

Abstract
Retinal photoreceptor degeneration takes many forms. Mutations in rhodopsin genes or disorders of the retinal pigment epithelium, defects in the adenosine triphosphate binding cassette transporter, ABCR gene defects, receptor tyrosine kinase defects, ciliopathies and transport defects, defects in both transducin and arrestin, defects in rod cyclic guanosine 3′,5′-monophosphate phosphodiesterase, peripherin defects, defects in metabotropic glutamate receptors, synthetic enzymatic defects, defects in genes associated with signaling, and many more can all result in retinal degenerative disease like retinitis pigmentosa (RP) or RP-like disorders. Age-related macular degeneration (AMD) and AMD-like disorders are possibly due to a constellation of potential gene targets and gene/gene interactions, while other defects result in diabetic retinopathy or glaucoma. However, all of these insults as well as traumatic insults to the retina result in retinal remodeling. Retinal remodeling is a universal finding subsequent to retinal degenerative disease that results in deafferentation of the neural retina from photoreceptor input as downstream neuronal elements respond to loss of input with negative plasticity. This negative plasticity is not passive in the face of photoreceptor degeneration, with a phased revision of retinal structure and function found at the molecular, synaptic, cell, and tissue levels involving all cell classes in the retina, including neurons and glia. Retinal remodeling has direct implications for the rescue of vision loss through bionic or biological approaches, as circuit revision in the retina corrupts any potential surrogate photoreceptor input to a remnant neural retina. However, there are a number of potential opportunities for intervention that are revealed through the study of retinal remodeling, including therapies that are designed to slow down photoreceptor loss, interventions that are designed to limit or arrest remodeling events, and oplogenetic approaches that target appropriate classes of neurons in the remnant neural retina.
Keywords: Retinal remodeling, Retina, Retinal degeneration, Retinitis pigmentosa, Macular degeneration
Introduction
Retinal remodeling as a consequence of retinal degenerative disease is an unavoidable phenomenon. A large number of investigators are engaged in exploring neural retinal remodeling as well as the therapeutic implications of retinal degenerations. The consensus is that the neural retina becomes reactive and is not passive in the face of photoreceptor degenerations. Furthermore, the substantial alterations now recognized as retinal remodeling occur from the molecular up through the synaptic, cellular, and tissue levels [1–24], involving not only photoreceptors but all of the cell populations in the neural retina [1, 7, 9, 13, 18, 20–23, 25, 26].
Photoreceptor degeneration takes many forms that occur naturally in disease [27] and in conditions as far apart as trauma [28], the myriad forms of retinitis pigmentosa (RP) and RP-like disorders that are manifested through mutations in rhodopsin genes [16, 27, 29, 30], disorders of the retinal pigment epithelium (RPE) in Leber’s congenital amaurosis [31–33], defects in the ATP-binding cassette transporter seen in Stargardt’s disease [34–36], ABCR gene defects [37, 38], receptor tyrosine kinase defects [32, 39, 40], ciliopathies and transport defects [41, 42], defects in both transducin and arrestin [43–46], defects in rod cGMP phosphodiesterase [47–49], peripherin defects [50], defects in metabotropic glutamate receptors (mGluRs) [51, 52], synthetic enzymatic defects [53, 54], defects in genes associated with signaling [55–58], age-related macular degeneration (AMD) disorders [34, 36, 59–70], defects in photoreceptor function incriminating photoreceptor degeneration in diabetic retinopathy [71], glaucoma [72], and others. The fundamental biological reality is that, regardless of the initiator of the retinal insult or degeneration, if afferent activity from the photoreceptors is lost, the neural retina responds in a dynamic fashion by altering the function and connectivity of the remaining cells at the tissue, cellular, and molecular levels.
Retinal remodeling
Defects that result in the loss of photoreceptor input to the neural retina initiate an event cascade that forever alters neural retinal structure as well as the pharmacologic response profiles of neurons in the retina and their connectivities. Retinal remodeling occurs in all forms of retinal degeneration, which is considered a true neural retinal deafferentation, passing through three phases of structural and functional revision. The first change is often subtle, but has severe implications for the physiology of the neural retina. During phase 1 remodeling in cone-sparing forms of retinal disease, the earliest forms of pathology are revealed by an initiation of photoreceptor stress that induces a cascade of events that culminates in molecular changes and eventual cell death. Cell stress is clinically occult and occurs prior to photoreceptor cell death, with one of the earliest histological indications revealed initially through both rod and cone opsin delocalization (Fig. 1). Rhodopsin delocalization in many cases extends from the inner segments of photoreceptors down to the cell membrane in processes that extend into the inner nuclear layer and ganglion cell layers [73]. Further, molecular alterations of bipolar cell dendritic glutamate receptor expression in phase 1 begins to alter the pharmacology of bipolar cells, shifting their functional phenotypes from ON responses to OFF responses [26, 74]. These earliest phases of retinal remodeling are likely clinically occult and occur prior to photoreceptor cell death [11].
Fig. 1.

Human retina from a male patient with retinitis pigmentosa. Rod opsins reveal dramatic shortening of outer segments in a. Rod and LWS cone opsins are shown in phase one of retinal degeneration a and b. demonstrating photoreceptor stress through opsin delocalization from where it should normally be found in the outer segments: it now extends to the inner segments and the inner plexiform layer. Scale bar 10 μm
Remodeling continues in phase 2 with photoreceptor death and phagocytic ablation of photoreceptor cell bodies. Additionally. Müller cell hypertrophy and the collapse of the distal scaffolding of the Müller cells in the absence of photoreceptor and bipolar cells form the Müller cell seal that isolates the neural retina from the RPE and choroid [7, 11, 24].
Bipolar cells are completely deafferented in phase 2: not only physiologically, through the elimination of glutamate receptors in the outer plexiform layer, but also anatomically, through the physical retraction of all bipolar cell dendrites, resulting in altered morphologies of bipolar cells (Figs. 2, 3). This results in the complete loss of glutamatergic input after the degeneration of rod photoreceptors, as shown in the rd mouse [75]. These changes in the rd/rd mouse also include the expression of aberrant ionotropic glutamate receptors (iGluR) on ON cone bipolar cells from postnatal day 15 (PND 15), poor functional activation of metabotropic glutamate receptors (mGluR) on both rod and ON cone bipolar cells throughout development, and the degenerative process and poor functional activation of N-methyl-D-aspartate (NMDA) receptors on amacrine cells from PND 15 animals [76].
Fig. 2.
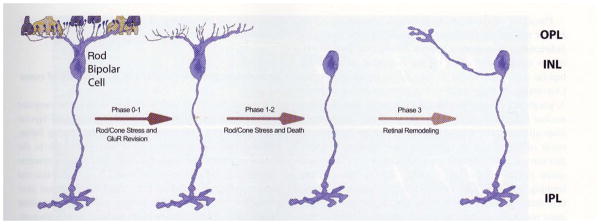
In models of RP where rods and cones die simultaneously, bipolar cells lose dendrites and all iGluR/mGluR responsivity. In phase 0–1, rod bipolar cells downregulate GluR expression in the dendrites. In phase 1–2, rod and cone photoreceptor stress and death occur while dendritic modules are lost. In phase 3, wider retinal remodeling ensues, resulting in sprouting and formation of new axonal modules
Fig. 3.

In models of RP where cones outlive rods, some bipolar cell dendrites switch targets. In normal rod bipolar cell architecture, dendrites from rod bipolar cells bypass cone pedicles. In phase 0–1, as in rod/cone dystrophies, rod bipolar- cells downregulate GluR expression in the dendrites. In phase 1–2 with rod stress and death, dendritic modules are lost. In late phase 2/phase 3, some rod bipolar cells form sprouts that contact cone pedicles, making peripheral contacts on cone pedicles
Retinitis pigmentosa disorders occur in both cone-sparing and cone-decimating forms. In models of RP where rods and cones die within similar time domains, retinas undergo large-scale remodeling events where bipolar cells lose dendrites and all iGluR and mGluR responsivity. Glial seal formation occurs, as does large-scale neuritogenesis leading to microneuroma formation, as well as neuronal migration [24]. In RP models where cones outlive rods (Fig. 3), some bipolar cell dendrites become involved in target switching, as shown by the fact that rod bipolar cells receive ectopic synapses from cones in the absence of rods [77], though cones may persist for some time, with Müller cells engaging in seal formation around them [24, 74]. As long as cones or remnant cone cell bodies are present, bipolar cells underneath the cone photoreceptors appear to persist and progression of the neural retina into phase 3 appears to be arrested or dramatically slowed. Other retinal changes in cone-sparing retinal degenerations in phase 2 involve the initiation of anomalous sprouting, which often coalesces into structures called microneuromas, with contributions from not only bipolar cells but also other neuronal cell classes, including amacrine cells and horizontal cells, which can be observed to extend processes down into the inner plexiform layer [20–22, 74, 78].
Phase 3 is characterized by persistent remodeling, which further revises the fundamental topology of the retina via bidirectional migration of neurons through the vertical axis of the retina, as evinced by the migration of surviving bipolar and amacrine cells into the ganglion cell layer. Conversely, in many advanced degenerate retinas in phase 3, ganglion cells can be observed migrating into the inner nuclear layer [7, 9, 11, 12, 24]. The evolution of processes from all remaining neuron types in the retina continues to occur in phase 3, with some processes forming fascicles that can run in bundles for great distances within the neural retina (>100 microns) [24], while others run together forming tangles or tufts of novel neuropils termed microneuromas, which form outside the normal lamination of the inner plexiform layer [7, 8, 11, 12, 24, 26, 79]. Pigmented bone spicules (Fig. 4) are a common finding in patients with RP, and are often translocated into the neural retina [80]. Pigmented bone spicules appear to co-segregate with Müller cell columns, which appear to mediate much of the gross topological restructuring and are responsible for the translocation of RPE invasion of the neural retina (Fig. 4), as well as the formation of the aforementioned pigmented bone spicules.
Fig. 4.
