

Abstract
Vast tracts of noncoding DNA contain elements that regulate gene expression in higher eukaryotes. Describing these regulatory elements and understanding how they evolve represent major challenges for biologists. Advances in the ability to survey genome-scale DNA sequence data are providing unprecedented opportunities to use evolutionary models and computational tools to identify functionally important elements and the mode of selection acting on them in multiple species. This chapter reviews some of the current methods that have been developed and applied on noncoding DNA, what they have shown us, and how they are limited. Results of several recent studies reveal that a significantly larger fraction of noncoding DNA in eukaryotic organisms is likely to be functional than previously believed, implying that the functional annotation of most noncoding DNA in these organisms is largely incomplete. In Drosophila, recent studies have further suggested that a large fraction of noncoding DNA divergence observed between species may be the product of recurrent adaptive substitution. Similar studies in humans have revealed a more complex pattern, with signatures of recurrent positive selection being largely concentrated in conserved noncoding DNA elements. Understanding these patterns and the extent to which they generalize to other organisms awaits the analysis of forthcoming genome-scale polymorphism and divergence data from more species.
Keywords: Adaptive evolution, Neutrality test, Selective constraint, Deleterious mutations, McDonald–Kreitman test, Population genetics
1. Introduction and Methods
The lion’s share of higher eukaryotic genomes comprises noncoding DNA, which encodes the information necessary to regulate the level, timing, and spatial organization of the expression of thousands of genes (1). A growing body of evidence supports the view that the evolution of gene expression regulation is the primary genetic mechanism behind the modular organization, functional diversification, and origin of novel traits in higher organisms (2–5). Historically, noncoding DNA has been little studied relative to proteins and the lack of knowledge about its function has led toit being viewed as mostly “junk.” More recently, technological advances have allowed researchers to probe noncoding DNA function in more detail, including the annotation of genomic elements that regulate levels of DNA transcription and translation (6). The complexity of regulation generally precludes the direct evaluation of all functions of regulatory elements in noncoding DNA, or an understanding of how genetic variation in regulation corresponds to organismal fitness. Nonetheless, even in the absence of this information, developments in evolutionary theory and computational biology, in conjunction with the increasing availability of genome-scale data, are providing unprecedented insights into the functional significance of noncoding DNA and its evolution. The emerging picture, in many eukaryotic organisms, is that a much larger fraction of noncoding DNA is functional and subject to both positive and negative natural selection than previously believed. These findings, in turn, have profound implications for our broader understanding of the evolutionary processes underlying patterns of genome evolution and how we should interpret patterns of genomic divergence between closely related species (7–10).
Here, we review some of the emerging evolutionary/computational methods for detecting and quantifying selection acting on noncoding DNA, and how these might be used to identify functionally important elements in genomes and the mode of selection acting on them. We focus on methods that have been developed or adapted specifically for application to noncoding DNA rather than approaches that can be more generically applied to genome sequences. For an overview of the latter approaches, including tests for selection based on genomic scans for high levels of population differentiation (e.g., Fst), linkage disequilibrium and haplotype structure, or reduced variation, Hahn (11), Oleksyk et al. (12), and Charlesworth and Charles-worth (13) offer recent reviews. In addition, our purpose here is to highlight seminal papers and recent good examples rather than exhaustively review what is quickly becoming a vast literature.
1.1. Phylogenetic Methods: Quantifying Functionality of Noncoding DNA via Constraint
What fraction of noncoding DNA in eukaryotic genomes isfunctional? Modern functional genomics approaches, like Chip-seq (14), RNA-seq (15), and DNAse I hypersensitivity mapping (16), will likely provide at least part of the answer to this question. However, the complete answer to this question is unlikely to come from direct functional studies alone because they lack sensitivity given the vast complexity of gene regulation (e.g., tissue or developmental specificity, environmental factors, context dependence, as yet undiscovered biology, etc.). A complementary guide to evaluating the functional significance of noncoding DNA is the notion of measuring “evolutionary constraint.” This notion is perhaps most familiar in its application to proteins. That is, codons defining a protein sequence can be divided into discrete functional classes of sites: nonsynonymous sites, at which a newly arising mutation will alter the protein sequence, and synonymous sites, at which a newly arising mutation will alter the codon used, but not the protein sequence. If nonsynonymous sites and synonymous sites were functionally equivalent, we would expect that the probability of a substitution at either class of sites, defined as dN and dS, respectively, would be the same. However, in comparisons of homologous proteins from related species in a phylogenetic context, it is clear that dN is usually considerably smaller than dS on average (17). If one considers that the vast majority of randomly occurring amino acid substitutions to a protein is detrimental to the protein’s function, dN < dS is expected and consistent with the removal of deleterious nonsynonymous mutations by natural selection. Thus, the measure “constraint” in the context of protein evolution is defined as the fraction of newly arising nonsynonymous mutations in a protein that are deleterious enough to be removed by natural selection and is measured as the deficit in divergence at nonsyonymous sites relative to expectations based on synonymous sites (18). If we are to assume that synonymous substitutions are neutral and that mutation rates to synonymous and nonsynonymous sites are equal, then a measure of constraint on protein sequences can be defined as 1 − (dN/dS). Even when reference sites are not truly neutral, such a comparative approach is a powerful way to detect purifying selection on a particular class of sites.
The same logic can be applied to comparisons of any class of functional sites in the genome, and has been used to identify conserved noncoding (CNC) sequences. That is, using a class of sites in the genome that can be regarded as neutral reference sites, assuming that differences in mutation rates can be accounted for and that all newly arising mutations are deleterious, one can use levels of divergence at these reference sites to estimate levels of constraint in noncoding DNA as a proxy for its functional significance. Several early applications of this approach suggested that the number of functionally important nucleotides in noncoding DNA equals or exceeds the number of functionally important coding nucleotide sites in the genomes of nematodes, Drosophila, and mammals (19–21) and more recent studies have generally pushed these estimates even higher (22–26). Looking at constraint in the context of larger phylogenies and varying phylogenetic distances (23, 27, 28) has sometimes been referred to as “phylogenetic footprinting” (29) or “phylogenetic shadowing” (30). Though the latter approaches use essentially the same principles, they are more often used to detect individual functional elements rather than to estimate genomic constraint in general.
Using “constraint” as a measure of functionality of noncoding DNA is not without its difficulties. Typically, synonymous sites, intronic DNA, or ancestral repeats are chosen as reference sites. However, recent studies of divergence in Arabidopsis and mammals have highlighted how the choice of reference sites can add considerable uncertainty to estimates of constraint in intergenic DNA (25, 26, 31). Of primary concern is the possibility that selection on reference sites themselves leads to underestimates of constraint. For example, selection on synonymous sites likely downwardly biases estimates of constraint in Drosophila and humans (24, 26). Further, there is no guarantee that ancient transposable element-derived DNA, another popular source of reference sites, has not been functionally co-opted (32, 33). A first difficulty, thus, becomes in identifying reliable reference sites in the genome. Halligan and Keightley (24) suggested using the fastest evolving intronic (FEI) sites in the Drosophila genome, bases 8–20 of short introns, to calibrate estimates of constraint, though the fact that they are the fastest evolving sites in the genome does not guarantee that they are the most neutral (see below).
