

Abstract
Herein, we have studied a consanguineous Egyptian family with two children diagnosed with severe autosomal recessive osteogenesis imperfecta (AR-OI) and a large umbilical hernia. Homozygosity mapping in this family showed lack of linkage to any of the previously known AR-OI genes, but revealed a 10.27 MB homozygous region on chromosome 8p in the two affected sibs, which comprised the procollagen I C-terminal propeptide (PICP) endopeptidase gene BMP1. Mutation analysis identified both patients with a Phe249Leu homozygous missense change within the BMP1 protease domain involving a residue, which is conserved in all members of the astacin group of metalloproteases. Type I procollagen analysis in supernatants from cultured fibroblasts demonstrated abnormal PICP processing in patient-derived cells consistent with the mutation causing decreased BMP1 function. This was further confirmed by overexpressing wild type and mutant BMP1 longer isoform (mammalian Tolloid protein [mTLD]) in NIH3T3 fibroblasts and human primary fibroblasts. While overproduction of normal mTLD resulted in a large proportion of proα1(I) in the culture media being C-terminally processed, proα1(I) cleavage was not enhanced by an excess of the mutant protein, proving that the Phe249Leu mutation leads to a BMP1/mTLD protein with deficient PICP proteolytic activity. We conclude that BMP1 is an additional gene mutated in AR-OI.
Keywords: osteogenesis imperfecta, BMP1, astacin-like metalloproteases, type I collagen
Introduction
Osteogenesis imperfecta (OI type I-XII; MIM#s 166200, 166210, 259420, 166220, 610967, 610968, 610682, 610915, 259440, 613848, 613849, 613982) is a rare genetic disorder primarily characterized by bone fragility leading to increased risk of fractures. The clinical manifestations of patients with OI vary considerably from severe bone deformities or perinatal lethality to individuals with nearly no fractures [Rauch and Glorieux, 2004; Sillence et al., 1979]. While the majority of OI cases are autosomal dominant and arise from heterozygous mutations in the structural genes coding for the two procollagen chains, proα1(I) COL1A1 (MIM# 120150) and proα2(I) COL1A2 (MIM# 120160), there is a less frequent group of autosomal recessive forms of OI (AR-OI), for which, the molecular bases are increasingly heterogeneous [Byers and Steiner, 1992]. So far, seven AR-OI loci have been identified, Prolyl 3-hydroxylase 1 (LEPRE1; MIM# 610339), cartilage-associated protein (CRTAP; MIM# 605497), and peptidylprolyl isomerase B (PPIB; MIM# 123841), which are the three components of the endoplasmic reticulum (ER) complex responsible for 3-hydroxylation of Pro 986 in the proα1(I) chain [Barnes et al., 2006; Cabral et al., 2007; Ishikawa et al., 2009; Morello et al., 2006; van Dijk et al., 2009]; SERPINH1 (Serpin peptidase inhibitor, clade H; MIM# 600943) and FKBP10 (Fk506-binding protein 10; MIM# 607063) that function as type I procollagen molecular chaperons [Alanay et al., 2010; Christiansen et al., 2010; Drogemuller et al., 2009]; the osteoblast specific transcription factor SP7/OSTERIX (MIM# 606633), implicated in the differentiation of osteoblasts [Lapunzina et al., 2010]; and the type I collagen interacting protein and antiangiogenic extracellular matrix factor SERPINF1 (Serpin peptidase inhibitor, clade F, member 1; MIM# 172860) [Becker et al., 2011]. Except for SERPINF1, whose molecular pathomechanism remains to be elucidated, the rest of AR-OI genes identified are involved in type I collagen synthesis, posttranslational modification, or secretion.
Type I collagen, which is the most abundant protein in bone and other connective tissues, is initially synthesized in the ER as a precursor molecule (type I procollagen) that combines two proα1(I) and one proα2(I) peptide chains in a triple helix. Prior to helix formation, procollagen chains undergo a series of posttranslational modifications including 3 and 4-hydroxylation of proline residues and lysine hydroxylations. These modifications are necessary for the correct folding and thermal stability of the triple helix and posterior crosslinking between collagen molecules. Procollagen trimers are then secreted into the extracellular space via the Golgi network, where they are processed into mature type I collagen molecules by proteolytic cleavage of the N and C-terminal propeptides, and self-assembled into collagen fibrils and fibers [Canty and Kadler, 2005; Makareeva et al., 2011]. Type I procollagen N-propeptide is removed by the protein product of ADAMTS-2 (A disintegrin and metalloproteinase with thrombospondin motifs 2; MIM# 604539) and mutations in this gene are associated with recessive Ehlers–Danlos syndrome type VIIC (EDS type VIIC; MIM# 225410) [Colige et al., 1999]. Two ADAMTS-2-related enzymes, ADAMTS-3 (MIM# 605011) and ADAMTS-14 (MIM# 607506), also display some amino-peptidase activity [Colige et al., 2002; Fernandes et al., 2001]. Processing of procollagen I C-terminal propeptide (PICP) is accomplished by the family of mammalian bone morphogenetic protein 1/Tolloid-like (BMP1/TLD-like) proteinases, which is composed of four secreted proteins encoded by three different loci [Hopkins et al., 2007; Kessler et al., 1996]. BMP1 and its longer isoform mammalian Tolloid protein (mTLD) are alternative spliced products of BMP1 (MIM# 112264), which is located on 8p21.3. The other two remaining BMP1/TLD-like members, Tolloid-like protein 1 (TLL1; MIM# 606742) and Tolloid-like protein 2 (TLL2; MIM# 606743), are transcribed from genes that lie on chromosomes 4 and 10, respectively [Scott et al., 1999; Takahara et al., 1994, 1996]. All four mammalian BMP1/TLD-like proteases share domain structure and sequence similarity with Drosophila tolloid (tld), a protein that is involved in dorsal–ventral patterning of the Drosophila embryo [Shimell et al., 1991]. BMP1/TLD-like family structure consists of a signal peptide, followed by an aminoterminal protease inhibitor prodomain, that is proteolytically removed by the subtilisin-like proprotein convertases in the mature proteins, a conserved astacin-like protease domain shared by a wider family of metalloproteases and a variable number of complement-uegf-BMP1 (CUB) domains and epidermal growth factor like domains, involved in protein–protein interactions and in certain cases in Ca+2 binding [Ge and Greenspan, 2006; Hopkins et al., 2007]. Although the four BMP1/TLD-like mammalian proteins can process the C-terminal propeptide of type I procollagen, they do not show the same cleavage efficiency, with BMP1 having the highest PICP-protease activity, followed by mTLD and TLL1, and TLL2 having the lowest procollagen I C-propeptidase efficiency [Pappano et al., 2003; Scott et al., 1999]. BMP1/TLD-like proteases are also known to be involved in type II, type III, and type V procollagen processing and in the activation of the lysyl oxidase zymogen (LOX; MIM# 153455) [Bonod-Bidaud et al., 2007; Ge and Greenspan, 2006]. LOX is an extracellular copper enzyme that initiates crosslinking of collagens. Furthermore, a variety of secreted factors and extracellular matrix proteins including the dorsalizing agent Chordin have been additionally described as substrates of the BMP1/TLD-like peptidases [Ge and Greenspan, 2006].