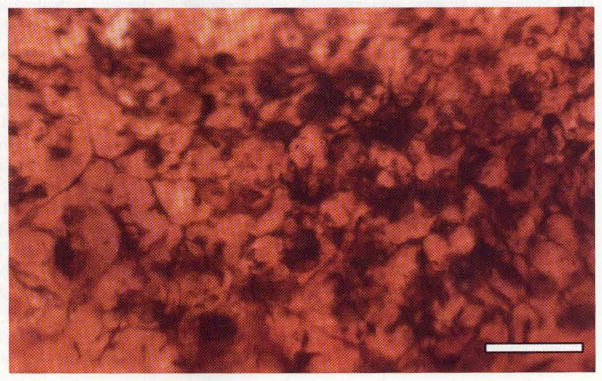
Retina from a human patient with advanced RP, illustrating pigmented bone spicules—accumulations of RPE pigment granules that derive from translocations of Müller cells, which alter the topology of the neural retina and cause the accumulation of pigment along clumps, lines, and grooves in the vertical axis of the neural retina. Scale bar 200 μm
As phase 3 remodeling progresses, breaks in Bruch’s membrane provide opportunities for some neurons to emigrate out of the neural retina proper and into the membrane choriocapillaris complex [13] (Fig. 5). Whether or not similar emigration events out of the neural retina appear in human diseases like AMD is not clear, though it seems likely in AMD and AMD-like disorders. Certainly in the late forms of AMD with vascular involvement, there are breaches of Bruch’s membrane, but other evidence indicates that Bruch’s membrane may become calcified following the breakdown of elastin and collagen in non-vascular or dry AMD [81].
Fig. 5.

Neural emigration in a light damage model of retinal degeneration in rat. Taurine, glutamine, glycine::r, g, b mapping in a demonstrates Müller glia with glycinergic amacrine cells embedded in them passing outside of the retina through a break (box) in Bruch’s membrane. GABA, glycine, glutamate::r, g, b mapping in b demonstrates both GABAergic and glycinergic amaerine cells in addition to bipolar cells escaping from the neural retina into the choroid (arrows). Scale bar 90 μm
Remodeling process variation in the presence of cones
While total loss of all photoreceptors leads to the complete loss of functional iGluR expression in ON and OFF bipolar cells, patches of cones may persist in cone-sparing forms, but they are substantially altered and embedded in the Müller cell seal. These altered cones still possess synaptic ribbons [82], but a more recent work [26] revealed that rod bipolar cells can withdraw their dendrites from rod photoreceptor terminals, subsequently elaborating transient ectopic non-ribbon contacts with surviving cone photoreceptors. Bipolar cells initiate these changes by retracting dendrites upon the loss of rod and cone contact [20–22], followed by a loss of signaling capacity in response to glutamate in regions of photoreceptor loss [75] (Figs. 2. 3). While RP disorders occur in both cone-sparing and cone-decimating forms, in cone-sparing RP. patches or isolated cones can persist for some time and appear to prolong the onset of gross topographic retinal remodeling. It is important to note that changes to network topology and iGluR/mGluR6 expression do occur in response to cone loss. Specifically, the retina underneath any remaining cone pedicles appears to be relatively preserved topologically, along with the seeming preservation of circuitry and iGluR expression in bipolar cell dendrites. However, proximal cones within 50 μm are fundamentally altered, with no iGluR signaling capacity remaining [26].
Neural information processing in the retina is immediately impacted by retinal photoreceptor degeneration and subsequent remodeling. The structural, biological, and information processing consequences of rod bipolar cell rewiring alone are substantial (Figs. 2, 6). The normal flow of information in the retina begins in the photoreceptors, where rod photoreceptor terminals bypass cone photoreceptor pedicles to make synapses on rod bipolar cells at their terminal spherules. Normally, hyperpolarizing signals from rod photoreceptors depolarize rod bipolar cells via sign-inverting mGluR6 receptors. Rod bipolar cells then “piggyback” onto the ON cone bipolar cell pathway through synaptic contact onto glycinergic amacrine cells that mediate signaling through to the ON cone bipolar cells via gap junctions, resulting in a sign-conserving signal, and to the OFF cone bipolar cell pathway via sign-inverting glycine receptors (Fig. 6). In cone-sparing RP, bipolar cell recordings reveal wholesale phenotype class switching from rod bipolar cells to OFF bipolar cell populations through alterations in the iGluR populations, effectively reprogramming the retina by inverting rod bipolar cell to amaerine cell to cone bipolar cell pathways, creating networks that are unable to maintain coherent signaling out of the retina [26] (Fig. 6). Additionally, from a circuit topology perspective, problems are obvious. In cone-sparing RP, the remaining cone pedicles are not numerous enough to accommodate all remaining rod bipolar cells. Furthermore, there aren’t enough ribbons to accommodate rod BC dendrites, and the ON cone BCs already occupy the existing ribbon contacts. Those ectopic contacts that are formed are too far away from the ribbon to activate the existing mGluR6 receptors. Therefore, each cell type expresses receptors that are matched to concentrations that prevail at a given distance from the ribbon in normal retinas at least. If rod BC mGluR6 receptors are not activated, then rod BCs should be persistently depolarized, which presents an additional problem for visual processing.
Fig. 6.

Circuit diagram: rods provide sign-inverting input to ON bipolar cells via mGluR6-mediated synaptic connections. OFF bipolar cells are driven by cones through sign-conserving KA-mediated iGluRs, while ON bipolar cells are driven by cones through sign-inverting mGluR6-mediated synapses. Cone bipolar cells also synapse upon ganglion cells with sign-conserving AMPA-mediated iGluRs that define ON and OFF channels of information flow out of the retina. Information flow through the retina in humans during scotopic light levels is driven by rods and rod bipolar cells that piggyback onto intermediary amacrine cells, which shunt the flow of information to ON cone bipolar cells via sign-conserving gap junctions. Signaling to OFF cone bipolar cells occurs through sign-inverting glycinergic conventional synapses. In sum, a flash of light hyperpolarizes rod photoreceptors, and the network preserves the ON and OFF channel polarities. However, in cone-sparing RP seen in human and animal models, network flows arc compromised through the formation of pathological networks that generate conflicting signaling driven by the remaining cones. With cone degeneration, this cone signaling is eventually lost, and retinas are driven by signaling intrinsic to the remnant neural retinal amacrine and ganglion cells
Human diseases and animal models of retinal degeneration that show remodeling
Human tissues from patients with late-stage RP show dramatic changes to the normal architecture of the retina (Fig. 7a, b). These samples demonstrate complete loss of rod and cone photoreceptors, bipolar cell loss, and subsequent topological restructuring of the retina. YGE > rgb imaging (Fig. 7a) reveals novel tufts of neuropils, termed microneuromas [24], with amaerine cells abutting the choroid and no barrier in-between due to the absent RPE. TQE > rgb signals demonstrate the formation and elaboration of the Müller cell seal isolating the neural retina (Fig. 7b). The subretinal space in these tissues is absent and impacts potential rescue strategies. In fact, because of the adherent nature of the glial seal, experimental retinal detachments of retinas in the regions of a Müller cell seal are almost impossible without some degree of trauma to the neural retina.
Fig. 7.

Imaging of human retina from late-stage RP. GABA, glycine, glutamate::r, g, b mapping in a reveals novel tufts of neuropil, termed microneuromas (box), with amacrine cells abutting Bruch’s membrane and the choroid. b Taurine, glutamine, glycine::r, g, b mapping of the same region demonstrates Müller cell revision and Müller cell seal formation (arrows), walling off the neural retina. Scale bar 90 μm
One might presume that since photoreceptors are lost in retinal degenerative diseases, the neural retina would be inactive in the absence of any afferent input. However, the sprouting of the neuronal processes and the alteration of the topology of the retina beg the question of function. It turns out that, even though photoreceptors are lost in regions of retinal atrophy, the neurons that are left in the retina continue to signal. Previous work examining the intrinsic activity of the rdcl mouse revealed dramatic levels of activity in the amacrine and ganglion cells, even though the bipolar cells were silent [26], while traditional electrophysiologic methods by Margolis and Detwiler [83] in the rdl mouse reveal substantial increases in oscillatory spike activity in the ganglion cells. Prior work by Pu et al. [17] in the RCS rat as well as Stasheff [19] in the rdl mouse and Sekirnjak et al. [84] in the P23H rat using single unit recording also shows substantial increases in the numbers of spontaneous ganglion cell spikes, despite the loss of photoreceptors to drive retinal activity. While results from single unit recording have yet to be correlated to retinas from human patients with RP, other recording methodologies were able to correlate retinal, specifically bipolar cell activity in both human and animal models of RP, which revealed identical disease mechanisms in both human and models of cone-sparing RP.
By combining excitation mapping with computational molecular phenotyping (CMP) [10], activated neurons (whether by ligands or light pathways) can be visualized ex vivo using small molecular probes. Simultaneous imaging of an activity probe, 1-amino-4-guanidobutane (AGB, functioning as a nonselective marker of ionotropic glutamatergic activity), combined with the visualization of other intrinsic small molecular signals [10], allows for the interrogation of neuronal cell classes that have been activated by either endogenous light drive or by pharmacologic activation (Fig. 8). Using CMP to segment images based upon their intrinsic small molecular signals allows the simultaneous extraction of excitation histograms revealed by AGB permeation for each class of bipolar cell, allowing the discrimination of OFF bipolar cell classes from ON bipolar cell classes. Additionally, ON bipolar cell classes can be segmented into rod and cone classes, allowing the elucidation of response profiles to AMPA [26, 74].
Fig. 8.