A second potential source of uncertainty is mutation bias (25, 31, 34) and these are particularly important when the reference and queried sites differ in base composition or, perhaps more problematically, genomic location. Thirdly, the very notion of “constraint” as an index of functionality depends on the assumption that newly arising beneficial mutations are exceedingly rare and contribute negligibly to divergence between species (18, 35). These assumptions have recently been challenged using other approaches and population genetic data from Drosophila (see below). Notably, if a substantial fraction of the divergence observed between species is positively selected, rather than neutral or slightly deleterious, “constraint” is difficult to interpret. Finally, the notion of “constraint” on noncoding DNA is usually thought of as a property of sites in the genome rather than, more correctly, a property of possible mutations that occur at these sites. For example, it is possible for a completely functionless piece of noncoding DNA to exhibit constraint if some fraction of the mutations that occur at these sites create spurious regulatory sites that result in the misexpression of genes (36, 37). Another example is that the functional status of some binding sites in an enhancer may depend on the state at other binding sites (38). Thus, while “constraint” may be a reasonable first approximation to functionality in noncoding DNA, its interpretation can sometimes be difficult. In addition, a lack of evidence for selection may be misleading about function, as suggested by the recent identification of functional transcriptional enhancers in the human genome with little evidence of constraint (39).
Recently, a number of methods have been introduced to detect noncoding sequences evolving faster than “neutral” reference sites (40–47), presumably due to the action of recurrent adaptive substitution. Generally, these approaches have focused on lineage-specific accelerations in the rate of substitution in CNC sequences. Lineage-specific changes in the rate of evolution can be caused by recurrent positive selection, but also a simple relaxation in selective constraint (e.g., loss of function). However, sequences exceeding the rate of evolution at neutral reference sites can be inferred to be the targets of recurrent positive selection (as for protein sequences—see ref. 48). Using this logic, Pollard et al. (40) identified 202 genomic regions that are highly conserved in most vertebrates but evolve more rapidly in humans. Interestingly, most of these regions (80.4%) localize to noncoding regions in the vicinity of genes involved in transcription and DNA binding. Another example is a similar study on Drosophila that identified 64 highly conserved genomic regions that exhibited a recent rate acceleration in the Drosophila melanogaster lineage (46). However, only a fraction of these regions (28%) are found in noncoding DNA. Kim and Pritchard (44) looked for heterogeneity in evolutionary rates for CNCs across vertebrates and estimated that 32% of CNC regions exhibit branch-specific rate changes. Prabhakar et al. (41) found that CNC regions with rate accelerations in human and chimpanzee are significantly enriched near genes with neurological functions and (42) showed that accelerated CNCs in the human lineage are associated with human-specific segmental duplications.
Using a similar approach, Hahn et al. (49) suggested comparing rates of substitution in putative functional sites (in this case, transcription factor-binding sites, Kb) to intervening, nonfunctional sites (Ki). They found a significant excess of fixations in putative binding sites in the 5′ noncoding region of the factor VII locus of humans (i.e., Kb/Ki > 1); however, it is difficult in such a test to rule out selective constraint on the intervening sites. Thus, using such an approach alone, it is difficult to distinguish a relaxation of selection from positive selection.
More generally, methods based on sequence divergence alone lack power to detect selection because they tend to assume that a given region of the genome is either negatively selected or positively selected, whereas in most cases positively and negatively selected sites may be interspersed. One notable exception is a study by Lunter et al. (50) that used the distribution of small insertion and deletion (indel) substitutions in putatively neutral reference sequences to identify functional noncoding DNA (i.e., regions resistant to indels were inferred to be under selective constraint). Of the noncoding DNA sequences inferred to be functional, based on the pattern of indel substitutions, those that evolve faster than neutral reference sites with respect to the rate of nucleotide substitution were identified to be under positive selection. Using this approach, Lunter et al. estimate that 2–3% of human genome is functional with 0.03% of sites being the targets of recent adaptive substitution. While the model of Lunter et al. (50) does allow for heterogeneous selective pressures on noncoding DNA (i.e., negative selection on indels and negative or positive selection on nucleotide substitutions), the model is still obviously limited in the way that it can accommodate this heterogeneity. That is, there is no reason to suppose that some fraction of indel substitutions is not positively selected or that a particular region of noncoding DNA must be either selectively constrained or positively selected at the nucleotide level. Indeed, recent analyses in Drosophila have revealed complex lineage-specific selection pressures on indel variation (51, 52). In addition, like inferences of constraint, inferences of recurrent positive selection on noncoding DNA using divergence-based approaches suffer from the limitation that it is difficult or sometimes impossible to rule out variation in mutation rates (or mutation bias) or selective constraint on the chosen reference sites themselves.
Another approach allowing for some degree of heterogeneity in selection pressures is that proposed by Moses (53) to look at the evolution of transcription factor-binding sites (TFBSs) in enhancers. The approach is to compute a null distribution of the effects of random substitutions on the strength of binding affinity in TFBSs. By comparing the effects of actual divergence to this distribution, one can identify TFBSs that show a larger change than expected under the null distribution, presumably due to negative or positive selection to either weaken or strengthen the binding affinity. At the moment, this method might be most successfully applied to well-characterized enhancers, where changes in binding site affinity lead to concrete predictions about the output of the system. However, the method may be difficult to apply to (or interpret) situations in which the effects of substitutions are highly context dependent (38) or to noncoding DNA with unknown function, as there may be as much or more selection in favor of reducing binding site affinity as increasing it.
Intricately tied to the issue of detecting and estimating selection based on patterns of substitution, whether single-nucleotide substitutions or indels, is the issue of uncertainty in alignment (54–58). The implicit assumption in an alignment, from which patterns of substitution are inferred, is that orthologous base positions are being compared. Pollard et al. (58) compared the performance of numerous tools that have been developed to align noncoding sequences and predictably found that the accuracy of alignments decreases with increasing divergence for all tools and declines faster in the presence of indel substitutions. Keightley and Johnson (57) proposed using empirical estimates of mutation parameters (e.g., the observed distribution of indel substitutions) to improve the quality of alignments, and a growing number of studies (54, 55, 59, 60) propose approaches to estimate the degree of certainty associated with particular alignments, which can in turn be used to appropriately weight estimates of evolutionary parameters (such as mutation and selection). Several recent advances in alignment algorithms (61, 62) are aimed at reducing errors associated with alignments by incorporating phylogenetic information.
1.2. Population Genetic Approaches: The Distribution of Polymorphism Frequencies
As defined above, the detection and quantification of “constraint due to negative selection” or “accelerated evolution due to positive selection” are intrinsically tied to the estimation of evolutionary distances. Doing this accurately can be challenging given differences in mutation rate or bias of nucleotides in different genomic contexts. An alternative population genetic approach is to compare the distribution of polymorphism frequencies (DPF) at a putatively selected class of sites with that at a putatively neutral class of reference sites (63–66). This approach relies on the fact that purifying selection tends to decrease the frequencies of polymorphisms at functional sites relative to neutral sites. This approach has the advantage of being robust to the details of the mutation process, provided that the method employed either does not depend on the ancestral state (for example, the folded distribution (35)) or that the ancestral state can be accurately reconstructed (67, 68).
