

Abstract
Excitatory amino acid transporters (EAATs) are a class of glutamate transporters that terminate glutamatergic synaptic transmission in the mammalian CNS. GltPh, an archeal EAAT homolog from Pyrococcus horikoshii, is currently the only member with a known 3D structure. Here, we studied the kinetics of substrate binding of a single tryptophan mutant (L130W) GltPh in detergent micelles. At low millimolar [Na+], the addition of l-aspartate resulted in complex time courses of W130 fluorescence changes over tens of seconds. With increasing [Na+], the kinetics were dominated by a fast component [kobs,fast; KD (Na+) = 22 ± 3 mM, nHill = 1.7 ± 0.3] with values of kobs,fast rising in a saturable manner to ≈500 s−1 (at 6 °C) with increasing [l-aspartate]. The binding kinetics of l-aspartate differed from the binding kinetics of two alternative substrates: l-cysteine sulfinic acid and d-aspartate. l-cysteine sulfinic acid bound with higher affinity than l-aspartate but involved lower saturating rates, whereas the saturating rates after d-aspartate binding were higher. Thus, after the association of two Na+ to the empty transporter, GltPh binds amino acids by induced fit. Cross-linking and proteolysis experiments suggest that the induced fit results from the closure of helical hairpin 2. This conformational change is faster for GltPh than for most mammalian homologues, whereas the amino acid association rates are similar. Our data reveal the importance of induced fit for substrate selection in EAATs and illustrate how high-affinity binding and the efficient transport of glutamate can be accomplished simultaneously by this class of transporters.
Keywords: sodium-coupled transport, stopped flow, uptake
In the CNS of vertebrates, excitatory amino acid transporters (EAATs) catalyze the energetically uphill movement of glutamate into glial and neuronal cells by coupling to the passage of sodium and potassium ions as well as protons along their respective electrochemical gradients (1). High-resolution crystal structures of a prokaryotic member of the EAAT family, the archeal aspartate transporter GltPh, in aspartate- or inhibitor-bound conformation showed the substrate binding site to be either occluded from or exposed to the extracellular solvent, depending on the position of one of the reentrant loops [helical hairpin 2 (HP2)] (2) (Fig. 1 A and B). Many secondary-active transporters use an alternating access mechanism of transport (3). In its simplest form, two gates open and close alternately and thereby, allow or prevent the entry of the transported substrate(s) from both membrane sides into an aqueous conduction pathway with a central binding site (single binding center gated pore) (4). Initially, a mechanism with HP2 acting as the extracellular gate was considered sufficient to explain the coupled transport by GltPh. However, the structural identification of the inward facing state of the transporter revealed a large, piston-like movement of the substrate binding domain as an essential rearrangement underlying substrate transport (5, 6) (Fig. 1C).
Fig. 1.

Spectral properties of WT and L130W GltPh. (A–C) A cartoon representation of a GltPh protomer in (A) the D,L-threo-β-benzyloxyaspartate-bound outward-facing conformation [Protein Data Bank (PDB) ID code 2NWW], (B) the aspartate-bound outward-facing conformation (PDB ID code 2NWX), or (C) the inward-facing conformation (PDB ID code 3KBC) with L130 (red; CPK). The Tl+ that were interpreted to occupy the Na+ binding sites (yellow) are shown as spheres, and the amino acid and inhibitor (both orange) are depicted as sticks. The transport domain is colored blue, with HP1 (below) and HP2 (above) in dark blue, and the trimerization domain is in gray. (D) The corrected emission spectra of WT and L130W GltPh after excitation at 295 nm. (E) The emission spectra of WT GltPh after excitation at 289 nm. The fluorescence intensities were corrected for differences in [protein] and normalized to the peak intensity of L130W GltPh in the absence of substrates, and they reflect the relative brightness. The buffers contained 500 mM choline chloride (dotted line), 500 mM choline chloride and 100 mM NaCl (dashed line), or 500 mM choline chloride, 100 mM NaCl, and 500 µM l-aspartate (solid line).
Many enzymes function according to an induced fit mechanism (7): after an initial loose association of the substrate, the enzyme proceeds to a state where the substrate is bound tightly, thereby releasing additional energy for catalytic function. For transporters, the induced transition fit concept proposes that this energy is used to drive the conformational changes that are necessary for the substrate translocation (8). The transition leading to the fit state can consist of only side chain rearrangements (9), or it can comprise conformational changes of the peptide backbone (10). Because different ligands induce fit states that differ in tightness and/or differently affect the function of the protein, induced fit contributes to selectivity (11). In GltPh, HP2 closes after the association of the substrate from the external membrane side and might be involved in substrate selection by induced fit.
Here, we performed stopped-flow experiments on detergent-solubilized GltPh while using the intrinsic fluorescence intensity variation of the tryptophan mutation L130W as well as the tyrosine fluorescence of the WT transporter. Our data clarify the order of the steps in the binding sequence and reveal the rates of both substrate binding and the associated conformational changes. Our findings permit a functional comparison of archeal and mammalian EAAT homologues and delineate a role for HP2 in substrate selection by induced fit in these transporters.