Excitation recording with KA and AGB in horizontal sections through the bipolar cell layer of a 12-week old TgP347L rabbit retina visualized with glycine, AGB, glutamate::r, g, b mapping, showing that most (~82 %)of the bipolar cells have phenotypically switched from rod (diamonds) and ON-like BC (squares) responses to OFF-like BC (triangles) responses, revealing a fundamental molecular reprogramming of response states. Scale bar 60 μm
In remodeling retinas, amacrine and ganglion cells continue to encode signals via iGluR receptor complexes. This is an important finding, as identical findings in the mouse, rabbit, and human [26, 74] in relation to cone-sparing RPs demonstrate that after CMP segmentation, the incidence of OFF-like iGluR-expressing bipolar cells doubles in cone-sparing RP, while the incidence of remnant rod bipolar cells drops nearly tenfold. However. there appears to be no detectable bipolar cell death. leading to the conclusion that most rod bipolar cells make new contacts and begin to express iGluRs, leading to significant implications for retinal wiring. In these cases where cones outlive rods, the rod BCs actively down-regulate the expression of dendritic glutamate receptor modules and retarget them to cone pedicles. The outcome is mGluR6 to iGluR phenotype switching and substantial corruption of cone pathway signaling. Conversely, in RP disorders where both rod and cones die concurrently, all BCs engage in the disassembly of iGluR modules in the dendrites, repress GluR expression, and form supernumerary axons in combination with substantial neuronal death. Mixed cone-decimating and cone-sparing zones are also possible in both human RP and animal models [26, 74].
The spectrum of diseases in humans that include retinal degeneration and subsequent remodeling is broader than widely considered. Studies show that RP and animal models of RP demonstrate and duplicate features of human retinal degeneration and remodeling. Mouse, rat, pig, dog, cat, and rabbit models of retinal degeneration are extensively documented [7–9, 11–13, 20–22, 24, 26, 74, 85–90]. Porcine P23H models of RP show a progressive loss of photoreceptors (Fig. 9a–d) with a concomitant loss of visual percepts as measured by ERG [91]. Additionally, these porcine models demonstrate aberrant Müller cell signatures (Fig. 9a) identical to those observed in human (Figs. 7b, 12b, inset), rodent, and rabbit (Fig. 10b) [74].
Fig. 9.

Early-stage porcine P23H model of retinal degeneration. a Taurine, glutamine, glycine::r, g, b showing reduced photoreceptor outer segment length and early stages of Müller cell stress and alteration of molecular signatures (green/yellow), b GABA, glycine, glutamate::r, g, b of the same region showing normal-appearing OPL, IPL, and neuronal signatures. c Glycine immunohistochemistry demonstrating early retinal remodeling/sprouting in the glycinergic amacrine cell populations. d GABA, glycine, glutamate::r, g, b mapping shows dramatically truncated photoreceptor outer and inner photoreceptor segments with the glycine signal shown in c in the green channel, demonstrating remodeling events. Scale bar 30 μm
Fig. 12.

a GABA, glycine, glutamate::r, g, b mapping in human geographic atrophy/AMD tissue demonstrates processes arising from both glycinergic and GABAergic amacrine cells (GABAergic processes extending into the outer plexiform layer in inset). These processes are the beginnings of microneuroma formation. b Taurine, glutamine, glutamate::r, g, b mapping demonstrates alterations in Müller cell signatures, notably an increase in the amount of taurine in subsets of Müller cells indicative of Müller cell stress (inset). Scale bar 90 μm
Fig. 10.

a GABA, glycine, glutamate::r, g, b mapping of a 746-day-old GHL rabbit, showing a glial column with migration of amacrine and bipolar cells into the ganglion cell layer. Microneuroma (rectangle) has also formed distal to the heavily depleted inner nuclear layer, b Taurine, glutamine. glycine::r, g, b mapping of the same P347L rabbit tissue, revealing normal and abnormal Müller cells (box) in the mid-stage degenerate retina. Scale bar 30 μm
During the relatively early stages of degeneration, porcine tissues also demonstrate early aberrant neuritic sprouting in the glycinergic and GABAergic amacrine cell populations (Fig. 9c, d), identical to that observed in rodent models [24] and human RP [26, 74]. Some of the more dramatic retinal remodeling revisions are held at bay so long as cone photoreceptors appear to be present. By the time cone photoreceptors are decreasing in number, Müller cell hypertrophy begins and forms the Müller cell seal, walling off the neural retina from the remnant RPE and choroid (Fig. 10b). During the later stages, when the cone photoreceptors have gone, the retina begins more dramatic revisions, including the formation of microneuromas and neuronal translocation through the axis of the retina (Fig. 11), duplicating findings observed in human (Fig. 7) [26, 74].
Fig. 11.

a GABA, glycine, gluiamate::r, g, b mapping of a 900-day-old RCS rat retina. This image shows three columns of neuronal translocation from ONL to GCL in which bipolar and amacrine cells are migrating through the retinal axis, b GABA, glycine, glutamate::r, g, b mapping of a 630-day-old rdl mouse, demonstrating a column of neurons bridging the depleted inner nuclear layer with bidircctionally migrating amacrine and bipolar cells. c GABA, glycine, glutamale::r, g, b mapping of a 746-day-old GHL mouse, showing a glial column with migration of amacrine and bipolar cells into the ganglion cell layer. A microneuroma has also formed distal to the heavily depleted inner nuclear layer. Scale bar 60 μm
The obvious question is whether other retinal degenerative diseases also show retinal remodeling, even though the principal mechanisms of retinal degeneration are different. Does retinal remodeling happen in age-related macular degeneration (AMD) for instance? Could retinal remodeling also exist in glaucoma, even though the retina is not deafferented in a top-down fashion?
The documentation on negative plasticity or aberrant remodeling in other retinal degenerative diseases is sparse, though some labs have demonstrated bipolar cell sprouting and synaptic abnormalities in human AMD [23] and in potential animal models of AMD [13]. Additional preliminary work in our lab revealed that remodeling events in both glycinergic and GABAergic amacrine cells in human geographic atrophy (GA), as shown in Fig. 12a, b, also reveal the earliest histologically observable signs of retinal remodeling. YGE > rgb imaging (Fig. 12a) also shows aberrant sprouting in both the glycine signals as well as in GABA signals (inset). TQE > rgb signals show some early indications of Müller cell variation in metabolism (inset), but no evidence at this time point of gross morphological alterations or responses in Müller cell populations.
These cells are tertiary network cells in the retina, representing critical interneurons with a complex network topology [92] that is fundamental to visual processing. Alterations of connectivities in these populations presumably dramatically disrupt visual processing, even in the presence of surviving afferent photoreceptors.
Retinal negative plasticity in glaucoma is also sparsely represented in the literature, though with a notable exception. Nico Cuenca demonstrated remodeling events in ON rod bipolar cells and horizontal cells in a model of glaucoma [93]. Work in our lab in collaboration with Monica Vetter and Alejandra Bosco demonstrated dramatic remodeling events of GABAergic amacrine cell processes in the DBA/2J mouse model of glaucoma (Fig. 13). Additional work with different disease models is beginning to show evidence for retinal remodeling in a number of animal models not traditionally associated with retinal disease, including spinocerebellar ataxia [94].
Fig. 13.

Retina from a 23-month-old male DBA/2J mouse labeled for GABA, demonstrating aberrant GABAergic amacrine cell remodeling with new neurites projecting upwards into the outer plexiform layer. Scale bar 30 μm
Mechanisms
Many mechanisms may be responsible for the various observed pathologies identified in retinal remodeling, including alterations in glutamate channel expression, changes in integrin expression/signaling, and other disparate molecular pathways that ultimately result in retinal circuit reprogramming and restructuring or topological revision of the surviving retina. In this review, we present three potential mechanisms that may be responsible for some of the sequelae identified in retinal remodeling: retinal cell reprogramming mediated by alterations in GluR expression, topological restructuring mediated by integrin expression or alterations in integrin expression, and neuritic sprouting found in remodeling retina mediated by retinoic acid receptors (RARs).
Retinal cell reprogramming (alterations in iGluR expression)
Retinal degenerative diseases result in functional reprogramming of the retina in at least three distinct ways. (1) Defined bipolar cell pathways of mGluR6- and iGluR-mediated ON and OFF responses may become corrupted. (2) Alternatively, in the absence of externally mediated drive, intrinsic drive from specific cell populations in the retina occurs, resulting in self signaling. (3) Cone-sparing forms of retinal degeneration alter iGluR expression, thus functionally changing phenotypic response profiles [26].
Retinal bipolar cell reprogramming is a universal feature in retinal degenerative diseases in humans, rodents, and rabbits [26, 74]. In retinal degenerative diseases and animal models of the same diseases that ablate both rods and cones, all evidence indicates that bipolar cell dendrites are lost [20–22], as are any responses mediated by glutamate [75]. Additionally, experiments with AGB (Fig. 8) demonstrate an absence of a cation current in these tissues [26], implying that the mGluR6-mediated signal transduction is defective through potential alteration or elimination of the receptors, or due to alterations in mGluR6 trafficking to other regions of the neuron [20–22].
Most notably, the presence of cones partially rescues the overall retinal structure, and the remaining bipolar cells underneath the surviving cones alter their response profiles so that the ratios of OFF cone to ON cone and ON rod bipolar cells shift from roughly 40:30:30 to ~ 80:15:05 [26, 74]. Normally, in primates, rodents, and rabbit, ON and OFF cone bipolar cells and rod bipolar cells each comprise about 33 % of all bipolar cells.
Unmasking of autoexcitatory retinal signaling also occurs in retinas bereft of photoreceptor input [26], perhaps through variations in intrinsic calcium levels that result in alterations of amacrine cell potentials [95]. These variations in responsivity or excitation may be due to oscillating inhibitory feedback from remaining glycinergic or GAB-Aergic inputs. An alternative mechanism could arise from an existing excitatory cell type in the retina that is spontaneously active and has extensive input to both ON and OFF pathways. The dopaminergic amacrine cell has considerable input into AII amacrine cell pathways [96, 97], demonstrates spontaneous spike activity in the absence of synaptic input [98]. and exhibits glutamatergic signatures. Additionally, dopaminergic amacrine cells are strongly activated by release from inhibition, and perhaps function by increasing tonic excitatory drive via synaptic glutamate release [99].
Light-induced retinal degeneration (LIRD) represents an effective tool for coherent photoreceptor loss that results in retinal remodeling and reprogramming [13]. Though the overall anatomy of the neural retina, and especially the inner nuclear layer, inner plexiform layer, and ganglion cell layer seem normal early in LIRD, key synaptic markers in the inner retina demonstrate rapid inner retina responses to photoreceptor stress that lead to fundamental reprogramming of neuronal responses and may represent an attempt to prevent excitotoxic damage and/or cell death.
