

Abstract
Progressive osseous heteroplasia (POH) is a rare developmental disorder of heterotopic ossification (HO) caused by heterozygous inactivating germline mutations in the paternal allele of the GNAS gene. Interestingly, POH lesions have a bewildering mosaic distribution. Using clinical, radiographic, and photographic documentation, we found that most of the 12 individuals studied had a lesional bias toward one side or the other, even showing exclusive sidedness. Most strikingly, all had a dermomyotomal distribution of HO lesions. We hypothesized that somatic mutations in a progenitor cell of somitic origin may act on a background of germline haploinsufficiency to cause loss of heterozygosity at the GNAS locus and lead to the unilateral distribution of POH lesions. Taking advantage of the chick system, we examined our hypothesis by mimicking loss of heterozygosity of GNAS expression using dominant-negative GNAS that was introduced into a subset of chick somites, the progenitors that give rise to dermis and muscle. We observed rapid ectopic cartilage and bone induction at the axial and lateral positions in a unilateral distribution corresponding to the injected somites, which suggests that blocking GNAS activity in a targeted population of progenitor cells can lead to mosaic ectopic ossification reminiscent of that seen in POH.
Introduction
Progressive osseous heteroplasia (POH; OMIM 166350) is a rare genetic condition of progressive heterotopic ossification (HO) that begins with cutaneous and subcutaneous ossification in infancy and progresses in childhood to involve deep connective tissues, including skeletal muscle and fascia (1). Eventually, extensive HO in the soft tissue leads to loss of mobility of affected areas (2). Most cases of POH are caused by heterozygous inactivating mutations in the paternally inherited allele of GNAS. The GNAS gene is a transcriptionally complex locus that encodes several transcripts, including Gsα, the most abundant product (3). Gsα is the α subunit of the stimulatory G protein, a protein associated with GPCRs (1, 4). Patients with POH have various heterozygous frameshift mutations that lead to inactivation of this protein (5). As a result of reduced Gsα levels, less adenylyl cyclase is activated upon ligand binding to receptors, leading to a reduced level of cAMP (5–7). Unlike other GNAS-inactivation disorders of HO, POH occurs in the absence of hormone resistance and multiple features of Albright hereditary osteodystrophy (AHO), including obesity (4, 8).
Although diagnostic criteria have been proposed (4), the extent and patterns of HO lesions in POH have not been characterized. In this study, we describe the mosaic distribution of patient lesions and show that they lateralized in a striking dermomyotomal pattern. Our observations that POH lesions were often lateralized and did not cross the midline suggested that the affected tissues are derived from a common progenitor cell population. This unique pattern of HO distribution led us to hypothesize that the common progenitor cell might be somitic in origin.
The somite is a transient mesoderm-derived structure formed during embryogenesis that contains the progenitors of dermis, muscle, and bone (9). The somite is first formed as an epithelial sphere from the unsegmented mesoderm that lies laterally to the neural tube. As development proceeds, ventral cells of the somite become de-epithelialized and form the sclerotome, which gives rise to the cartilage and bone of the vertebrae and ribs. Dorsal cells of the somite remain epithelialized, and are designated as the dermomyotome. Eventually, the dermomyotome is separated into the dermatome and the myotome, which form the dermis of the trunk and the muscle of the trunk and limbs, respectively (9, 10).
The identity of the somite is tightly controlled (11, 12). Early-formed somites give rise to progenitors of the dermis and musculoskeletal structures at the cervical spine level, while later-formed somites give rise to more posterior skeletal structures at the thoracic and lumbosacral levels. Therefore, tissues of the body wall at each axial level arise from descendants of a subset of somite progenitors. After the initial specification, the somitic cells continue to remain as a clearly defined group in the trunk with very little mixing from adjacent somites (13). Trunk neural crest cells migrate laterally between these structures, forming a segmented arrangement of the spinal nerves (14). Accordingly, the somitic progenitor cells give rise to tissues that follow the horizontal stripes of the trunk of the body, which are also referred to as dermomyotomes in clinical analysis (15).