Results
Fluorescence Spectra of L130W and WT GltPh Reflect the Binding of Na+ and Aspartate.
L130W GltPh contains a single tryptophan at the junction between the transmembrane domain (TM) 4 and the TM3–TM4 loop (Fig. 1C). W130 reports on the coupled binding of aspartate and Na+ by a fluorescence increase and allows for the quantification of binding by monitoring intrinsic protein fluorescence (2). We expressed and purified WT and L130W GltPh and measured the spectral properties of proteins in detergent micelles. The emission spectra of L130W GltPh peaked at ∼318 nm (Fig. 1D), which suggests an exposition of W130 to a hydrophobic environment (12). On the addition of l-aspartate to L130W GltPh in the presence of 100 mM Na+, the emission increased without a change in spectral shape. The sole addition of 100 mM Na+ resulted in a relatively small increase that was limited to lower wavelengths, which indicates an origin other than the fluorescence increase on the coupled binding of Na+ and aspartate. The addition of aspartate in the absence of Na+ was previously reported to have no effect on fluorescence when monitored at a single wavelength (2).
Exciting WT GltPh at 295 nm results in fluorescence with a similar emission maximum to L130W GltPh. This fluorescence is most likely caused by tyrosine, because WT GltPh contains 18 tyrosine residues and no tryptophan (SI Results). The addition of Na+ results in a small shift to a higher intensity that is limited to lower wavelengths. The extent of the increase becomes considerable when a shorter excitation wavelength is used (Fig. 1E). On the addition of aspartate, an additional smaller increase is detectable.
These results show that L130W GltPh fluorescence is well-suited to report on the coupled binding of aspartate and Na+, whereas the tyrosine fluorescence of WT GltPh permits the monitoring of Na+ binding to the empty transporter.
Rapid Substrate Addition Reveals Slow Processes That Precede Aspartate Association.
Fig. 2A shows the L130W GltPh fluorescence changes on the simultaneous rapid addition of l-aspartate and Na+ compared with the addition of l-aspartate to protein that was previously incubated with Na+ at a high millimolar concentration. After mixing the Na+- and aspartate-free protein with both substrates, we observed a complex time course of fluorescence increase on timescales ranging from milliseconds to seconds. In contrast, on the addition of l-aspartate to Na+-preincubated protein, almost all of the fluorescence change occurred within a few milliseconds. We conclude that the Na+-bound state is a prerequisite for fast aspartate binding. The observed slow transitions represent processes that lead to the state from which aspartate binding occurs, such as the initial Na+ binding and either preceding or consecutive events. In accordance with this conclusion, the overall rate of the fluorescence increase after the simultaneous addition of the substrates is Na+-dependent (Fig. S1).
Fig. 2.

Fast aspartate binding after slow processes associated with the binding of two Na+. (A) The tryptophan fluorescence transients upon the rapid addition of 100 µM l-aspartate to L130W GltPh preequilibrated with 100 mM NaCl (upper curve) or mixing 100 µM l-aspartate and 250 mM NaCl with empty L130W GltPh (lower curve). NaCl (500 mM) was equimolarly substituted with choline chloride. The black lines represent experimental data, and the fits to the sums of exponential functions as well as a linear function to account for photobleaching are in gray. (B) Fluorescence transients and corresponding exponential fits after mixing 100 µM l-aspartate with L130W GltPh preincubated with three different [Na+] (the fit results of all experiments are in Table S1). (C) The relative amplitudes of the highest rates obtained from the fits (kobs,fast) as a function of [Na+]. The continuous line represents the fit to a Hill function with nHill = 1.7 ± 0.3 and KD = 22 ± 3 mM. For comparison, the result of a fit with nHill fixed to one is shown as a broken line. (D) The Na+ titration curve of WT GltPh emission at 308 nm after excitation at 289 nm in 500 mM choline chloride fitted with a Hill function (nHill = 1.9 ± 0.2, KD = 25 ± 2 mM) is shown as a continuous line. The fraction of bound protein is calculated by dividing the relative fluorescence change caused by each addition by the maximum change. All error bars represent ±SEM of three experiments.
Two Na+ Bind Before Aspartate.
To determine the [Na+] dependence of relative amplitudes and the observed rates (kobs,fast) of the fast process, we fitted sums of exponential functions to the fluorescence transients resulting from the addition of l-aspartate after preincubation with various [Na+] (Table S1 shows the results). When using protein preincubated with 5 mM Na+, most of the fluorescence change after l-aspartate addition was slow, with only a small fraction of the amplitude assigned to kobs,fast (Fig. 2B and Table S1). Increasing [Na+] resulted in higher relative amplitudes of kobs,fast, whereas the fitted values of kobs,fast were around 400 s−1 and did not depend on Na+. At 400 mM Na+, the time course was entirely dominated by kobs,fast. Fig. 2C shows the relative amplitudes of kobs,fast plotted against the [Na+]. A fit of this relationship with a Hill function yields a Hill coefficient greater than one (nHill = 1.7 ± 0.3, KD = 22 ± 3 mM; n = 3), which is in agreement with at least two Na+ binding before aspartate. Given that one Na+ has to bind after aspartate (2) and the total number of transported Na+ is three (13), we conclude that two Na+ bind before aspartate binding.