In LIRD, GluR2 subunits of AMPA receptors predominantly associated with inner retinal processing display rapid and significant alterations in protein levels and may reflect the same pathways used in CNS for neuroprotection [100]. Examination of GluR expression in LIRD demonstrates alterations in GluR expression over 60 days post light damage. Specifically, low-conductance AMPA receptor GluR2 subunits increase by 65 % (measured by protein level), high-conductance KA receptor GluR5 sub-units decrease by 50 %, while AMPA receptor GluR1 subunits show no significant change in expression (Lin et al., submitted). A 65 % increase in GluR2 subunit availability is sufficient to stochastically add one subunit to every AMPA receptor that does not already express one, potentially performing three protective actions: (1) decreasing the Ca2+ permeability of the entire channel unitary conductance by 10×, (2) decreasing the conductance of the entire channel by over 50 %, and (3) preventing GluR1 phosphorylation-dependent increases in channel conductance. Combined, these effects potentially play a powerful role in neuroprotection by decreasing Ca2+ loads in neurons (Lin et al., submitted).
Topological restructuring via alterations in integrin expression
Other potential mechanisms involved in the observed topological restructuring of degenerate retina lie in alterations of integrin-mediated signaling. It is thought that virtually all adult tissue remodeling and cell migration involves integrin–integrin receptor signaling [101, 102]. It could be argued that the activation or reconfiguration of integrins might change cell-cell recognition or contact programming without substantial alterations in gene expression. If this is the case, clinical interventions that are designed to modulate integrin arginyl-glycyl-aspartic acid (RGD) motif binding in the remnant retina may work to modify or attenuate retinal remodeling without the need to resort to gene therapies. These studies in vivo are complicated by the slow progression in many models of retinal degeneration, particularly those that mimic human retinal degenerative diseases more precisely in their inhomogeneity or patchy degeneration. One possibility is to perform the modeling of integrin-mediated alterations in in vitro systems, but substantial evidence is present to suggest that cell–cell adhesion mechanisms are very different in in vitro systems as opposed to in vivo systems [103].
Evidence from Usher syndrome studies reveals links between the Usher protein complex and cadherins/catenins in junction-associated complexes which may play substantial roles in specific static or developmental cell polarity and tissue organization [104]. It also begs the question of whether these pathways and other integrin-mediated pathways are altered in retinal degenerative diseases.
Neuritic sprouting mediated by retinoic acid receptors (RARs)
Other pathways, including retinoic acid (RA) mediated signaling pathways, are likely involved in retinal remodeling in the adult retina. RA-mediated signaling is activated in the LIRD model, where coherent photoreceptor loss and early dendritic remodeling occur. Additionally, exogenous application of RA leads to robust neuritic growth in primary cultured rod bipolar cells [105]. We believe that RA signaling displays large alterations early in retinal degeneration, suggesting that RA signaling pathways may be responsible for survival-related neuritogenesis and subsequent plasticity. Deficiencies in RA signaling may contribute to neurite degeneration, and retinoic acid (RA) and RA/retinoid receptor (RAR/RXR)-dependent pathways in retina incur the activation of neuritogenesis, resulting in the formation and elaboration of complex circuits and structures comprising all of the remaining retinal neuronal classes.
We demonstrated that RAR/RXR signaling is a mechanism driving pathological neuritogenesis. Even more compellingly, RAR/RXR antagonists appear to attenuate neuritogenesis from BC populations, while CaMKII inhibitors block neuritogenesis [105–107]. RAR/RXR signaling in phase 3 remodeling appears to induce anomalous neuronal sprouting and outgrowth, forming fascicles and microneuromas. This sometimes leads to retinas becoming so completely topologically restructured that the tissue no longer resembles retina. The model is that loss of photoreceptors eliminates retinoid buffering capacity (opsins) and increases retinoid precursor availability, leading to the generation of RA. RAR/RXR transcription subsequently induces sprouting and neuritogenesis. This occurs simultaneously with the loss of photoreceptors, which reduces glutamate-mediated neurotransmission and, by extension, decreases Ca2+-mediated signaling. βCaMKII then detects the decrease in Ca2+ flux and contributes to RAR/RXR-mediated neuritogenesis through RXR binding.
Circuit outcomes of retinal remodeling
By hybridizing light microscopy with TEM analysis, phenotypic identity can be established for neurons, glia, and processes in ultrastructural datasets (Fig. 14) [24, 108], and circuitry can be defined for those phenotyped neurons [92. 109]. Analysis of over 30 animal models of RP and human RP samples indicates that, as remodeling progresses, the retinal circuitry substrate is altered. Beginning with bipolar cell dendritic truncation and polyaxonal growth early in the disease process [26] through to amacrine and ganglion cell neuritogenesis [24], evolving into anomalous fascicles and microneuromas, the retina is restructuring its connectivities. Whether or not those connectivities are proper or improper is currently unknown and will only be answered through complete connectomic approaches [92, 109]. This is work that is underway in the P347L rabbit [110], the RCS rat [111], and the light-induced retinal degeneration mouse model [26], and it is so far demonstrating that these novel structures are not synaptically silent. Microneuromas exhibit numerous synaptic structures from both ribbon and conventional synapses, but it is still too early to judge their viability as functional or nonfunctional visual circuits.
Fig. 14.

GABA, taurine, glutamate::r, g, b overlay on a TEM image of a peptidergic GABAergic amacrinc cell (asterisk) adjacent to a forming microneuroma in the RCS rat retina. Overlay CMP/TEM imaging enables connectomics and pathoconnectomics projects that elucidate precise, ultrastructural reconstruction of neuronal circuitry. Scale bar 6 μm
Implications for retinal vision rescue
Retinal remodeling as a phenomenon introduces substantial barriers to any therapy designed to recover vision loss. Interventions in the remaining populations of neurons are complicated by the loss of BCs during the remodeling process [26]. More importantly, as discussed above, the initial response of the neural retina likely induces a substantial change to the retinal circuitry prior to cell loss, with initial alterations in BC connectivities and programming eliminating targets for photoreceptor progenitor cell or fetal retina transplants.
Retinas are reactive when deafferented and demonstrate enhanced remodeling in response to direct intervention. Retinal transplants, for example, remodel more aggressively than the host degenerate retina [112]. Prevention of neurite/dendritic loss appears to be critical to holding off phase 3 remodeling events, yet that loss appears to be one of the earliest indicators of neural disease in retina [20], cortex [113], and midbrain [114], with dendrites appearing to be far more sensitive to pathophysiological alterations than axons. Early changes include the aforementioned mislocalization of mGluR6 receptors [20–22], followed by bipolar cell regression and loss of iGluR expression in the dendrites, which effectively reprograms the physiologic response profiles of these bipolar cells and—one would presume—all subsequent downstream elements. Bipolar cells do not recover iGluR expression and thus the ability to signal through iGluR-mediated circuitry [26], making them poor targets for intervention strategies. Once downstream circuit revision occurs, there is persistent loss of iGluR expression in other cell populations as well [26], which leads to irrevocable alterations of the topology and circuitry of the retina [7–9, 11–13, 24, 26, 79].
Failing the prevention of dendritic loss of bipolar cells, secondary targets might prove successful by preventing horizontal, amacrine, and ganglion cells from sprouting aberrantly. A number of approaches aimed at reducing aberrant neuronal sprouting have been attempted, including electrical stimulation, which appears to enhance sprouting (data not published), tetrodotoxin (TTX) exposure in posttraumatic epilepsy models [115], and manipulation of integrins and retinoid X receptors [105], as discussed above. These approaches appear promising, as the loss of photoreceptors removes retinoid binding pools, thus increasing the availability of retinoid precursors. RA becomes available [106, 116], which subsequently triggers neuritogenesis through RAR/RXR transcriptional activity [117]. Simultaneously, photoreceptor loss reduces neural Ca2+ influx, activating the βCaMKII and driving neuritogenesis [107, 118]. This opens the door to therapeutic interventions that are designed to antagonize CaMKII, among other therapies.
Another approach we are working on in collaboration with Wolfgang Baehr is an early intervention with AAV-rRho-shRNA vectors to suppress rod opsin synthesis while patients still have photoreceptors, in order to arrest or delay photoreceptor cell death and prolong vision preservation by decreasing the continued retinal stress and bystander killing effects of dying rods on cones. If one were to presume that rod opsin mediated toxicity, either via misfolding or mistrafficked opsins, is responsible for rod photoreceptor stress and subsequent cell death, it might be appropriate to reduce total overall rod opsin expression levels. The fundamental idea is to prolong rod photoreceptor life and limit the bystander killing effect on cone photoreceptors. This strategy has shown promise in GCAP1-Y99C transgenic mice, delaying vision loss through the expression of a GCAP1 shRNA by recombinant scAAV2/8 [119].
Other molecular changes occur in degenerate retina, and there is no evidence to suggest that retinal remodeling has an end point or arrest point [7, 24, 26]. More needs to be done in these areas, and possibly also in controlling or manipulating integrin-mediated contact on cell surfaces, which brings up the possibility of other forms of reprogramming in neural systems that have been deafferented. Other molecular changes are unquestionably occurring in the degenerate retina, and substantial evidence indicates that remodeling does not have an arrest point [7, 24, 26]. Fundamentally, these approaches are designed to slow the progression of retinal degeneration and not to recover vision that has already been lost.
A number of approaches are proposed that are designed to recover lost vision, but these approaches all have the same number of problems. The fundamental problem in rescuing vision is to define windows of opportunity or when to intervene. Current therapeutic interventions are designed for those patients that have already lost some, if not all, of their vision. These patients often present at a late stage with advanced retinal degeneration (Fig. 4), and are likely to exhibit early profound alterations to the retinal circuitry that corrupt any surrogate inputs. Any visual prosthetic design will depend upon the stage of retinal degeneration of the patient. Specifically, intervening in a highly remodeled retina to recover vision will represent an entirely different set of bioengineering or bionic engineering hurdles than intervening in a retina that has most or all of its circuitry still intact. It might be supposed that unless one actually knows what the fundamental circuitry is in the normal retina, one cannot predict what the output of the neural retina will be. Additionally, early evidence indicates that synaptic contacts in the remodeling retina are inappropriate, with some models [26] predicting that circuits may generate oscillatory ringing, which is incompatible with normal visual processing. Therefore, predicting what the ganglion cell output of the retina will be is difficult at best.