Analysis of the distribution of polymorphism frequencies has been used to demonstrate negative selection on amino acid variants in a variety of plant and animal species (22, 63, 69–72) and certain classes of synonymous codon changes relative to others in Drosophila (64, 73). The approach has also been extended to demonstrate evidence for selective constraint on noncoding DNA in Drosophila (22, 74–77), humans (69, 78–81), and Arabidopsis (72). Ronald and Akey (82) and Emerson et al. (83) extended this approach to look at the frequencies of polymorphisms underlying expression variation in yeast and were able to infer that most polymorphisms affecting expression in cis and trans are under purifying selection.
Recently, Kern and Haussler (84) developed a Hidden Markov model (popGenHMM), similar to that developed by Siepel et al. (23), that uses the distribution of polymorphism frequencies (instead of divergence) to detect genomic regions experiencing negative or positive selection. In a scan of a 7 Mb of the D. melanogaster genome, Kern and Haussler estimate that approximately 75% of sites in untranslated-transcribed regions (UTRs) are under negative selection, which is comparable to estimates based on levels of constraint (22). Kern and Haussler’s method does come with a number of important caveats. In particular, the assumption of independence among sites and the assumption of an equilibrium panmictic population similarly lead to high false-positive rates. The authors recommend simulations of the genealogies with recombination and demography (85) to be used to generate appropriate null distributions. Perhaps more problematic, like similar methods based on divergence (23), this method assumes that negatively and positively selected sites cluster into discreet “elements” rather than being interspersed. Studies in both Drosophila and humans suggest that, while more and less constrained elements can be identified, constraint appears to be widely dispersed throughout noncoding DNA in both genomes (22, 24, 79), and constrained and positively selected sites may often be interdigitated. These caveats are likely to seriously limit the power and accuracy of this approach in both detecting and quantifying selection in noncoding DNA (see Fig. 1).
Fig. 1.

The effect of directional selection on the distribution of polymorphism frequencies (DPFs). Plotted are expected proportion of polymorphisms on the y-axis and frequency in a sample of 20 chromosomes based on equations in Bustamante et al. (90). Selected variants are assumed to have additive effects on fitness. In brown is a mixture model that posits 50% of newly arising mutations being neutral, 40% being negatively selected, and 10% positively selected. The similarity of this mixture model to neutral expectations implies that it may be difficult to detect positive or negative selection in regions of the genome with pluralistic selective pressures based on the shape of the DPF alone.
1.3. Population Genetic Approaches: Using Polymorphism and Divergence
The interdigitation of positively and negatively selected sites in genomes limits the power of approaches that assume a particular form of selection acting on a genomic region. McDonald–Kreitman (MK) (86) proposed a statistical test to detect selection by utilizing information on both divergence and polymorphism. The method works by comparing two ways to estimate constraint at a class of putatively selected sites (X)—one based on polymorphism within species (pX/pneutral) and one based on divergence between species (dX/dneutral). Under Kimura’s neutral hypothesis (17), which assumes that all mutations are either neutral or strongly negatively selected, these two ratios should be equal. Departures from equality can be informative about the direction and intensity of selection on a class of putatively selected sites. That is, a divergence deficit relative to polymorphism at putatively selected sites suggests that some polymorphism is deleterious enough that it does not contribute to divergence. Conversely, an excess of divergence relative to polymorphism at putatively selected sites is consistent with recurrent adaptive substitution (86, 87) or a relaxation in the intensity of negative selection in the past (88). Several statistical approaches based on this framework have been developed to quantify the intensity of selection (65, 87, 89, 90), and the fraction of divergence in excess of the neutral model predictions (77, 89, 91–94). As these are based on essentially the same statistical framework as first proposed by McDonald and Kreitman (86), we refer to these collectively as “McDonald–Kreitman” approaches.
Though the McDonald–Kreitman test was originally applied to proteins (i.e., comparing nonsynonymous to putatively neutral synonymous sites), several authors have also applied modified versions of this test to noncoding DNA. Generally, this has been applied in two ways. First, the test has been used to detect selection at individual elements in the genome, for example, by comparing “functional” noncoding DNA, such as TFBSs, to “nonfunctional” noncoding DNA (95, 96). However, given high levels of constraint found in noncoding DNA currently lacking annotated function (see above), this approach is expected to lack power because “nonfunctional” noncoding DNA may in fact be functional. This has prompted others to modify the approach to use synonymous sites as a neutral reference to detect selection at individual noncoding DNA elements (97).
Second, a variety of MK approaches have been used in more broad-scale comparisons of classes of sites to infer the mode of selection acting on noncoding DNA throughout the genome (22, 75–77, 98, 99). Using this approach, Andolfatto (22) used polymorphism data from D. melanogaster, and divergence to its closest relative D. simulans, to show that there is a significant divergence excess relative to polymorphism for almost all classes of noncoding sequence, consistent with widespread recurrent adaptive substitution in noncoding DNA. In particular, Andolfatto estimated that ~20% of nucleotide divergence in introns and inter-genic regions and ~60% of divergence in UTRs are in excess of neutral theory predictions. Similar conclusions are reached when using polymorphism from D. simulans rather than D. melanogaster, and lineage-specific estimates of divergence (75). Casillas et al. (76) noted that purifying selection appears to be stronger in conserved noncoding sequences in Drosophila while the inferred divergence excess appears to be larger in less constrained sequences. In mice and humans, the Drosophila-like patterns of widespread constraint and a divergence excess relative to neutral expectations are not generally observed (77), though there is some evidence for negative and positive selection in CNCs (99). This might be expected given the size of mammalian genomes. That is, regulatory elements may be much more diffuse in noncoding DNA of mammals than in organisms like Drosophila, making recurrent positive selection difficult to detect in most noncoding DNA, but easier to detect in regions of the genome enriched for functional sites (such as CNCs in mammals). In support of this view, Kousathanas et al. (100) estimate similar numbers of adaptive substitutions in coding regions and upstream/downstream noncoding DNA in mice, though the latter estimates are not significantly different than zero. Little evidence for constraint and positive selection has also been documented in yeast, despite the expectation of a highly streamlined genome. This said, sample sizes from yeast populations have been very small (71) which limits the power of population genetic approaches. In addition, yeast populations appear to be highly structured and population sizes within demes appear to be quite small (101), which may render many mutations that would be deleterious in Drosophila effectively neutral in yeast.
Though MK approaches are expected to be more informative about the direction and intensity of selection than divergence-alone or polymorphism-alone methods, they also can be biased by several factors. First, the approach is limited by an appropriate choice of neutral reference sites. While synonymous sites are often chosen for this purpose, weak purifying selection on these sites (which has been documented in numerous taxa) can be expected to bias the MK test in favor of detecting positive selection (22, 102), and bias estimates of the divergence excess at putatively selected sites upward (22, 92). Alternative choices of neutral reference sites, such as the fastest evolving sites of short introns (24), have been proposed, though levels of polymorphism and divergence at these sites appear to be quite similar to synonymous sites, at least in D. melanogaster (52).
