

Abstract
Exogenous dopamine inhibits insulin secretion from pancreatic β-cells, but the lack of dopaminergic neurons in pancreatic islets has led to controversy regarding the importance of this effect. Recent data, however, suggest a plausible physiologic role for dopamine in the regulation of insulin secretion. We review the literature underlying our current understanding of dopaminergic signaling that can down-regulate glucose-stimulated insulin secretion from pancreatic islets. In this negative feedback loop, dopamine is synthesized in the β-cells from circulating l-dopa, serves as an autocrine signal that is cosecreted with insulin, and causes a tonic inhibition on glucose-stimulated insulin secretion. On the whole animal scale, l-dopa is produced by cells in the gastrointestinal tract, and its concentration in the blood plasma increases following a mixed meal. By reviewing the outcome of certain types of bariatric surgery that result in rapid amelioration of glucose tolerance, we hypothesize that dopamine serves as an “antiincretin” signal that counterbalances the stimulatory effect of glucagon-like peptide 1.
After the discovery of norepinephrine in 1901 by Takemine (1) and Aldrich (2), dopamine was long considered as only a chemical intermediate toward the synthesis of epinephrine in the adrenal gland, devoid of any other physiologic role. Carlsson et al (3) first showed in 1957 that dopamine had physiologic functions in the brain, and we know today that dopamine is a critical neurotransmitter in the central nervous system (CNS). In this minireview, we present our current view on the role of dopamine as a paracrine/autocrine regulator of insulin secretion from the β-cell in the pancreatic islet. Although the role of dopamine in endocrine cells has been controversial, there is now strong evidence demonstrating both the source and function of dopamine in pancreatic islets. These data lead to a putative physiologic role for dopamine in modulating insulin secretion by the existence of a dopaminergic negative feedback loop acting on the endocrine pancreas.
Insulin secretion from pancreatic β-cells is a fundamental process that is required to maintain glucose homeostasis in a healthy individual. During glucose-stimulated insulin secretion from pancreatic β-cells, glucose is metabolized and the ATP:ADP ratio is increased, inhibiting ATP-sensitive inward rectifying potassium (KATP) channel activity. The β-cell is subsequently depolarized, activating voltage-gated calcium channels and stimulating insulin secretion. In addition to glucose stimulation, however, multiple G-protein coupled receptor (GPCR) ligands also play a large role in the modulation of insulin release (4). Impairments in the mechanisms that control insulin secretion result in complications ranging from glucose intolerance to overt type 2 diabetes (5). GPCRs are common therapeutic targets and constitute about 50% of drugs on the market. Thus, understanding of the mechanisms by which these ligands, in general, and dopamine, in particular, modulate insulin release is of increasing importance.
Nonneuronal Sources of Dopamine
In the CNS, dopamine functions as a neurotransmitter in which neurons in the substantia nigra and the ventral tegmental area produce and store dopamine. These neurons extend fibers to the basal ganglia, nucleus accumbens, and prefrontal cortex where dopamine is released during synaptic transmission. Dopaminergic neurons are also present in the hypothalamus, where they modulate the secretion of the hormone prolactin from the anterior pituitary gland (6). These dopaminergic pathways control crucial functions such as motor coordination, motivation, reward, and working memory (7, 8). Dysfunction of the dopaminergic neurons can cause Parkinson's disease and is thought to be the cause of schizophrenia and attention deficit hyperactivity disorder (8). In addition to these neuronal functions, dopamine has other regulatory functions outside the nervous system where it is synthesized by, and released from, nonneuronal tissues. As a result of this peripheral production, dopamine and its precursor l-dopa are present in the plasma at picomolar and nanomolar concentrations, respectively (9). Peripheral dopamine production does not interfere with the brain dopamine activity because dopamine does not cross the blood-brain barrier, but it exerts a paracrine role in the tissues involved in its synthesis. Extensive literature exists on the role of dopamine in kidney function (10). The intrarenal dopaminergic system regulates Na+ and water reabsorption, thus affecting blood pressure (11). The dopaminergic system in the lungs affects liquid clearance through the alveolar epithelium (12). Among the mesenteric organs, substantial dopamine synthesis has been shown in the exocrine pancreas (13) and in upper gastrointestinal tract where it has been suggested to have a protective role on the intestinal mucosa (14). Fluctuations in plasma levels of dopamine and l-dopa after a meal have been reported (9), and they have been attributed to synthetic activity of dopaminergic cells in the intestine.
The endocrine pancreas itself has dopaminergic properties. The anatomy of this endocrine gland consists of clusters of endocrine cells, the pancreatic islets, which are interspersed in the exocrine pancreas (15). Although the first mention of biogenic amines in pancreatic islets dates back to 1963 by Falck and Hellman (16), publications that followed this first report offered contradicting results and conclusions. In the first decade following Falck and Hellman's brief communication, the scientific consensus was that mouse islets were devoid of biogenic amines. Therefore, most of the studies that followed were performed in golden hamster, guinea pig, and rabbit (17–20). The results of these studies proved that islets from different species show great differences in their responses to experimental treatments (21). Therefore, that knowledge was difficult to extend widely to other species used as biological models (mouse, rat), or to human islet physiology.
Dopamine Receptor Signaling
In 1988, the first cDNA for a dopamine receptor was isolated (22) and, soon after, the other isotypes of the dopamine receptors were identified. Dopamine receptors are members of the rhodopsin family of GPCRs, and they include 5 different receptor subtypes named D1, D2, D3, D4, and D5. These 5 proteins are the products of 5 genes from different chromosomal loci, but they display significant homology in their protein structure and function (23). The D1 and D5 receptor genes do not have introns, and they show 80% homology in their transmembrane domains. They usually display a stimulatory function on adenylate cyclase activity and, for this reason, they are classified as D1-like receptors (23). The D2, D3, and D4 receptors are encoded by genes that do have introns. The D2 and D3 receptors show 75% homology in their transmembrane domain, whereas D2 and D4 exhibit 54% homology. These 3 receptors are classified as D2-like receptors, and they each have a long third intracellular loop that is a common feature of GPCRs that interact with Gi/o proteins. In fact, the D2-like receptors generally show inhibitory function on adenylate cyclase activity (24).
The type of dopamine receptor that is expressed in a tissue determines the tissue-specific effect of dopamine. As we discuss below, the D2-like receptors are found in pancreatic β-cells. D2-like receptors can activate a variety of signaling cascades in each cell type (25). They can decrease protein kinase A activity by negatively regulating cAMP production via activation of the Gα subunit upon dopamine binding (24). Consequently, the decreased kinase activity can affect multiple targets including cAMP response element-binding protein, ionotropic glutamate receptors, and ion channels (26). D2-like receptors can also activate signaling cascades that are mediated by the Gβγ subunit. In medium spiny neurons, for example, they activate phospholipase C and increase intracellular Ca2+ levels by affecting both intracellular stores and L-type calcium channels (27, 28). They have also been shown to regulate N-type calcium channels in striatal interneurons (29). Additionally D2-like receptors can regulate G protein-coupled inwardly rectifying K+ channels (30). Dopamine receptors are expressed in blood vessels (31), in the adrenal gland (32), in the kidneys (33), and in the pancreatic islets (34–36).