Other approaches, including stem cell transplants or embryonic sheet transplants, will have to overcome a number of issues, such as (and not limited to) improper wiring, cell fusion, and the rather scary possibility of transplant “rescues” being co-opted into Frankenstein-like assemblies of defective or nonfunctional cell populations by resident neurons and glia. A number of reports of transplanted stem cells that co-opt the phenotypes of host cells are documented as instances of cell fusion [120]. In addition to the documented circuit revision that occurs in retinal remodeling, there is a question of patterning, as remodeling retinas likely have altered spacing and visual functions that are related to alterations in cell–cell spacing. Cell populations that are introduced into an existing retina, albeit remodeled, do not have a history of normal developmental structuring such as that which occurs during retinal maturation, and no proposed research has ever discussed re-patterning the retina to recapitulate spacing rules. So, if introduced cell populations are to be successful, properly phenotyping transplanted cells [121] becomes a critical task in order to track identity and any positional issues that might impact visual field processing, though most efforts to date document a rejection of transplant cells [122] or a loss of the transplant cells’ own mature phenotypes at various time points post-transplantation.
Fundamentally, most bionic or biological transplant/implant approaches presume or are engineered on the belief that the underlying neural retina is normal. The degenerate retina, on the other hand, is not normal, and most of its cell types show abnormality. The basic assumptions of transplant technologies (intactness, receptivity, and capacity of the host neural retina to carry the signals offered by the implant/transplant) are false.
However, we believe that there is a way forward using approaches pioneered in the optogenetics community, utilizing existing, remnant cell populations with intervention prior to large-scale restructuring of the retina. These genetic, therapeutic approaches using AAV-mediated channelrhodopsins/halorhodopsin vectors that target appropriate classes of retinal neurons could conceivably happen as a relatively late intervention when patients have lost functional photoreceptors [123–125]. This approach has been used before in ON bipolar cells in mouse to deliver channelrhodopsin2 [125]. The key here will be to target the right classes of neurons, as photoreceptor loss in RP effectively deafferents approximately 20 different ganglion cell pathways [10, 126]. Targeting broad ganglion cell classes with channelrhodopsins [127] is tricky, as separate ganglion cell outflow channels will then be generating the same signals to the cortex and other regions simultaneously. This is likely to set up errors in higher visual signal processing, so the decision about which specific retinal cell classes to target will be critical.
Human tissues
Tissue from patients with advanced RP and early GA/AMD were obtained from the Lion’s Eye Bank donor pool at the University of Utah within 3 h of death. The Lion’s Eye Bank donor procurement and distribution complies with the Declaration of Helsinki. All data were de-identified in accordance with the HIPAA Privacy Rule.
Acknowledgments
We would like to thank Carl B. Watt for his work in electron microscopy, imaging, and manuscript review. William Drew Ferrell was invaluable in data preparation and in reviewing the manuscript. Kevin Rapp assisted with confocal microscopy, which resulted in figure construction. Maggie Shaw and Jia-Hui Yang assisted with data acquisition, tissue preparation, immunocytochemistry, and ultramicrotomy. James R. Anderson assisted in ultrastructural data assembly. Monica Vetter and Alejandra Bosco helped provide the DBA/2J mouse tissues, provided guidance, and helped to review the manuscript. Support: NIH EY015128 (RM), NIH EY02576 (RM), EYO14800 Vision Core, an unrestricted grant from Research to Prevent Blindness to the Moran Eye Center; Edward N. and Della L. Thome Memorial Foundation grant for Age-Related Macular Degeneration Research (BWJ), a Research to Prevent Blindness Career Development Award (BWJ), Moran Eye Center Tiger Team Translational Medicine Award (BWJ), Sciences Research Grant H16-sensory-001 from the Ministry of Health, Labor and Welfare, Japan (MK).
Footnotes
The content of this invited review article was presented at the ARVO-JOS joint symposium on April 15, 2010, held during the 114th Annual Meeting of the Japanese Ophthalmological Society.
Contributor Information
B. W. Jones, Email: bryan.jones@m.cc.utah.edu, Department of Ophthalmology, Moran Eye Center, University of Utah, 65 Mario Capecchi Dr., Salt Lake City, UT 84132, USA
M. Kondo, Department of Ophthalmology, Nagoya University, Graduate School of Medicine, Nagoya, Japan
H. Terasaki, Department of Ophthalmology, Nagoya University, Graduate School of Medicine, Nagoya, Japan
Y. Lin, Department of Ophthalmology, Moran Eye Center, University of Utah, 65 Mario Capecchi Dr., Salt Lake City, UT 84132, USA
M. McCall, Department of Ophthalmology and Visual Sciences, University of Louisville, Louisville, KY, USA
R. E. Marc, Department of Ophthalmology, Moran Eye Center, University of Utah, 65 Mario Capecchi Dr., Salt Lake City, UT 84132, USA
References
- 1.Aleman TS, Cideciyan AV, Sumaroka A, Schwartz SB, Roman AJ, Windsor EAM, et al. Inner retinal abnormalities in X-linked retinitis pigmentosa with RPGR mutations. Invest Ophthalmol Vis Sci. 2007;48:4759–65. doi: 10.1167/iovs.07-0453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cuenca N, Pinilla I, Sauvé Y, Lu B, Wang S, Lund RD. Regressive and reactive changes in the connectivity patterns of rod and cone pathways of P23H transgenic rat retina. Neuroscience. 2004;127:301–17. doi: 10.1016/j.neuroscience.2004.04.042. [DOI] [PubMed] [Google Scholar]
- 3.Cuenca N, Pinilla I, Sauvé Y, Lund RD. Early changes in synaptic connectivity following progressive photoreceptor degeneration in RCS rats. Eur J Neurosci. 2005;22:1057–72. doi: 10.1111/j.1460-9568.2005.04300.x. [DOI] [PubMed] [Google Scholar]
- 4.de Raad S, Szczesny PJ, Munz K, Remé CE. Light damage in the rat retina: glial fibrillary acidic protein accumulates in Müller cells in correlation with photoreceptor damage. Ophthalmic Res. 1996;28:99–107. doi: 10.1159/000267881. [DOI] [PubMed] [Google Scholar]
- 5.Fariss RN, Li ZY, Milam AH. Abnormalities in rod photoreceptors, amacrine cells, and horizontal cells in human retinas with retinitis pigmentosa. Am J Ophthalmol. 2000;129:215–23. doi: 10.1016/s0002-9394(99)00401-8. [DOI] [PubMed] [Google Scholar]
- 6.Fletcher EL, Kalloniatis M. Neurochemical architecture of the normal and degenerating rat retina. J Comp Neurol. 1996;376:343–60. doi: 10.1002/(SICI)1096-9861(19961216)376:3<343::AID-CNE1>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 7.Jones BW, Marc RE. Retinal remodeling during retinal degeneration. Exp Eye Res. 2005;81:123–37. doi: 10.1016/j.exer.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 8.Jones BW, Marc RE, Watt CB, Vaughan DK, Organisciak DT. Neural plasticity revealed by light-induced photoreceptor lesions. Adv Exp Med Biol. 2006;572:405–10. doi: 10.1007/0-387-32442-9_57. [DOI] [PubMed] [Google Scholar]
- 9.Jones BW, Watt CB, Marc RE. Retinal remodelling. Clin Exp Optom. 2005;88:282–91. doi: 10.1111/j.1444-0938.2005.tb06712.x. [DOI] [PubMed] [Google Scholar]
- 10.Marc RE, Jones BW. Molecular phenotyping of retinal ganglion cells. J Neurosci. 2002;22:413–27. doi: 10.1523/JNEUROSCI.22-02-00413.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marc RE, Jones BW. Retinal remodeling in inherited photoreceptor degenerations. Mol Neurobiol. 2003;28:139–47. doi: 10.1385/MN:28:2:139. [DOI] [PubMed] [Google Scholar]
- 12.Marc RE, Jones BW, Watt CB, Strettoi E. Neural remodeling in retinal degeneration. Prog Retin Eye Res. 2003;22:607–55. doi: 10.1016/s1350-9462(03)00039-9. [DOI] [PubMed] [Google Scholar]
- 13.Marc RE, Jones BW, Vazquez-Chona F, Vaughan DK, Organisciak DT. Extreme retinal remodeling triggered by light damage: implications for age related macular degeneration. Mol Vis. 2008;14:782–805. [PMC free article] [PubMed] [Google Scholar]
- 14.Kolb H, Gouras P. Electron microscopic observations of human retinitis pigmentosa, dominantly inherited. Invest Ophthalmol. 1974;13:487–98. [PubMed] [Google Scholar]
- 15.Li ZY, Kljavin IJ, Milam AH. Rod photoreceptor neurite sprouting in retinitis pigmentosa. J Neurosci. 1995;15:5429–38. doi: 10.1523/JNEUROSCI.15-08-05429.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Machida S, Kondo M, Jamison JA, Khan NW, Kononen LT, Sugawara T, et al. P23H rhodopsin transgenic rat: correlation of retinal function with histopathology. Invest Ophthalmol Vis Sci. 2000;41:3200–9. [PubMed] [Google Scholar]