A second concern is the presence of appreciable numbers of weakly deleterious polymorphisms in the putatively selected class of sites, which tend to limit the power of the MK test to detect a divergence excess due to positive selection (103). To circumvent this problem, it has been proposed that a frequency filter be used (on both neutral and selected sites) to exclude low-frequency polymorphisms, which are enriched for substitutions that contribute to polymorphism but not divergence (91, 104). An alternative approach is to estimate the distribution of selective effects of deleterious mutations and use this estimate to infer the fraction of divergence in excess of neutral expectations (Fig. 2) (66, 77, 99, 105). Importantly, these latter methods assume a particular distribution of fitness effects of newly arising mutations (e.g., normal, exponential, gamma, etc.), which may or may not be biologically meaningful. A subset of the methods above (66, 77) also co-estimate a demographic model, the purpose of which is discussed below.
Fig. 2.
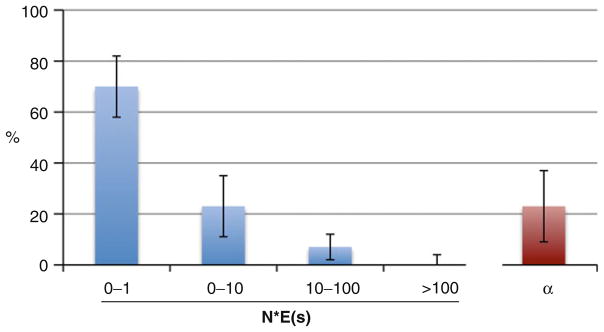
Selective constraint and positive selection on noncoding DNA inferred using polymorphism and divergence. Shown is the inferred distribution of fitness effects of newly arising mutations and the fraction of divergence in excess of expectations (α) for a sample of intronic sites in D. melanogaster (from Table 6 of 77). The method uses the DPF for synonymous sites to estimate parameters of a population size change model. The method then uses this demographic model, with the DPF and divergence at synonymous and intronic sites, to estimate selection on the latter class of sites. The implication is that 30% of newly arising mutations in these introns are subject to deterministic negative selection and that 20% of the nucleotide divergence observed between species is in excess of expectations under the neutral model. The error bars indicate standard errors on the estimates.
A third concern is that in comparisons of putatively selected and neutral reference sites, the assumption of the MK test is that these sites share the same genealogical history (86, 106). In general, this assumption works when there is either no recombination between neutral and selected sites or selected and neutral sites are close to evenly interdigitated. This assumption is rarely met in comparisons involving noncoding DNA potentially leading to underestimates of confidence intervals on estimates of the divergence excess (22) or false positives in tests for selection at individual genomic regions (106). This issue can be corrected by establishing the appropriate significance level using parametric coalescent simulations to generate null distributions of the test statistic. A similar issue stems from the practice of pooling sites across the genome, which can induce biased estimates of adaptive evolution if there is a negative correlation between levels of diversity and the extent of divergence at putatively selected sites (107, 108). In fact, such a correlation has been observed in patterns of polymorphism and divergence for protein coding (108–113) and noncoding DNA sequences in humans (112).
A final concern stems from the assumption that the current level of selective constraint on a genomic region (recorded in levels of polymorphism) has either remained constant over time or is not different than the average level of constraint in the past history of the species (recorded in levels of divergence). The relative contribution of deleterious mutations to divergence is determined by the distribution of deleterious selective effects of mutations and the effective population size of the species (92, 114, 115). If the effective population size of a species changes over time, as one might expect due to bottlenecks and expansions, levels of constraint on selected sites could change over time, leading to genome-wide biases in estimates of negative and positive selection (91, 116). The observation of positive selection in noncoding DNA in Drosophila and mice appears to be robust to recent population expansion (77, 117). However, it may be difficult to rule out the possibility of ancient bottlenecks that were more severe. The extent of shared polymorphism in two species (due to shared ancestry) may put useful limits on the severity of past bottlenecks, as suggested by Andolfatto et al. (117).
A related issue is the possibility of shifting constraints on noncoding DNA over time. Such changes in constraint over time may arise by a period of relaxed selection due to, for example, duplication (creating a period of functional redundancy) or changes in the environment. Another example is binding site turnover expected under simple models of stabilizing selection for a regulatory element, which can cause levels of selective constraint to shift within the element over time (38). The extent to which these issues cause a problem for inferences of positive and negative selection on noncoding elements using MK approaches is in need of further investigation.
1.4. Prospects
Our understanding of the function of noncoding DNA and the population-level processes shaping its evolution is in its infancy. Many approaches that have been applied to detect and quantify selection on noncoding DNA are derivatives of approaches first formulated for protein-coding genes (e.g., dN/dS, the MK test, etc.); thus, many of the same limitations of these methods apply equally to coding and noncoding DNA. The study of noncoding DNA is also fraught with its own additional specific challenges. Paramount among these is the comparative lack of functional annotation of sites. Apart from knowledge of the putative binding sites for a handful of transcription factors and regulatory RNAs, the function of most noncoding DNA is unknown. The finding of widespread selective constraint across the genomes of many eukaryotes suggests that we have much to learn about the functional significance of most noncoding DNA in eukaryotic genomes. Some of this constraint may be due to protein-coding and RNA genes yet to be discovered (118, 119), though it is unclear to what extent this can account for the widespread constraint patterns in unannotated noncoding DNA of many organisms. The inability to form prior hypotheses about function in noncoding DNA is a key factor limiting the power of statistical methods to detect and quantify selection. For example, where should we look for selection in noncoding DNA and what sites in the genome constitute appropriate neutral reference sites? The answer to the latter question in organisms with highly streamlined genomes and large population sizes (which determines the efficacy of selection), like Drosophila or Arabidopsis, might be very few sites indeed.
Much of the evidence for selection on noncoding DNA currently comes from generalized genomic studies that benefit from the statistical power afforded by looking at many sites in the genome. One of the outstanding questions in this area of investigation is whether the inferences of selection being made are robust to past changes in population size and structure. Another is how general these findings are across different organisms—notably, signatures of positive selection observed in Drosophila noncoding DNA (albeit multiple species) are not obvious in other organisms, such as yeast, Arabidopsis, mice, and humans. Part of the explanation for this might be that functional sites in noncoding DNA are more diffuse in very large genomes. However, these species also differ in many other aspects of biology that may play an important role in determining patterns of selection in noncoding DNA, including population size, population structure, and mating system (8, 120). Population genomic data from more species should shed light on the generality of this pattern and perhaps point to important factors determining our ability to detect positive and negative selection.
A second challenge is the ability to use any of the approaches outlined above to reliably detect positive and negative selection at individual regulatory elements in the genome. Genome-wide scans for selection based on genetic hitchhiking patterns (e.g., haplotype structure, reduced variation, etc.) are typically likely to lack the resolution to definitively identify specific targets of positive selection in noncoding DNA (but see ref. 121). Another issue is that power to detect selection at a single locus is typically limited by the number of informative substitutions and confidence in their frequencies (i.e., sample size). To date, polymorphism data has been quite limited, particularly those involving samples of individuals that are large enough to meaningfully estimate allele frequencies. Forthcoming genome projects of large samples of genomes for some organisms (e.g., http://browser.1000genomes.org; http://www.1001genomes.org) should usher in a new era of progress in detecting selection in the noncoding genome.
2. Exercises
Download the coding and noncoding polymorphism data of Andolfatto (22)—http://genomics.princeton.edu/AndolfattoLab/link_nature2005.html. The first sequence in each file is the sequence for D. simulans (an appropriate outgroup). The next 12 sequences are from a Zimbabwean population of D. melanogaster. You will need a script to extract polymorphism and divergence statistics from this data.