For lower [Na+], the fits with sums of exponentials did not provide an unambiguous quantitative description of the remaining, slower processes. The fit results were satisfying for individual traces but occasionally provided inconsistent rate constants on repetition of the experiment under the same experimental conditions, presumably because the signal-to-noise ratio of the data was not always sufficient to detect all underlying processes. Thus, we only used the values and the amplitudes of kobs,fast for additional analysis.
L130W might have affected substrate binding, and we, therefore, performed additional experiments exploiting the tyrosine emission of WT GltPh (Fig. 2D). The increase in fluorescence intensity with [Na+] could be fitted with a Hill function yielding parameters that, within the experimental error, did not differ from the results obtained with rapid aspartate binding to L130W GltPh (nHill = 1.9 ± 0.2, KD = 25 ± 2 mM; n = 3). In addition, the kinetics of Na+ binding to WT GltPh are comparable with the kinetics observed after mixing l-aspartate and Na+ with L130W GltPh. (Fig. S2). Thus, L130W leaves the initial sodium binding largely unperturbed.
Induced Fit Mechanism Allows for Substrate Selectivity.
Fig. 3A shows the fluorescence time course of Na+-bound L130W GltPh on addition of various concentrations of l-aspartate. The rate constant of this process, kobs,fast, increases in a saturable manner with [l-aspartate] (Fig. 3B), which is expected for kobs,fast reflecting l-aspartate binding. The observed deviation from a linear [l-aspartate] dependence—predicted by a one-step association of the substrate and transporter (14)—indicates an additional unimolecular reaction that limits the overall rate at high [l-aspartate].
Fig. 3.
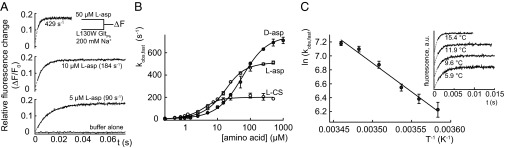
Concentration and temperature dependence of amino acid binding rates to L130W GltPh. (A) The tryptophan fluorescence transients after the rapid mixing of L130W GltPh in 200 mM NaCl with various [l-aspartate]. Gray traces represent fits of the sum of two exponential functions to the experimental data (black), which were performed with rates and amplitudes of the slower component fixed to a value that was previously assessed from a longer recording. The parameters were (relative amplitudes in parentheses) 429 s−1 (0.17) and 0.5 s−1 (0.018) for 50 µM l-aspartate, 184 s−1 (0.18) and 0.5 s−1 (0.017) for 10 µM l-aspartate, and 89 s−1 (0.17) and 0.5 s−1 (0.017) for 10 µM l-aspartate. The data during the dead time of the apparatus (2.3 ms) are represented as a broken line. For each trace, the fitted value for kobs,fast is presented. (B) The [amino acid] dependence of kobs,fast. The continuous lines represent the fits of Eq. 1 to the data. (C) An Arrhenius plot of kobs,fast because of the addition of 100 µM l-aspartate in 500 mM NaCl according to the linearization of the Arrhenius equation, 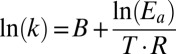 , where B represents the frequency factor, Ea represents the Arrhenius activation energy, T represents the absolute temperature, and R represents the universal gas constant. The continuous line represents the fit of a linear function to the data, which results in Ea = 68 ± 6 kJ mol−1. (Inset) Fluorescence transients at different temperatures that were used to generate the Arrhenius plot. All error bars represent ±SEM of at least three experiments.
, where B represents the frequency factor, Ea represents the Arrhenius activation energy, T represents the absolute temperature, and R represents the universal gas constant. The continuous line represents the fit of a linear function to the data, which results in Ea = 68 ± 6 kJ mol−1. (Inset) Fluorescence transients at different temperatures that were used to generate the Arrhenius plot. All error bars represent ±SEM of at least three experiments.
The temperature dependence of the aspartate association kinetics at saturating [l-aspartate] (500 µM) (Fig. 3B) provides an activation energy (Ea) of 68 ± 6 kJ/mol, which is equivalent to a Q10 = 2.7 for the rate limiting step (Fig. 3C). These values correspond to an energy barrier too high to be assigned to a diffusion-controlled reaction and therefore, provide additional evidence for a conformational change associated with l-aspartate binding.
This conformational change could occur after the association of the substrate to the binding site, a scenario that is found in an induced fit mechanism (7) (Scheme 1). The alternative mechanism, where a conformational change is necessary to permit subsequent substrate association (Scheme 2), is commonly referred to as conformational selection (15):
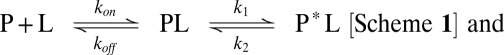 |
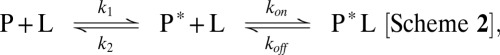 |
where L is the ligand and P or P* denote distinct conformations of the protein.