- 17.Pu M, Xu L, Zhang H. Visual response properties of retinal ganglion cells in the Royal College of Surgeons dystrophic rat. Invest Ophthalmol Vis Sci. 2006;47:3579–85. doi: 10.1167/iovs.05-1450. [DOI] [PubMed] [Google Scholar]
- 18.Specht D, Tom Dieck S, Ammermüller J, Regus-Leidig H, Gundelfinger ED, Brandstätter JH. Structural and functional remodeling in the retina of a mouse with a photoreceptor synaptopathy: plasticity in the rod and degeneration in the cone system. Eur J Neurosci. 2007;26:2506–15. doi: 10.1111/j.1460-9568.2007.05886.x. [DOI] [PubMed] [Google Scholar]
- 19.Stasheff SF. Emergence of sustained spontaneous hyperactivity and temporary preservation of OFF responses in ganglion cells of the retinal degeneration (rd1) mouse. J Neurophysiol. 2008;99:1408–21. doi: 10.1152/jn.00144.2007. [DOI] [PubMed] [Google Scholar]
- 20.Strettoi E, Pignatelli V. Modifications of retinal neurons in a mouse model of retinitis pigmentosa. Proc Natl Acad Sci USA. 2000;97:11020–5. doi: 10.1073/pnas.190291097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strettoi E, Pignatelli V, Rossi C, Porciatti V, Falsini B. Remodeling of second-order neurons in the retina of rd/rd mutant mice. Vision Res. 2003;43:867–77. doi: 10.1016/s0042-6989(02)00594-1. [DOI] [PubMed] [Google Scholar]
- 22.Strettoi E, Porciatti V, Falsini B, Pignatelli V, Rossi C. Morphological and functional abnormalities in the inner retina of the rd/rd mouse. J Neurosci. 2002;22:5492–504. doi: 10.1523/JNEUROSCI.22-13-05492.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sullivan RK, Woldemussie E, Pow DV. Dendritic and synaptic plasticity of neurons in the human age-related macular degeneration retina. Invest Ophthalmol Vis Sci. 2007;48:2782–9. doi: 10.1167/iovs.06-1283. [DOI] [PubMed] [Google Scholar]
- 24.Jones BW, Watt CB, Frederick JM, Baehr W, Chen CK, Levine EM, et al. Retinal remodeling triggered by photoreceptor degenerations. J Comp Neurol. 2003;464:1–16. doi: 10.1002/cne.10703. [DOI] [PubMed] [Google Scholar]
- 25.Sullivan R, Penfold P, Pow DV. Neuronal migration and glial remodeling in degenerating retinas of aged rats and in nonneovascular AMD. Invest Ophthalmol Vis Sci. 2003;44:856–65. doi: 10.1167/iovs.02-0416. [DOI] [PubMed] [Google Scholar]
- 26.Marc RE, Jones BW, Anderson JR, Kinard K, Marshak DW, Wilson JH, et al. Neural reprogramming in retinal degeneration. Invest Ophthalmol Vis Sci. 2007;48:3364–71. doi: 10.1167/iovs.07-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baehr W, Frederick JM. Naturally occurring animal models with outer retina phenotypes. Vision Res. 2009;49:2636–52. doi: 10.1016/j.visres.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang CJ, Lai WW, Edward DP, Tso MOM. Apoptotic photoreceptor cell death after traumatic retinal detachment in humans. Arch Ophthalmol. 1995;113:880–6. doi: 10.1001/archopht.1995.01100070054025. [DOI] [PubMed] [Google Scholar]
- 29.Frederick JM, Krasnoperova NV, Hoffmann K, Church-Kopish J, Rüther K, Howes K, et al. Mutant rhodopsin transgene expression on a null background. Invest Ophthalmol Vis Sci. 2001;42:826–33. [PubMed] [Google Scholar]
- 30.Humphries MM, Rancourt D, Farrar GJ, Kenna P, Hazel M, Bush RA, et al. Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nat Genet. 1997;15:216–9. doi: 10.1038/ng0297-216. [DOI] [PubMed] [Google Scholar]
- 31.Aguirre GD, Baldwin V, Pearce-Kelling S, Narfström K, Ray K, Acland GM. Congenital stationary night blindness in the dog: common mutation in the RPE65 gene indicates founder effect. Mol Vis. 1998;4:23. [PubMed] [Google Scholar]
- 32.Gu SM, Thompson DA, Srikumari CR, Lorenz B, Finckh U, Nicoletti A, et al. Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat Genet. 1997;17:194–7. doi: 10.1038/ng1097-194. [DOI] [PubMed] [Google Scholar]
- 33.Morimura H, Fishman GA, Grover SA, Fulton AB, Berson EL, Dryja TP. Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or Leber congenital amaurosis. Proc Natl Acad Sci USA. 1998;95:3088–93. doi: 10.1073/pnas.95.6.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15:236–46. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- 35.Allikmets R. Simple and complex ABCR: genetic predisposition to retinal disease. Am J Hum Genet. 2000;67:793–9. doi: 10.1086/303100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Allikmets R, Shroyer NF, Singh N, Seddon JM, Lewis RA, Bernstein PS, et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science. 1997;277:1805–7. doi: 10.1126/science.277.5333.1805. [DOI] [PubMed] [Google Scholar]
- 37.Cremers FP, van de Pol DJ, van Driel M, den Hollander AI, van Haren FJ, Knoers NV, et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum Mol Genet. 1998;7:355–62. doi: 10.1093/hmg/7.3.355. [DOI] [PubMed] [Google Scholar]
- 38.Molday LL, Rabin AR, Molday RS. ABCR expression in foveal cone photoreceptors and its role in Stargardt macular dystrophy. Nat Genet. 2000;25:257–8. doi: 10.1038/77004. [DOI] [PubMed] [Google Scholar]
- 39.D’Cruz PM, Yasumura D, Weir J, Matthes MT, Abderrahim H, LaVail MM, et al. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Genet. 2000;9:645–51. doi: 10.1093/hmg/9.4.645. [DOI] [PubMed] [Google Scholar]
- 40.Duncan JL, Yang H, Vollrath D, Yasumura D, Matthes MT, Trautmann N, et al. Inherited retinal dystrophy in Mer knockout mice. Adv Exp Med Biol. 2003;533:165–72. doi: 10.1007/978-1-4615-0067-4_21. [DOI] [PubMed] [Google Scholar]
- 41.Li JB, Gerdes JM, Haycraft CJ, Fan Y, Teslovich TM, May-Simera H, et al. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell. 2004;117:541–52. doi: 10.1016/s0092-8674(04)00450-7. [DOI] [PubMed] [Google Scholar]
- 42.Yen HJ, Tayeh MK, Mullins RF, Stone EM, Sheffield VC, Slusarski DC. Bardet-Biedl syndrome genes are important in retrograde intracellular trafficking and Kupffer’s vesicle cilia function. Hum Mol Genet. 2006;15:667–77. doi: 10.1093/hmg/ddi468. [DOI] [PubMed] [Google Scholar]
- 43.Dryja TP, Berson EL, Rao VR, Oprian DD. Heterozygous missense mutation in the rhodopsin gene as a cause of congenital stationary night blindness. Nat Genet. 1993;4:280–3. doi: 10.1038/ng0793-280. [DOI] [PubMed] [Google Scholar]
- 44.Zeitz C, Gross AK, Leifert D, Kloeckener-Gruissem B, McAlear SD, Lemke J, et al. Identification and functional characterization of a novel rhodopsin mutation associated with autosomal dominant CSNB. Invest Ophthalmol Vis Sci. 2008;49:4105–14. doi: 10.1167/iovs.08-1717. [DOI] [PubMed] [Google Scholar]
- 45.Sommer ME, Farrens DL. Arrestin can act as a regulator of rhodopsin photochemistry. Vision Res. 2006;46:4532–46. doi: 10.1016/j.visres.2006.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sommer ME, Smith WC, Farrens DL. Dynamics of arrestin–rhodopsin interactions: acidic phospholipids enable binding of arrestin to purified rhodopsin in detergent. J Biol Chem. 2006;281:9407–17. doi: 10.1074/jbc.M510037200. [DOI] [PubMed] [Google Scholar]
- 47.Huang SH, Pittler SJ, Huang X, Oliveira L, Berson EL, Dryja TP. Autosomal recessive retinitis pigmentosa caused by mutations in the alpha subunit of rod cGMP phosphodiesterase. Nat Genet. 1995;11:468–71. doi: 10.1038/ng1295-468. [DOI] [PubMed] [Google Scholar]
- 48.McLaughlin ME, Ehrhart TL, Berson EL, Dryja TP. Mutation spectrum of the gene encoding the beta subunit of rod phosphodiesterase among patients with autosomal recessive retinitis pigmentosa. Proc Natl Acad Sci USA. 1995;92:3249–53. doi: 10.1073/pnas.92.8.3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McLaughlin ME, Sandberg MA, Berson EL, Dryja TP. Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat Genet. 1993;4:130–4. doi: 10.1038/ng0693-130. [DOI] [PubMed] [Google Scholar]
- 50.Clarke G, Goldberg AF, Vidgen D, Collins L, Ploder L, Schwarz L, et al. Rom-1 is required for rod photoreceptor viability and the regulation of disk morphogenesis. Nat Genet. 2000;25:67–73. doi: 10.1038/75621. [DOI] [PubMed] [Google Scholar]
- 51.Dryja TP, McGee TL, Berson EL, Fishman GA, Sandberg MA, Alexander KR, et al. Night blindness and abnormal cone electroretinogram ON responses in patients with mutations in the GRM6 gene encoding mGluR6. Proc Natl Acad Sci USA. 2005;102:4884–9. doi: 10.1073/pnas.0501233102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zeitz C, van Genderen M, Neidhardt J, Luhmann UF, Hoeben F, Forster U, et al. Mutations in GRM6 cause autosomal recessive congenital stationary night blindness with a distinctive scotopic 15-Hz flicker electroretinogram. Invest Ophthalmol Vis Sci. 2005;46:4328–35. doi: 10.1167/iovs.05-0526. [DOI] [PubMed] [Google Scholar]