Herein, we report the identification of a homozygous missense mutation in a highly conserved residue of the protease domain of BMP1/mTLD in two siblings diagnosed with AR-OI type III and show that this change inhibits the proteolytic activity of BMP1, thus causing deficient PICP processing in patients. We propose that this mutation is the most likely cause of AR-OI in this family.
Material and Methods
Array Hybridization
DNA samples were genotyped applying 250 ng of genomic DNA obtained from peripheral blood to the Human 610-Quad chip (Illumina, San Diego, CA) following the manufacturer’s instructions. Single nucleotide polymorphisms (SNPs) with call rates <0.95 were excluded. Image data were analyzed using the Chromosome Viewer tool contained in GenomeStudio V2010.3 (Illumina). Genomic positions were based upon human build GRCh37.
Genomic Sequencing
BMP1 coding exons and at least 100 bp of the flanking introns were amplified by standard PCR (primers available on request). Prior to sequencing, the PCR products were treated with shrimp alkaline phosphatase and exonuclease I (ExoSapit; GE Healthcare, Piscataway, NJ) according to the manufacturers instructions. Sequencing reactions were carried out using a dye terminator cycle sequencing kit and run on an ABI 3730 sequencer (Applied Biosystems, Foster City, CA). Chromatograms were aligned and compared with the mRNA reference nucleotide sequence of BMP1 (NM_001199.3 and NM_006129.4) and with the corresponding genomic sequence obtained from the Ensembl genome browser using Sequencher (Gene Codes Corp, Ann Arbor, MI). Mutation name was checked using Mutalizer http://www.mutalyzer.nl/2.0/ [Wildeman et al., 2008]. Sequencing of COL1A1 and COL1A2 coding exons was performed following standard procedures.
Cell Culture and Western Blotting Conditions
Human primary fibroblasts generated from skin biopsies were seeded in 60-mm Petri dishes (3.5 × 105 cells/dish) in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% foetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin B (Invitrogen, Carlsbad, CA), and 50μg/ml ascorbic acid for 24 hours at 37°C, 5% CO2. After that, we took off the media, washed the cells three times in phosphate buffered saline (PBS), and kept the cultures in serum free DMEM at 37 °C for 15 minutes. Cells were then washed twice in PBS and incubated in 2 ml of DMEM with 50 μg/ml of ascorbic acid and 40 μg/ml of soybean trypsin inhibitor for 24 hours before cell culture supernatants were collected and protease inhibitors added as described previously [Pappano et al., 2003]. Supernatants were subjected to SDS-PAGE in 5% or 7% acrylamide gels to separate the high molecular weight type I procollagen variants, except for free PICP analysis that because of its small molecular size, we used 12% acrylamide gels. For cell layer analysis, cells were lysed during 10 minutes on ice in RIPA buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1mM EDTA, 1%Triton X-100, 0.1% SDS, 1% Sodium Deoxycholate) containing 1 mM PMSF (Sigma (P7626), St. Louis, MO) and protease and phosphatase inhibitors (Sigma, P8340, P5726 and P0044). Lysates were cleared at 13,000 rpm for 15 minutes at 4°C and proteins quantified using the BCA protein assay (Pierce, Rockford, IL). SDS-PAGE gels of supernatants or cell extracts were transferred onto nitrocellulose membrane (GE Healthcare) and processed with ECL-Western Blotting Detection Reagents (GE Healthcare). LF antibodies were used at 1:1,500 (LF-68) or 1:3,000 (LF-42 and LF-9) dilutions, respectively. BMP1 and mTLD were detected with an anti-BMP1 (0,2μg/ml) antibody from R&D Systems (AF1927; R&D Systems, Minneapolis, MN). Anti α-Tubulin (1:15,000) obtained from Sigma (T9026) served as loading control. Horseradish peroxidase-conjugated secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PA).
Biochemical Analyses
Collagen biochemical analysis was performed by standard methods [Steinmann et al., 1984]. Briefly, fibroblasts were grown to confluence in DMEM and 10% foetal calf serum, and then plated in 35-mm dishes. After 48 hours, medium was changed for DMEM (lacking serum, proline, and glycine) and preincubated with ascorbic acid (50 μg/ml), before overnight labeling with 20 μCi [2,3,4,5-3H]-proline and 20 μCi [2-3H]-glycine. The medium and cell layers were harvested separately for each sample, and a portion of them was digested with pepsin (50 μg/ml) for 2 hours at 20°C. Purified procollagens and collagens were separated on 5% SDS-PAGE with 0.05 M dithiothreitol (DTT) as reducing agent, processed for fluorography and exposed to X-ray films.
Plasmid Constructions and Retroviral Infections
Wild type and mutant mTLD cDNAs were generated by PCR using pfx50TM DNA polymerase (Invitrogen) and cloned into pBABE-puro. Absence of nucleotide differences between normal and mutant mTLD cDNAs other than the c.747C>G mutation was verified by sequencing. Packaging of pBABE, pBABE-mTLD wild type, or pBABE-mTLD-mutant was performed by cotransfecting 70% confluent HEK293T cells with each one of these vectors individually and the packaging plasmid pCL-Eco (for NIH3T3 cells) or pCL-Amphos (for human primary fibroblats) using calcium phosphate. Supernatants containing retroviral particles were collected at 36, 48, and 60 hours posttransfection, passed through a 0.45 μm nitrocellulose filter, diluted 1:1 in growth medium, and applied to the cells of interest with polybrene. Three consecutive infections were performed. Twenty-four hours after the last infection, cells were selected with puromycin (2 μg/ml) for 2–3 days before they were plated at adequate density (3.5 × 105 cells/p60 for human fibroblasts or 8 × 105 cells/p60 for NIH3T3 cells) and assayed as described above for human primary fibroblasts.