Both mechanisms can result in saturating behavior, because the overall reaction is limited by the unimolecular reaction. One way to discriminate kinetically between Schemes 1 and 2 is to use alternative substrates (16, 17). In the case of an induced fit mechanism, the ligands are already associated to the protein and may modulate the unimolecular reaction rate (i.e., the nature of the ligand can affect k1 and k2, which results in distinct saturation levels for different ligands). If conformational selection was operating, the unimolecular step would occur in the absence of the ligand, and k1 and k2 would be independent of the properties of the ligand.
We determined kobs,fast for different concentrations of two additional ligands, d-aspartate and l-cysteine sulfinic acid (l-CS). l-CS is a substrate with comparable transport activity with l-aspartate (Fig. S3C) but lower affinity (2) (Fig. S3 A and B). To date, it has been unclear whether GltPh distinguishes between d- and l-aspartate (2, 18). The saturating values of kobs,fast provide a clear distinction between l-aspartate and the tested alternative ligands, because they were larger for d-aspartate and less than one-half as large for l-CS (Fig. 3B), which is in agreement with induced fit (Scheme 1).
Scheme 1 predicts fluorescence kinetics with two exponential components. Our experiments were performed under pseudo-first order conditions, with [L] in large excess over [P]. This case leads to two nonzero solutions for the observed rate constants (SI Results shows their derivation), of which only the solution representing the slower of the two components accounts for the saturating behavior of kobs,fast (Fig. 3B):
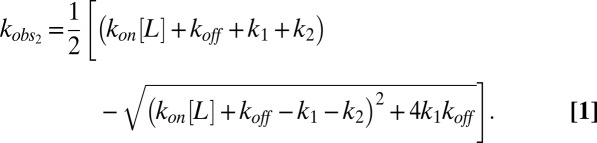 |
We fitted Eq. 1 to the [amino acid] dependencies of kobs,fast to determine the substrate association/dissociation rates (kon, koff) as well as the forward rate constant of the conformational change (k1) (Table 1). These fits reliably constrain k1, which results in distinct values for all tested substrates as anticipated above. For the bimolecular step, we find similar association rate constants (kon), whereas the koff values differ among substrates. The comparison of the l-/d-aspartate and l-CS data illustrates a crucial role for the induced fit mechanism for substrate selection. l-CS exhibits a considerably smaller koff value, which results in tighter initial binding. However, the higher forward rates of the subsequent conformational change promote the higher apparent affinities of the transporter for d- or l-aspartate (2). Preliminary fits resulted in a large error for the values of the backward rate constant, k2. We, therefore, had to fix this parameter during the fits and chose a common value from the interval between the initial results for the three tested substrates (10 s−1), with little effect on the results for the remaining parameters. Although it was not possible to determine k2 values by fitting, these values can be estimated from the apparent binding affinities measured at 20 °C (2) according to 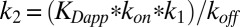 , when assuming that there are no differences in the temperature dependencies for the diverse substrates. The resulting k2 values are smaller for l- or d-aspartate (0.1 or 0.2 s−1) than for l-CS (3.7 s−1). Thus, aspartate exhibits higher rates of establishing induced fit, and the lower rates back to the loosely bound state. The simulated binding time courses based on the obtained values of the rate constants show consistent agreement with experimental traces and sensitivity to parameter changes (Fig. S4), showing the accuracy of our model.
, when assuming that there are no differences in the temperature dependencies for the diverse substrates. The resulting k2 values are smaller for l- or d-aspartate (0.1 or 0.2 s−1) than for l-CS (3.7 s−1). Thus, aspartate exhibits higher rates of establishing induced fit, and the lower rates back to the loosely bound state. The simulated binding time courses based on the obtained values of the rate constants show consistent agreement with experimental traces and sensitivity to parameter changes (Fig. S4), showing the accuracy of our model.
Table 1.
Best fit parameters of the kobs,fast substrate dependence
| Amino acid | kon (107 M−1 s−1) | koff (s−1) | k1 (s−1) | k2 | KD (µM)* |
| l-aspartate | 3.0 ± 0.2† | 250 ± 40 | 506 ± 4 | n.d.§ | 8.3 ± 2.0 |
| (2.5; 4.1)‡ | (150; 400) | (490; 530) | (4.7; 11.8) | ||
| d-aspartate | 2.8 ± 0.5 | 1,200 ± 320 | 750 ± 10 | n.d. | 43 ± 20 |
| (1.6; 3.9) | (500; 1,900) | (730; 760) | (8.0; 78) | ||
| l-CS | 3.9 ± 0.6 | 41 ± 19 | 194 ± 5 | n.d. | 1.1 ± 0.7 |
| (2.6; 5.1) | (0; 82) | (184; 204) | (0; 2.2) |
n.d., not determined.
Intrinsic dissociation constant calculated from KD = koff/kon.
The SE of the fit is indicated.
Parentheses contain 95% confidence intervals for the measured rate constants and 90% confidence intervals for calculated KD values.
k2 was fixed to 10 s−1 in all fits.
W130 Fluorescence Changes Do Not Require Isomerization to the Inward-Facing State.
Crystallographic studies revealed that HP2 alternately exposes and occludes the substrate binding site (2), which makes the latter movement a perfect candidate for a conformational change involved in an induced fit mechanism. However, there are additional transitions within the transport cycle that might contribute to the observed fluorescence changes.