- 53.Vasireddy V, Uchida Y, Salem N, Jr, Kim SY, Mandal MNA, Reddy GB, et al. Loss of functional ELOVL4 depletes very long-chain fatty acids (≥C28) and the unique ω-O-acylceramides in skin leading to neonatal death. Hum Mol Genet. 2007;16:471–82. doi: 10.1093/hmg/ddl480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang K, Kniazeva M, Han M, Li W, Yu Z, Yang Z, et al. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat Genet. 2001;27:89–93. doi: 10.1038/83817. [DOI] [PubMed] [Google Scholar]
- 55.Chen CK, Burns ME, He W, Wensel TG, Baylor DA, Simon MI. Slowed recovery of rod photoresponse in mice lacking the GTPase accelerating protein RGS9-1. Nature. 2000;403:557–60. doi: 10.1038/35000601. [DOI] [PubMed] [Google Scholar]
- 56.Hu G, Wensel TG. R9AP, a membrane anchor for the photoreceptor GTPase accelerating protein. RGS9-1. Proc Natl Acad Sci USA. 2002;99:9755–60. doi: 10.1073/pnas.152094799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu G, Zhang Z, Wensel TG. Activation of RGS9-1 GTPase acceleration by its membrane anchor, R9AP. J Biol Chem. 2003;278:14550–4. doi: 10.1074/jbc.M212046200. [DOI] [PubMed] [Google Scholar]
- 58.Wensel TG. Signal transducing membrane complexes of photoreceptor outer segments. Vision Res. 2008;48:2052–61. doi: 10.1016/j.visres.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boon CJF, Klevering BJ, Hoyng CB, Zonneveld-Vrieling MN, Nabuurs SB, Blokland E, et al. Basal laminar drusen caused by compound heterozygous variants in the CFH gene. Am J Hum Genet. 2008;82:516–23. doi: 10.1016/j.ajhg.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cameron DJ, Yang Z, Gibbs D, Chen H, Kaminoh Y, Jorgensen A, et al. HTRA1 variant confers similar risks to geographic atrophy and neovascular age-related macular degeneration. Cell Cycle. 2007;6:1122–5. doi: 10.4161/cc.6.9.4157. [DOI] [PubMed] [Google Scholar]
- 61.Chen H, Yang Z, Gibbs D, Yang X, Hau V, Zhao P, et al. Association of HTRA1 polymorphism and bilaterality in advanced age-related macular degeneration. Vision Res. 2008;48:690–4. doi: 10.1016/j.visres.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 62.Dewan A, Liu M, Hartman S, Zhang SS, Liu DT, Zhao C, et al. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science. 2006;314:989–92. doi: 10.1126/science.1133807. [DOI] [PubMed] [Google Scholar]
- 63.Edwards AO, Ritter R, III, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–4. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 64.Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, Gehrs K, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–62. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA. 2005;102:7227–32. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jakobsdottir J, Conley YP, Weeks DE, Mah TS, Ferrell RE, Gorin MB. Susceptibility genes for age-related maculopathy on chromosome 10q26. Am J Hum Genet. 2005;77:389–407. doi: 10.1086/444437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kaplan J, Gerber S, Larget-Piet D, Rozet JM, Dollfus H, Dufier JL, et al. A gene for Stargardt’s disease (fundus flavimaculatus) maps to the short arm of chromosome 1. Nat Genet. 1993;5:308–11. doi: 10.1038/ng1193-308. [DOI] [PubMed] [Google Scholar]
- 68.Maller JB, Fagerness JA, Reynolds RC, Neale BM, Daly MJ, Seddon JM. Variation in complement factor 3 is associated with risk of age-related macular degeneration. Nat Genet. 2007;39:1200–1. doi: 10.1038/ng2131. [DOI] [PubMed] [Google Scholar]
- 69.Stone EM, Braun TA, Russell SR, Kuehn MH, Lotery AJ, Moore PA, et al. Missense variations in the fibulin 5 gene and age-related macular degeneration. N Engl J Med. 2004;351:346–53. doi: 10.1056/NEJMoa040833. [DOI] [PubMed] [Google Scholar]
- 70.Yates JRW, Sepp T, Matharu BK, Khan JC, Thurlby DA, Shahid H, et al. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007;357:553–61. doi: 10.1056/NEJMoa072618. [DOI] [PubMed] [Google Scholar]
- 71.Phipps JA, Yee P, Fletcher EL, Vingrys AJ. Rod photoreceptor dysfunction in diabetes: activation, deactivation, and dark adaptation. Invest Ophthalmol Vis Sci. 2006;47:3187–94. doi: 10.1167/iovs.05-1493. [DOI] [PubMed] [Google Scholar]
- 72.Choi SS, Zawadzki RJ, Lim MC, Brandt JD, Keltner JL, Doble N, et al. Evidence of outer retinal changes in glaucoma patients as revealed by ultrahigh-resolution in vivo retinal imaging. Br J Ophthalmol. 2011;95:131–41. doi: 10.1136/bjo.2010.183756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Milam AH, Li ZY, Fariss RN. Histopathology of the human retina in retinitis pigmentosa. Prog Retin Eye Res. 1998;17:175–205. doi: 10.1016/s1350-9462(97)00012-8. [DOI] [PubMed] [Google Scholar]
- 74.Jones BW, Kondo M, Terasaki H, Watt CB, Rapp K, Anderson J, et al. Retinal remodeling in the Tg p3471 rabbit, a large-eye model of retinal degeneration. J Comp Neurol. 2011;519:2713–33. doi: 10.1002/cne.22703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Varela C, Igartua I, De la Rosa EJ, De la Villa P. Functional modifications in rod bipolar cells in a mouse model of retinitis pigmentosa. Vision Res. 2003;43:879–85. doi: 10.1016/s0042-6989(02)00493-5. [DOI] [PubMed] [Google Scholar]
- 76.Chua J, Fletcher EL, Kalloniatis M. Functional remodeling of glutamate receptors by inner retinal neurons occurs from an early stage of retinal degeneration. J Comp Neurol. 2009;514:473–91. doi: 10.1002/cne.22029. [DOI] [PubMed] [Google Scholar]
- 77.Peng YW, Hao Y, Petters RM, Wong F. Ectopic synaptogenesis in the mammalian retina caused by rod photoreceptor-specific mutations. Nat Neurosci. 2000;3:1121–7. doi: 10.1038/80639. [DOI] [PubMed] [Google Scholar]
- 78.Park SJ, Kim IB, Choi KR, Moon JI, Oh SJ, Chung JW, et al. Reorganization of horizontal cell processes in the developing FVB/N mouse retina. Cell Tissue Res. 2001;306:341–6. doi: 10.1007/s004410100453. [DOI] [PubMed] [Google Scholar]
- 79.Marc RE, Jones BW, Watt CB. Retinal remodeling: circuitry revisions triggered by photoreceptor degeneration. In: Pinaud R, Tremere L, De Weerd P, editors. Plasticity in the visual system: from genes to circuits. New York: Springer; 2006. pp. 33–54. [Google Scholar]
- 80.Li ZY, Possin DE, Milam AH. Histopathology of bone spicule pigmentation in retinitis pigmentosa. Ophthalmology. 1995;102:805–16. doi: 10.1016/s0161-6420(95)30953-0. [DOI] [PubMed] [Google Scholar]
- 81.Spraul CW, Lang GE, Grossniklaus HE, Lang GK. Histologic and morphometric analysis of the choroid, Bruch’s membrane, and retinal pigment epithelium in postmortem eyes with age-related macular degeneration and histologic examination of surgically excised choroidal neovascular membranes. Surv Ophthalmol. 1999;44(Suppl 1):S10–32. doi: 10.1016/s0039-6257(99)00086-7. [DOI] [PubMed] [Google Scholar]
- 82.Carter-Dawson LD, LaVail MM, Sidman RL. Differential effect of the rd mutation on rods and cones in the mouse retina. Invest Ophthalmol Vis Sci. 1978;17:489–98. [PubMed] [Google Scholar]
- 83.Margolis DJ, Detwiler PB. Cellular origin of spontaneous ganglion cell spike activity in animal models of retinitis pigmentosa. J Ophthalmol. 2011 doi: 10.1155/2011/507037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sekirnjak C, Hulse C, Jepson LH, Hottowy P, Sher A, Dabrowski W, et al. Loss of responses to visual but not electrical stimulation in ganglion cells of rats with severe photoreceptor degeneration. J Neurophysiol. 2009;102:3260–9. doi: 10.1152/jn.00663.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lewis GP, Linberg KA, Fisher SK. Neurite outgrowth from bipolar and horizontal cells after experimental retinal detachment. Invest Ophthalmol Vis Sci. 1998;39:424–34. [PubMed] [Google Scholar]
- 86.Linberg KA, Lewis GP, Matsumoto B, Fisher SK. Immunocytochemical evidence that rod-connected horizontal cell axon terminals remodel in response to experimental retinal detachment in the cat. Mol Vis. 2006;12:1674–86. [PubMed] [Google Scholar]
- 87.Wong RO, Herrmann K, Shatz CJ. Remodeling of retinal ganglion cell dendrites in the absence of action potential activity. J Neurobiol. 1991;22:685–97. doi: 10.1002/neu.480220704. [DOI] [PubMed] [Google Scholar]
- 88.Beltran WA, Hammond P, Acland GM, Aguirre GD. A frame-shift mutation in RPGR exon ORF15 causes photoreceptor degeneration and inner retina remodeling in a model of X-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2006;47:1669–81. doi: 10.1167/iovs.05-0845. [DOI] [PubMed] [Google Scholar]
- 89.Ray A, Sun GJ, Chan L, Grzywacz NM, Weiland J, Lee EJ. Morphological alterations in retinal neurons in the S334ter-line3 transgenic rat. Cell Tissue Res. 2010;339:481–91. doi: 10.1007/s00441-009-0916-5. [DOI] [PubMed] [Google Scholar]
- 90.Iandiev I, Uckermann O, Pannicke T, Wurm A, Tenckhoff S, Pietsch UC, et al. Glial cell reactivity in a porcine model of retinal detachment. Invest Ophthalmol Vis Sci. 2006;47:2161–71. doi: 10.1167/iovs.05-0595. [DOI] [PubMed] [Google Scholar]