Compare the distribution of polymorphism frequencies for non-coding sites and fourfold synonymous sites of the D. melanogaster sequences. Since both demography and selection can influence polymorphism frequencies, how can you distinguish between these processes based on this comparison? Katzman et al. (80) compared the distribution of polymorphism frequencies in coding regions to CNCs, but used different population samples for these two classes of sites. What is the danger of comparing the distribution of polymorphism frequencies in this context?
Perform a McDonald–Kreitman test for each UTR locus using pooled synonymous sites as a neutral reference and obtain a distribution of p-values. What kinds of factors influence the type-I error of this test when used in this way? Describe how you might correct p-values for these factors.
Pooling UTR loci and using pooled synonymous sites as a neutral reference, estimate the fraction of UTR divergence in excess of neutral expectations (α) using the estimators of Fay et al. (91) and Eyre-Walker and Keightley (77) (see the DFE-alpha server http://homepages.ed.ac.uk/eang33/). According to the Eyre-Walker and Keightley approach, what fraction of newly arising mutations in noncoding sites is subject to weak negative selection? What factors make these two estimators of (α) different?
Acknowledgments
Thanks to Stephen Wright, Molly Przeworski, Kevin Bullaughey, and anonymous reviewers for helpful discussion and comments on the manuscript. This work was supported in part by NIH grant R01-GM083228.
References
- 1.Lewin B. Genes IX. Oxford University Press; 2007. p. 892. [Google Scholar]
- 2.Stern DL. Evolution, development and the predictable genome. Roberts and Co. Publishing; 2010. p. 264. [Google Scholar]
- 3.Wray G, Hahn M, Abouheif E, Balhoff J, Pizer M, Rockman M, Romano L. The evolution of transcriptional regulation in eukaryotes. Mol Biol Evol. 2003;20:1377–1419. doi: 10.1093/molbev/msg140. [DOI] [PubMed] [Google Scholar]
- 4.Davidson EH. Genomic regulatory systems: development and evolution. Academic Press; San Diego: 2001. [Google Scholar]
- 5.Carroll SB. Endless forms: the evolution of gene regulation and morphological diversity. Cell. 2000;101:577–580. doi: 10.1016/s0092-8674(00)80868-5. [DOI] [PubMed] [Google Scholar]
- 6.Sakabe NJ, Nobrega MA. Genome-wide maps of transcription regulatory elements. Wiley Interdiscip Rev Syst Biol Med. 2010;2:422–437. doi: 10.1002/wsbm.70. [DOI] [PubMed] [Google Scholar]
- 7.Charlesworth B, Betancourt AJ, Kaiser VB, Gordo I. Genetic recombination and molecular evolution. Cold Spring Harb Symp Quant Biol. 2009;74:177–186. doi: 10.1101/sqb.2009.74.015. [DOI] [PubMed] [Google Scholar]
- 8.Wright S, Andolfatto P. The impact of natural selection on the genome: emerging patterns in drosophila and arabidopsis. Annu Rev Ecol Evol Syst. 2008;39:193–213. [Google Scholar]
- 9.Keightley PD, Eyre-Walker A. Terumi Mukai and the riddle of deleterious mutation rates. Genetics. 1999;153:515–523. doi: 10.1093/genetics/153.2.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kondrashov AS. Deleterious mutations and the evolution of sexual reproduction. Nature. 1988;336:435–440. doi: 10.1038/336435a0. [DOI] [PubMed] [Google Scholar]
- 11.Hahn M. Detecting natural selection on cis-regulatory DNA. Genetica. 2007;129:7–18. doi: 10.1007/s10709-006-0029-y. [DOI] [PubMed] [Google Scholar]
- 12.Oleksyk TK, Smith MW, O’Brien SJ. Genome-wide scans for footprints of natural selection. Phil Trans Roy Soc B. 2010;365:185–205. doi: 10.1098/rstb.2009.0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charlesworth B, Charlesworth D. Elements of evolutionary genetics. Roberts and Co. Publishers; 2010. [Google Scholar]
- 14.Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shibata Y, Crawford GE. Mapping regulatory elements by DNaseI hypersensitivity chip (DNase-Chip) Methods Mol Biol. 2009;556:177–190. doi: 10.1007/978-1-60327-192-9_13. [DOI] [PubMed] [Google Scholar]
- 17.Kimura M. The neutral theory of molecular evolution. Cambridge University Press; Cambridge: 1983. [Google Scholar]
- 18.Kondrashov AS, Crow JF. A molecular approach to estimating the human deleterious mutation rate. Hum Mutat. 1993;2:229–234. doi: 10.1002/humu.1380020312. [DOI] [PubMed] [Google Scholar]
- 19.Shabalina S, Kondrashov A. Pattern of selective constraint in C-elegans and C-briggsae genomes. Genet Res. 1999;74:23–30. doi: 10.1017/s0016672399003821. [DOI] [PubMed] [Google Scholar]
- 20.Shabalina S, Ogurtsov A, Kondrashov V, Kondrashov A. Selective constraint in intergenic regions of human and mouse genomes. Trends in Genetics. 2001;17:373–376. doi: 10.1016/s0168-9525(01)02344-7. [DOI] [PubMed] [Google Scholar]
- 21.Bergman C, Kreitman M. Analysis of conserved noncoding DNA in Drosophila reveals similar constraints in intergenic and intronic sequences. Genome Res. 2001;11:1335–1345. doi: 10.1101/gr.178701. [DOI] [PubMed] [Google Scholar]
- 22.Andolfatto P. Adaptive evolution of non-coding DNA Drosophila. Nature. 2005;437:1149–1152. doi: 10.1038/nature04107. [DOI] [PubMed] [Google Scholar]
- 23.Siepel A, Bejerano G, Pedersen J, Hinrichs A, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier L, Richards S, Weinstock G, Wilson R, Gibbs R, Kent W, Miller W, Haussler D. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Halligan DL, Keightley PD. Ubiquitous selective constraints in the Drosophila genome revealed by a genome-wide interspecies comparison. Genome Res. 2006;16:875–884. doi: 10.1101/gr.5022906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaffney DJ, Keightley PD. Genomic selective constraints in murid non-coding DNA. PLoS Genetics. 2006;2:1912–1923. doi: 10.1371/journal.pgen.0020204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eory L, Halligan DL, Keightley PD. Distributions of Selectively Constrained Sites and Deleterious Mutation Rates in the Hominid and Murid Genomes. Mol Biol Evol. 2010;27:177–192. doi: 10.1093/molbev/msp219. [DOI] [PubMed] [Google Scholar]
- 27.Consortium. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- 28.Cooper G, Stone E, Asimenos G, Green E, Batzoglou S, Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duret L, Bucher P. Searching for regulatory elements in human noncoding sequences. Curr Opin Struc Biol. 1997;7:399–406. doi: 10.1016/s0959-440x(97)80058-9. [DOI] [PubMed] [Google Scholar]
- 30.Boffelli D, McAuliffe J, Ovcharenko D, Lewis K, Ovcharenko I, Pachter L, Rubin E. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science. 2003;299:1391–1394. doi: 10.1126/science.1081331. [DOI] [PubMed] [Google Scholar]
- 31.DeRose-Wilson LJ, Gaut BS. Transcription-related mutations and GC content drive variation in nucleotide substitution rates across the genomes of Arabidopsis thaliana and Arabidopsis lyrata. BMC Evol Biol. 2007;7:66. doi: 10.1186/1471-2148-7-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Britten R. Cases of ancient mobile element DNA insertions that now affect gene regulation. Mol Phylogenet Evol. 1996;5:13–17. doi: 10.1006/mpev.1996.0003. [DOI] [PubMed] [Google Scholar]