Results
Clinical Characterization of Patients
Patients in this study include two siblings diagnosed with AR-OI (Fig. 1A). The proband of the family (II-3) is a 15-year-old female, the offspring of first cousin normal Egyptian parents, who was referred to us with history of recurrent fractures and bone deformities. She has a younger similarly affected brother, two normal sibs, and an older deaf mute sister who was not available for examination. After normal vaginal delivery at full term, fracture of the left tibia was noted. Since then, fractures occurred frequently and spontaneously with an average number of 10–15 fractures/year affecting both upper and lower limbs. On clinical examination, she had triangular face, broad forehead, wide palpebral fissures, long eyelashes, faint blue sclera, long philtrum, thin lips, and prominent ears. Gross motor development was delayed and she was not able to stand unsupported. Her mental development was normal. No dentinogenesis imperfecta was present. Her thorax was relatively large with increased both anteroposterior and transverse diameters compressing the abdomen. She had a large umbilical hernia, 5-cm wide and 7-cm long with abdominal content, generalized hypotonia, muscle wasting, and nocturnal enuresis. Her skin had normal stretchability with no bruising or scars. Generalized hirsutism was noted. Skeletal examination showed severe generalized deformities of all bones with consistent pain on touch, including deformed clavicles; bilateral bowed angulated humerus, radius, and ulna; arachnodactyly; and hyperextensibility of elbow, wrist, and interphalangeal (IP) joints. Lower limbs showed bowing of femora, severely angulated deformed leg bones and limited movements of the knee joints. She also had kyphoscoliosis and pectus carinatum. Radiological examination revealed deformed long bones with multiple fractures and callus formation, lack of bone modeling with wide distal metaphyses of femora, serpentine thin tibiae and fibulae in addition to S-curve scoliosis of thoracic and lumbar spine with platyspondyly, and generalized decreased bone density (Fig. 1B–G). Skull X-ray showed wormian bones. Anthropometric measurements at 13 years of age were below normal for weight (−3.8 SD), length (−11.5 SD) and head circumference (−3.0 SD). Bone densitometry (DEXA) at 15 years revealed borderline osteoporosis at the hip and spine (Z-score −2.22, −2.13, respectively).
Figure 1.
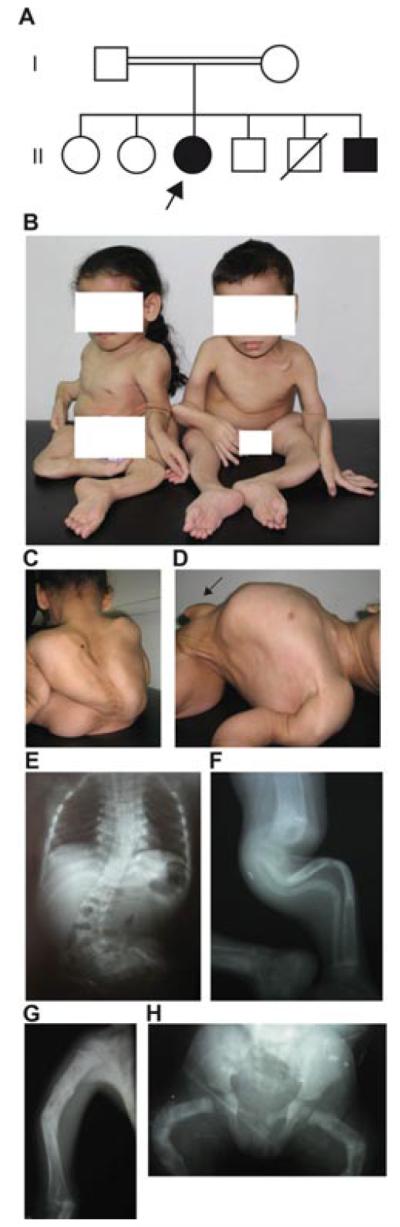
Clinical and radiological features of the patients studied in this report. A: Family pedigree showing parental consanguinity. The proband is indicated with an arrow. B: Severe bone deformities in both affected sibs. C–D: Panels illustrate severe kyphoscoliosis (C) and relatively large thorax, small abdomen and large umbilical hernia (arrow) in the proband (D). E: X-ray of the spine of the proband showing S-curve scoliosis of thoracic and lumbar spine and platyspondyly. F: X-ray of the left leg of the proband showing serpentine bowing and osteoporosis of tibia and fibula. G: X-ray of the right humerus showing healed fractures, angulations, and lack of bone modeling in the proband. H: X-ray of the pelvis and both femora of patient II-6 showing multiple healed fractures, with bowing, and lack of bone modeling.
Patient II-6 is the 5-year-old younger brother of patient II-3. Bilateral femoral bowing was detected intrauterine by fetal ultra-sound and fracture of both femora was noted at birth. Recurrent fractures of both upper and lower limbs occurred with an average rate of 15/year. He had facial features, no dentinogenesis imperfecta, and normal mentality similar to his sister. He was not able to stand unsupported. Skeletal examination revealed generalized bone deformities; hyperextensibility of elbows, wrists, and IP joints; and arachnodactyly. He also had a relatively large thorax compressing the abdomen with a large umbilical hernia (4-cm wide and 5-cm long with abdominal contents). X-ray survey revealed wormian bones of skull, deformed long bones with lack of modeling, fractures and callus formation, scoliosis, platyspondyly, and wide symphyses pubis (Fig. 1B and H). Anthropometric measurements were below normal for weight (−3.7 SD) and length (−7.2 SD) with normal head circumference (−0.8 SD). Bone densitometry (DEXA) at 5 years revealed borderline osteoporosis at the spine (Z-score −1.63) and osteoporosis at the left femur (Z-score −2.75). Audiological and cardiovascular examinations of both patients were unremarkable. Serum calcium and phosphate were normal, while alkaline phosphatase levels were slightly high. Both patients were diagnosed as OI Sillence type III, and cyclic IV biphosphonate injections were started. All studies and investigations involving the proband and other members of her family were performed in accordance with the ethical standards of the Medical Research Ethics Committee of the National Research Centre (Egypt), and all DNA samples were obtained with appropriate informed consent.