Dopaminergic Regulation of Insulin Secretion
The idea that dopamine could affect insulin secretion has been considered repeatedly for more than 40 years, but neither the origin of dopamine nor the mechanism of action was clearly identified until recently. In 1967, it was reported that an injection of l-dopa and dopamine produced a hyperglycemic response in mice, but the contribution to this effect of epinephrine and norepinephrine release from adrenergic nerve fibers could not be excluded (17). Dopamine was first detected in freshly isolated mouse islet homogenates in 1977 by Hansen and Hedeskov (37), but again they could not exclude that dopamine was coming from adrenergic nerve fibers fragment in the islets. In fact, they simultaneously detected dopamine, epinephrine, norepinephrine, and serotonin in the homogenate. Later, the work of Mahony and Feldman (38) and Zern et al (39) expanded the knowledge on monoamine uptake and action in the islets of the golden hamster, but they emphasized that islets from different species show great differences in their responses to experimental treatments (21). At the same time the work by Ericson et al. (40) showed that dopamine could be detected in mouse β-cell secretory granules following the injection of radiolabeled l-dopa. They reported a partial inhibition of glucose-stimulated insulin secretion following the injection, but in this and further studies, they suggested that dopamine synthesis could have taken place in other tissues (41). In follow-up studies, they concluded that the l-dopa-induced inhibition of glucose-stimulated insulin secretion was independent from dopamine accumulation but rather related to a direct effect of l-dopa (42).
A significant amount of work has shown that the components necessary for dopamine synthesis and secretion are all present in the β-cell. For instance, aromatic L-amino acid decarboxylase and monoamine oxidases activities have been characterized in mouse islets homogenates (42–44). The vesicular monoamine transporter type 2 (VMAT2) is a key element in β cell dopamine metabolism because it sequesters dopamine within vesicles away from catabolic enzymes such as monoamine oxidases. The expression of VMAT2 in human β-cells (45) seems to be broadly accepted, but there is some disagreement as to whether rodent β-cells express VMAT2. Some investigators find no evidence that VMAT2 is expressed in β-cells of the rodent pancreas (46) using techniques such as immunohistochemistry or RT-PCR. Other investigators have found ample evidence for expression of VMAT2 in β-cells of the endocrine pancreas. The experimental evidence falls into 3 broad categories. Most evidence is based on the binding (or inhibition of binding) of the specific VMAT2 inhibitor dihydrotetrabenazine (47) including positron emission tomography studies of the pancreas (48). It has been shown that dihydrotetrabenazine can enhance insulin secretion in vitro in cultures of purified rodent islets (49). Likewise, using purified rodent islets and the techniques of western blot or PCR, VMAT2 expression has been demonstrated in rodent islet cells (49). Because VMAT2 is believed to be responsible for the loading of β-cells, vesicles with monoamines, another proof of VMAT2 expression in rodent cells, takes form in the demonstration of serotonin and dopamine release by rodent β-cells. There is ample evidence of rodent β-cell secretion of dopamine as covered in this review: the recent report by Ustione and Piston (35) and previous publications documenting rodent β-cell vesicular storage and elaboration of serotonin (50–52).
In 2005, Rubí et al (36) showed that exogenous dopamine can inhibit glucose-stimulated insulin secretion from isolated islets. They also showed the expression of D2-like receptors in the islets, providing a candidate mediator of the observed inhibition, although the immunohistochemistry results were interpreted to show only an intracellular localization of the receptor (36). A role for D2-like receptors in regulating insulin secretion was suggested by 2 other studies; in one, D2 receptors were knocked out in the mammalian cell line INS-1 832/13 by using small interfering RNA, resulting in an increased insulin secretion (53). In the other, the authors used a global D2 knockout mouse and came to the opposite conclusion: that disruption of the D2 receptor impairs insulin secretion and causes glucose intolerance (54). Despite the presence of dopaminergic machinery in the β-cells, until recently, it was not known where dopamine could originate to stimulate islets in a living mouse. Whereas there is a high degree of innervation in the islets, there are no reports of dopaminergic neurons innervating them (55). Although there are peripheral sources of dopamine in the body (9, 13, 14, 56), circulating dopamine levels in the plasma are too low to activate its receptors (9, 57, 58) in the absence of mechanisms that store and concentrate dopamine.
The subject was further investigated independently by both our laboratories, resulting in publications that came to a similar conclusion: a dopaminergic negative feedback loop regulates insulin secretion from human and murine pancreatic islets (34, 35) (Figure 1). In both studies, we showed that dopamine is stored in the β-cells and that it is cosecreted with insulin, by using crono-amperometry and ELISA to measure dopamine. This endogenous dopamine acts in an autocrine/paracrine manner on the insulin-secreting β-cells that express D2-like receptors. In the mouse study, both D2 and D3 expression was confirmed by SDS-PAGE and Western blot, whereas the study on human islets tested the expression of the D2 receptor (34). Interestingly, selective pharmacologic inhibition of each receptor showed that D3 is the only mediator of dopaminergic inhibition of insulin secretion in the mouse, and D3 inhibition resulted in increased insulin secretion (35). In human islets, the D2 was the only receptor tested, but because the antagonist used has affinity for both D2 and D3 receptors, the specific roles of the D2 and D3 receptors cannot be disentangled. For both species, expression of the dopamine transporter was confirmed, so that dopamine transporter can uptake dopamine from the bloodstream and function to terminate the dopaminergic signaling by clearance of the secreted dopamine.
Figure 1.

Dopaminergic Negative Feedback Regulating Insulin Secretion Schematic diagram of a β-cell expressing the molecular machinery for dopamine synthesis, storage, and secretion. The dopamine receptor D3 mediates the inhibition of glucose- stimulated insulin secretion. AADC, l-amino acid decarboxylase; DA, dopamine; GSIS, glucose-stimulated insulin secretion; INS, insulin; MAO, monoamine oxidase; LAAT, l-type amino acide transporter; DRD3, dopamine receptor D3; dopac, 3,4-dihydroxyphenylacetic acid.
At this time, the molecular mechanism(s) responsible for the inhibition of insulin secretion in mouse islets remain unknown. There is no observed effect of D3 receptor activation on the redox state of the β-cells, but there is a significant reduction of the frequency of the intracellular [Ca2+] oscillations (35). As was shown by Rubí et al. (36), D2-like receptor activation does not affect cAMP levels. Thus, we proposed that D3 receptors signal mainly via the Gβγ subunit to affect the L-type calcium channels directly (28), or indirectly by activating the potassium channels (30). Given the high degree of overlap in dopaminergic inhibition of insulin secretion between the mouse and human islet data, it is reasonable to speculate that a similar mechanism is responsible for this effect in the human β-cells as well.
Does Dopamine Oppose Incretin Signals?
Might this dopaminergic circuit offer a mechanism to explain the rapid resolution of type 2 diabetes hyperglycemia observed after certain types of gastrointestinal bypass surgery? The foregut (including the stomach) is the major source of circulating dopamine (14). Tyrosine hydroxylase, the rate-limiting dopamine biosynthetic enzyme, is expressed in parietal cells, Lieberkühn crypts, ileal epithelial cells, and throughout the lamina propria of the small intestines (59). Further, VMAT1, which is responsible for transport of serotonin or dopamine into storage vesicles, is expressed by enterochromaffin cells and the parietal cells of the oxyntic stomach (13, 14, 59). Following ingestion of a standard mixed meal, healthy volunteers show increased plasma levels of l-dopa and dopamine (60, 61). The postprandial rise of plasma GLP-1 (glucagon-like peptide 1) appears to precede the production and release of dopamine and l-dopa from intestinal stores. Postprandial arterial dopamine and l-dopa excursions (up to 15 nmol/L) represent significant amounts with regard to regulating β-cell insulin secretion, given the ability of β-cells to take up dopamine (via dopamine transporter [DAT]) and l-dopa (via l-type amino acid transporter system) (34, 35) and synthesize and concentrate dopamine for vesicular release. β-Cells are in intimate contact with other β-cells in the human islet (62). This is perhaps analogous to synapses in the CNS. In the CNS, vesicular dopamine release and response to dopamine by D2 receptors occur at high concentrations and in limited volumes, as governed by the presence of DAT and the amount and concentration of vesicular release. The local concentration of dopamine here has been estimated to be about 100 to 0.5 μM within a 5-μm radius from the release site (63). The kinetics of release of GLP-1 and dopamine suggests the existence of a second (gut-based) layer of regulation of glucose homeostasis.