- 33.Nishihara H, Smit AFA, Okada N. Functional noncoding sequences derived from SINEs in the mammalian genome. Genome Res. 2006;16:864–874. doi: 10.1101/gr.5255506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haddrill P, Charlesworth B, Halligan D, Andolfatto P. Patterns of intron sequence evolution in Drosophila are dependent upon length and GC content. Genome Biology. 2005;6:r67. doi: 10.1186/gb-2005-6-8-r67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hahn M, Stajich J, Wray G. The effects of selection against spurious transcription factor binding sites. Mol Biol Evol. 2003;20:901–906. doi: 10.1093/molbev/msg096. [DOI] [PubMed] [Google Scholar]
- 37.Clop A, Marcq F, Takeda H, Pirottin D, Tordoir X, Bibe B, Bouix J, Caiment F, Elsen J, Eychenne F, Larzul C, Laville E, Meish F, Milenkovic D, Tobin J, Charlier C, Georges M. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nature Genetics. 2006;38:813–818. doi: 10.1038/ng1810. [DOI] [PubMed] [Google Scholar]
- 38.Bullaughey K. Changes in selective effects over time facilitate turnover of enhancer sequences. Genetics. 2011;187:567–82. doi: 10.1534/genetics.110.121590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blow MJ, McCulley DJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, Afzal V, Bristow J, Ren B, Black BL, Rubin EM, Visel A, Pennacchio LA. ChIP-Seq identification of weakly conserved heart enhancers. Nat Genet. 2010;42:806–810. doi: 10.1038/ng.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pollard KS, Salama SR, King B, Kern AD, Dreszer T, Katzman S, Siepel A, Pedersen JS, Bejerano G, Baertsch R, Rosenbloom KR, Kent J, Haussler D. Forces shaping the fastest evolving regions in the human genome. PLos Genetics. 2006;2:1599–1611. doi: 10.1371/journal.pgen.0020168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prabhakar S, Noonan JP, Paabo S, Rubin EM. Accelerated evolution of conserved noncoding sequences in humans. Science. 2006;314:786–786. doi: 10.1126/science.1130738. [DOI] [PubMed] [Google Scholar]
- 42.Bird C, Stranger B, Liu M, Thomas D, Ingle C, Beazley C, Miller OW, Hurles M, Dermitzakis E. Fast-evolving noncoding sequences in the human genome. Genome Biol. 2007;8:R118. doi: 10.1186/gb-2007-8-6-r118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haygood R, Fedrigo O, Hanson B, Yokoyama K-D, Awray G. Promoter regions of many neural- and nutrition-related genes have experienced positive selection during human evolution. Nature Genetics. 2007;39:1140–1144. doi: 10.1038/ng2104. [DOI] [PubMed] [Google Scholar]
- 44.Kim SY, Pritchard JK. Adaptive evolution of conserved noncoding elements in mammals. PLos Genetics. 2007;3:1572–1586. doi: 10.1371/journal.pgen.0030147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wong W, Nielsen R. Detecting selection in noncoding regions of nucleotide sequences. Genetics. 2004;167:949–958. doi: 10.1534/genetics.102.010959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Holloway AK, Begun DJ, Siepel A, Pollard KS. Accelerated sequence divergence of conserved genomic elements in Drosophila melanogaster. Genome Res. 2008;18:1592–1601. doi: 10.1101/gr.077131.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of non-neutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hurst L. The Ka/Ks ratio: diagnosing the form of sequence evolution. 2002;18:486–487. doi: 10.1016/s0168-9525(02)02722-1. [DOI] [PubMed] [Google Scholar]
- 49.Hahn M, Rockman M, Soranzo N, Goldstein D, Wray G. Population genetic and phylogenetic evidence for positive selection on regulatory mutations at the Factor VII locus in humans. Genetics. 2004;167:867–877. doi: 10.1534/genetics.103.025726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lunter G, Ponting CP, Hein J. Genome-wide identification of human functional DNA using a neutral indel model. PLoS Comp BIol. 2006;2:2–12. doi: 10.1371/journal.pcbi.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Presgraves DC. Intron length evolution in drosophila. Mol Biol Evol. 2006;23:2203–2213. doi: 10.1093/molbev/msl094. [DOI] [PubMed] [Google Scholar]
- 52.Parsch J, Novozhilov S, Saminadin-Peter S, Wong K, Andolfatto P. On the utility of short intron sequences as a reference for the detection of positive and negative Selection in Drosophila. Mol Biol Evol. 2010;27:1226–1234. doi: 10.1093/molbev/msq046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moses AM. Statistical tests for natural selection on regulatory regions based on the strength of transcription factor binding sites. BMC Evol Biol. 2009;9:286. doi: 10.1186/1471-2148-9-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Satija R, Pachter L, Hein J. Combining statistical alignment and phylogenetic footprinting to detect regulatory elements. Bioinformatics. 2008;24:1236–1242. doi: 10.1093/bioinformatics/btn104. [DOI] [PubMed] [Google Scholar]
- 55.Lunter G, Rocco A, Mimouni N, Heger A, Caldeira A, Hein J. Uncertainty in homology inferences: assessing and improving genomic sequence alignment. Genome Res. 2008;18:298–309. doi: 10.1101/gr.6725608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang J, Keightley PD, Johnson T. MCALIGN2: faster, accurate global pairwise alignment of non-coding DNA sequences based on explicit models of in-del evolution. BMC Bioinformatics. 2006;7:292. doi: 10.1186/1471-2105-7-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Keightley PD, Johnson T. MCALIGN: stochastic alignment of noncoding DNA sequences based on an evolutionary model of sequence evolution. Genome Res. 2004;14:442–450. doi: 10.1101/gr.1571904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pollard DA, Bergman CM, Stoye J, Celniker SE, Eisen MB. Benchmarking tools for the alignment of functional noncoding DNA. BMC Bioinformatics. 2004;5:6. doi: 10.1186/1471-2105-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Landan G, Graur D. Heads or tails: a simple reliability check for multiple sequence alignments. Mol Biol Evol. 2007;24:1380–1383. doi: 10.1093/molbev/msm060. [DOI] [PubMed] [Google Scholar]
- 60.Satija R, Hein J, Lunter GA. Genome-wide functional element detection using pairwise statistical alignment outperforms multiple genome footprinting techniques. Bioinformatics. 2010;26:2116–2120. doi: 10.1093/bioinformatics/btq360. [DOI] [PubMed] [Google Scholar]
- 61.Liu K, Raghavan S, Nelesen S, Linder CR, Warnow T. Rapid and accurate large-scale coestimation of sequence alignments and phylogenetic trees. Science. 2009;324:1561–1564. doi: 10.1126/science.1171243. [DOI] [PubMed] [Google Scholar]