Genetic Analysis
Genetic studies in this family were performed by genome-wide homozygosity mapping in the affected children and in two normal sibs. To do this genomic DNA from each individual was hybridized to whole-genome high-density SNP arrays and the four resulting SNP genotypes were analyzed with Illumina software. The result of this study showed none of the patients to be homozygous at the CRTAP, PPIB, SERPINH1, SP7, and SERPINF1 loci, while only one of the affected children was homozygous for LEPRE1 and FKBP10, thus indicating further genetic heterogeneity in AR-OI. Sequencing of COL1A1 and COL1A2 coding exons in patient II-6 revealed no mutations in these genes (Fig. 1A). This prompted us to search for the causative gene, and so, we used the genotypes generated by the array hybridizations to look for blocks of homozygosity shared by both individuals with OI. Taking an arbitrary cutoff of 1 MB of consecutive homozygous SNPs, we identified two large homozygous DNA regions on chromosomes 3 and 8p, which were present in the patients but absent in the other two normal siblings. The homozygous region on chromosome 3 expanded 35.4 MB around the centromere and contained 151 genes. The 8p region consisted of 10.27 MB and contained 84 genes.
Identification of a Missense Mutation in BMP1
Analysis of candidate genes comprised in each DNA fragment revealed the presence of BMP1 within the chromosome 8 region. Given the nature of the disease, we considered this gene to be the best functional candidate. Moreover, a recent report has shown that dominant missense mutations in the C-terminal cleavage site of COL1A1 or COL1A2, which disrupt PICP processing, cause OI [Lindahl et al., 2011]. Hence, we direct sequenced all 21 coding exons (including those that are alternatively spliced) and exon–intron boundaries of BMP1 in the proband of the family and found a homozygous c.747C>G nucleotide change in exon 6 that causes the substitution of phenylalanine at position 249 to leucine (p.Phe249Leu) (Fig. 2A and 2B). The mutation was numbered with respect to the NM_006129.4 reference sequence taking the A of the first ATG as nucleotide +1, following the current nomenclature guidelines [den Dunnen and Antonarakis, 2000; den Dunnen and Paalman, 2003]. Phe249 is a residue contained within the peptidase domain of BMP1/mTLD that is conserved in all members of the astacin family of metallo-proteases independently of their substrate and whether they are of a vertebrate or invertebrate origin (Fig. 2C). Functionally, this amino acid is thought to be involved in internal bonds within the astacin protease module [Bond and Beynon, 1995; Guevara et al., 2010]. Segregation analysis of the mutation within the family showed the two affected children to be homozygous for the c.747C>G nucleotide change, while the normal sister (II-2) and brother (II-4) from whom DNA was available and the parents carried the mutation in the heterozygous state. To exclude the possibility of c.747C>G being a rare polymorphism, we checked normal controls for the presence of the c.747C>G change by direct sequencing. The mutation was not found in 100 control chromosomes of the same ethnic origin or in 884 normal chromosomes from controls of European ethnicity. c.747C>G is not listed as a polymorphism in any of the public databases.
Figure 2.

Identification of a missense mutation in BMP1 in individuals with AR-OI. A: Exon–intron structure of the two alternative spliced isoforms (BMP1 and mTLD) transcribed from the BMP1 locus. B: Sequence analysis of genomic DNA from a control (ctrl), a heterozygous individual (ht), and one of the patients (p) showing the c.747C>G transversion in BMP1 exon 6 (red and squared nucleotide). C: Multiple sequence alignment of a fragment from the catalytic domain of different astacin-like metalloproteases from diverse organisms showing conservation of the F249 residue labeled in red. The lower cluster of sequences corresponds to protein members of the BMP1/tolloid-like subgroup of astacin-like metalloproteases both from vertebrate and invertebrate organisms. Aaa_Asta, astacin from the crayfish A. Astacus (P07584), Tpa_My01 myosinase from giant squid Todarodes pacificus (Q8IU46), Cel_Nas13 metalloproteinase from Caenorhabditis elegans, (Q20191), Qu_Cam-I from quail (AAA20842.1), Hu_Ovast, human ovastacin (Q6HA08), Hu_MEPα (Q16819) and Hu_MEPβ (Q16820), human meprins α and β, Hvu_Hmp1 (AAA92361.2) and Hvu_Hmp2 (AAD33860.1) from Hydra vulgaris, Spu_Span from sea urchin (P98068), Lpo_Astm from the horseshoe crab Limulus polyphemus (B4F320), Ola_Hce1 (NP_001188427.1) and Ola_Lce1 (NP_001098292.1) from medaka fish Oryzias latipes, Dr_tld, Drosophila tolloid (NP_524487.2), Ate_tll, tolloid-like peptidase from the spider Achaearanea tepidariorum (Q75UQ6), Ci_Bmp1 from Ciona intestinalis (NP_001071840.1), Da_Bmp1a from Danio rerio (NP_001035126.1), Xe_Bmp1 from Xenopus laevis (AAI70427.1), Mu_Bmp1 from Mus musculus (NP_033885.2), Hu_BMP1/mTLD, Hu_TLL1 and Hu_TLL2 human BMP1/TLD-like proteases (NP_001190.1/NP_006120.1, NP_036596.3 and NP_036597.1). For most of the proteins shown here, a wider sequence alignment of the catalytic domain and information about substrate specificity have been described previously [Bond and Beynon, 1995; Guevara et al., 2010].
