rather than RNase III, is the target for EB. As a further test to determine whether RNA is the site of ethidium action, AD was substituted for EB in a cleavage reaction. AD is an intercalating agent that binds DNA but not dsRNA (55,56). Thus, an inhibitory effect of AD would suggest the presence of a binding site on RNase III for planar, apolar compounds. The assay results (Fig. 2C) reveal that cleavage of R1.1[WC] RNA is not inhibited by AD, even at 40 µM concentration. We also carried out cleavage assays using two additional RNase III substrates: T7 R4.7 RNA (23) and the rrnB operon T1 transcription terminator hairpin (59). The assay results (data not shown) reveal that EB in the low micromolar concentration range is a general inhibitor of RNase III.
Figure 2.

Ethidium inhibition of substrate cleavage by RNase III. Cleavage assays were performed as described in Materials and Methods using 5′-32P-labeled substrate. Therefore, the only observable cleavage product is the one containing the substrate 5′-end [indicated by 5′ on the left side of (A) and (B)]. EB was combined with substrate in assay buffer, followed by RNase III (6 nM for R1.1 RNA and 4 nM for R1.1[WC] RNA). MgCl2 was added to initiate cleavage and the reaction time was 30 s. Reactions were stopped and electrophoresed in a 15% polyacrylamide, 7 M urea gel. Reactions were visualized and quantitated by autoradiography and phosphorimaging, respectively (see Materials and Methods). (A) EB inhibition of R1.1 RNA cleavage. Lane 1, no Mg2+; lane 2, no EB; lane 3, 4 µM EB; lane 4, 12 µM EB; lane 5, 20 µM EB; lane 6, 40 µM EB. (B) EB inhibition of R1.1[WC] RNA cleavage. Lane 1, incubation of substrate with RNase III in the absence of MgCl2; lane 2, no EB; lane 3, 4 µM EB; lane 4, 12 µM EB; lane 5, 20 µM EB; lane 6, 40 µM EB. (C) Effect of AD on R1.1[WC] RNA cleavage. The experiment was performed as described in (A). Lane 1, no AD; lane 2, 4.1 µM AD; lane 3, 12.4 µM AD; lane 4, 20.6 µM AD; lane 5, 41.2 µM AD. (D) Comparison of the EB inhibition profiles for R1.1 RNA and R1.1[WC] RNA. The triangles indicate EB inhibition of R1.1[WC] RNA cleavage, while the squares indicate EB inhibition of R1.1 RNA cleavage. The 100% cleavage value represents the amount of cleavage occurring in 30 s in the absence of EB. For R1.1 RNA each point represents the average of two experiments. The value at 4 µM EB is 77 ± 47%; at 12 µM EB, 38 ± 22%; at 20 µM EB, 21 ± 16%; at 40 µM EB, 4 ± 2%. For R1.1[WC] RNA each point represents the average of three experiments. The value at 4 µM EB is 52 ± 10%; at 12 µM EB, 13 ± 8%; at 20 µM EB, 8 ± 5%; at 40 µM EB, 5 ± 1%.
Ethidium is a reversible, apparently competitive inhibitor of RNase III
Ethidium intercalation into double-helical structures is a reversible process (60). It would be expected that EB inhibition of RNase III is also reversible and that derepression of cleavage would occur following addition of a nucleic acid that can competitively bind EB but which does not interact with RNase III. One such species is tRNA, which contains an EB-binding site in the acceptor helix (61,62; see also below) and does not significantly affect RNase III action (15). Adding increasing amounts of tRNA to a cleavage reaction containing R1.1 RNA and 20 µM EB causes a corresponding increase in the fraction of substrate cleaved, such that at the highest concentration of tRNA (38 µM) the extent of R1.1 RNA cleavage was the same in the presence or absence of EB (Table 1). In other experiments (data not shown) either increasing the reaction time or the RNase III concentration at a fixed EB concentration also provides an increasing amount of substrate cleavage. In summary, these results indicate the reversibility of ethidium inhibition.
Table 1. Effect of tRNA on EB inhibition of substrate cleavage.
| |
–tRNA |
+tRNA (38 µM) |
| No EB | [100%] | 66% |
| 20 µM EB | 19% | 68% |
Substrate cleavage assays were carried out as described in Materials and Methods and used 5′-32P-labeled R1.1[WC] RNA. Reactions were quantitated by phosphorimaging. The reported values are the averages of two experiments. The amount of substrate cleavage obtained in the absence of tRNA and EB (4.1 ± 2.7%) was normalized to 100% (indicated by the brackets). A nominal amount of tRNA (estimated to be ∼0.02 µM) is already present in the cleavage reactions, which derived from the substrate purification procedure. Note that there is a slight inhibition of cleavage (34% reduction) conferred by 38 µM tRNA in the absence of EB.
To determine the kinetic parameters for EB inhibition the initial rate of R1.1 RNA cleavage was measured as a function of substrate concentration at two EB concentrations (10 and 15 µM), as well as in the absence of EB. To provide steady-state conditions, substrate was in excess of enzyme. The dependence of initial rate on substrate and inhibitor concentrations was assessed by a double reciprocal analysis (Fig. 3), which shows that apparent Km increases with increasing amounts of EB, while Vmax is unaffected. This behavior indicates an apparent competitive inhibition by EB. The calculated Ki for EB (see Fig. 3 legend) is 1.7 ± 0.3 µM. We show below that the apparent competitive inhibition is due to formation of an ethidium–substrate complex that is resistant to cleavage but which can compete with free substrate for RNase III.