- 62.Loytynoja A, Goldman N. web-PRANK: a phylogeny-aware multiple sequence aligner with interactive alignment browser. BMC Bioinformatics. 2010;11:579. doi: 10.1186/1471-2105-11-579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sawyer SA, Dykhuizen DE, Hartl DL. Confidence interval for the number of selectively neutral amino acid polymorphisms. Proc Natl Acad Sci U S A. 1987;84:6225–6228. doi: 10.1073/pnas.84.17.6225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Akashi H, Schaeffer S. Natural selection and the frequency distributions of “silent” DNA polymorphism in Drosophila. Genetics. 1997;146:295–307. doi: 10.1093/genetics/146.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Keightley PD, Eyre-Walker A. Joint inference of the distribution of fitness effects of deleterious mutations and population demography based on nucleotide polymorphism frequencies. Genetics. 2007;177:2251–2261. doi: 10.1534/genetics.107.080663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boyko AR, Williamson SH, Indap AR, Degenhardt JD, Hernandez RD, Lohmueller KE, Adams MD, Schmidt S, Sninsky JJ, Sunyaev SR, White TJ, Nielsen R, Clark AG, Bustamante CD. Assessing the evolutionary impact of amino acid mutations in the human genome. PLoS Genetics. 2008;30:e1000083. doi: 10.1371/journal.pgen.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hernandez RD, Williamson SH, Bustamante CD. Context dependence, ancestral misidentification, and spurious signatures of natural selection. Mol Biol Evol. 2007;24:1792–1800. doi: 10.1093/molbev/msm108. [DOI] [PubMed] [Google Scholar]
- 68.Baudry E, Depaulis F. Effect of misoriented sites on neutrality tests with outgroup. Genetics. 2003;165:1619–1622. doi: 10.1093/genetics/165.3.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kryukov G, Schmidt S, Sunyaev S. Small fitness effect of mutations in highly conserved non-coding regions. Human Molecular Genetics. 2005;14:2221–2229. doi: 10.1093/hmg/ddi226. [DOI] [PubMed] [Google Scholar]
- 70.Foxe JP, Dar V-u-N, Zheng H, Nordborg M, Gaut BS, Wright SI. Selection on amino acid substitutions in Arabidopsis. Mol Biol Evol. 2008;25:1375–1383. doi: 10.1093/molbev/msn079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Doniger SW, Kim HS, Swain D, Corcuera D, Williams M, Yang SP, Fay JC. A catalog of neutral and deleterious polymorphism in yeast. PLoS Genet. 2008;4:e1000183. doi: 10.1371/journal.pgen.1000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim S, Plagnol V, Hu TT, Toomajian C, Clark RM, Ossowski S, Ecker JR, Weigel D, Nordborg M. Recombination and linkage disequilibrium in Arabidopsis thaliana. Nat Genet. 2007;39:1151–1155. doi: 10.1038/ng2115. [DOI] [PubMed] [Google Scholar]
- 73.Zeng K, Charlesworth B. Studying patterns of recent evolution at synonymous sites and intronic sites in Drosophila melanogaster. J Mol Evol. 2010;70:116–128. doi: 10.1007/s00239-009-9314-6. [DOI] [PubMed] [Google Scholar]
- 74.Bachtrog D, Andolfatto P. Selection, recombination and demographic history in Drosophila miranda. Genetics. 2006;174:2045–2059. doi: 10.1534/genetics.106.062760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Haddrill P, Bachtrog D, Andolfatto P. Positive and negative selection on noncoding DNA in Drosophila simulans. Mol Biol Evol. 2008;25:1825–1834. doi: 10.1093/molbev/msn125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Casillas S, Barbadilla A, Bergman C. Purifying selection maintains highly conserved Noncoding sequences in Drosophila. Mol Biol Evol. 2007;24:2222–2234. doi: 10.1093/molbev/msm150. [DOI] [PubMed] [Google Scholar]
- 77.Eyre-Walker A, Keightley PD. Estimating the rate of adaptive molecular evolution in the presence of slightly deleterious mutations and population size change. Mol Biol Evol. 2009;26:2097–2108. doi: 10.1093/molbev/msp119. [DOI] [PubMed] [Google Scholar]
- 78.Drake J, Bird C, Nemesh J, Thomas D, Newton-Cheh C, Reymond A, Excoffier L, Attar H, Antonarakis S, Dermitzakis E, Hirschhorn J. Conserved noncoding sequences are selectively constrained and not mutation cold spots. Nature Genetics. 2006;38:223–227. doi: 10.1038/ng1710. [DOI] [PubMed] [Google Scholar]
- 79.Asthana S, Noble W, Kryukov G, Grantt C, Sunyaev S, Stamatoyannopoulos J. Widely distributed noncoding purifying selection in the human genome. Proc Natl Acad Sci USA. 2007;104:12410–12415. doi: 10.1073/pnas.0705140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Katzman S, Kern AD, Bejerano G, Fewell G, Fulton L, Wilson RK, Salama SR, Haussler D. Human genome ultraconserved elements are ultraselected. Science. 2007;317:915. doi: 10.1126/science.1142430. [DOI] [PubMed] [Google Scholar]
- 81.Chen K, Rajewsky N. Natural selection on human microRNA binding sites inferred from SNP data. Nat Genet. 2006;38:1452–1456. doi: 10.1038/ng1910. [DOI] [PubMed] [Google Scholar]
- 82.Ronald J, Akey JM. The evolution of gene expression QTL in Saccharomyces cerevisiae. PLoS One. 2007;2:e678. doi: 10.1371/journal.pone.0000678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Emerson JJ, Hsieh LC, Sung HM, Wang TY, Huang CJ, Lu HH, Lu MY, Wu SH, Li WH. Natural selection on cis and trans regulation in yeasts. Genome Res. 2010;20:826–836. doi: 10.1101/gr.101576.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kern A, Haussler D. A population genetic Hidden Markov Model for detecting genomic regions under selection. Mol Biol Evol. 2010;27:1673–85. doi: 10.1093/molbev/msq053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hudson RR. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- 86.McDonald JH, Kreitman M. Adaptive Protein Evolution at the Adh Locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- 87.Sawyer SA, Hartl DL. Population genetics of polymorphism and divergence. Genetics. 1992;132:1161–1176. doi: 10.1093/genetics/132.4.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ohta T. Amino acid substitution at the Adh locus of Drosophila is facilitated by small population size. Proc Natl Acad Sci U S A. 1993;90:4548–4551. doi: 10.1073/pnas.90.10.4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sawyer SA, Parsch J, Zhang Z, Hartl DL. Prevalence of positive selection among nearly neutral amino acid replacements in Drosophila. Proc Natl Acad Sci U S A. 2007;104:6504–6510. doi: 10.1073/pnas.0701572104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bustamante CD, Wakeley J, Sawyer S, Hartl DL. Directional selection and the site-frequency spectrum. Genetics. 2001;159:1779–1788. doi: 10.1093/genetics/159.4.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fay JC, Wyckoff GJ, Wu CI. Positive and negative selection on the human genome. Genetics. 2001;158:1227–1234. doi: 10.1093/genetics/158.3.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Eyre-Walker A, Keightley PD, Smith NG, Gaffney D. Quantifying the slightly deleterious mutation model of molecular evolution. Mol Biol Evol. 2002;19:2142–2149. doi: 10.1093/oxfordjournals.molbev.a004039. [DOI] [PubMed] [Google Scholar]