To test for a possible contribution of movements of the transport domain relative to the trimerization domain (5, 6, 19, 20), we studied the effect of cross-linking the transporter in either the inward- or outward-facing conformation on the observed fluorescence changes. The oxidative cross-linking of two inserted cysteines, C55 and C364, traps GltPh in the inward-facing conformation (5) (Fig. 4A). After cross-linking L130W K55C C321S A364C GltPh with Cu(II)(1,10-phenanthroline)3 (CuPh), W130 fluorescence is independent of [l-aspartate] (Fig. 4C). When cross-linking is prevented by the addition of N-ethyl-maleimide before CuPh, the mutant transporters show a significant fluorescence increase on aspartate addition. These data show that the observed fluorescence changes rely on conformations other than an inward-facing GltPh. We noticed a lower relative fluorescence change in the mutant compared with L130W GltPh (Fig. 4F), which may have resulted from N-ethyl-maleimide treatment or other incubation conditions by an unknown mechanism.
Fig. 4.

Structural basis for W130 fluorescence changes. (A and D) The sites of cross-linking mutations in cartoon representations of a single GltPh protomer in (A) the inward-facing conformation (K55C/A364C) or (D) the aspartate-bound outward-facing conformation (V216C/A391C). The amino acids mutated to cysteines are in magenta or green, and the protein backbone is in wheat. (B and E) Nonreducing SDS/PAGE of (B) K55C L130W C321S A364C GltPh or (E) L130W V216C C321S A391C GltPh. The SDS/PAGE sample buffer was with or without DTT as indicated. (C and F) Relative l-aspartate–induced fluorescence changes for (C) K55C L130W C321S A364C GltPh or (F) L130W V216C C321S A391C GltPh. (F) As a reference for the relative l-aspartate effects on fluorescence, the data from untreated L130W GltPh (±SEM; n = 5) are shown. In the double cysteine mutants, the error bars represent the SEM of (C) three or (F) four experiments. (G) The factor Xa cleavage site in a ribbon presentation of the aspartate-bound L130W GltPh protomer with the N- and C-terminal cleavage products in green and wheat, respectively. (H) An SDS/PAGE of the factor Xa cleavage products of AGIH111-114IDGR L130W GltPh. (I) The mean relative fluorescence response to the addition of 500 µM l-aspartate to the cleaved and uncleaved mutant in 500 mM NaCl (bars). The values of individual experiments are shown as filled circles.
To test whether the fluorescence signals report on conformational changes of GltPh in the outward-facing conformation, we sought to trap the transporter in the outward-facing conformation with the help of suitable cysteine insertions. V216 and A391 show sufficient proximity for cysteine cross-linking in the outward-facing conformation (Fig. 4D) (Cα distance: 6.6 Å) but not the inward-facing conformation (Cα distance: 16.4 Å). SDS/PAGE analysis shows two electrophoretic species of L130W V216C C321S A391C GltPh (Fig. 4E). Whereas CuPh treatment does not change the proportions of these two electrophoretic species, the apparent lower molecular weight species disappears after treatment with DTT and therefore, corresponds to an intramolecularly linked (outward-facing) conformation. This species accounts for 75–80% of the transporters after CuPh treatment (Fig. S5B), and a fluorescence change of ∼15% is observed on l-aspartate addition, which is roughly identical to 75% of the fluorescence change that is observed after amino acid binding to L130W GltPh (Fig. 4F). The identity of the remaining 25% remains unclear. GltPh trapped in the outward-facing state displays similar fluorescence signals as compared to when it is free to move.
A recent study showed substrate induced changes of the TM3–TM4 loop in susceptibility to protease cleavage and that cutting either end of this loop abolishes radioactive aspartate uptake by GltPh (21). We inserted a factor Xa site at the end of the TM3–TM4 loop that is remote from W130 into L130W GltPh (Fig. 4G). Fig. 4I shows that cleavage at this site does not interfere with the W130 fluorescence changes on aspartate binding.
Another possible explanation for the observed W130 fluorescence variation is large collective motions that bring the transport domains of neighboring EAAT subunits into close proximity (22). However, amino acid binding to GltPh detected by W130 fluorescence does not show cooperativity (2), whereas the reported large collective motions are cooperative in nature. Furthermore, the TM3–TM4 loop is engaged in all three modes of these collective motions, whereas the conformational change that we report is independent of an intact TM3–TM4 loop (Fig. 4 G–I).
Taken together, our experimental results show that neither the transitions between the outward- and inward-facing transporters nor the TM3–TM4 loop is necessary for the observed fluorescence signals. These findings strongly suggest that HP2 movement is the determinant of these signals.