Figure 3.

Ethidium bromide exerts apparent competitive inhibitory kinetics. The initial rate of cleavage of internally 32P-labeled R1.1 RNA was determined as a function of substrate concentration at two EB concentrations. The concentration of RNase III in the assays was 12 nM (dimer concentration). The data were analyzed by plotting the reciprocal of the initial cleavage rate versus the reciprocal of the substrate concentration and generating best fit lines according to Michaelis–Menten kinetics (50). The lines shown share a common y intercept, determined by the average value of the y intercepts for each of the experiments. The Km and kcat values for R1.1 RNA cleavage in the absence of EB are 325 µM and 28 min–1. The Ki value (1.7 ± 0.3 µM) was determined as described (50).
Ethidium uncouples RNase III binding and cleavage of substrate
Intercalation can block protein–RNA recognition (see for example 63). To determine whether EB inhibits RNase III binding to substrate 5′-32P-labeled R1.1[WC] RNA was combined with specific amounts of EB, followed by RNase III and the RNA–protein complex resolved by non-denaturing gel electrophoresis. So that the assay would be maximally sensitive to EB inhibition, the RNase III concentration was chosen to provide only partial binding of substrate. In addition, Mg2+ was replaced with Ca2+, which promotes substrate binding while preventing cleavage (37). The results of a representative assay are presented in Figure 4A, which reveals that increasing the EB concentration to 100 µM causes only a minor inhibition of RNase III binding to R1.1[WC] RNA. Essentially the same result was obtained using 5′-32P-labeled R1.1 RNA (Fig. 4B).
Figure 4.

Ethidium does not inhibit RNase III binding to substrate. Gel shift assays were carried out as described in Materials and Methods. (A) RNase III binding to R1.1[WC] RNA. 5′-32P-labeled RNA was combined with EB (concentrations given below), then RNase III was added (5 nM dimer concentration) and the sample electrophoresed in a non-denaturing polyacrylamide gel (see Materials and Methods). The concentration of RNase III was chosen to provide only a partial shift, in order to provide maximal sensitivity to any inhibitory effect of EB. Higher concentrations of RNase III provide a complete shift of the free RNA to the bound form (data not shown). CaCl2 (10 mM) was included in the binding reactions and gel and electrophoresis buffers. The positions of bound and free R1.1[WC] RNA are indicated. The smear of radioactivity between free and bound RNA represents partial dissociation of the RNA–protein complex during electrophoresis, which has been noted elsewhere (37). Lane 1, no RNase III; lane 2, no EB; lane 3, 4 µM EB; lane 4, 12 µM EB; lane 5, 20 µM EB; lane 6, 40 µM EB; lane 7, 100 µM EB. (B) RNase III binding to R1.1 RNA. The same conditions as described above were used (10 mM CaCl2). Lane 1, no protein added; lane 2, no EB, lane 3, 4 µM EB; lane 4, 12 µM EB; lane 5, 20 µM EB; lane 6, 40 µM EB; lane 7, 80 µM EB.
Figure 5 presents a quantitative analysis of EB inhibition of RNase III binding and cleavage of R1.1 RNA and R1.1[WC] RNA. For both substrates there occurred a strong suppression of cleavage with only a slight inhibition of binding. The binding inhibition can be attributed to a non-specific effect of the salt, since the same minor drop in binding was observed when EB was replaced by sodium bromide (dotted line in Fig. 5). Protein titration–gel shift experiments were performed to obtain the apparent dissociation constant (Kd) for RNase III binding to R1.1[WC] RNA in the presence of EB. In this experiment the RNase III[Glu117Gln] mutant was used, which can bind substrate in the presence of Mg2+ but cannot catalyze cleavage (47). The Kd value in the absence of EB was 14.4 ± 6.6 µM, while in the presence of 40 µM EB it was 20.7 ± 0.9 µM (Table 2). We conclude that EB has only a minor destabilizing effect on the RNase III–R1.1[WC] RNA complex. Interestingly, an EB concentration of 5 µM stimulates RNase III binding to R1.1 RNA, which is not observed with R1.1[WC] RNA (Fig. 5). We believe that this enhancement of RNase III binding is due to EB binding to a site in the R1.1 RNA internal loop (see below).
Figure 5.

Ethidium-dependent uncoupling of substrate binding and cleavage by RNase III. The cleavage inhibition curves (filled squares and triangles) are from Figure 2D. Data from four gel shift assays were averaged to generate the points for R1.1[WC] RNA (open triangles): at 4 µM EB, 91 ± 35%; at 12 µM EB, 76 ± 24%; at 20 µM EB, 97 ± 48%; at 40 µM EB, 77 ± 16%. Data from two gel shift assays were used to generate the points for R1.1 RNA (open squares): at 4 µM EB, 165 ± 32%; at 12 µM EB, 89 ± 21%; at 20 µM EB, 91 ± 13%; at 40 µM EB, 87 ± 8%. See text for an explanation for the enhancement of R1.1 RNA binding at 4 µM EB. The relatively greater maximum error values at low EB concentrations is discussed in Materials and Methods. The effect of NaBr on RNase III binding to R1.1[WC] RNA is indicated by the dotted line. The relative percent binding at 40 µM NaBr was 85 ± 27% (average of two experiments, shown by the filled circle). At 100 µM NaBr (data not shown) the relative percent binding was 61 ± 13%.