We hypothesized that GNAS inactivation in a subpopulation of somite progenitor cells during embryogenesis would result in a mosaic distribution of subcutaneous ectopic cartilage and bone formation in the progeny of these affected cells. The chick embryo system allows the flexibility of introducing genes at specific locations and developmental stages (16); thus, we tested this hypothesis by introducing a dominant-negative GNAS mutant (referred to herein as DN-GNAS) into a subset of chicken somites that give rise to structures at the lumbosacral level. Our data revealed that DN-GNAS expression in vivo induced rapid ectopic cartilage growth that protruded outward from the skin, as well as ossification in localized areas independent of existing skeletal elements. The phenotype was only observed on the injected side and at the expected lumbosacral axial level, exhibiting unilateral distribution and supporting the notion of a localized effect of GNAS inactivation.
Results
POH patients exhibit sidedness of HO lesions with a dermomyotomal distribution.
We evaluated 12 consecutive patients with POH who had severe musculoskeletal complaints with joint involvement that limited activities of daily living. All patients had GNAS mutations, except patients 1 and 2 (Supplemental Table 1; supplemental material available online with this article; doi: 10.1172/JCI69746DS1). The number and location of regional body areas affected by HO were determined in order to assess the presence of preferential sidedness (Figure 1). 10 of 12 patients (83%) had a lesional bias toward either the left (n = 6, patients 1–6) or the right (n = 4, patients 9–12) side.
Figure 1. Lateralization of POH lesions.

The number of affected anatomic sites was scored as the presence of HO in any of 30 regional body areas. Patients are ranked in order of percent sidedness, from the leftmost to the rightmost. Note the exclusive lateralization in 7 of the 12 patients (83%) examined.
7 of the 12 patients (58%; patients 1–4 and 10–12) showed exclusive lateralization, in which the extent of HO lesions never crossed the midline (Figure 2). Based on direct observation, plain radiographs, CT scans, and clinical photographs, we characterized the distribution of lesions by plotting them onto surface body maps. The pattern of lesion extension followed a dermomyotomal distribution (Figure 3). Most lesions did not cross the midline, but when they did, they continued along the same or adjacent dermatomes (e.g., patients 6–9; Figure 3).
Figure 2. Sidedness in POH.
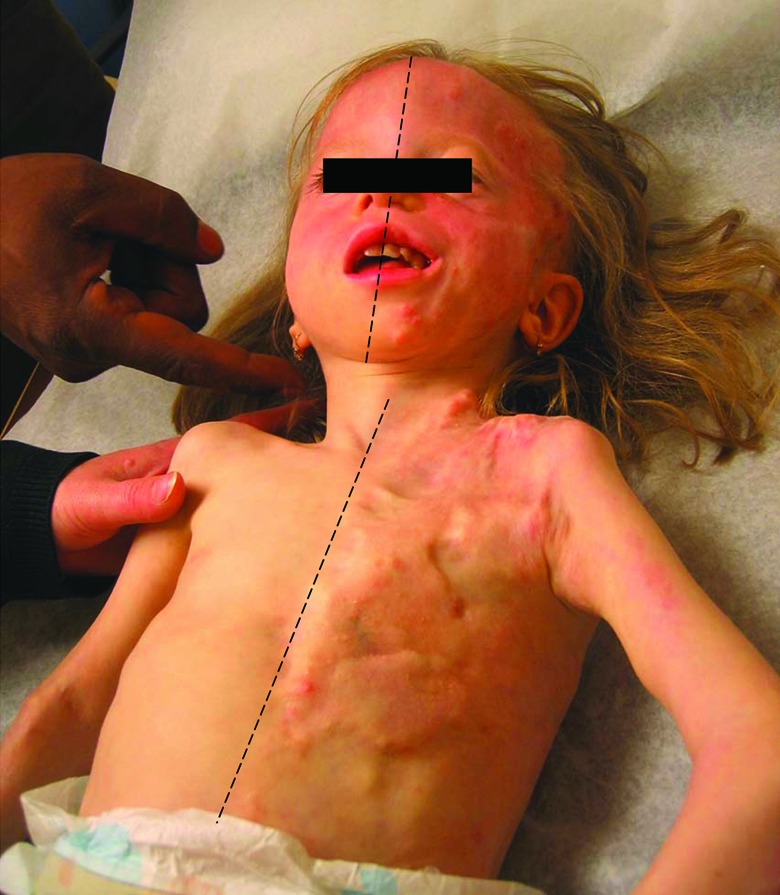
Left-sided lesions of HO on the head, face, chest, and abdomen of patient 5 did not cross the midline.