Deficient Procollagen I C-Terminal Processing in Patient Fibroblasts
Subsequently, we established dermal fibroblast cultures from skin biopsies of the two siblings with OI, their heterozygous normal sister (II-2), and an unrelated control individual to test whether PICP processing was altered in patients. Serum-free supernatants from cultured fibroblasts treated with ascorbic acid were analyzed by Western blotting using LF-68, LF-42, and LF-9 proα1(I) antisera. These antibodies were a generous gift from Dr. Larry Fisher (National Institute of Dental and Craniofacial Research, NIH, Bethesda, MD) and were raised specifically against proα1(I) C-telopeptide, proα1(I) C-propeptide, and proα1(I) N-propeptide, respectively [Fisher et al., 1987, 1995]. Thus, LF-68 can detect α1(I) and all forms of proα1(I), while LF-9 and LF-42 distinguish between proα1(I) chains containing the N-propeptide or C-propeptide, respectively (Fig. 3A). Western Blot analysis of supernatants from control cells using LF-68 showed a major proα1(I) band and two minor intermediately processed proα1(I) forms referred as pCα1(I) and pNα1(I), each one of these being exclusively recognized by LF-42 or LF-9 (Fig. 3B and 3C). pCα1(I) has lost the aminopropeptide but retains the larger C-terminal propeptide, whereas pN1α(I) lacks the C-terminal propeptide and retains the N-propeptide (Fig. 3A). Compared to control cells, LF-68 analysis of supernatants from patient-derived cells demonstrated a significant decrease in the amount of pNα1(I) with respect to pCα1(I) in both patients, thus indicating diminished PICP-peptidase activity in homozygous mutant cells (Fig. 3B and C). In agreement with this, we also detected decreased concentration of free PICP in the supernatants of patient cells after probing an equivalent loaded gel made at a higher percentage of acrylamide with LF-42 (Fig. 3B). Nevertheless, pNα1(I) and free PICP were both detected in patient fibroblasts, and so there is residual PICP-peptidase activity from the mutant enzyme or from other proteases in the culture. Cell cultures derived from heterozygous individual II-2 were found slightly impaired in procollagen I C-terminal processing as the pCα1(I)/pNα1(I) ratio in LF-68 blots was increased in these cells (1.7) with respect to control fibroblasts (0.9) (Fig. 3B). Steady-state procollagen processing was additionally analyzed in fibroblast cultures. This showed diminished pNα1(I), pNα2(I) and fully processed α1(I) and α2(I) in the supernatants of patient fibroblasts compared to the heterozygous control culture, hence corroborating impaired processing of type I procollagen in patient cells (Supp. Figure S1A). Pepsin-treated procollagens purified from the medium and cell layers of both patient and heterozygous fibroblasts showed a normal migration patterns of collagen type I, III, and V, indicating lack of collagen overmodification (Supp. Figure S1B). Next, we checked whether the Phe249Leu mutation could trigger BMP1/mTLD degradation due to possible missfolding, however no significant differences in the intensity of bands corresponding to mTLD or BMP1 were detected between cell extracts from control and mutant fibroblasts on western blot analysis using an anti-BMP1 antibody (Supp. Figure S2).
Figure 3.

Phe249Leu impairs type I procollagen C-propeptide protease activity of BMP1/mTLD. A: Diagram representing unprocessed type I procollagen and intermediate variants following removal of the N or C-terminal propeptides. LF antibodies are shown underneath their corresponding epitope. B: Western blot analysis of supernatants from fibroblasts derived from patients II-3 and II-6, their heterozygous sibling (II-2), and an unrelated normal control (C) incubated with the indicated LF antibodies to monitor proteolytic processing of proα1(I). For each sample the volume of supernatant loaded per total amount of protein in the cell extracts of the corresponding culture was the same (15 μl, 14 μl, 15 μl, and 18 μl were loaded for samples C, II-3, II-6, and II-2, respectively). Analysis of proα1(I) variants is shown in the upper panel and free PICP analysis with LF-42 is in the lower panel. Compared to controls, patients have lower levels of pNα(I) and free PICP, while pCα1(I) is accumulated. Slight impairment of PICP-processing is detected in the heterozygous individual II-2. Quantification of the pCα1(I)/pNα1(I) ratio in each lane using densitometry data from two independent LF-68 blots is shown in the graph on the right. C: Western blot analysis of supernatants run in parallel and probed with the indicated antibodies demonstrating the propeptide composition of each of the intermediately processed proα1(I) bands. D: Western blot analysis of supernatants from NIH3T3 cell cultures overexpressing wild type mTLD (mTLD-wt) or Phe249Leu mTLD (mTLD-mut). Cells infected with the empty retroviral vector (pBABE-puro) were used as control (Control). 30 μl of supernatant from each culture with the same number of cells were loaded in each lane. Both LF-68 and LF-42 antibodies detected a dramatic decrease in unprocessed proα1(I) that is converted into a smaller size C-terminally processed variant when mTLD-wt is overexpressed. Since LF-9 does not crossreact with the murine N-propeptide, we could not use this antibody, and hence, the processed variant detected by LF-68 is assumed to correspond to pNα1(I) because of its size and because it is not recognized by LF-42. The amount of mTLD-wt and mTLD-mut in the media of the corresponding culture was demonstrated by reprobing the upper LF-blots with anti-BMP1. E: Western blot of supernatants from a control individual (C) and patient II-3 fibroblast cultures infected with retroviral vectors as in panel D and probed against LF antibodies. Only expression of mTLD-wt strongly improves PICP-protease activity both in patient and control cells as demonstrated by the augmented pNα1(I)/pCα1(I) ratio and reduction of unprocessed proα1(I). Levels of mTLD variants in the media were monitored with anti-BMP1.
Mutant mTLD Shows Impaired Proteolytic Activity
To confirm that the Phe249Leu substitution inhibits BMP1/mTLD-mediated PICP cleavage, we used puromycin-resistant retroviral vectors to deliver normal or mutant mTLD cDNA under control of a constitutive promoter into NIH3T3 mouse fibroblasts. Following viral infection, puromycin was added to the media to generate homogenous cultures, in which all surviving cells carry the recombinant vector integrated into their genome. After antibiotic selection, cells were plated at 8 × 105/p60 dish density and treated in the same way as described for human primary fibroblasts. Western blotting of serum-free supernatants using LF-68 and LF-42 showed that the vast majority of proα1(I) secreted into the media by NIH3T3 fibroblasts overexpressing wild-type mTLD, had lost the C-terminal propeptide and had been processed into a smaller size proα1(I) variant. In contrast, the supernatants of cells overexpressing mutant mTLD were found with similar levels of proα1(I) C-terminal processing as the control cells transduced with the empty retroviral vector, thus indicating that the Phe249Leu mutation causes strong reduction of the BMP1/mTLD proteolytic function and confirming the pathogenicity of the mutation (Fig. 3D). The presence of equivalent levels of normal or mutant mTLD in the media of NIH3T3 cultures was verified by reprobing the LF-68 and LF-42 blots with anti-BMP1 (Fig. 3D). We repeated the same experiment using retroviral-mediated expression of normal and mutant mTLD in human primary fibroblasts and found the same result with only expression of the wild-type protein being able to rescue the phenotype of mutant fibroblasts (Fig. 3E).