Table 2. Effect of EB on RNase III–R1.1 RNA complex stability.
| |
–EB |
+EB (40 µM) |
| Kd (nM) | 14.4 ± 6.9 | 20.7 ± 0.9 |
Gel shift assays used 5′-32P-labeled R1.1 RNA (see Materials and Methods). The reported values are the averages of two experiments, along with the maximum error.
RNA binding by the RNase III dsRBD is not inhibited by ethidium
The dsRBD of RNase III is important for substrate binding in vitro and in vivo (A.K.Amarasinghe, S.Su, W.Sun, R.W.Simons and A.W.Nicholson, manuscript in preparation). One possibility is that the EB resistance of the RNase III–substrate complex reflects the behavior of the dsRBD. To test this the dsRBD was purified as an ∼10 kDa polypeptide and gel shift assays were performed using 5′-32P-labeled R1.1[WC] RNA. To provide maximum sensitivity to any inhibitory effect of EB the amount of dsRBD was chosen to provide only a partial shift of substrate. The assay results (Fig. 6) show that dsRBD binding to R1.1[WC] RNA is resistant to EB, up to 100 µM concentration. We conclude that the EB resistance of the RNase III–substrate complex in part reflects a sustained interaction of the dsRBD with substrate in the presence of the drug.
Figure 6.
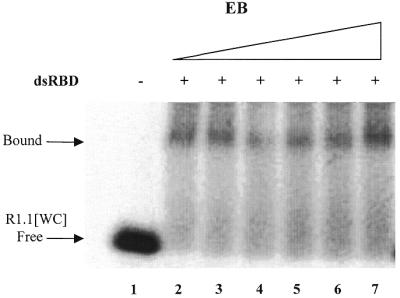
Binding of the RNase III dsRBD to R1.1[WC] RNA is resistant to EB. A gel shift assay was performed using 5′-32P-labeled R1.1[WC] RNA and purified dsRBD, as described in Materials and Methods. We have shown elsewhere that the RNase III dsRBD binds R1.1[WC] RNA with a Kd of ∼800 nM (A.K.Amarasinghe, S.Su, W.Sun, R.W.Simons and A.W.Nicholson, manuscript in preparation). CaCl2 (10 mM) was included in the binding reaction and gel and running buffers. The reaction was analyzed by phosphorimaging. The dsRBD concentration was 800 nM. The positions of bound and free R1.1[WC] RNA are indicated. The smear of radioactivity reflects dissociation of the dsRBD–R1.1[WC] RNA complex during electrophoresis. Lane 1, no dsRBD; lane 2, no EB; lane 3, 4 µM EB; lane 4, 12 µM EB; lane 5, 20 µM EB; lane 6, 40 µM EB; lane 7, 100 µM EB.
Photochemical mapping of ethidium-binding sites in R1.1 RNA
The substrate cleavage assays point to RNA as the target for EB inhibition. To map the ethidium-binding site(s) we took advantage of the ability of photoexcited ethidium to cleave nucleic acid chains at intercalation sites (52,53). Although the mechanism of cleavage is not fully understood, the process probably involves hydrogen atom abstraction from the ribose sugar by the photoexcited ethidium, followed by loss of the base and chain breakage (53). The light source was a 532 nm laser line and RT–PE was used to identify the cleavage sites (see Materials and Methods). Using this assay an ethidium-binding site was identified by an RT stop which was both EB and light dependent.
Since RT–PE has not been used previously to map sites of ethidium-dependent photocleavage, we first applied the assay to a well-characterized RNA which contains a known binding site for ethidium. NMR spectroscopic studies have shown that E.coli tRNAVal contains a single ethidium intercalation site near the base of the acceptor helix, between base pairs A6–U67 and U7–A66 (61,62). We prepared an in vitro transcript of E.coli tRNAVal, combined it with EB and irradiated the complex with 532 nm light. RT–PE analysis of the experiment reveals an RT stop at U67, which maps to the primary intercalation site (61). A second RT stop occurs at U64, which is engaged in a U-G wobble pair in the T stem and therefore probably represents a second intercalation site. A third RT stop was also observed at A58, which may represent non-intercalative cleavage within the T loop, which has also been seen in another RNA stem–hairpin (51). Given the concordance of RT–PE with the NMR characterization of the ethidium-binding site in the tRNAVal acceptor stem, we conclude that RT–PE is a valid approach to map EB intercalation sites.
We next identified the EB-binding sites in R1.1 RNA. The transcript contains a 3′ sequence extension in order to allow binding of the RT primer (Fig. 7B). The additional sequence does not affect RNase III cleavage of the RNA (data not shown). RT–PE analysis of an irradiation experiment indicates the presence of two specific ethidium-binding sites (Fig. 7A, lanes 3 and 4). One site occurs in the lower stem between nucleotides A62 and G61 and a second site is between A58 and G57, which is within the internal loop and adjacent to the scissile bond (Fig. 7B). The occurrence of an ethidium-binding site adjacent to the scissile phosphodiester suggests a mechanism for cleavage inhibition (see Discussion). Each ethidium-binding site is formed by a CG pair stacked on an A-A mismatch and for both sites the base that is lost upon irradiation is a purine (G). The preferential loss of a purine during irradiation of EB–nucleic acid complexes has been noted elsewhere (52). In summary, the laser photocleavage experiments show that ethidium binds to a RNase III substrate in a site-specific manner and also identify a RNA motif preferentially recognized by ethidium.