Figure 3. Dermomyotomal distribution of HO in POH.

Stippled areas depict lesion formation along the indicated dermomyotomes of patients 1–12. F, female; M, male.
Ectopic expression of DN-GNAS in adipose-derived stromal cells leads to increased mineralization.
This unique pattern of dermomyotomal distribution in POH patients suggested that GNAS inactivation in a subpopulation of somite progenitor cells during embryogenesis would result in a mosaic distribution of subcutaneous ectopic cartilage and bone formation in the progeny of these affected cells. To test this hypothesis, we first selected a mutant form of GNAS that acts in a dominant-negative manner and thus causes a dramatic reduction of cAMP levels. G proteins form a heterotrimeric complex consisting of 3 subunits: α, β, and γ. Upon ligand binding to a GPCR, Gsα becomes activated, as it dissociates GDP and binds to GTP, an event that induces adenylate cyclase, which converts ATP to cAMP (17). We characterized a mutant construct of GNAS, GNAS-long-Q227L/D295N, where double amino acid changes (glutamine [Q] to leucine [L] and aspartate [D] to asparagine [N]) resulted in a high affinity for GDP as well as a marked reduction in GTPase activity, thereby inhibiting signaling downstream of the GPCR (17). Thus, these mutant proteins should behave in a dominant-negative manner by competing with endogenous GNAS and blocking its function (18–20). As it is not clear which GPCRs mediate the signaling that causes the POH phenotype, we tested the effect of this mutant using prostaglandin 1 (PGE1), which acts through GPCRs, as an established reagent to raise cAMP level (21). Transfection of GNAS-long-Q227L/D295N strongly prevented PGE1-mediated cAMP induction (Supplemental Figure 1). Furthermore, transfection of GNAS-long-Q227L/D295N in the presence of forskolin, a reagent that induces cAMP independently of a GPCR (22), did not prevent cAMP induction (data not shown). Together, these results indicated that GNAS-long-Q227L/D295N inhibits GPCR-mediated signaling at the receptor level by acting in a dominant-negative fashion. Hence, GNAS-long-Q227L/D295N, which we termed DN-GNAS, was used for our subsequent experiments.
Because POH lesions have been observed to initiate in the dermal and subcutaneous fat (1), we directly assessed whether DN-GNAS could cause increased mineralization in chicken adipose-derived stromal cells cultured in DMEM with 10% FBS. Consistent with reduced cAMP levels, DN-GNAS inhibited the expression of PPARG in in vitro–cultured cells (Figure 4A). DN-GNAS led to a significant increase in the mRNA expression of the early bone marker alkaline phosphatase (ALPL) in these cells at day 5, which correlated with ALPL activity; no differences in ALPL mRNA expression in day 7 and 14 cultures were detected (Figure 4, A and B). Consistent with ALPL expression at day 5, we observed a strong increase in Alizarin red staining at day 14 (Figure 4C). As these adipose-derived stromal cells were grown in standard growth media, this result suggests that DN-GNAS is sufficient to promote osteogenesis and mineralization, even in the absence of osteogenic induction factors.
Figure 4. Ectopic expression of DN-GNAS increased mineralization of adipose-derived stromal cells.

(A) RT-PCR analysis of PPARG and ALPL in adipose-derived stromal cells after culturing for 5, 7, and 14 days in DMEM with 10% FBS. Data represent mean ± SD. (B) ALPL activity assay in adipose-derived stromal cells after 5 days of culture. (C) Alizarin red staining of adipose-derived stromal cells after 14 days of culture. Original magnification, ×10 (B and C).