The idea of a second layer of regulation of glucose homeostasis originates in the foregut and hindgut hypotheses (as reviewed by Rubino et al [Ref. 64]) posited to explain the effects of bariatric surgery on type 2 diabetes. According to the hindgut hypothesis, the rapid delivery of nutrients to the distal intestine results in the secretion of “incretins” which enhance insulin release and/or action, resulting in a subsequent decrease in blood glucose levels. An alternate, but not mutually exclusive, foregut hypothesis was also proposed; here bypass of the upper small intestine resulted in the removal or diminution of a hyperglycemia-promoting signal (ie, an “antiincretin”). The major factor responsible for this incretin effect is now recognized as GLP-1 derived from the L-cells (found in the ileum and colon) (65). As described above, dopamine and/or l-dopa fulfill many of the requirements of such an antiincretin posited by the foregut hypothesis (66, 67). Overall, it is plausible that bypass and/or partial resection of organs (eg, in Roux-en-Y gastric bypass [RYGB] duodenal jeunal bypass, sleeve gastrectomy), known to store and produce significant amounts of an insulin secretion inhibitor (ie, dopamine) from nutritional sources would move the organism to a more glucose-tolerant state. In this fashion, surgical manipulation of total body dopamine content, along with stimulation of GLP-1 secretion, might be responsible for the rapid improvement in glucose tolerance and enhanced insulin secretion observed before weight loss following RYGB surgery.
Meta-analyses have reported improved glycemic control and remission of type 2 diabetes in 83.8% of patients following gastric bypass vs 47.8% following gastric banding (a procedure that only restricts the stomach to produce weight loss) (68–70). It is well accepted that the remission of type 2 diabetes seen following gastric banding is related to weight loss (70, 71). However, in procedures such as RYGB, there are more immediate improvements in glycemia and insulin resistance, seen as early as the first week after surgery (72–74). If these surgeries do indeed decrease circulating levels of l-dopa and dopamine, then β-cells would lack the substrate to synthesize and accumulate dopamine and, consequently, their glucose-stimulated insulin secretion would increase. Moreover, it is relevant to note that l-dopa regulates glycogen concentration, glycogen synthase (GS) activity, and insulin-stimulated glucose transport in rat skeletal muscle (75). Furthermore, skeletal muscle, a major insulin-responsive tissue, responds to dopamine by elevating endogenous cAMP (76). In these cells, cAMP has been implicated as a negative regulator of insulin action (77, 78). The rate-limiting enzyme for the storage of intracellular glucose as glycogen in skeletal muscle is GS. GS is activated in the presence of insulin and inhibited by agents that increase intracellular cAMP concentrations (79). The insulin resistance and hyperglycemia observed in type 2 diabetes have been associated with impaired activation of GS (80–82) Under conditions in which GS activity is dampened, nonoxidative glucose disposal (ie, glycogen synthesis) and glucose accumulation in skeletal muscle is also down-regulated, and it has been suggested that impaired glycogen metabolism is an important defect in the pathogenesis of type 2 diabetes (83).
More recently, intramyocellular accumulation of triglycerides and other lipid metabolites and decreased glucose transport have been linked to poor glucose disposal rates seen in type 2 diabetes (84). Nevertheless, some tonic negative regulation of GS activity and glycogen accumulation in muscle cells by dopamine (via a β-adrenergic pathway) might be expected to rapidly reduce some level of insulin resistance following surgical removal of organs contributing to circulating dopamine levels. Likewise, surgical reduction of circulating dopamine levels, particularly the postprandial elevations in dopamine observed after a mixed-meal tolerance test, might also result in rapid gains of β-cell function (85, 86). This is consistent with the model we have put forward, because these gains are observed after gastric bypass surgery such as RYGB, but not seen in other gastrointestinal surgeries that do not manipulate the dopamine-producing organs (ie, gastric banding alone) (87, 88).
If dopamine acts as an antiincretin, regulating β-cell insulin secretion by opposing the insulin secretion-enhancing effects of GLP-1, then D2-like receptor activation by dopamine might result in specific β-cell signaling events that oppose those initiated by GLP-1 receptor activation. AKT/PKB is a serine-threonine kinase known to be activated in β-cell lines by GLP-1 through phosphorylation at serine 473 (89). In rodent neuronal cell line models, AKT/PKB is also serine phosphorylated following agonism at the D2 receptor (90, 91) Using the rodent β-cell line INS-1E (92), we examined the activation of AKT/PKB by glucose and GLP-1 with or without dopamine (Figure 2). We found that the enhanced activation of AKT/PKB mediated by GLP-1 was partially reversed by the addition of exogenous dopamine. The multiple possible sites for cross talk between the D2-like receptor signaling and the GLP-1 receptor signaling are shown in Table 1.
Figure 2.
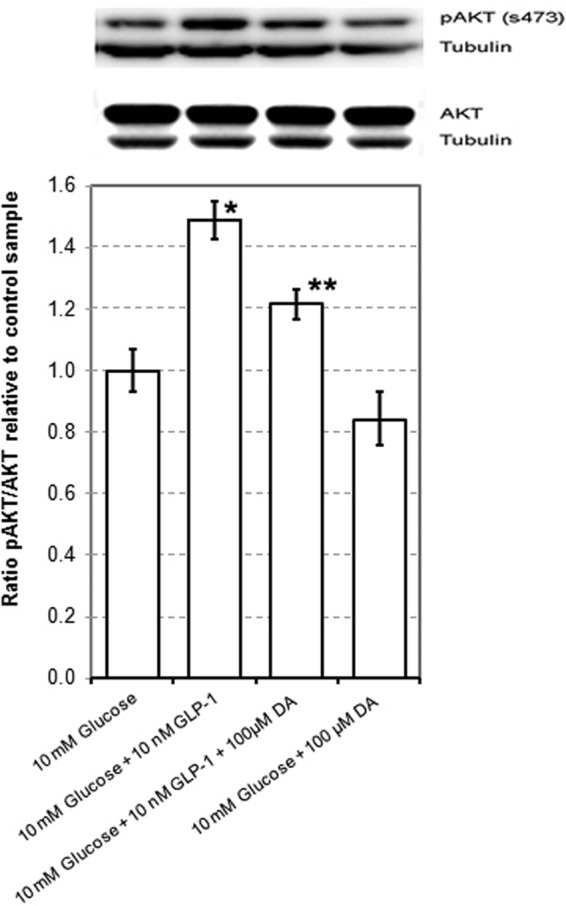
Dopamine Dampens GLP-1-Enhanced Glucose-Stimulated AKT Phosphorylation. INS-1E cells were grown to 80% confluence in 100-cm2 flasks, washed in PBS, and then rested for 1 hour in glucose-free RPMI supplemented with 2 mM glucose and 0.5% BSA. Cells were then incubated for 30 minutes in glucose-free RPMI containing 10 mM glucose, 10 nM GLP-1 and/or 100 μM dopamine. Protein lysates were prepared from cell monolayers, and equal amounts of protein were separated by reducing SDS-PAGE and analyzed by Western blot using anti-AKT and pAKT antibodies from CST. As a loading control, the amount of β-tubulin was also measured. The amount of immunoreactive protein on each blot was quantitated by the horseradish peroxidase-enhanced chemiluminescence reaction using a Flurochem M imaging station and associated imaging software. Western blot photograph from a representative experiment in a series of 4. The data from quantitation of immunodetected proteins are mean ± SE values from the same series of 4 experiments. The single asterisk denotes a statistically significant difference (P < .05) from the mean of 10 mM glucose control, and the double asterisk denotes a statistically significant difference from the 10 mM glucose plus GLP-1 group as determined by Student's t test. DA, dopamine.