DISCUSSION
This report has described the inhibitory action of EB on E.coli RNase III cleavage of small model substrates in vitro. RNA is the target for EB inhibition and intercalation is the mode of binding, since the EB concentration which confers half-maximal inhibition is similar to the dissociation constants of other well-characterized EB–RNA intercalation complexes, which are in the low micromolar range (64,65). However, we cannot rigorously rule out an interaction of ethidium with RNase III. In this regard it was reported that propidium, an intercalating agent structurally similar to ethidium, inhibits pancreatic RNase by binding to a non-specific, apolar site on the enzyme (66). However, if such a site were present on RNase III the inhibitory action of ethidium would be insensitive to substrate structure and AD most likely would have inhibited cleavage, neither of which were observed. Ethidium intercalation creates a substrate–inhibitor (S·I) complex which can bind RNase III but which is resistant to cleavage. The behavior of the S·I complex rationalizes the apparent competitive inhibitory kinetics. Thus, it is the S·I complex rather than ethidium which competes with free substrate for RNase III (Fig. 8). As the substrate concentration was increased at fixed EB concentration the S·I concentration became small relative to free substrate and the same Vmax was obtained.
Figure 8.

Scheme for ethidium inhibition of RNase III. The scheme includes R1.1 RNA as substrate, with the internal loop indicated by the absence of formal Watson–Crick base pairing (indicated by the horizontal bars). RNase III is shown as a dimer of ovals. Ethidium is represented by the solid bars. The inset shows the kinetic scheme for inhibition: S, E and I refer to substrate, enzyme (RNase III) and inhibitor (ethidium), respectively. Since it is not yet known whether ethidium can directly dissociate from the E·S·I complex to allow cleavage, this step is not included in the scheme (see also Results and Discussion).
The two ethidium-binding sites in R1.1 RNA are formed by a CG pair stacked on an A·A pair (a 5′-purine–pyrimidine-3′ motif). In contrast, structural analyses of dinucleoside monophosphate–ethidium complexes suggest a preference of ethidium for 5′-pyrimidine–purine-3′ Watson–Crick base pair dinucleotide ‘steps’ (67,68). Why does the AC/GA motif apparently provide a preferred binding site? First, the A·A mismatch may locally destabilize the helix and lessen the thermodynamic cost of intercalation. In this regard it was shown that bulged nucleotides in model RNA hairpins significantly enhance ethidium binding and it was proposed that the additional sugar–phosphate linkage provided by the bulged nucleotide can accommodate the torsional strain accompanying ethidium binding (52). Secondly, the CG pair provides an energetically optimal stacking interaction with the bound ethidium (52,67,68). We also found that low concentrations of ethidium can stimulate RNase III binding to R1.1 RNA (Fig. 4). An imino proton NMR analysis of R1.1 RNA revealed that the internal loop C27 and G57 residues are not engaged in a Watson–Crick pair (57). Ethidium binding may promote formation of this Watson–Crick pair, which in turn would confer greater double-helical character to the internal loop and enhance RNase III binding. In this regard it is known that RNase III binds more tightly to an R1.1 RNA variant containing a fully Watson–Crick base paired internal loop (37).
This study has shown that RNase III binding and cleavage of substrate can be uncoupled by a ligand-induced alteration in RNA structure. Although it is not known whether inhibition requires ethidium binding to both sites in R1.1 RNA, we note that the two sites are far enough apart not to be constrained by the nearest neighbor exclusion principle for intercalation (55,56,69). We believe that ethidium binding to the R1.1 internal loop site is sufficient to cause uncoupling. First, the lower stem of R1.1 RNA can be substantially shortened without strongly affecting substrate reactivity (48,57). The dispensibility of the lower stem would, therefore, indicate the functional inconsequence of ethidium binding to the lower stem site. Secondly, ethidium binding to the internal loop places the drug adjacent to the scissile bond and, therefore, also at (or near) the enzyme active site. The bound ethidium could block placement of the scissile phosphodiester in the active site or inhibit a conformational change in the E·S complex required to reach the transition state. The first mechanism corresponds to inhibition of a ‘lock and key’ interaction, while the second mechanism would correspond to inhibition of an ‘induced fit’ process (52,53). A precise description of how EB inhibits RNase III awaits a structural analysis of the E·S·I complex, however, the ability of intercalated ethidium to reduce local helical motion and increase helical stability (70) provides a basis for inhibition of either mechanism.