Discussion
The archeal GltPh is currently the only glutamate transporter with a known 3D structure. To date, no high-resolution functional data on GltPh have been reported, which makes the comparison between mammalian and prokaryotic transporters and the correlation of structural and functional data difficult. Here, we used stopped-flow substrate addition and tryptophan fluorescence measurements on purified and solubilized GltPh to obtain information. Our data are explained by a kinetic model (Fig. 5) that predicts the binding of two Na+ to the unliganded transporter that is required for subsequent aspartate binding (Fig. 2). The kinetics of the initial Na+ binding were found to be considerably slower than the conformational transition to the induced fit after the association of aspartate (Fig. 3 and Figs. S1 and S2). For this fast transition, we were able to rule out a contribution of the isomerization between the inward- and outward-facing transporter (Fig. 4 A–F) and conformational changes of the TM3–TM4 loop (Fig. 4 G–I). Thus, the induced fit, which was shown in this study, is most likely mediated by the closure of HP2, which is known to result in an occluded state (2) (Fig. 1B). In contrast, it is unclear from which state the HP2 closure proceeds, because it is not known where the initial association of the amino acid to the transporter takes place. Intuitively, one may assume that the interaction occurs in the binding pocket. However, a recent molecular dynamics study suggests that HP2 itself initiates the substrate interaction (23). Thus, the substrate might be directed to its final position by HP2 as it is closing down over the binding pocket.
Fig. 5.
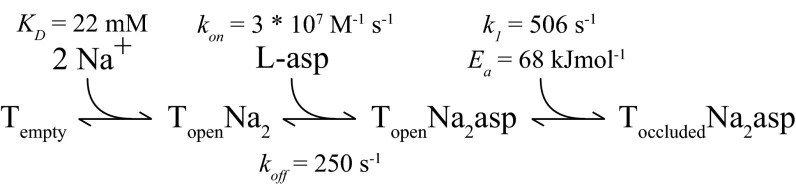
Kinetic model of l-aspartate association to GltPh with kinetic and thermodynamic parameters determined at 6 °C.
While this article was in the review process, a study was published (24) that used a voltage-sensitive dye to measure substrate binding to GltPh. In agreement with our results, Reyes et al. (24) report a weak initial binding of Na+ to the empty transporter and suggest, like we show here, that two Na+ are required for the binding of aspartate.
At ambient temperatures, GltPh aspartate transport rates (0.29 min−1 at 30 °C) (18) are significantly slower than the glutamate transport rates of mammalian EAATs, which raises the question of which steps of the uptake cycle of prokaryotic and eukaryotic glutamate transporters differ. In electrophysiological studies combining patch-clamp and fast substrate addition, Mim et al. (25) interpreted the rapidly decaying phase of the transport currents and the rising phase of the anion currents to report on the substrate occluding movements of HP2. The corresponding rates for EAAT1 (1,000 s−1) (26), EAAC1/EAAT3 (1,200 s−1) (27), and EAAT4 (340 s−1) (28) are slower than the rates of GltPh (kobs at high [aspartate] as extrapolated to 22 °C is ∼2,700 s−1), whereas EAAT2 (∼4,000 s−1) (29) exhibits faster rates. Thus, HP2 closure by GltPh resembles its mammalian counterparts. In experiments exploiting the temperature dependence of the above rates, an enthalpy of activation for EAAC1/EAAT3 was determined that is considerably larger (121 kJ/mol) (25) than our results for the Arrhenius energy of activation (68 kJ/mol) on GltPh. This difference might be because of isoform-specific variations. Alternatively, although the activation energy of EAAC1/EAAT3 was assigned to the closure of HP2, the electrophysiological approach might reflect more extensive conformational changes, including the transition from the outward- to inward-facing state. When comparing the association of the amino acid and transporter that precedes the conformational change, we find similar rates among GltPh (kon: ∼3 × 107 M−1 s−1) and the mammalian glutamate transporters (EAAT3: 2 × 107 M−1 s−1) (30). In conclusion, our data define GltPh as an accurate model for EAAT substrate recognition.
The small transport rates of GltPh must, therefore, originate from processes that either precede or follow substrate association. Slow transitions associated with Na+ binding show an increase in the overall rate with increasing [Na+] (Fig. S1), which indicates that conformational change(s) occur after the association of Na+. Whereas Na+ has, at most, only a minor impact on the distribution of inward- and outward-facing transporters (20), the opening of HP2 has been shown to rely on the binding of Na+ (31). Furthermore, large heat capacity changes on Na+ binding were reported that cannot be attributed to HP2 opening alone (24). Therefore, the slow conformational change(s) after Na+ binding might represent slow HP2 opening and/or more extensive rearrangements, which can explain the small transport rates of GltPh. However, L130W GltPh displays significantly slowed aspartate transport compared with WT (2), whereas the kinetics of Na+ binding are similar (Fig. 2 and Fig. S1). Therefore, the processes associated with Na+ binding cannot be rate-limiting in L130W GltPh.
Carrier-mediated transport requires tight interaction on one membrane side—to enable selection among various transport substrates—as well as fast release on the other side to support effective transport. GltPh exhibits similar binding affinities for outward- and inward-facing conformations (5), and substrates with low unbinding rates will, thus, stay bound in the binding pocket and reduce transport rates. Our data illustrate how an induced fit mechanism can overcome these antagonistic effects. The main substrate of GltPh—l-aspartate—initially binds with lower affinity to the binding pocket than l-CS but promotes induced fit with a higher forward rate (k1) than the alternative ligand. Thus, the induced fit enables l-aspartate to displace the competing l-CS from the binding site (Fig. S3A) without reducing the transport activity (Fig. S3C).