Table 1.
Selected References to Studies Supporting the Integration of D2-Like Receptor Signaling with GLP-1 Receptor Signaling.
| Possible Phosphor-proteome Sites of Integration of D2-Like Receptor Signaling into β-Cell Signaling | References to β-Cell/GLP-1 Receptor Signaling | References to D2-Like Receptor Signaling |
|---|---|---|
| AKT/PKB /p-AKT Ser 473, Thr 308 | (89, 105) | (90, 91, 106) |
| GSK-3/p-GSK-3 (Ser 9, Ser 21, Thr-390) | (107, 108) | (91, 109–111) |
| 14–3–3 / p-14–3–3 | (112) | (112) |
| p70 S6 Kinase / p-p70 S6K | (113) | (106) |
| PKCζ/p-PKC (Thr410) | (114, 115) | (116, 117) |
| ERK/p-ERK 1/2 (Thr202/Tyr204) | (118) | (119) |
| CaMKII/p-CaMKII (Tyr231 or 286) | (120, 121) | (120–122) |
| DARPP-32/p-DARPP-32 (Thr 34 75) | (123, 124) | (106, 125) |
| CREB/ p-CREB (Ser133) | (126) | (119) |
| CDK5/ p-CDK5 | (124) | (125) |
| Rap1/p-Rap1 | (127) | (128) |
| Munc18/ p-Munc18 | (129) | (130) |
AKT/PKB, protein kinase B; CaMK, calmodulin-dependent kinase; CDK, cyclin-dependent kinase; CREB, cAMP response element binding protein; DARPP, dopamine- and cAMP-regulated phospho-protein; GSK, glycogen synthase kinase; p-ERK, phospho-ERK; pGSK, phospho-GSK; p-PKC, phospho-protein kinase C.
The Islet Dopaminergic System and Its Links to Neurologic Diseases
Our current view shows that a dopaminergic negative feedback acting on insulin secretion is active in β-cells. Importantly, blocking this dopaminergic feedback increases insulin secretion. Therefore the D3 receptor, or one of the steps downstream of its activation, is a potential target for new drugs to treat type 2 diabetes. At the same time, the existence of a dopaminergic inhibition of glucose-stimulated insulin secretion allows speculation regarding the high prevalence of abnormal glucose tolerance in 50%–80% of Parkinson patients (93). Prospective studies have suggested that diabetes is not a preceding risk factor for Parkinson's disease, yet the 2 diseases show a significant positive association, possibly explained by a common underlying biologic mechanism (94, 95). It can be reasoned that dopaminergic regulation could be one such common mechanism. Because β-cells share the dopaminergic system with the dopaminergic neurons of the substantia nigra, it is possible that the still-unknown cause of dopaminergic neuron loss underlying Parkinson's disease could be related to the specific loss of β-cell function that results in type 2 diabetes. Another interesting consideration involves the therapy for Parkinson's disease, which consists of administration of l-dopa and benserazide. Whereas l-dopa inhibits glucose-stimulated insulin secretion, benserazide blocks the effect of l-dopa by preventing its conversion into dopamine. The combined action of these drugs increases dopamine concentration in dopaminergic neurons and prevent peripheral conversion of l-dopa and its consequent side effects (96). However, some patients still experience hyperglycemia and hyperinsulinemia as side effects of the treatment (96, 97). From in vitro experiments, benserazide did not completely prevent the conversion of l-dopa into dopamine in isolated islets when the 2 drugs were used in combination, and dopamine accumulation was only reduced by 60% in comparison with the effect of l-dopa alone (35). Therefore, it is possible that this Parkinson's disease treatment regimen could partially inhibit glucose-stimulated insulin secretion, and thus, put a chronic stress on islet function that exacerbates an underlying associative cause of type-2 diabetes and Parkinson's disease (93, 94, 96). Although we suggest that dopaminergic regulation of insulin secretion is a possible link between Parkinson's disease and type 2 diabetes, other mechanisms can also be responsible for the association between these 2 diseases; these include the following: the loss of neuroprotection by insulin in patients with impaired insulin secretion, the existence of common genetic susceptibility factors that would affect neuronal and β-cell survival/function concomitantly, and the presence of systemic low-level inflammation (98).
The expression of the DAT in the pancreatic islet provides another possible link relating potential environmental causes of type 2 diabetes and Parkinson's disease. It is possible that the brain and endocrine pancreas are responding to the same exogenous insult and that both tissues suffer the same type of damage (ie, loss of dopaminergic cells). If substance(s) that enter neurons via DAT is a cause of Parkinson's disease, then the same substance(s) could have equally deleterious effects on islet cells (99, 100).
Finally, the presence of the D3 dopamine receptor makes the β-cell a potential undesired target of antipsychotic drugs. This could be particularly relevant considering the number of studies showing associations between metabolic syndrome and atypical-antipsychotic therapy (101–103). The results on this subject are difficult to interpret, because these atypical antipsychotic drugs act on multiple receptors. Still, the action on D2-like dopamine receptors remains a single unifying property of these drugs (104). Thus, it is not unreasonable to expect that some of their metabolic side effects are independent from their action on the CNS, but instead are related to direct action on insulin secretion. A better knowledge of their mechanism of action on the D2-like receptors, and consequently on glucose-stimulated insulin secretion from the islet, would be helpful in designing antipsychotic drugs with fewer metabolic side effects, or improved therapeutic regimens that minimize these side effects with the current available drugs.
Acknowledgments
This work was supported by National Institutes of Health Grant DK085064 (to A.U. and D.W.P.) and a grant from The Helmsley Charitable Trust (to P.E.H.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- CNS
- central nervous system
- DAT
- dopamine transporter
- GLP-1
- glucagon-like peptide 1
- GPCR
- G protein-coupled receptor
- GS
- glycogen synthase
- RYGB
- Roux-en-Y gastric bypass
- VMAT2
- vesicular monoamine transporter type 2.