Expression of DN-GNAS in somites induces unilateral outgrowth of ectopic skeletal elements at the respective axial levels.
To determine whether inactivation of GNAS-mediated signaling in a subset of somite progenitor cells leads to ossification in a spatially restricted manner, we injected retroviruses encoding DN-GNAS into 4 consecutive caudal somites that lie on one side of the neural tube in 2.5-day-old chick embryos (Supplemental Figure 2A). We chose this stage (stage 17) because no endogenous (skeletal) ossification has yet taken place, allowing sufficient time for the virus to act on somite progenitor cells during cartilage and bone formation (16, 23). During embryonic development, the caudal somites of stage-17 embryos give rise to dermis, muscle, and bone tissues of the trunk at the lumbosacral level as well as the limb (11, 12).
The effect of ectopic expression of DN-GNAS was examined after 10 days of in ovo incubation after somite injections. All embryos injected with control GFP retrovirus (n = 17) showed a limited unilateral GFP protein distribution in the trunk and limbs at the expected axial level and had no anatomical defects (Supplemental Figure 2, B and C). We found that 16 of 19 DN-GNAS–injected embryos showed a “hardened ridge,” which gave a shiny appearance to the skin surface and was devoid of feathers (Figure 5, A and B). The outgrowth was evaluated histologically by H&E staining and was found to consist of a skeletal-like structure that was contiguous with the pelvic girdle and extended to the epidermal layer of the skin. No apparent dermal or epidermal structures, including feather buds, were present at that location (Figure 5C). The uninjected contralateral side showed normal dermal and skeletal structures (Figure 5C). Pan-keratin, an epithelial cell marker, showed the expected continuous expression in the skin of the uninjected side, while DN-GNAS injection caused a complete loss of pan-keratin expression at the skeletal-like outgrowth (Figure 5D and ref. 24). Alcian blue staining indicated that the outgrowth was cartilaginous (Figure 5E). Immunohistochemistry using an antibody against the viral coat protein GAG indicated that the virus had caused widespread infection in the injected side, with stronger staining present in the soft tissues surrounding the cartilage element (Figure 5F). These observations suggest that DN-GNAS inhibits the epidermal or dermal cell fate in cells adjacent to the ectopic cartilage tissue, but does not directly activate the chondrogenic program in these cells.
Figure 5. DN-GNAS viral expression in the somites induces skin protrusions.

(A) “Hardened” outgrowth (arrow) in the lumbosacral region of the embryo on the DN-GNAS–injected side. Note the shiny appearance of the outgrowth, which was devoid of feather structures. (B) The outgrowth on the DN-GNAS–injected side (arrow) was excised for histological analysis. (C) Histology showing DN-GNAS–induced extension of the pelvic girdle through the dermis; (D) absence of the epithelial cell marker pan-keratin (arrow) in areas near the outgrowth; (E) and extension of cartilage elements of the pelvic girdle (Alcian blue [arrow]; the area immediately left of the cartilage element was devoid of staining for Alcian blue as well as pan-keratin [arrowhead]). (F) Immunohistochemistry detection of viral coat protein GAG. Original magnification, ×1 (A and B); ×10 (C, low mag); ×20 (C, high mag, and D–F).
Ectopic expression of DN-GNAS induces HO.
The finding of excess cartilage growth of the pelvic girdle in GNAS-injected embryos prompted us to perform Alcian blue staining analysis on the entire animal. To visualize the distribution of the injected viruses, GFP virus was coinjected with DN-GNAS. We found that DN-GNAS induced substantial thickening of the ribs and pelvic girdle (Figure 6A), corresponding to the area of DN-GNAS virus expression (18 of 19 embryos). To visualize mineralization, we included Alizarin red along with Alcian blue in our staining solution and found cartilaginous nodules in already-mineralized tissues (Figure 6, B and C), suggestive of delayed or altered endochondral ossification. In addition, we observed thickened mineralized tissues in response to DN-GNAS (Figure 6C), consistent with the observed thickened cartilage template.