Discussion
Two OI patients have been recently reported by Lindahl et al., one with a dominant missense mutation in the COL1A1 C-propeptide cleavage site and another one with a dominant missense mutation in the C-propeptide cleavage site of COL1A2. Both patients were described with a mild phenotype including fractures, radiographic osteopenia, and wormian bones. Mild tibial bowing was found in one of the cases. Biochemical analysis of collagens in cells derived from these patients showed minimal alteration of the triple helix folding process, but delayed processing of type I procollagen secreted into the media, hence indicating that abnormal PICP processing is a cause of OI in humans [Lindahl et al., 2011]. As PICP is critical for the initiation of chain folding, these mutations differ from those placed in the C-terminal propeptide region that interferes with the formation of the triple helix [Chessler et al., 1993; Khoshnoodi et al., 2006]. The patients we report here have a more severe phenotype than the cleavage site mutation patients described by Lindahl et al. [2011]. This could be explained by the fact that deficient BMP1/mTLD function alters C-terminal processing of both procollagen (I) chains simultaneously leading to low levels of fully processed type I collagen, which in our patients according to the negligible BMP1/mTLD activity detected in the overexpression experiments, will be essentially depending on the activity of TLD-like proteins. Furthermore, since the Phe249Leu change lies within the BMP1/mTLD catalytic protease domain, it is highly probable that processing of other BMP1/mTLD substrates including fibrillar type II and III procollagens, activation of LOX zymogen, as well as processing of other secreted factors and extracellular matrix proteins is compromised in our patients [Ge and Greenspan, 2006]. In addition, the possibility of other genome variants contributing to the phenotype spectrum in this family cannot be ruled out.
Our data indicate that the phenotype resulting from diminished function of BMP1 in human diverges from that of Bmp1−/− mice. This is similar to the SERPINF1 situation, as no features resembling OI were described in SerpinF1-deficient mice [Becker et al., 2011; Doll et al., 2003]. Bmp1 homozygous knockout mice are perinatal lethal and although it is unknown whether they could have developed OI later in life, they do not show gross skeletal abnormalities at the time of birth excepting for reduced ossification of some skull bones [Suzuki et al., 1996]. In contrast, patient II-6 was detected with femoral bowing during intrauterine examination and bone fractures were noted at birth in both patients. C-terminal processing of type I procollagen secreted into the media by Bmp1−/− mouse embryonic fibroblasts (MEFs) was demonstrated to be reduced, nevertheless these cells were still producing fully processed type I collagen chains [Suzuki et al., 1996]. We also detected residual PICP-protease activity in the fibroblasts from our patients, however further experiments comparing human and mouse cells under the same methodology are necessary to understand whether the remaining PICP-protease activity in mutant human fibroblasts is lower than that of the Bmp1 knockout cells. PICP processing was nearly undetectable in double knockout Bmp1−/−;Tll1−/− fibroblasts and thus Tll1 is the protein designated to be accountable for the remaining PICP activity of Bmp1−/− MEFs. Bmp1−/−;Tll1−/− mice are embryonic lethal owing to cardiac failure and evaluation of skeletal abnormalities in this genotype was not achieved [Pappano et al., 2003]. A possible explanation for the human/mouse phenotypic differences could be a more essential role for BMP1/mTLD in protein processing in the human skeletal system than in mice, or in other words TLD-like proteases could compensate less in the human skeleton. It is also possible that the process of collagen fibril assembly in mice is more resilient to the presence of unprocessed type I procollagen molecules. There is however one phenotypic aspect in common between Bmp1 knockout mice and the human phenotype. Bmp1−/− mice were reported with consistent herniation of the gut and both the proband and her affected brother have large umbilical hernias with intestinal content. It was suggested that this mouse phenotype could be due to lack of tensile strength in the ventral body wall secondary to abnormal collagen fibril formation. Additionally, since BMP1 is involved in processing dorso-ventral patterning factors such as Chordin, this could also play a role in this particular phenotypic feature [Ge and Greenspan, 2006; Suzuki et al., 1996]. The broad function of BMP1 includes processing of type III and type V procollagens that are associated with specific forms of EDS ([EDS type III–IV; MIM#s 130020, 130050] and [EDS type I–II; MIM#s 130000, 130010] linked to collagen III and V, respectively). Our patients lack the skin increased stretchability and fragility and the joint hyperextensibility characteristic of EDS but have in common blue sclera, kyphoscoliosis, and presence of hernia that in EDS is usually inguinal but umbilical in our cases.
In summary, we describe a mutation associated to severe AR-OI (OI type III) in an additional gene, BMP1, which extends further the list of genes that are mutated in this heterogeneous disorder.
Supplementary Material
Acknowledgments
This work was supported by the Spanish Ministry of Science and Innovation SAF-17901 and the CIBERER Programa de Investigación de Enfermedades Pediátricas. We are very grateful to our patients and their family who were very cooperative during this study. We also wish to thank the National Society of Human Genetics in Egypt, which provided the biphosphonate injections for the patients. We thank Dr. Larry Fisher (NIH, Bethesda, MD) for the kind gift of LF-9, LF-42, and LF-68 antibodies that were so useful in this study and Dr. Ignacio Palmero (IIB, CSIC-UAM) for helping with the retroviral infection technique. The technical assistance of Angelika Schwarze is kindly acknowledged. Biochemical analysis of collagens was supported by a grant from the Gottfried und Julia Bangerter-Rhyner Stiftung to C.G. and M.R. M.A. and S.T. analyzed data, performed diagnoses, and phenotyping, V. M-G., J.T., V.P, P.L., and V.L.R-P. conducted genetic and mutational studies, J.A.C-M., M.V., and V.L.R-P performed procollagen functional analysis on primary fibroblasts and retroviral experiments; D.E. and M.R contributed to the discussion and interpretation of experiments; and C.G. and U.L. carried out procollagen biochemical analyses. P.L. and V.L.R.-P. led the group. All authors contributed to the writing of the manuscript.
Contract grant sponsors: Spanish Ministry of Science and Innovation (SAF-17901); CIBERER Programa de Investigación de Enfermedades Pediátricas; Gottfried und Julia Bangerter-Rhyner Stiftung (to C.G. and M.R.).