Although it is known that specific mutations in RNase III can block cleavage without affecting substrate binding (37,38), speculation has persisted whether catalytically active RNase III can act as a RNA-binding protein. A genetic study suggested that RNase III can bind RNA without concomitant cleavage (71), however, there was no confirmatory biochemical evidence. The ability of a small, RNA-directed ligand to uncouple RNase III action suggests that specific RNA structures may exist which can ‘redirect’ RNase III as a RNA-binding protein and provide an alternative mechanism of gene regulation. Based on these results, we predict that EB inhibits cleavage of the bacterial 30S rRNA precursor by allowing RNase III recognition of the 16S and 23S processing sites but blocking the cleavage step. The known ability of EB to block in vivo maturation of a eukaryotic pre-rRNA (72) could also reflect inhibition of a RNase III-dependent step. In this regard we have determined (I.Calin-Jageman and A.W.Nicholson, unpublished results) that EB blocks yeast RNase III (Rnt1p) cleavage in vitro of the cognate 35S pre-rRNA processing signal, which is present within the 3′-ETS. Since the obligatory first step in the yeast rRNA maturation pathway is performed by RNase III (73,74), ethidium may also block all downstream processing reactions. How ethidium inhibits eukaryotic rRNA maturation awaits further analyses of eukaryotic RNase III homolog interactions with their cognate substrates.
The sustained ability of E.coli RNase III to bind substrate in the presence of EB is reflected by the behavior of the dsRBD. Thus, even though ethidium intercalation causes partial unwinding, lengthening and localized structural distortion of the double helix (55,56), these changes are tolerated by the dsRBD. This is perhaps not unexpected, as it has been shown that the dsRBDs of other proteins can recognize dsRNA structures containing base mismatches, bulges, loops and coaxially stacked helices (6,75–77). The behavior of the dsRBD may be relevant to the effect of EB on other dsRBD-containing proteins. It was reported that EB inhibits the dsRNA-dependent protein kinase PKR (78). However, EB did not block the ability of pre-activated (i.e. autophosphorylated) PKR to phosphorylate eIF2. Although this is consistent with the proposal that EB blocks dsRNA binding by PKR (77), an alternative possibility is that EB allows PKR binding to dsRNA but instead blocks autophosphorylation. Since PKR recognizes dsRNA by employing a tandem dsRBD set (4), the latter mechanism would be consistent with the ethidium insensitivity of the dsRBD.
Acknowledgments
ACKNOWLEDGEMENTS
The authors thank Dr Ahmed Harmouch for providing purified T7 RNA polymerase and Dr Weimei Sun for providing purified RNase III and the RNase III[Glu117Gln] mutant. We also thank Dr Jack Horowitz (Iowa State University) for providing plasmid pFVAL119 and Drs Nils Walter and Zoe Chen (Department of Chemistry, University of Michigan) for providing the laser light source and assistance with the irradiation experiments. We gratefully acknowledge comments on the manuscript by Drs Ashok Bhagwat and Philip Cunningham and we also thank Dr Robert Simons for insightful discussions on this project. This project was funded by grants from the NIH (1-RO1-GM56457 and 1-RO1-GM56772).
References
- 1.Nicholson A.W. (1996) Structure, reactivity and biology of double-stranded RNA. Prog. Nucleic Acid Res. Mol. Biol., 52, 1–65. [DOI] [PubMed] [Google Scholar]
- 2.Jacobs B.L. and Langland,J.O. (1996) When two strands are better than one: the mediators and modulators of the cellular responses to double-stranded RNA. Virology, 219, 339–349. [DOI] [PubMed] [Google Scholar]
- 3.Kumar M. and Carmichael,G.G. (1998) Antisense RNA: function and fate of duplex RNA in cells of higher eukaryotes. Microbiol. Mol. Biol. Rev., 62, 1415–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fierro-Monti I. and Mathews,M.B. (2000) Trends Biochem. Sci., 25, 241–246. [DOI] [PubMed] [Google Scholar]
- 5.St Johnston D., Brown,N.H., Gall,J.G. and Jantsch,M. (1992) A conserved double-stranded RNA-binding domain. Proc. Natl Acad. Sci. USA, 89, 10979–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramos A., Grunert,S., Adams,J., Micklem,D.R., Proctor,M.R., Freund,S., Bycroft,M., St Johnston,D. and Varani,G. (2000) RNA recognition by a staufen double-stranded RNA-binding domain. EMBO J., 19, 997–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bass B.L. (1997) RNA editing and hypermutation by adenosine deamination. Trends Biochem. Sci., 22, 157–162. [DOI] [PubMed] [Google Scholar]
- 8.Baulcombe D.C. (1999) Gene silencing: RNA makes RNA makes no protein. Curr. Biol., 26, 599–601. [DOI] [PubMed] [Google Scholar]
- 9.Fire A. (1999) RNA-triggered gene silencing. Trends Genet., 15, 358–363. [DOI] [PubMed] [Google Scholar]
- 10.Bosher J.M. and Labouesse,M. (2000) RNA interference: genetic wand and watchdog. Nat. Cell Biol., 2, 31–36. [DOI] [PubMed] [Google Scholar]
- 11.Tuschl T., Zamore,P.D., Lehmann,R., Bartel,D.P. and Sharp,P.A. (1999) Targeted mRNA degradation by double-stranded RNA in vitro. Genes Dev., 13, 3191–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernstein E., Caudy,A.A., Hammond,S.M. and Hannon,G.J. (2001) Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature, 409, 363–366. [DOI] [PubMed] [Google Scholar]
- 13.Rotondo G. and Frendewey,D. (1996) Purification and characterization of the Pac1 ribonuclease of Schizosaccharomyces pombe. Nucleic Acids Res., 24, 2377–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mian I.S. (1997) Comparative sequence analysis of ribonucleases HII, III, PH and D. Nucleic Acids Res., 25, 3187–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robertson H.D., Webster,R.E. and Zinder,N.D. (1968) Purification and properties of ribonuclease III from Escherichia coli. J. Biol. Chem., 243, 82–91. [PubMed] [Google Scholar]
- 16.Dunn J.J. (1982) Ribonuclease III. In Boyer,P. (ed.) The Enzymes. Academic Press, New York, NY, pp. 485–499.