Figure 6. DN-GNAS expression in the somites causes skeletal abnormalities.

(A) Whole-mount Alcian blue images of retrovirus GFP and DN-GNAS coinjected embryos. GFP encoding avian retrovirus vector RCAS-B was coinjected with DN-GNAS encoding retrovirus RCAS-A to allow visualization of viral expression. Thickening of the cartilaginous ribs (arrow) as well as the pelvic girdle (arrowhead) was observed. DN-GNAS virus injection alone yielded the same results as DN-GNAS coinjection with GFP virus. (B) Alcian blue and Alizarin red staining showing a cartilage nodule in the forelimb of DN-GNAS–injected side (arrow), while the contralateral side showed uniform mineralization. (C) An ectopic cartilage element was present in the hindlimb of the DN-GNAS–injected side (arrow) of a representative embryo. Note that the mineralized femur was also thicker on the injected side (asterisk) than on the contralateral side. (D) μCT analysis of GFP-injected embryos, indicating normal mineralization. (E) A DN-GNAS–injected embryo showing altered mineralization during endochondral bone formation in the ribs (arrowheads). (F) A DN-GNAS–injected embryo showing thickened mineralized ribs after endochondral bone formation (arrows). Original magnification, ×0.71 (A, low mag); ×2.0 (A, high mag); ×1.6 (B); ×1.25 (C).
The mineralization pattern of the embryos was confirmed by μCT analysis (Figure 6, D–F). DN-GNAS injection clearly showed rib abnormalities that ranged from impaired ossification (Figure 6E), which corresponded to enhanced Alcian blue staining, to increased thickness and irregularity of mature rib bones (Figure 6F). It is possible that phenotype variation with respect to degrees of mineralization at the time of harvest is due to embryos that might be at slightly different stages in development when DN-GNAS was injected or that they exhibited variable growth rates. Thus, the embryo shown in Figure 6F might be slightly more mature at the time of harvest, giving the expanded cartilage tissue sufficient time to become mineralized bone.
In addition to the rib phenotype, we observed distinct islands of HO between the skeletal elements and in the muscle of the hindlimb (Figure 7, A–C). μCT images also identified multiple areas of heterotopic bone in the area around the pelvic girdle (Figure 7, D and E). H&E staining showed that this ectopic mineralized zone was independent of preexisting skeletal elements, and no remnants of cartilage matrix were present (Figure 7F). At this stage of embryonic development, cartilage matrix should not be completely absent if the ossification process uses an endochondral ossification mechanism; therefore, the absence of cartilage matrix in the mineralized tissue supports the notion that the observed HO was formed by intramembraneous ossification.
Figure 7. DN-GNAS expression in somites induces HO independently of the skeletal elements.

(A) Alcian blue and Alizarin red staining showing patches of Alizarin red–positive tissues in between the ribs and the pelvic girdle (arrows). (B) Alcian blue and Alizarin red staining of sections of the protruding pelvic girdle. Note islands of Alizarin red stained areas outside the cartilaginous pelvic girdle (arrows). (C) Alcian blue and Alizarin red staining showing patches of mineralized tissues in the musculature of the hindlimb (arrow). (D) μCT analysis showing HO (arrows). (E) Enlarged images showing islands of mineralized zones. (F) H&E staining of heterotopic bone from the boxed region (red arrow) in E. Original magnification, ×1.6 (A and C); ×20 (B); ×10 (F).
Discussion
POH criteria redefined.
POH, a rare autosomal-dominant disease associated with inactivation of GNAS, is characterized by infantile ectopic bone formation in the dermis, which progresses into the deep connective tissue and skeletal muscle during childhood (1, 2, 5). Histological analysis revealed that the HO in the dermis is closely associated with subcutaneous adipose tissue and can arise by intramembranous ossification, endochondral ossification, or both (1, 4, 5). We previously established diagnostic criteria for POH to include the presence of superficial HO that progresses to deeper tissues in the absence of multiple other AHO features and without hormone resistance (4). It is important to note that while progressive deep HO is an obligate feature of POH, even superficial HO is a variable feature of AHO, with some but not all patients with AHO/pseudopseudohypoparathyroidism presenting with HO. In this study, we provide evidence that supports refinement of diagnostic criteria for POH to include progressive HO along a predominantly dermomyotomal distribution that often has a lateralization bias.