- 93.Bierne N, Eyre-Walker A. The genomic rate of adaptive amino acid substitution in Drosophila. Mol Biol Evol. 2004;21:1350–1360. doi: 10.1093/molbev/msh134. [DOI] [PubMed] [Google Scholar]
- 94.Welch JJ. Estimating the genome-wide rate of adaptive protein evolution in Drosophila. Genetics. 2006;173:821–837. doi: 10.1534/genetics.106.056911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jenkins DL, Ortori CA, Brookfield JF. A test for adaptive change in DNA sequences controlling transcription. Proc Biol Sci. 1995;261:203–207. doi: 10.1098/rspb.1995.0137. [DOI] [PubMed] [Google Scholar]
- 96.Ludwig MZ, Kreitman M. Evolutionary dynamics of the enhancer region of even-skipped in Drosophila. Mol Biol Evol. 1995;12:1002–1011. doi: 10.1093/oxfordjournals.molbev.a040277. [DOI] [PubMed] [Google Scholar]
- 97.Holloway A, Lawniczak M, Mezey J, Begun D, Jones C. Adaptive gene expression divergence inferred from population genomics. PLoS Genetics. 2007;3:2007–2013. doi: 10.1371/journal.pgen.0030187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kohn M, Fang S, Wu C. Inference of positive and negative selection on the 5′ regulatory regions of Drosophila genes. Mol Biol Evol. 2004;21:374–383. doi: 10.1093/molbev/msh026. [DOI] [PubMed] [Google Scholar]
- 99.Torgerson D, Boyko A, Hernandez R, Indap A, Hu X, White T, Sninsky J, Cargill M, Adams M, Bustamante C, Clark A. Evolutionary Processes Acting on Candidate cis-Regulatory Regions in Humans Inferred from Patterns of Polymorphism and Divergence. PLoS Genetics. 2009;5:e1000592. doi: 10.1371/journal.pgen.1000592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kousathanas A, Oliver F, Halligan DL, Keightley PD. Positive and negative selection on non-coding DNA close to protein-coding genes in wild house mice. Mol Biol Evol. 2010;28:1183–91. doi: 10.1093/molbev/msq299. [DOI] [PubMed] [Google Scholar]
- 101.Elyashiv E, Bullaughey K, Sattath S, Rinott Y, Przeworski M, Sella G. Shifts in the intensity of purifying selection: An analysis of genome-wide polymorphism data from two closely related yeast species. Genome Res. 2010;20:1558–1573. doi: 10.1101/gr.108993.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Akashi H. Inferring Weak Selection from Patterns of Polymorphism and Divergence at Silent Sites in Drosophila DNA. Genetics. 1995;139:1067–1076. doi: 10.1093/genetics/139.2.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Templeton AR. Contingency tests of neutrality using intra/interspecific gene trees: the rejection of neutrality for the evolution of the mitochondrial cytochrome oxidase II gene in the hominoid primates. Genetics. 1996;144:1263–1270. doi: 10.1093/genetics/144.3.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Charlesworth J, Eyre-Walker A. The rate of adaptive evolution in enteric bacteria. Mol Biol Evol. 2006;23:1348–1356. doi: 10.1093/molbev/msk025. [DOI] [PubMed] [Google Scholar]
- 105.Sawyer SA, Kulathinal RJ, Bustamante CD, Hartl DL. Bayesian analysis suggests that most amino acid replacements in Drosophila are driven by positive selection. J Mol Evol. 2003;57(Suppl 1):S154–164. doi: 10.1007/s00239-003-0022-3. [DOI] [PubMed] [Google Scholar]
- 106.Andolfatto P. Controlling type-I error of the McDonald-Kreitman test in genome wide scans for selection on noncoding DNA. Genetics. 2008;180:1767–1771. doi: 10.1534/genetics.108.091850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Smith NG, Eyre-Walker A. Adaptive protein evolution in Drosophila. Nature. 2002;415:1022–1024. doi: 10.1038/4151022a. [DOI] [PubMed] [Google Scholar]
- 108.Shapiro JA, Huang W, Zhang C, Hubisz MJ, Lu J, Turissini DA, Fang S, Wang HY, Hudson RR, Nielsen R, Chen Z, Wu CI. Adaptive genic evolution in the Drosophila genomes. Proc Natl Acad Sci U S A. 2007;104:2271–2276. doi: 10.1073/pnas.0610385104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Andolfatto P. Hitchhiking effects of recurrent beneficial amino acid substitutions in the Drosophila melanogaster genome. Genome Res. 2007;17:1755–1762. doi: 10.1101/gr.6691007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Macpherson J, Sella G, Davis J, Petrov D. Genomewide spatial correspondence between nonsynonymous divergence and neutral polymorphism reveals extensive adaptation in drosophila. Genetics. 2007;177:2083–2099. doi: 10.1534/genetics.107.080226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bachtrog D. Similar rates of protein adaptation in Drosophila miranda and D. melanogaster, two species with different current effective population sizes. BMC Evol Biol. 2008;8:334. doi: 10.1186/1471-2148-8-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cai J, Macpherson J, Sella G, Petrov D. Pervasive Hitchhiking at Coding and Regulatory Sites in Humans. PLoS Genetics. 2009;5:e1000336. doi: 10.1371/journal.pgen.1000336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ingvarsson PK. Natural selection on synonymous and nonsynonymous mutations shapes patterns of polymorphism in Populus tremula. Mol Biol Evol. 2009;27:650–660. doi: 10.1093/molbev/msp255. [DOI] [PubMed] [Google Scholar]
- 114.Fay JC, Wu CI. The neutral theory in the genomic era. Curr Opin Genet Dev. 2001;11:642–646. doi: 10.1016/s0959-437x(00)00247-1. [DOI] [PubMed] [Google Scholar]
- 115.Eyre-Walker A, Keightley PD. The distribution of fitness effects of new mutations. Nature Reviews Genetics. 2007;8:610–618. doi: 10.1038/nrg2146. [DOI] [PubMed] [Google Scholar]
- 116.Eyre-Walker A. Changing effective population size and the McDonald-Kreitman test. Genetics. 2002;162:2017–2024. doi: 10.1093/genetics/162.4.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Andolfatto P, Wong KM, Bachtrog D. Effective population size and the efficacy of selection on the X chromosomes of two closely related Drosophila species. Genome Biol Evol. 2011;3:114–128. doi: 10.1093/gbe/evq086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hanada K, Zhang X, Borevitz JO, Li WH, Shiu SH. A large number of novel coding small open reading frames in the intergenic regions of the Arabidopsis thaliana genome are transcribed and/or under purifying selection. Genome Res. 2007;17:632–640. doi: 10.1101/gr.5836207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, Veyrieras JB, Stephens M, Gilad Y, Pritchard JK. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464:768–772. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sella G, Petrov D, Przeworski M, Andolfatto P. Pervasive Natural Selection in the Drosophila Genome? PLoS Genetics. 2009;5:e1000495. doi: 10.1371/journal.pgen.1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kudaravalli S, Veyrieras JB, Stranger BE, Dermitzakis ET, Pritchard JK. Gene expression levels are a target of recent natural selection in the human genome. Mol Biol Evol. 2009;26:649–658. doi: 10.1093/molbev/msn289. [DOI] [PMC free article] [PubMed] [Google Scholar]