- 17.Court D. (1993) RNA processing and degradation by RNase III. In Belasco,J.G. and Brawerman,G. (eds) Control of Messenger RNA Stability. Academic Press, New York, NY, pp. 71–116.
- 18.Nicholson A.W. (1999) Function, mechanism and regulation of bacterial ribonucleases. FEMS Microbiol. Rev., 23, 371–390. [DOI] [PubMed] [Google Scholar]
- 19.Srivastava A.K. and Schlessinger,D. (1990) Mechanism and regulation of bacterial ribosomal RNA processing. Annu. Rev. Microbiol., 44, 105–129. [DOI] [PubMed] [Google Scholar]
- 20.Abouelela ,S, Igel,H. and Ares,M. (1996) RNase III cleaves eukaryotic preribosomal RNA at a U3 snoRNP-dependent site. Cell, 85, 115–124. [DOI] [PubMed] [Google Scholar]
- 21.Kufel J., Dichtl,B. and Tollervey,D. (1999) Yeast Rnt1p is required for cleavage of the pre-ribosomal RNA in the 3′ ETS but not the 5′ ETS. RNA, 5, 909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu H., Xu,H., Miraglia,L.J. and Crooke,S.T. (2000) Human RNase III is a 160 kDa protein involved in preribosomal RNA processing. J. Biol. Chem., 275, 36957–36965. [DOI] [PubMed] [Google Scholar]
- 23.Dunn J.J. and Studier,F.W. (1983) Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J. Mol. Biol., 166, 477–535. [DOI] [PubMed] [Google Scholar]
- 24.Kameyama ,L, Fernandez,L., Court,D.L. and Guarneros,G. (1991) RNase III activation of bacteriophage λ N synthesis. Mol. Microbiol., 5, 2953–2963. [DOI] [PubMed] [Google Scholar]
- 25.Artistarkhov A., Mikulskis,A., Belasco,J.G. and Lin,E.C.C. (1996) Translation of the adhE transcript to produce ethanol dehydrogenase requires RNase III cleavage in Escherichia coli. J. Bacteriol ., 178, 4327–4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koraimann G., Schroller,C., Graus,H., Angerer,D., Teferle,K. and Hogenauer,G. (1993) Expression of gene 19 of the conjugative plasmid R1 is controlled by RNase III. Mol. Microbiol., 9, 717–727. [DOI] [PubMed] [Google Scholar]
- 27.Wagner E.G.H. and Simons,R.W. (1994) Antisense RNA control in bacteria, phages and plasmids. Annu. Rev. Biochem., 48, 713–742. [DOI] [PubMed] [Google Scholar]
- 28.Bardwell J.C.A., Regnier, P,, Chen,S.M., Nakamura,Y., Grunberg-Manago,M. and Court,D.L. (1989) Autoregulation of the RNase III operon by mRNA processing. EMBO J., 8, 3401–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsunaga J., Simons,E.L. and Simons,R.W. (1996) E. coli RNase III autoregulation: structure and function of rncO, the posttranscriptional ‘operator’. RNA, 2, 1228–1240. [PMC free article] [PubMed] [Google Scholar]
- 30.Matsunaga J., Simons,E.L. and Simons,R.W. (1997) Escherichia coli RNase III (rnc) autoregulation occurs independently of rnc gene translation. Mol. Microbiol., 26, 1125–1135. [DOI] [PubMed] [Google Scholar]
- 31.Nicholson A.W., Niebling,K.R., McOsker,P.L. and Robertson,H.D. (1988) Accurate in vitro cleavage by RNase III of phosphorothioate-substituted RNA processing signals in bacteriophage T7 early mRNA. Nucleic Acids Res., 16, 1577–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robertson H.D. (1990) Escherichia coli ribonuclease III. Methods Enzymol., 181, 189–202. [DOI] [PubMed] [Google Scholar]
- 33.Chelladurai B.S., Li,H. and Nicholson,A.W. (1991) A conserved sequence element in ribonuclease III processing signals is not required for accurate in vitro enzymatic cleavage. Nucleic Acids Res., 19, 1759–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dunn J.J. (1976) RNase III cleavage of single-stranded RNA: effect of ionic strength on the fidelity of cleavage. J. Biol. Chem., 251, 3807–3814. [PubMed] [Google Scholar]
- 35.Gross G. and Dunn,J.J., (1987) Structure of secondary cleavage sites of Escherichia coli RNase III in A3t RNA from bacteriophage T7. Nucleic Acids Res., 15, 431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kharratt A., Macia,M.J., Gibson,T.J., Nilges,M. and Pastore,A. (1995) Structure of the dsRNA-binding domain of E. coli RNase III. EMBO J., 14, 3572–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H. and Nicholson,A.W. (1996) Defining the enzyme binding domain of a ribonuclease III processing signal. Ethylation interference and hydroxyl radical footprinting using catalytically inactive RNase III mutants. EMBO J., 15, 1421–1433. [PMC free article] [PubMed] [Google Scholar]