References
- 1. Takamine J. Adrenalin the active principle of the supra-renal glands and its mode of preparation. Amer J Pharm. 1901;73:523–531 [Google Scholar]
- 2. Aldrich T. A preliminary report on the active principle of the suprarenal gland. Am J Physiol. 1901;5:457–461 [Google Scholar]
- 3. Carlsson A, Lindqvist M, Magnusson T. 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature. 1957;180:1200. [DOI] [PubMed] [Google Scholar]
- 4. Winzell MS, Ahrén B. G-protein-coupled receptors and islet function-implications for treatment of type 2 diabetes. Pharmacol Ther. 2007;116:437–448 [DOI] [PubMed] [Google Scholar]
- 5. Weyer C, Bogardus C, Mott DM, Pratley RE. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest. 1999;104:787–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Björklund A, Dunnett SB. Dopamine neuron systems in the brain: an update. Trends Neurosci. 2007;30:194–202 [DOI] [PubMed] [Google Scholar]
- 7. Shultz MD, Reveles JU, Khanna SN, Carpenter EE. Reactive nature of dopamine as a surface functionalization agent in iron oxide nanoparticles. J Am Chem Soc. 2007;129:2482–2487 [DOI] [PubMed] [Google Scholar]
- 8. Iversen SD, Iversen LL. Dopamine: 50 years in perspective. Trends Neurosci. 2007;30:188–193 [DOI] [PubMed] [Google Scholar]
- 9. Goldstein DS, Eisenhofer G, Kopin IJ. Sources and significance of plasma levels of catechols and their metabolites in humans. J Pharmacol Exp Ther. 2003;305:800–811 [DOI] [PubMed] [Google Scholar]
- 10. Harris RC, Zhang MZ. Dopamine, the kidney, and hypertension. Curr Hypertens Rep. 2012;14:138–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang X, Luo Y, Escano CS, et al. Upregulation of renal sodium transporters in D5 dopamine receptor-deficient mice. Hypertension. 2010;55:1431–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Adir Y, Azzam ZS, Lecuona E, et al. Augmentation of endogenous dopamine production increases lung liquid clearance. Am J Respir Crit Care Med. 2004;169:757–763 [DOI] [PubMed] [Google Scholar]
- 13. Mezey E, Eisenhofer G, Harta G, et al. A novel nonneuronal catecholaminergic system: exocrine pancreas synthesizes and releases dopamine. Proc Natl Acad Sci USA. 1996;93:10377–10382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eisenhofer G, Aneman A, Friberg P, et al. Substantial production of dopamine in the human gastrointestinal tract. J Clin Endocrinol Metab. 1997;82:3864–3871 [DOI] [PubMed] [Google Scholar]
- 15. Unger RH, Dobbs RE, Orci L. Insulin, glucagon, and somatostatin secretion in the regulation of metabolism. Annu Rev Physiol. 1978;40:307–343 [DOI] [PubMed] [Google Scholar]
- 16. Falck B, Hellman B. Evidence for the presence of biogenic amines in pancreatic islets. Cell Mol Life Sci. 1963;19:139–140 [Google Scholar]
- 17. Hákanson R, Lundquist I, Rerup C. On the hyperglycaemic effect of DOPA and dopamine. Eur J Pharmacol. 1967;1:114–119 [DOI] [PubMed] [Google Scholar]
- 18. Cegrell L. The occurrence of biogenic monoamines in the mammalian endocrine pancreas. Acta Physiol Scand Suppl. 1968;314:1–60 [PubMed] [Google Scholar]
- 19. Feldman JM, Lebovitz HE. The nature of the interaction of amines with the pancreatic β cell to influence insulin secretion. J Pharmacol Exp Ther. 1971;179:56–65 [PubMed] [Google Scholar]
- 20. Wilson JP, Downs RW, Feldman JM, Lebovitz HE. β-Cell monoamines: further evidence for their role in modulating insulin secretion. Am J Physiol. 1974;227:305–312 [DOI] [PubMed] [Google Scholar]
- 21. Feldman JM. Species variation in the islets of Langerhans. Diabetologia. 1979;16:1–4 [DOI] [PubMed] [Google Scholar]
- 22. Bunzow JR, Van Tol HH, Grandy DK, et al. Cloning and expression of a rat D2 dopamine receptor cDNA. Nature. 1988;336:783–787 [DOI] [PubMed] [Google Scholar]
- 23. Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. 1998;78:189–225 [DOI] [PubMed] [Google Scholar]
- 24. Enjalbert A, Bockaert J. Pharmacological characterization of the D2 dopamine receptor negatively coupled with adenylate cyclase in rat anterior pituitary. Mol Pharmacol. 1983;23:576–584 [PubMed] [Google Scholar]
- 25. Tritsch NX, Sabatini BL. Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron. 2012;76:33–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Greengard P. The neurobiology of slow synaptic transmission. Science. 2001;294:1024–1030 [DOI] [PubMed] [Google Scholar]
- 27. Hernandez-Lopez S, Tkatch T, Perez-Garci E, et al. D2 dopamine receptors in striatal medium spiny neurons reduce L-type Ca2+ currents and excitability via a novel PLC[beta]1-IP3-calcineurin-signaling cascade. J Neurosci. 2000;20:8987–8995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ivanina T, Blumenstein Y, Shistik E, Barzilai R, Dascal N. Modulation of L-type Ca2+ channels by gβ γ and calmodulin via interactions with N and C termini of α 1C. J Biol Chem. 2000;275:39846–39854 [DOI] [PubMed] [Google Scholar]
- 29. Yan Z, Song WJ, Surmeier J. D2 dopamine receptors reduce N-type Ca2+ currents in rat neostriatal cholinergic interneurons through a membrane-delimited, protein-kinase-C-insensitive pathway. J Neurophysiol. 1997;77:1003–1015 [DOI] [PubMed] [Google Scholar]
- 30. Kuzhikandathil EV, Yu W, Oxford GS. Human dopamine D3 and D2L receptors couple to inward rectifier potassium channels in mammalian cell lines. Mol Cell Neurosci. 1998;12:390–402 [DOI] [PubMed] [Google Scholar]
- 31. Missale C, Castelletti L, Memo M, Carruba MO, Spano PF. Identification and characterization of postsynaptic D1- and D2-dopamine receptors in the cardiovascular system. J Cardiovasc Pharmacol. 1988;11:643–650 [DOI] [PubMed] [Google Scholar]
- 32. Pupilli C, Lanzillotti R, Fiorelli G, et al. Dopamine D2 receptor gene expression and binding sites in adrenal medulla and pheochromocytoma. J Clin Endocrinol Metab. 1994;79:56–61 [DOI] [PubMed] [Google Scholar]
- 33. O'Connell DP, Botkin SJ, Ramos SI, Sibley DR, Ariano MA, Felder RA, Carey RM. Localization of dopamine D1A receptor protein in rat kidneys. Am J Physiol. 1995;268:F1185–F1197 [DOI] [PubMed] [Google Scholar]
- 34. Simpson N, Maffei A, Freeby M, et al. Dopamine-mediated autocrine inhibitory circuit regulating human insulin secretion in vitro. Mol Endocrinol. 2012;26:1757–1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ustione A, Piston DW. Dopamine synthesis and D3 receptor activation in pancreatic β-cells regulates insulin secretion and intracellular [Ca(2+)] oscillations. Mol Endocrinol. 2012;26:1928–1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rubí B, Ljubicic S, Pournourmohammadi S, et al. Dopamine D2-like receptors are expressed in pancreatic β cells and mediate inhibition of insulin secretion. J Biol Chem. 2005;280:36824–36832 [DOI] [PubMed] [Google Scholar]