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- Alanay Y, Avaygan H, Camacho N, Utine GE, Boduroglu K, Aktas D, Alikasifoglu M, Tuncbilek E, Orhan D, Bakar FT, Zabel B, Superti-Furga A, Bruckner-Tuderman L, Curry CJ, Pyott S, Byers PH, Eyre DR, Baldridge D, Lee B, Merrill AE, Davis EC, Cohn DH, Akarsu N, Krakow D. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. 2010;86:551–559. doi: 10.1016/j.ajhg.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes AM, Chang W, Morello R, Cabral WA, Weis M, Eyre DR, Leikin S, Makareeva E, Kuznetsova N, Uveges TE, Ashok A, Flor AW, Mulvihill JJ, Wilson PL, Sundaram UT, Lee B, Marini JC. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N Engl J Med. 2006;355:2757–2764. doi: 10.1056/NEJMoa063804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker J, Semler O, Gilissen C, Li Y, Bolz HJ, Giunta C, Bergmann C, Rohrbach M, Koerber F, Zimmermann K, de Vries P, Wirth B, Schoenau E, Wollnik B, Veltman JA, Hoischen A, Netzer C. Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. 2011;88:362–371. doi: 10.1016/j.ajhg.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond JS, Beynon RJ. The astacin family of metalloendopeptidases. Protein Sci. 1995;4:1247–1261. doi: 10.1002/pro.5560040701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonod-Bidaud C, Beraud M, Vaganay E, Delacoux F, Font B, Hulmes DJ, Ruggiero F. Enzymatic cleavage specificity of the proalpha1(V) chain processing analysed by site-directed mutagenesis. Biochem J. 2007;405:299–306. doi: 10.1042/BJ20070051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers PH, Steiner RD. Osteogenesis imperfecta. Annu Rev Med. 1992;43:269–282. doi: 10.1146/annurev.me.43.020192.001413. [DOI] [PubMed] [Google Scholar]
- Cabral WA, Chang W, Barnes AM, Weis M, Scott MA, Leikin S, Makareeva E, Kuznetsova NV, Rosenbaum KN, Tifft CJ, Bulas DI, Kozma C, Smith PA, Eyre DR, Marini JC. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet. 2007;39:359–365. doi: 10.1038/ng1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canty EG, Kadler KE. Procollagen trafficking, processing and fibrillogenesis. J Cell Sci. 2005;118(Pt 7):1341–1353. doi: 10.1242/jcs.01731. [DOI] [PubMed] [Google Scholar]
- Colige A, Sieron AL, Li SW, Schwarze U, Petty E, Wertelecki W, Wilcox W, Krakow D, Cohn DH, Reardon W, Byers PH, Lapière CM, Prockop DJ, Nusgens BV. Human Ehlers-Danlos syndrome type VII C and bovine dermatosparaxis are caused by mutations in the procollagen I N-proteinase gene. Am J Hum Genet. 1999;65:308–317. doi: 10.1086/302504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colige A, Vandenberghe I, Thiry M, Lambert CA, Van Beeumen J, Li SW, Prockop DJ, Lapiere CM, Nusgens BV. Cloning and characterization of ADAMTS-14, a novel ADAMTS displaying high homology with ADAMTS-2 and ADAMTS-3. J Biol Chem. 2002;277:5756–5766. doi: 10.1074/jbc.M105601200. [DOI] [PubMed] [Google Scholar]
- Chessler SD, Wallis GA, Byers PH. Mutations in the carboxyl-terminal propeptide of the pro alpha 1(I) chain of type I collagen result in defective chain association and produce lethal osteogenesis imperfecta. J Biol Chem. 1993;268(24):18218–18225. [PubMed] [Google Scholar]
- Christiansen HE, Schwarze U, Pyott SM, AlSwaid A, Al Balwi M, Alrasheed S, Pepin MG, Weis MA, Eyre DR, Byers PH. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am J Hum Genet. 2010;86:389–398. doi: 10.1016/j.ajhg.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15:7–12. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- den Dunnen JT, Paalman MH. Standardizing mutation nomenclature: why bother? Hum Mutat. 2003;22:181–182. doi: 10.1002/humu.10262. [DOI] [PubMed] [Google Scholar]
- Doll JA, Stellmach VM, Bouck NP, Bergh AR, Lee C, Abramson LP, Cornwell ML, Pins MR, Borensztajn J, Crawford SE. Pigment epithelium-derived factor regulates the vasculature and mass of the prostate and pancreas. Nat Med. 2003;9:774–780. doi: 10.1038/nm870. [DOI] [PubMed] [Google Scholar]
- Drogemuller C, Becker D, Brunner A, Haase B, Kircher P, Seeliger F, Fehr M, Baumann U, Lindblad-Toh K, Leeb T. A missense mutation in the SERPINH1 gene in Dachshunds with osteogenesis imperfecta. PLoS Genet. 2009;5:e1000579. doi: 10.1371/journal.pgen.1000579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes RJ, Hirohata S, Engle JM, Colige A, Cohn DH, Eyre DR, Apte SS. Procollagen II amino propeptide processing by ADAMTS-3. Insights on dermatosparaxis. J Biol Chem. 2001;276:31502–31509. doi: 10.1074/jbc.M103466200. [DOI] [PubMed] [Google Scholar]
- Fisher LW, Hawkins GR, Tuross N, Termine JD. Purification and partial characterization of small proteoglycans I and II, bone sialoproteins I and II, and osteonectin from the mineral compartment of developing human bone. J Biol Chem. 1987;262:9702–9708. [PubMed] [Google Scholar]
- Fisher LW, Stubbs JT, 3rd, Young MF. Antisera and cDNA probes to human and certain animal model bone matrix noncollagenous proteins. Acta Orthop Scand Suppl. 1995;266:61–65. [PubMed] [Google Scholar]
- Ge G, Greenspan DS. Developmental roles of the BMP1/TLD metalloproteinases. Birth Defects Res C Embryo Today. 2006;78:47–68. doi: 10.1002/bdrc.20060. [DOI] [PubMed] [Google Scholar]