- 38.Dasgupta S., Fernandez,L., Kameyama,L., Inada,T., Nakamura,Y., Pappas,A. and Court,D.L. (1998) Genetic uncoupling of the dsRNA-binding and RNA cleavage activities of the Escherichia coli endoribonuclease RNase III—the effect of dsRNA binding on gene expression. Mol. Microbiol., 28, 629–640. [DOI] [PubMed] [Google Scholar]
- 39.Lienhard G.E., Secemski,I.I., Koehler,K.A. and Lindquist,R.N. (1971) Enzymatic catalysis and the transition state theory of reaction rates: transition state analogs. Cold Spring Harbor Symp. Quant. Biol., 36, 45–51. [DOI] [PubMed] [Google Scholar]
- 40.Russo N. and Shapiro,R. (1999) Potent inhibition of mammalian ribonucleases by 3′-5′-pyrophosphate-linked nucleotides. J. Biol. Chem., 274, 14902–14908. [DOI] [PubMed] [Google Scholar]
- 41.Nikolaev N., Birge,C.H., Gotoh,S., Glazier,K. and Schlessinger,D. (1975) Primary processing of high molecular weight preribosomal RNA in Escherichia coli and HeLa cells. Brookhaven Symp. Biol., 26, 175–193. [PubMed] [Google Scholar]
- 42.Grodberg J. and Dunn,J.J. (1990) OmpT encodes the Escherichia coli outer membrane protease that cleaves T7 RNA polymerase during purification. J. Bacteriol ., 170, 1245–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He B., Rong,M., Lyakhov,D., Gartenstein,H., Diaz,G., Castagna,R., McAllister,W.T. and Durbin,R.K. (1997) Rapid mutagenesis and purification of phage RNA polymerases. Protein Expr. Purif., 9, 142–151. [DOI] [PubMed] [Google Scholar]
- 44.LePecq J.B. (1971) Use of ethidium bromide for separation and determination of nucleic acids of various conformational forms and measurement of their associated enzymes. Methods Biochem. Anal., 20, 41–86. [DOI] [PubMed] [Google Scholar]
- 45.Meienhofer J. (1970) Synthesis of actinomycin and analogs. III. A total synthesis of actinomycin D (C1) via peptide cyclization between proline and sarcosine. J. Am. Chem. Soc., 92, 3771–3777. [DOI] [PubMed] [Google Scholar]
- 46.Amarasinghe A.K., Calin-Jageman,I., Harmouch,A., Sun,W. and Nicholson,A.W. (2001) Escherichia coli ribonuclease III. Affinity purification of (His)6-tagged enzyme and assays for substrate binding and cleavage. Methods Enzymol., in press. [DOI] [PubMed] [Google Scholar]
- 47.Sun W. and Nicholson,A.W. (2001) Mechanism of action of Escherichia coli ribonuclease III. Stringent chemical requirement for a glutamic acid side chain at position 117 and Mn2+ rescue of the Glu117Asp mutant. Biochemistry, in press. [DOI] [PubMed] [Google Scholar]
- 48.Chelladurai B.S., Li,H., Zhang,K. and Nicholson,A.W. (1993) Mutational analysis of a ribonuclease III processing signal. Biochemistry, 32, 7549–7558. [DOI] [PubMed] [Google Scholar]
- 49.Zhang K. and Nicholson,A.W. (1997) Regulation of ribonuclease III processing by double-helical sequence antideterminants. Proc. Natl Acad. Sci. USA, 94, 13437–13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fersht A. (1985) Enzyme Structure and Mechanism. W.H. Freeman and Co., New York, NY.
- 51.Tanner N.K. and Cech,T.R. (1985) Self-catalyzed cyclization of the intervening sequence RNA of Tetrahymena: inhibition by intercalating dyes. Nucleic Acids Res., 13, 7741–7758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.White S.A. and Draper,D.E. (1987) Single base bulges in small RNA hairpins enhance ethidium binding and promote an allosteric transition. Nucleic Acids Res., 15, 4049–4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krishnamurthy G., Polte,T., Rooney,T. and Hogan,M.E. (1990) A photochemical method to map ethidium bromide binding sites on DNA: application to a bent DNA fragment. Biochemistry, 29, 981–988. [DOI] [PubMed] [Google Scholar]
- 54.Liu M. and Horowitz,J. (1993) In vitro transcription of transfer RNAs with 3′-end modifications. Biotechniques, 15, 264–266. [PubMed] [Google Scholar]
- 55.Berman H.M. and Young,P.R. (1981) The interaction of intercalating drugs with nucleic acids. Annu. Rev. Biophys. Bioeng., 10, 87–114. [DOI] [PubMed] [Google Scholar]
- 56.Saenger W. (1984) Principles of Nucleic Acid Structure. Springer-Verlag, New York, NY.