- 37. Hansen SE, Hedeskov CJ. Simultaneous determination of the content of serotonin, dopamine, noradrenaline and adrenaline in pancreatic islets isolated from fed and starved mice. Acta Endocrinol (Copenh). 1977;86:820–832 [DOI] [PubMed] [Google Scholar]
- 38. Mahony C, Feldman JM. Species variation in pancreatic islet monoamine uptake and action. Diabetes. 1977;26:257–261 [DOI] [PubMed] [Google Scholar]
- 39. Zern RT, Bird JL, Feldman JM. Effect of increased pancreatic islet norepinephrine, dopamine and serotonin concentration on insulin secretion in the golden hamster. Diabetologia. 1980;18:341–346 [DOI] [PubMed] [Google Scholar]
- 40. Ericson LE, Håkanson R, Lundquist I. Accumulation of dopamine in mouse pancreatic B-cells following injection of L-DOPA. Localization to secretory granules and inhibition of insulin secretion. Diabetologia. 1977;13:117–124 [DOI] [PubMed] [Google Scholar]
- 41. Lundquist I, Ahrén B, Hansson C, Håkanson R. Monoamines in pancreatic islets of guinea pig, hamster, rat, and mouse determined by high performance liquid chromatography. Pancreas. 1989;4:662–667 [DOI] [PubMed] [Google Scholar]
- 42. Lundquist I, Panagiotidis G, Stenström A. Effect of L-DOPA Administration on Islet Monoamine Oxidase Activity and Glucose-Induced Insulin Release in the Mouse. Pancreas. 1991;6:522–527 [DOI] [PubMed] [Google Scholar]
- 43. Lindström P. Aromatic-L-amino-acid decarboxylase activity in mouse pancreatic islets. Biochim Biophys Acta. 1986;884:276–281 [DOI] [PubMed] [Google Scholar]
- 44. Teitelman G, Joh TH, Reis DJ. Transformation of catecholaminergic precursors into glucagon (A) cells in mouse embryonic pancreas. Proc Natl Acad Sci USA. 1981;78:5225–5229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Saisho Y, Harris PE, Butler AE, et al. Relationship between pancreatic vesicular monoamine transporter 2 (VMAT2) and insulin expression in human pancreas. J Mol Histol. 2008;39:543–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schafer MK, Hartwig NR, Kalmbach N, et al. Species-specific vesicular monoamine transporter 2 (VMAT2) expression in mammalian pancreatic β cells: implications for optimising radioligand-based human beta cell mass (BCM) imaging in animal models. Diabetologia. 2013;56:1047–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Scherman D, Jaudon P, Henry JP. Characterization of the monoamine carrier of chromaffin granule membrane by binding of [2–3H]dihydrotetrabenazine. Proc Natl Acad Sci USA. 1983;80:584–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Singhal T, Ding YS, Weinzimmer D, et al. Pancreatic beta cell mass PET imaging and quantification with [11C]DTBZ and [18F]FP-(+)-DTBZ in rodent models of diabetes. Mol Imaging Biol. 2011;13:973–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Raffo A, Hancock K, Polito T, et al. Role of vesicular monoamine transporter type 2 in rodent insulin secretion and glucose metabolism revealed by its specific antagonist tetrabenazine. J Endocrinol. 2008;198:41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rosario LM, Barbosa RM, Antunes CM, et al. Regulation by glucose of oscillatory electrical activity and 5-HT/insulin release from single mouse pancreatic islets in absence of functional K(ATP) channels. Endocr J. 2008;55:639–650 [DOI] [PubMed] [Google Scholar]
- 51. Deeney JT, Bränström R, Corkey BE, Larsson O, Berggren PO. 3H-serotonin as a marker of oscillatory insulin secretion in clonal β-cells (INS-1). FEBS Lett. 2007;581:4080–4084 [DOI] [PubMed] [Google Scholar]
- 52. Braun M, Wendt A, Karanauskaite J, et al. Corelease and differential exit via the fusion pore of GABA, serotonin, and ATP from LDCV in rat pancreatic beta cells. J Gen Physiol. 2007;129:221–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wu W, Shang J, Feng Y, et al. Identification of glucose-dependant insulin secretion targets in pancreatic β cells by combining defined-mechanism compound library screening and siRNA gene silencing. J Biomol Screen. 2008;13:128–134 [DOI] [PubMed] [Google Scholar]
- 54. García-Tornadú I, Ornstein AM, Chamson-Reig A, et al. Disruption of the dopamine d2 receptor impairs insulin secretion and causes glucose intolerance. Endocrinology. 2010;151:1441–1450 [DOI] [PubMed] [Google Scholar]
- 55. Miller RE. Pancreatic neuroendocrinology: peripheral neural mechanisms in the regulation of the Islets of Langerhans. Endocr Rev. 1981;2:471–494 [DOI] [PubMed] [Google Scholar]
- 56. Eisenhofer G, Kopin IJ, Goldstein DS. Catecholamine metabolism: A contemporary view with implications for physiology and medicine. Pharmacol Rev. 2004;56:331–349 [DOI] [PubMed] [Google Scholar]
- 57. Eldrup E. Significance and origin of DOPA, DOPAC, and dopamine-sulphate in plasma, tissues and cerebrospinal fluid. Dan Med Bull. 2004;51:34–62 [PubMed] [Google Scholar]
- 58. Keller NR, Diedrich A, Appalsamy M, et al. Norepinephrine transporter-deficient mice exhibit excessive tachycardia and elevated blood pressure with wakefulness and activity. Circulation. 2004;110:1191–1196 [DOI] [PubMed] [Google Scholar]
- 59. Kozicz T, Arimura A. Distribution of urocortin in the rat's gastrointestinal tract and its colocalization with tyrosine hydroxylase. Peptides. 2002;23:515–521 [DOI] [PubMed] [Google Scholar]
- 60. Goldstein DS, Swoboda KJ, Miles JM, et al. Sources and physiological significance of plasma dopamine sulfate. J Clin Endocrinol Metab. 1999;84:2523–2531 [DOI] [PubMed] [Google Scholar]
- 61. Blum I, Vered Y, Graff E, et al. The influence of meal composition on plasma serotonin and norepinephrine concentrations. Metabolism. 1992;41:137–140 [DOI] [PubMed] [Google Scholar]
- 62. Cabrera O, Berman DM, Kenyon NS, Ricordi C, Berggren PO, Caicedo A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc Natl Acad Sci USA. 2006;103:2334–2339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cragg SJ, Rice ME. DAncing past the DAT at a DA synapse. Trends Neurosci. 2004;27:270–277 [DOI] [PubMed] [Google Scholar]
- 64. Rubino F, R'bibo SL, del Genio F, Mazumdar M, McGraw TE. Metabolic surgery: the role of the gastrointestinal tract in diabetes mellitus. Nat Rev Endocrinol. 2010;6:102–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tolhurst G, Reimann F, Gribble FM. Nutritional regulation of glucagon-like peptide-1 secretion. J Physiol. 2009;587:27–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pories WJ, Albrecht RJ. Etiology of type II diabetes mellitus: role of the foregut. World J Surg. 2001;25:527–531 [DOI] [PubMed] [Google Scholar]
- 67. Cummings DE. Endocrine mechanisms mediating remission of diabetes after gastric bypass surgery. Int J Obes (Lond). 2009;33(Suppl 1):S33–S40 [DOI] [PubMed] [Google Scholar]
- 68. Buchwald H, Avidor Y, Braunwald E, et al. Bariatric surgery: a systematic review and meta-analysis. JAMA. 2004;292:1724–1737 [DOI] [PubMed] [Google Scholar]