- Guevara T, Yiallouros I, Kappelhoff R, Bissdorf S, Stocker W, Gomis-Ruth FX. Proenzyme structure and activation of astacin metallopeptidase. J Biol Chem. 2010;285:13958–13965. doi: 10.1074/jbc.M109.097436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins DR, Keles S, Greenspan DS. The bone morphogenetic protein 1/Tolloid-like metalloproteinases. Matrix Biol. 2007;26(7):508–523. doi: 10.1016/j.matbio.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa Y, Wirz J, Vranka JA, Nagata K, Bachinger HP. Biochemical characterization of the prolyl 3-hydroxylase 1.cartilage-associated protein.cyclophilin B complex. J Biol Chem. 2009;284:17641–17647. doi: 10.1074/jbc.M109.007070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler E, Takahara K, Biniaminov L, Brusel M, Greenspan DS. Bone morphogenetic protein-1: the type I procollagen C-proteinase. Science. 1996;271:360–362. doi: 10.1126/science.271.5247.360. [DOI] [PubMed] [Google Scholar]
- Khoshnoodi J, Cartailler JP, Alvares K, Veis A, Hudson BG. Molecular recognition in the assembly of collagens: terminal noncollagenous domains are key recognition modules in the formation of triple helical protomers. J Biol Chem. 2006;281:38117–38121. doi: 10.1074/jbc.R600025200. [DOI] [PubMed] [Google Scholar]
- Lapunzina P, Aglan M, Temtamy S, Caparros-Martin JA, Valencia M, Leton R, Martinez-Glez V, Elhossini R, Amr K, Vilaboa N, Ruiz-Perez VL. Identification of a frameshift mutation in Osterix in a patient with recessive osteogenesis imperfecta. Am J Hum Genet. 2010;87:110–114. doi: 10.1016/j.ajhg.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl K, Barnes AM, Fratzl-Zelman N, Whyte MP, Hefferan TE, Makareeva E, Brusel M, Yaszemski MJ, Rubin CJ, Kindmark A, Roschger P, Klaushofer K, McAlister WH, Mumm S, Leikin S, Kessler E, Boskey AL, Ljunggren O, Marini JC. COL1 C-propeptide cleavage site mutations cause high bone mass osteogenesis imperfecta. Hum Mutat. 2011;32:598–609. doi: 10.1002/humu.21475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makareeva E, Aviles NA, Leikin S. Chaperoning osteogenesis: new protein-folding disease paradigms. Trends Cell Biol. 2011;21:168–176. doi: 10.1016/j.tcb.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morello R, Bertin TK, Chen Y, Hicks J, Tonachini L, Monticone M, Castagnola P, Rauch F, Glorieux FH, Vranka J, Bächinger HP, Pace JM, Schwarze U, Byers PH, Weis M, Fernandes RJ, Eyre DR, Yao Z, Boyce BF, Lee B. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127:291–304. doi: 10.1016/j.cell.2006.08.039. [DOI] [PubMed] [Google Scholar]
- Pappano WN, Steiglitz BM, Scott IC, Keene DR, Greenspan DS. Use of Bmp1/Tll1 doubly homozygous null mice and proteomics to identify and validate in vivo substrates of bone morphogenetic protein 1/tolloid-like metalloproteinases. Mol Cell Biol. 2003;23:4428–4438. doi: 10.1128/MCB.23.13.4428-4438.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch F, Glorieux FH. Osteogenesis imperfecta. Lancet. 2004;363:1377–1385. doi: 10.1016/S0140-6736(04)16051-0. [DOI] [PubMed] [Google Scholar]
- Scott IC, Blitz IL, Pappano WN, Imamura Y, Clark TG, Steiglitz BM, Thomas CL, Maas SA, Takahara K, Cho KW, Greenspan DS. Mammalian BMP-1/Tolloid-related metalloproteinases, including novel family member mammalian Tolloid-like 2, have differential enzymatic activities and distributions of expression relevant to patterning and skeletogenesis. Dev Biol. 1999;213:283–300. doi: 10.1006/dbio.1999.9383. [DOI] [PubMed] [Google Scholar]
- Shimell MJ, Ferguson EL, Childs SR, O’Connor MB. The Drosophila dorsalventral patterning gene tolloid is related to human bone morphogenetic protein 1. Cell. 1991;67:469–481. doi: 10.1016/0092-8674(91)90522-z. [DOI] [PubMed] [Google Scholar]
- Sillence DO, Rimoin DL, Danks DM. Clinical variability in osteogenesis imperfecta-variable expressivity or genetic heterogeneity. Birth Defects Orig Artic Ser. 1979;15:113–129. [PubMed] [Google Scholar]
- Steinmann B, Rao VH, Vogel A, Bruckner P, Gitzelmann R, Byers PH. Cysteine in the triple-helical domain of one allelic product of the alpha 1(I) gene of type I collagen produces a lethal form of osteogenesis imperfecta. J Biol Chem. 1984;259:11129–11138. [PubMed] [Google Scholar]
- Suzuki N, Labosky PA, Furuta Y, Hargett L, Dunn R, Fogo AB, Takahara K, Peters DM, Greenspan DS, Hogan BL. Failure of ventral body wall closure in mouse embryos lacking a procollagen C-proteinase encoded by Bmp1, a mammalian gene related to Drosophila tolloid. Development. 1996;122:3587–3595. doi: 10.1242/dev.122.11.3587. [DOI] [PubMed] [Google Scholar]
- Takahara K, Brevard R, Hoffman GG, Suzuki N, Greenspan DS. Characterization of a novel gene product (mammalian tolloid-like) with high sequence similarity to mammalian tolloid/bone morphogenetic protein-1. Genomics. 1996;34:157–165. doi: 10.1006/geno.1996.0260. [DOI] [PubMed] [Google Scholar]
- Takahara K, Lyons GE, Greenspan DS. Bone morphogenetic protein-1 and a mammalian tolloid homologue (mTld) are encoded by alternatively spliced transcripts which are differentially expressed in some tissues. J Biol Chem. 1994;269:32572–32578. [PubMed] [Google Scholar]
- van Dijk FS, Nesbitt IM, Zwikstra EH, Nikkels PG, Piersma SR, Fratantoni SA, Jimenez CR, Huizer M, Morsman AC, Cobben JM, van Roij MH, Elting MW, Verbeke JI, Wijnaendts LC, Shaw NJ, Högler W, McKeown C, Sistermans EA, Dalton A, Meijers-Heijboer H, Pals G. PPIB mutations cause severe osteogenesis imperfecta. Am J Hum Genet. 2009;85:521–527. doi: 10.1016/j.ajhg.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildeman M, van Ophuizen E, den Dunnen JT, Taschner PE. Improving sequence variant descriptions in mutation databases and literature using the mutalyzer sequence variation nomenclature checker. Hum Mutat. 2008;29:6–13. doi: 10.1002/humu.20654. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.