- 57.Schweisguth D.C., Chelladurai,B.S., Nicholson,A.W. and Moore,P.B. (1994) Structural characterization of a ribonuclease III processing signal. Nucleic Acids Res., 22, 604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robertson H.D. (1982) Escherichia coli ribonuclease III cleavage sites. Cell, 30, 669–672. [DOI] [PubMed] [Google Scholar]
- 59.Szeberenyi J., Roy,M.K., Vaidya,H.C. and Apirion,D. (1984) 7S RNA, containing 5S ribosomal RNA and the termination stem, is a specific substrate for the two RNA processing enzymes RNase III and RNase E. Biochemistry, 23, 2952–2957. [DOI] [PubMed] [Google Scholar]
- 60.Meyer-Almes F.J. and Porschke,D. (1993) Mechanism of intercalation into the DNA double helix by ethidium. Biochemistry, 32, 4246–4253. [DOI] [PubMed] [Google Scholar]
- 61.Jones C.R., Bolton,P.H. and Kearns,D.R. (1978) Ethidium bromide binding to transfer RNA: transfer RNA as a model system for studying drug–RNA interactions. Biochemistry, 17, 601–607. [DOI] [PubMed] [Google Scholar]
- 62.Chu W.-Y., Liu,J.C.-H. and Horowitz,J. (1997) Localization of the major ethidium binding site on tRNA. Nucleic Acids Res., 25, 3944–3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hamy F., Brondani,V., Florsheimer,A., Stark,W., Blommers,M.J.J. and Klimkait,T. (1998) A new class of HIV-1 antagonist acting through Tat-TAR inhibition. Biochemistry, 37, 5086–5095. [DOI] [PubMed] [Google Scholar]
- 64.Torgerson P.M., Drickamer,H.G. and Weber,G. (1980) Effect of hydrostatic pressure upon ethidium bromide association with transfer ribonucleic acid. Biochemistry, 19, 3957–3960. [DOI] [PubMed] [Google Scholar]
- 65.Kean J.M., White,S.A. and Draper,D.E. (1985) Detection of high-affinity intercalator sites in a ribosomal RNA fragment by the affinity cleavage intercalator methidiumpropyl-EDTA-iron(II). Biochemistry, 24, 5062–5070. [DOI] [PubMed] [Google Scholar]
- 66.McGrath M., Cascio,D., Williams,R., Johnson,D., Greene,M. and McPherson,A. (1987) Propidium binding to a ribonuclease–DNA complex: x-ray and fluorescence studies. Mol. Pharmacol., 32, 600–605. [PubMed] [Google Scholar]
- 67.Waring M.J. (1965) Complex formation between ethidium bromide and nucleic acids. J. Mol. Biol., 13, 269–282. [DOI] [PubMed] [Google Scholar]
- 68.Jain S.C. and Sobell,H.M. (1984) Visualization of drug–nucleic acid interactions at atomic resolution. VII. Structure of an ethidium/dinucleoside monophosphate crystalline complex, ethidium:uridylyl (3′-5′) adenosine. J. Biomol. Struct. Dyn., 1, 1161–1177. [DOI] [PubMed] [Google Scholar]
- 69.Jain S.C. and Sobell,H.M. (1984) Visualization of drug–nucleic acid interactions at atomic resolution. VIII. Structures of two ethidium/dinucleoside monophosphate crystalline complexes containing ethidium:cytidylyl (3′-5′) guanosine. J. Biomol. Struct. Dyn., 1, 1179–1193. [DOI] [PubMed] [Google Scholar]
- 70.Altuvia S., Locker-Giladi,H., Koby,S., Ben-Nun,O. and Oppenheim,A.B. (1987) RNase III stimulates the translation of the cIII gene of bacteriophage λ. Proc. Natl Acad. Sci. USA, 84, 6511–6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hogan M.E. and Jardetsky,O. (1980) Effect of ethidium bromide on deoxyribonucleic acid internal motions. Biochemistry, 19, 2079–2085. [DOI] [PubMed] [Google Scholar]
- 72.Snyder A.L., Kann,H.E. and Kohn,K.W. (1971) Inhibition of the processing of ribosomal precursor RNA by intercalating agents. J. Mol. Biol., 58, 555–565. [DOI] [PubMed] [Google Scholar]
- 73.Allmang C. and Tollervey,D. (1998) The role of the 3′ external transcribed spacer in yeast pre-rRNA processing. J. Mol. Biol., 278, 67–78. [DOI] [PubMed] [Google Scholar]
- 74.Reeder R.H., Guevara,P. and Roan,J.G. (1999) Saccharomyces cerevisiae RNA polymerase I terminates transcription at the Reb1 terminator in vivo. Mol. Cell. Biol., 19, 7369–7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bevilacqua P.C., George,C.X., Samuel,C.E. and Cech,T.R. (1998) Binding of the protein kinase PKR to RNAs with secondary structure defects: role of the tandem A-G mismatch and noncontiguous helixes. Biochemistry, 37, 6303–6316. [DOI] [PubMed] [Google Scholar]
- 76.Ryter J.M. and Schultz,S.C. (1998) Molecular basis of double-stranded RNA–protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J., 17, 7505–7513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nagel R. and Ares,M. (2000) Substrate recognition by a eukaryotic RNase III: the double-stranded RNA-binding domain of Rnt1p binds RNA containing a 5′-AGNN-3′ tetraloop. RNA, 6, 1142–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Baglioni C. and Maroney,P.A. (1981) Inhibition of double-stranded ribonucleic acid activated protein kinase and 2′,5′-oligo(adenylic acid) polymerase by ethidium bromide. Biochemistry, 20, 758–762. [DOI] [PubMed] [Google Scholar]