- 91.Ross JW, Fernandez de Castro JP, Zhao J, Samuel M, Walters E, Rios C, et al. Generation of an inbred miniature pig model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2012;53:501–7. doi: 10.1167/iovs.11-8784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Anderson JR, Bones JW, Watt CB, Shaw MV, Yang JH, Demill D, et al. Exploring the retinal connectome. Mol Vis. 2011;17:355–79. [PMC free article] [PubMed] [Google Scholar]
- 93.Cuenca N, Pinilla I, Fernández-Sánchez L, Salinas-Navarro M, Alarcón-Martínez L, Avilés-Trigueros M, et al. Changes in the inner and outer retinal layers after acute increase of the intraocular pressure in adult albino Swiss mice. Exp Eye Res. 2010;91:273–85. doi: 10.1016/j.exer.2010.05.020. [DOI] [PubMed] [Google Scholar]
- 94.Yefimova MG, Messaddeq N, Karam A, Jacquard C, Weber C, Jonet L, et al. Polyglutamine toxicity induces rod photoreceptor division, morphological transformation or death in spinocerebellar ataxia 7 mouse retina. Neurobiol Dis. 2010;40:311–24. doi: 10.1016/j.nbd.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 95.Firth SI, Feller MB. Dissociated GABAergic retinal interneurons exhibit spontaneous increases in intracellular calcium. Vis Neurosci. 2006;23:807–14. doi: 10.1017/S095252380623013X. [DOI] [PubMed] [Google Scholar]
- 96.Voigt T, Wassle H. Dopaminergic innervation of A II amacrine cells in mammalian retina. J Neurosci. 1987;7:4115–28. doi: 10.1523/JNEUROSCI.07-12-04115.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kolb H, Cuenca N, Dekorver L. Postembedding immunocyto-chemistry for GABA and glycine reveals the synaptic relationships of the dopaminergic amacrine cell of the cat retina. J Comp Neurol. 1991;310:267–84. doi: 10.1002/cne.903100210. [DOI] [PubMed] [Google Scholar]
- 98.Feigenspan A, Gustincich S, Bean BP, Raviola E. Spontaneous activity of solitary dopaminergic cells of the retina. J Neurosci. 1998;18:6776–89. doi: 10.1523/JNEUROSCI.18-17-06776.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jones BW, Marc RE, Watt CB. Dopaminergic amacrine and interplexiform cells exhibit glutamatergic signatures. Invest Ophthalmol Vis Sci. 2004;45 E (abstract 5435) [Google Scholar]
- 100.VanLeeuwen JE, Petzinger GM, Walsh JP, Akopian GK, Vuckovic M, Jakowec MW. Altered AMPA receptor expression with treadmill exercise in the 1-methyl-4-phenyl-1,2,3,6-tetra-hydropyridine-lesioned mouse model of basal ganglia injury. J Neurosci Res. 2010;88:650–68. doi: 10.1002/jnr.22216. [DOI] [PubMed] [Google Scholar]
- 101.Kim M, Carman CV, Springer TA. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science. 2003;301:1720–5. doi: 10.1126/science.1084174. [DOI] [PubMed] [Google Scholar]
- 102.Shimaoka M, Takagi J, Springer TA. Conformational regulation of integrin structure and function. Annu Rev Biophys Biomol Struct. 2002;31:485–516. doi: 10.1146/annurev.biophys.31.101101.140922. [DOI] [PubMed] [Google Scholar]
- 103.Cukierman E, Pankov R, Stevens DR, Yamada KM. Taking cell-matrix adhesions to the third dimension. Science. 2001;294:1708–12. doi: 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- 104.Kremer H, van Wijk E, Märker T, Wolfrum U, Roepman R. Usher syndrome: molecular links of pathogenesis, proteins and pathways. Hum Mol Genet. 2006;15(Spec2):R262–70. doi: 10.1093/hmg/ddl205. [DOI] [PubMed] [Google Scholar]
- 105.Lin Y, Jones BW, Liu A, Tucker JF, Rapp K, Luo L, et al. Retinoid receptors trigger neuritogenesis in retinal degenerations. FASEB J. 2011;36:81–92. doi: 10.1096/fj.11-192914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lin Y, Jones BW, Rapp K, Shaw MV, Yang J-H, Watt CB, et al. Roles of retinoic acid signaling in neuritogenesis during light-induced retinal degeneration. ARVO Meeting Abstracts. 2010;51:5592. [Google Scholar]
- 107.Lin Y, Jones BW, Rapp K, Shaw MV, Yang J-H, Watt CB, et al. CaMKII signaling is contributive to neuritogenesis in light-induced retinal degeneration. ARVO Meeting Abstracts. 2011;52:1846. [Google Scholar]
- 108.Marc RE, Liu W. Fundamental GABAergic amacrine cell circuitries in the retina: nested feedback, concatenated inhibition, and axosomatic synapses. J Comp Neurol. 2000;425:560–82. doi: 10.1002/1096-9861(20001002)425:4<560::aid-cne7>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 109.Anderson JR, Jones BW, Yang J-H, Shaw MV, Watt CB, Koshevoy P, et al. A computational framework for ultrastructural mapping of neural circuitry. PLoS Biol. 2009;7:e1000074. doi: 10.1371/journal.pbio.1000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kondo M, Sakai T, Komeima K, Kurimoto Y, Ueno S, Nishizawa Y, et al. Generation of a transgenic rabbit model of retinal degeneration. Invest Ophthalmol Vis Sci. 2009;50:1371–7. doi: 10.1167/iovs.08-2863. [DOI] [PubMed] [Google Scholar]
- 111.Gal A, Li Y, Thompson DA, Weir J, Orth U, Jacobson SG, et al. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat Genet. 2000;26:270–1. doi: 10.1038/81555. [DOI] [PubMed] [Google Scholar]
- 112.Seiler MJ, Aramant RB, Thomas BB, Peng Q, Sadda SR, Keirstead HS. Visual restoration and transplant connectivity in degenerate rats implanted with retinal progenitor sheets. Eur J Neurosci. 2010;31:508–20. doi: 10.1111/j.1460-9568.2010.07085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Baloyannis SJ. Dendritic pathology in Alzheimer’s disease. J Neurol Sci. 2009;283:153–7. doi: 10.1016/j.jns.2009.02.370. [DOI] [PubMed] [Google Scholar]
- 114.Bywood PT, Johnson SM. Dendrite loss is a characteristic early indicator of toxin-induced neurodegeneration in rat midbrain slices. Exp Neurol. 2000;161:306–16. doi: 10.1006/exnr.1999.7259. [DOI] [PubMed] [Google Scholar]
- 115.Prince DA, Parada I, Scalise K, Graber K, Shen F. Epilepsy following cortical injury: cellular and molecular mechanisms as targets for potential prophylaxis. Epilepsia. 2009;50(Suppl 2):30–40. doi: 10.1111/j.1528-1167.2008.02008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Duncan T, Wiggert B, Whittaker N, Darrow R, Organisciak DT. Effect of visible light on normal and p23h-3 transgenic rat retinas: characterization of a novel retinoic acid derivative present in the p23h-3 retina. Photochem Photobiol. 2006;82:741–5. doi: 10.1562/2005-10-05-RA-712. [DOI] [PubMed] [Google Scholar]
- 117.Clagett-Dame M, McNeill E, Muley P. Role of all-trans retinoic acid in neurite outgrowth and axonal elongation. J Neurobiol. 2006;66:739–56. doi: 10.1002/neu.20241. [DOI] [PubMed] [Google Scholar]
- 118.Thiagarajan TC, Piedras-Renteria ES, Tsien RW. Alpha and beta CAMKII: inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron. 2002;36:1103–14. doi: 10.1016/s0896-6273(02)01049-8. [DOI] [PubMed] [Google Scholar]
- 119.Jiang L, Boye SL, Dizhoor A, Hauswirth WW, Baehr W. Knock-down of GCAP1 by RNA interference delays photoreceptor degeneration in GCAP1-Y99C transgenic mice. Invest Ophthalmol Vis Sci. 2011;51:4488. [Google Scholar]
- 120.Terada N, Hamazaki T, Oka M, Hoki M, Mastalerz DM, Nakano Y, et al. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature. 2002;416:542–5. doi: 10.1038/nature730. [DOI] [PubMed] [Google Scholar]
- 121.Canola K, Angénieux B, Tekaya M, Quiambao A, Naash MI, Munier FL, et al. Retinal stem cells transplanted into models of late stages of retinitis pigmentosa preferentially adopt a glial or a retinal ganglion cell fate. Invest Ophthalmol Vis Sci. 2007;48:446–54. doi: 10.1167/iovs.06-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bull ND, Limb GA, Martin KR. Human Muller stem cell (MIO-M1) transplantation in a rat model of glaucoma: survival, differentiation, and integration. Invest Ophthalmol Vis Sci. 2008;49:3449–56. doi: 10.1167/iovs.08-1770. [DOI] [PubMed] [Google Scholar]
- 123.Busskamp V, Duebel J, Balya D, Fradot M, Viney TJ, Siegert S, et al. Genetic reactivation of cone photoreceptors restores visual responses in retinitis pigmentosa. Science. 2010;329:413–7. doi: 10.1126/science.1190897. [DOI] [PubMed] [Google Scholar]
- 124.Lagali P, Balya D, Awatramani GB, Münch TA, Kim DS, Busskamp V, et al. Light-activated channels targeted to ON bipolar cells restore visual function in retinal degeneration. Nat Neurosci. 2008;11:667–75. doi: 10.1038/nn.2117. [DOI] [PubMed] [Google Scholar]
- 125.Doroudchi MM, Greenberg KP, Liu J, Silka KA, Boyden ES, Lockridge JA, et al. Virally delivered channelrhodopsin-2 safely and effectively restores visual function in multiple mouse models of blindness. Mol Ther. 2011;19:1220–9. doi: 10.1038/mt.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Marc R. Injury and repair: retinal remodeling. In: Dana R, editor. Encyclopedia of the eye. Amsterdam: Elsevier; 2009. [Google Scholar]
- 127.Ivanova E, Pan Z-H. Evaluation of the adeno-associated virus mediated long-term expression of channelrhodopsin-2 in the mouse retina. Mol Vis. 2009;15:1680–9. [PMC free article] [PubMed] [Google Scholar]