- 69. O'Brien PE, Dixon JB, Laurie C, Anderson M. A prospective randomized trial of placement of the laparoscopic adjustable gastric band: comparison of the perigastric and pars flaccida pathways. Obes Surg. 2005;15:820–826 [DOI] [PubMed] [Google Scholar]
- 70. Dixon JB, O'Brien PE, Playfair J, et al. Adjustable gastric banding and conventional therapy for type 2 diabetes: a randomized controlled trial. JAMA. 2008;299:316–323 [DOI] [PubMed] [Google Scholar]
- 71. Ponce J, Haynes B, Paynter S, et al. Effect of Lap-Band-induced weight loss on type 2 diabetes mellitus and hypertension. Obes Surg. 2004;14:1335–1342 [DOI] [PubMed] [Google Scholar]
- 72. Pournaras DJ, Osborne A, Hawkins SC, et al. Remission of type 2 diabetes after gastric bypass and banding: mechanisms and 2 year outcomes. Ann Surg. 2010;252:966–971 [DOI] [PubMed] [Google Scholar]
- 73. Pories WJ, Swanson MS, MacDonald KG, et al. Who would have thought it? An operation proves to be the most effective therapy for adult-onset diabetes mellitus. Ann Surg. 1995;222:339–350; discussion 350–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Schauer PR, Burguera B, Ikramuddin S, et al. Effect of laparoscopic Roux-en Y gastric bypass on type 2 diabetes mellitus. Ann Surg. 2003;238:467–484; discussion 484–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Smith JL, Ju JS, Saha BM, Racette BA, Fisher JS. Levodopa with carbidopa diminishes glycogen concentration, glycogen synthase activity, and insulin-stimulated glucose transport in rat skeletal muscle. J Appl Physiol. 2004;97:2339–2346 [DOI] [PubMed] [Google Scholar]
- 76. Schubert D, Tarikas H, LaCorbiere M. Neurotransmitter regulation of adenosine 3′,5′-monophosphate in clonal nerve, glia, and muscle cell lines. Science. 1976;192:471–473 [DOI] [PubMed] [Google Scholar]
- 77. Hunt DG, Ding Z, Ivy JL. Clenbuterol prevents epinephrine from antagonizing insulin-stimulated muscle glucose uptake. J Appl Physiol. 2002;92:1285–1292 [DOI] [PubMed] [Google Scholar]
- 78. Lee AD, Hansen PA, Schluter J, Gulve EA, Gao J, Holloszy JO. Effects of epinephrine on insulin-stimulated glucose uptake and GLUT-4 phosphorylation in muscle. Am J Physiol. 1997;273:C1082–1087 [DOI] [PubMed] [Google Scholar]
- 79. Lawrence JC, Jr, Roach PJ. New insights into the role and mechanism of glycogen synthase activation by insulin. Diabetes. 1997;46:541–547 [DOI] [PubMed] [Google Scholar]
- 80. Vind BF, Birk JB, Vienberg SG, et al. Hyperglycaemia normalises insulin action on glucose metabolism but not the impaired activation of AKT and glycogen synthase in the skeletal muscle of patients with type 2 diabetes. Diabetologia. 2012;55:1435–1445 [DOI] [PubMed] [Google Scholar]
- 81. Iozzo P, Pratipanawatr T, Pijl H, et al. Physiological hyperinsulinemia impairs insulin-stimulated glycogen synthase activity and glycogen synthesis. Am J Physiol Endocrinol Metab. 2001;280:E712–E719 [DOI] [PubMed] [Google Scholar]
- 82. Beck-Nielsen H. The role of glycogen synthase in the development of hyperglycemia in type 2 diabetes: 'To store or not to store glucose, that's the question'. Diabetes Metab Res Rev. 2012;28:635–644 [DOI] [PubMed] [Google Scholar]
- 83. Shulman GI, Rothman DL, Jue T, Stein P, DeFronzo RA, Shulman RG. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N Engl J Med. 1990;322:223–228 [DOI] [PubMed] [Google Scholar]
- 84. Petersen KF, Shulman GI. Pathogenesis of skeletal muscle insulin resistance in type 2 diabetes mellitus. Am J Cardiol. 2002;90:11G–18G [DOI] [PubMed] [Google Scholar]
- 85. Falkén Y, Hellström PM, Holst JJ, Näslund E. Changes in glucose homeostasis after Roux-en-Y gastric bypass surgery for obesity at day three, two months, and one year after surgery: role of gut peptides. J Clin Endocrinol Metab. 2011;96:2227–2235 [DOI] [PubMed] [Google Scholar]
- 86. Korner J, Inabnet W, Febres G, et al. Prospective study of gut hormone and metabolic changes after adjustable gastric banding and Roux-en-Y gastric bypass. Int J Obes. 2009;33:786–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lin E, Davis SS, Srinivasan J, Sweeney JF, Ziegler TR, Phillips L, Gletsu-Miller N. Dual mechanism for type-2 diabetes resolution after Roux-en-Y gastric bypass. Am surgeon. 2009;75:498–502; discussion 502–493 [PMC free article] [PubMed] [Google Scholar]
- 88. Rodieux F, Giusti V, D'Alessio DA, Suter M, Tappy L. Effects of gastric bypass and gastric banding on glucose kinetics and gut hormone release. Obesity. 2008;16:298–305 [DOI] [PubMed] [Google Scholar]
- 89. Wang Q, Li L, Xu E, Wong V, Rhodes C, Brubaker PL. Glucagon-like peptide-1 regulates proliferation and apoptosis via activation of protein kinase B in pancreatic INS-1 beta cells. Diabetologia. 2004;47:478–487 [DOI] [PubMed] [Google Scholar]
- 90. Nair VD, Sealfon SC. Agonist-specific transactivation of phosphoinositide 3-kinase signaling pathway mediated by the dopamine D2 receptor. J Biol Chem. 2003;278:47053–47061 [DOI] [PubMed] [Google Scholar]
- 91. Nair VD, Olanow CW. Differential modulation of Akt/glycogen synthase kinase-3β pathway regulates apoptotic and cytoprotective signaling responses. J Biol Chem. 2008;283:15469–15478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Merglen A, Theander S, Rubi B, Chaffard G, Wollheim CB, Maechler P. Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology. 2004;145:667–678 [DOI] [PubMed] [Google Scholar]
- 93. Sandyk R. The relationship between diabetes mellitus and Parkinson's disease. Int J Neurosci. 1993;69:125–130 [DOI] [PubMed] [Google Scholar]
- 94. Driver JA, Smith A, Buring JE, Gaziano JM, Kurth T, Logroscino G. Prospective cohort study of type 2 diabetes and the risk of Parkinson's disease. Diabetes Care. 2008;31:2003–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 2004;3:169–178 [DOI] [PubMed] [Google Scholar]
- 96. Sirtori CR, Bolme P, Azarnoff DL. Metabolic responses to acute and chronic L-dopa administration in patients with parkinsonism. N Engl J Med. 1972;287:729–733 [DOI] [PubMed] [Google Scholar]
- 97. Van Woert MH, Mueller PS. Glucose, insulin, and free fatty acid metabolism in Parkinson's disease treated with levodopa. Clin Pharmacol Ther. 1971;12:360–367 [DOI] [PubMed] [Google Scholar]
- 98. Aviles-Olmos I, Limousin P, Lees A, Foltynie T. Parkinson's disease, insulin resistance and novel agents of neuroprotection. Brain. 2013;136:374–384 [DOI] [PubMed] [Google Scholar]
- 99. Sanyal J, Chakraborty DP, Sarkar B, et al. Environmental and familial risk factors of Parkinsons disease: case-control study. Can J Neurol Sci. 2010;37:637–642 [DOI] [PubMed] [Google Scholar]
- 100. Vance JM, Ali S, Bradley WG, Singer C, Di Monte DA. Gene-environment interactions in Parkinson's disease and other forms of parkinsonism. Neurotoxicology. 2010;31:598–602 [DOI] [PubMed] [Google Scholar]