Materials and Methods
Mutagenesis, Expression, and Purification of GltPh.
The expression constructs were generated using the QuikChange Kit and WT GltPh with a C-terminal 8× histidine tag cloned into a pBAD24 vector (provided by Eric Gouaux, Oregon Health and Science University, Portland, OR). Heterologous expression was induced by the addition of 1% l-arabinose to Escherichia coli Top10 or BL21-AI cells at OD500 = 0.5–0.7. The purification of GltPh was performed as previously described (32) using 1 mM n-dodecyl-β-D-maltopyranoside (DDM) and omitting sodium glutamate. If necessary, the protein was concentrated using ultrafiltration cartridges. Na+ and potentially bound amino acids were removed by replacing NaCl with choline chloride using disposable salt exchange columns followed by an additional salt exchange to an NaCl-containing buffer if necessary. All buffers contained 20 mM Tris (pH 7.4), 1 mM DDM, and choline chloride or NaCl as noted in the figures.
Fluorescence Measurements.
The [GltPh] was between 0.05 and 4 µM. The steady state fluorescence intensities of the WT and mutant GltPh were determined in a Quantamaster 4 spectrofluorometer (Photon Technology Inc.). The solutions were stirred in a 1-cm cuvette thermostated to 22 °C. The changes in GltPh fluorescence on the rapid addition of Na+ or amino acids were measured in a stopped-flow apparatus (HITECH SF-61SX2/s; TgK Scientific). Excitation was performed at 297 nm, and emission was selected using long-pass filters with cut on wavelengths of 320 or 306 nm; the latter resulted in a higher signal-to-noise ratio. If not otherwise specified, the mixing system and observation cell were at 6 °C. The measured dead time of the system was 2.3 ± 0.1 ms (n = 4) at this temperature. To determine the rates at higher temperatures, a smaller observation cell with a reaction volume of 5 µL (dead time = 0.9 ± 0.1 ms; n = 5) instead of 22.5 µL was used. The [amino acid] was maintained in at least fivefold excess over [GltPh] to fulfill pseudo-first order conditions (14).
Cysteine Cross-Linking and Factor Xa-Mediated Proteolysis.
CuPh-mediated cross-linking was performed as previously described (5). Cleavage of the L130W GltPh backbone at the N-terminal border of the TM3–TM4 loop was essentially performed as previously described (21).
Supplementary Material
Acknowledgments
We thank Patricia Hidalgo for advice on protein expression and purification, Manuel Taft and Georgios Tsiavaliaris for advice on the stopped-flow experiments, and Arne Franzen for technical assistance. We also thank Alexi Alekov, Martin Fischer, Nadine Havenstein, Jasmin Hotzy, Peter Kovermann, Nicole Schneider, Yunping Song, and Gabriel Stölting for discussing the manuscript. These studies were supported by Deutsche Forschungsgemeinschaft Grant FA301/9 (to Ch.F.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1300772110/-/DCSupplemental.
References
- 1.Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65(1):1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 2.Boudker O, Ryan RM, Yernool D, Shimamoto K, Gouaux E. Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature. 2007;445(7126):387–393. doi: 10.1038/nature05455. [DOI] [PubMed] [Google Scholar]
- 3.Jardetzky O. Simple allosteric model for membrane pumps. Nature. 1966;211(5052):969–970. doi: 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- 4.Klingenberg M. The ADP,ATP shuttle of the mitochondrion. Trends Biochem Sci. 1979;4(7):249–252. [Google Scholar]
- 5.Reyes N, Ginter C, Boudker O. Transport mechanism of a bacterial homologue of glutamate transporters. Nature. 2009;462(7275):880–885. doi: 10.1038/nature08616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crisman TJ, Qu S, Kanner BI, Forrest LR. Inward-facing conformation of glutamate transporters as revealed by their inverted-topology structural repeats. Proc Natl Acad Sci USA. 2009;106(49):20752–20757. doi: 10.1073/pnas.0908570106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koshland DE. Application of a theory of enzyme specificity to protein synthesis. Proc Natl Acad Sci USA. 1958;44(2):98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klingenberg M. Ligand-protein interaction in biomembrane carriers. The induced transition fit of transport catalysis. Biochemistry. 2005;44(24):8563–8570. doi: 10.1021/bi050543r. [DOI] [PubMed] [Google Scholar]
- 9.Mirza O, Guan L, Verner G, Iwata S, Kaback HR. Structural evidence for induced fit and a mechanism for sugar/H+ symport in LacY. EMBO J. 2006;25(6):1177–1183. doi: 10.1038/sj.emboj.7601028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loo TW, Bartlett MC, Clarke DM. Substrate-induced conformational changes in the transmembrane segments of human P-glycoprotein. Direct evidence for the substrate-induced fit mechanism for drug binding. J Biol Chem. 2003;278(16):13603–13606. doi: 10.1074/jbc.C300073200. [DOI] [PubMed] [Google Scholar]