- 101. Girgis RR, Javitch JA, Lieberman JA. Antipsychotic drug mechanisms: links between therapeutic effects, metabolic side effects and the insulin signaling pathway. Mol Psychiatry. 2008;13:918–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Melkersson K, Dahl ML. Adverse metabolic effects associated with atypical antipsychotics: literature review and clinical implications. Drugs. 2004;64:701–723 [DOI] [PubMed] [Google Scholar]
- 103. Pramyothin P, Khaodhiar L. Metabolic syndrome with the atypical antipsychotics. Curr Opin Endocrinol Diabetes Obes. 2010;17:460–466 [DOI] [PubMed] [Google Scholar]
- 104. Masri B, Salahpour A, Didriksen M, et al. Antagonism of dopamine D2 receptor/β-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc Natl Acad Sci USA. 2008;105:13656–13661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Trümper K, Trümper A, Trusheim H, Arnold R, Göke B, Hörsch D. Integrative mitogenic role of protein kinase B/Akt in β-cells. Ann NY Acad Sci. 2000;921:242–250 [DOI] [PubMed] [Google Scholar]
- 106. Valjent E, Bertran-Gonzalez J, Bowling H, et al. Haloperidol regulates the state of phosphorylation of ribosomal protein S6 via activation of PKA and phosphorylation of DARPP-32. Neuropsychopharmacology. 2011;36:2561–2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Liu Y, Tanabe K, Baronnier D, et al. Conditional ablation of Gsk-3β in islet β cells results in expanded mass and resistance to fat feeding-induced diabetes in mice. Diabetologia. 2010;53:2600–2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Mussmann R, Geese M, Harder F, Kegel S, Andag U, Lomow A, Burk U, Onichtchouk D, Dohrmann C, Austen M. Inhibition of GSK3 promotes replication and survival of pancreatic β cells. J Biol Chem. 2007;282:12030–12037 [DOI] [PubMed] [Google Scholar]
- 109. Doyle ME, Egan JM. Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacol Ther. 2007;113:546–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Sutton LP, Rushlow WJ. The dopamine D2 receptor regulates Akt and GSK-3 via Dvl-3. Int J Neuropsychopharmacol. 2012;15:965–979 [DOI] [PubMed] [Google Scholar]
- 111. Beaulieu JM, Sotnikova TD, Yao WD, et al. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci USA. 2004;101:5099–5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Aitken A. Post-translational modification of 14–3-3 isoforms and regulation of cellular function. Semin Cell Dev Biol. 2011;22:673–680 [DOI] [PubMed] [Google Scholar]
- 113. Kelly P, Bailey CL, Fueger PT, Newgard CB, Casey PJ, Kimple ME. Rap1 promotes multiple pancreatic islet cell functions and signals through mammalian target of rapamycin complex 1 to enhance proliferation. J Biol Chem. 2010;285:15777–15785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Suzuki Y, Zhang H, Saito N, Kojima I, Urano T, Mogami H. Glucagon-like peptide 1 activates protein kinase C through Ca2+-dependent activation of phospholipase C in insulin-secreting cells. J Biol Chem. 2006;281:28499–28507 [DOI] [PubMed] [Google Scholar]
- 115. Buteau J, Foisy S, Rhodes CJ, Carpenter L, Biden TJ, Prentki M. Protein kinase Czeta activation mediates glucagon-like peptide-1-induced pancreatic beta-cell proliferation. Diabetes. 2001;50:2237–2243 [DOI] [PubMed] [Google Scholar]
- 116. Thibault D, Albert PR, Pineyro G, Trudeau LÉ. Neurotensin triggers dopamine D2 receptor desensitization through a protein kinase C and beta-arrestin1-dependent mechanism. J Biol Chem. 2011;286:9174–9184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Namkung Y, Sibley DR. Protein kinase C mediates phosphorylation, desensitization, and trafficking of the D2 dopamine receptor. J Biol Chem. 2004;279:49533–49541 [DOI] [PubMed] [Google Scholar]
- 118. Quoyer J, Longuet C, Broca C, et al. GLP-1 mediates antiapoptotic effect by phosphorylating Bad through a beta-arrestin 1-mediated ERK1/2 activation in pancreatic beta-cells. J Biol Chem. 2010;285:1989–2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Hoffmann HM, Nadal R, Vignes M, Ortiz J. Chronic cocaine self-administration modulates ERK1/2 and CREB responses to dopamine receptor agonists in striatal slices. Addiction Biol. 2012;17:565–575 [DOI] [PubMed] [Google Scholar]
- 120. Norling LL, Colca JR, Kelly PT, McDaniel ML, Landt M. Activation of calcium and calmodulin dependent protein kinase II during stimulation of insulin secretion. Cell Calcium. 1994;16:137–150 [DOI] [PubMed] [Google Scholar]
- 121. Wenham RM, Landt M, Easom RA. Glucose activates the multifunctional Ca2+/calmodulin-dependent protein kinase II in isolated rat pancreatic islets. J Biol Chem. 1994;269:4947–4952 [PubMed] [Google Scholar]
- 122. Hou XY, Zhang GY. Inhibitory effect of dopamine on Ca(2+)-calmodulin-dependent protein kinase II activity in rat hippocampal slices. Zhongguo yao li xue bao = Acta pharmacologica Sinica. 1999;20:902–906 [PubMed] [Google Scholar]
- 123. Lilja L, Meister B, Berggren PO, Bark C. DARPP-32 and inhibitor-1 are expressed in pancreatic β-cells. Biochem Biophys Res Commun. 2005;329:673–677 [DOI] [PubMed] [Google Scholar]
- 124. Lilja L, Yang SN, Webb DL, Juntti-Berggren L, Berggren PO, Bark C. Cyclin-dependent kinase 5 promotes insulin exocytosis. J Biol Chem. 2001;276:34199–34205 [DOI] [PubMed] [Google Scholar]
- 125. Nishi A, Bibb JA, Matsuyama S, et al. Regulation of DARPP-32 dephosphorylation at PKA- and Cdk5-sites by NMDA and AMPA receptors: distinct roles of calcineurin and protein phosphatase-2A. J Neurochem. 2002;81:832–841 [DOI] [PubMed] [Google Scholar]
- 126. Sonoda N, Imamura T, Yoshizaki T, Babendure JL, Lu JC, Olefsky JM. β-Arrestin-1 mediates glucagon-like peptide-1 signaling to insulin secretion in cultured pancreatic β cells. Proc Natl Acad Sci USA. 2008;105:6614–6619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Leech CA, Chepurny OG, Holz GG. Epac2-dependent rap1 activation and the control of islet insulin secretion by glucagon-like peptide-1. Vitam Horm. 2010;84:279–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. McAvoy T, Zhou MM, Greengard P, Nairn AC. Phosphorylation of Rap1GAP, a striatally enriched protein, by protein kinase A controls Rap1 activity and dendritic spine morphology. Proc Natl Acad Sci USA. 2009;106:3531–3536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zhang W, Efanov A, Yang SN, Fried G, Kolare S, Brown H, Zaitsev S, Berggren PO, Meister B. Munc-18 associates with syntaxin and serves as a negative regulator of exocytosis in the pancreatic β-cell. J Biol Chem. 2000;275:41521–41527 [DOI] [PubMed] [Google Scholar]
- 130. Kost GC, Selvaraj S, Lee YB, Kim DJ, Ahn CH, Singh BB. Clavulanic acid increases dopamine release in neuronal cells through a mechanism involving enhanced vesicle trafficking. Neurosci Lett. 2011;504:170–175 [DOI] [PMC free article] [PubMed] [Google Scholar]