- 11.Johnson KA. Role of induced fit in enzyme specificity: A molecular forward/reverse switch. J Biol Chem. 2008;283(39):26297–26301. doi: 10.1074/jbc.R800034200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lakowicz J. Principles of Fluorescence Spectroscopy. Boston: Springer US; 2006. [Google Scholar]
- 13.Groeneveld M, Slotboom DJ. Na+:aspartate coupling stoichiometry in the glutamate transporter homologue GltPh. Biochemistry. 2010;49(17):3511–3513. doi: 10.1021/bi100430s. [DOI] [PubMed] [Google Scholar]
- 14.Fersht AR. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. New York: Freeman; 1999. [Google Scholar]
- 15.Tsai CJ, Kumar S, Ma B, Nussinov R. Folding funnels, binding funnels, and protein function. Protein Sci. 1999;8(6):1181–1190. doi: 10.1110/ps.8.6.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strickland S, Palmer G, Massey V. Determination of dissociation constants and specific rate constants of enzyme-substrate (or protein-ligand) interactions from rapid reaction kinetic data. J Biol Chem. 1975;250(11):4048–4052. [PubMed] [Google Scholar]
- 17.Vogt AD, Di Cera E. Conformational selection or induced fit? A critical appraisal of the kinetic mechanism. Biochemistry. 2012;51(30):5894–5902. doi: 10.1021/bi3006913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ryan RM, Compton ELR, Mindell JA. Functional characterization of a Na+-dependent aspartate transporter from Pyrococcus horikoshii. J Biol Chem. 2009;284(26):17540–17548. doi: 10.1074/jbc.M109.005926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Georgieva ER, Borbat PP, Ginter C, Freed JH, Boudker O. Conformational ensemble of the sodium-coupled aspartate transporter. Nat Struct Mol Biol. 2013;20(2):215–221. doi: 10.1038/nsmb.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hänelt I, Wunnicke D, Bordignon E, Steinhoff HJ, Slotboom DJ. Conformational heterogeneity of the aspartate transporter GltPh. Nat Struct Mol Biol. 2013;20(2):210–214. doi: 10.1038/nsmb.2471. [DOI] [PubMed] [Google Scholar]
- 21.Compton ELR, Taylor EM, Mindell JA. The 3-4 loop of an archaeal glutamate transporter homolog experiences ligand-induced structural changes and is essential for transport. Proc Natl Acad Sci USA. 2010;107(29):12840–12845. doi: 10.1073/pnas.1003046107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang J, Shrivastava IH, Watts SD, Bahar I, Amara SG. Large collective motions regulate the functional properties of glutamate transporter trimers. Proc Natl Acad Sci USA. 2011;108(37):15141–15146. doi: 10.1073/pnas.1112216108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shrivastava IH, Jiang J, Amara SG, Bahar I. Time-resolved mechanism of extracellular gate opening and substrate binding in a glutamate transporter. J Biol Chem. 2008;283(42):28680–28690. doi: 10.1074/jbc.M800889200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reyes N, Oh S, Boudker O. Binding thermodynamics of a glutamate transporter homolog. Nat Struct Mol Biol. 2013;20(5):634–640. doi: 10.1038/nsmb.2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mim C, Tao Z, Grewer C. Two conformational changes are associated with glutamate translocation by the glutamate transporter EAAC1. Biochemistry. 2007;46(31):9007–9018. doi: 10.1021/bi7005465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wadiche JI, Kavanaugh MP. Macroscopic and microscopic properties of a cloned glutamate transporter/chloride channel. J Neurosci. 1998;18(19):7650–7661. doi: 10.1523/JNEUROSCI.18-19-07650.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watzke N, Bamberg E, Grewer C. Early intermediates in the transport cycle of the neuronal excitatory amino acid carrier EAAC1. J Gen Physiol. 2001;117(6):547–562. doi: 10.1085/jgp.117.6.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mim C, Balani P, Rauen T, Grewer C. The glutamate transporter subtypes EAAT4 and EAATs 1-3 transport glutamate with dramatically different kinetics and voltage dependence but share a common uptake mechanism. J Gen Physiol. 2005;126(6):571–589. doi: 10.1085/jgp.200509365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bergles DE, Tzingounis AV, Jahr CE. Comparison of coupled and uncoupled currents during glutamate uptake by GLT-1 transporters. J Neurosci. 2002;22(23):10153–10162. doi: 10.1523/JNEUROSCI.22-23-10153.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grewer C, Watzke N, Wiessner M, Rauen T. Glutamate translocation of the neuronal glutamate transporter EAAC1 occurs within milliseconds. Proc Natl Acad Sci USA. 2000;97(17):9706–9711. doi: 10.1073/pnas.160170397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Focke PJ, Moenne-Loccoz P, Larsson HP. Opposite movement of the external gate of a glutamate transporter homolog upon binding cotransported sodium compared with substrate. J Neurosci. 2011;31(16):6255–6262. doi: 10.1523/JNEUROSCI.6096-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yernool D, Boudker O, Folta-Stogniew E, Gouaux E. Trimeric subunit stoichiometry of the glutamate transporters from Bacillus caldotenax and Bacillus stearothermophilus. Biochemistry. 2003;42(44):12981–12988. doi: 10.1021/bi030161q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.