Mosaicism in POH.
Despite the association of germline GNAS mutation with POH, the resulting HO that defines this disease manifests in a mosaic pattern. Mosaicism has been described in many monogenic skin disorders and occurs in various patterns, with and without lateralization (25). For example, epidermolytic hyperkeratosis (EHK), a disease characterized by patches of red skin and blistering, has been shown to exhibit mosaicism in the skin along Blaschko’s lines, which are V-shaped patterns along the middle back that are believed to trace pathways of the migration of ectodermal cells during development (26, 27). As another example, cutaneous periarticular ossification in melorheostosis can occur in a mosaic distribution and may be related to originating in a corresponding myotome (28). With respect to POH, HO in the dermis shows a seemingly random distribution of affected areas. We have characterized the mosaic distribution of lesions in POH and found that affected areas of heterotopic bone lateralized in a distinct dermomyotomal pattern that is present in very few conditions. In some patients, dermomyotomal distribution was partial, which suggests that lesion progression was incomplete or delayed at the time of presentation. Despite these observations, the manner in which this mosaicism occurs with regard to the associated GNAS mutation is currently unclear.
Pathogenesis of POH and relation to GNAS.
Heterozygous Gsα-specific exon 1 inactivation of Gnas in a mouse knockout model manifests as subcutaneous HO by an intramembranous process (29). While this model successfully demonstrates HO in the dermis, the lesions do not progress into deeper tissues, nor do they develop in the unique pattern we observed in POH patients in the present study. A possible explanation is that the modulation of GNAS expression levels may be critical to set the threshold for disease initiation. As such, the pathophysiology of GNAS inactivation leading to POH could plausibly behave in a manner similar to that of a tumor suppressor gene, with one inactivated allele insufficient to cause HO but a “second hit” enabling the process.
The etiology of classical genetic mosaicism is the presence of at least 2 genotypically different cell populations derived from a single zygote (30), which result from mutation during development and are retained by a finite number of cells in the postnatal state. Possible mechanisms include the presence of somatic mutations or random inactivation of the second GNAS allele in progenitor cells, a de novo mutation in a gene that normally functions in a GNAS-interacting pathway, or epigenetic changes in somatic cells. Revertant mosaicism in uninvolved dermomyotomes, or in patients with GNAS mutations and no apparent or very limited disease, cannot be excluded. Although the distribution of POH lesions was reminiscent of those found in herpes zoster, by clinical criteria, it is unlikely that viral reactivation serves as a precipitating event in HO formation. However, this does not rule out the possibility of peripheral nerve or nerve root involvement in POH.
Widespread monoallelic expression has recently been described and invokes the possibility that gene inactivation may occur via nonmutational mechanisms (31). Random inactivation of GNAS has been reported in tissues where it is biallelically expressed, and thus mosaic distribution of lesions could result from random GNAS inactivation in target stem cells (32). Such random inactivation could lead to a random distribution of disease-related lesions if target stem cell number was high; however, if relevant stem cells were rare, then random inactivation of a putative gene might lead to a nonrandom distribution, as in POH.
Most POH patients have an average age of onset less than 1 year; however, a subset of patients have much later ages of onset, ranging 4–30 years (4), which suggests that the timing of a potential second hit is variable. This does not exclude the possibility that concomitant epigenetic and/or environmental factors also play a role in clinical heterogeneity. For example, we cannot exclude a role for factors that may trigger secondary changes in stem cells (such as viral infection or trauma); however, such intercurrent events, at least during very early development, usually lead to more severe (multisystem) morphogenetic defects or embryopathy (33). In McCune-Albright syndrome, in which variable combinations of fibrous dysplasia of bone, endocrine hyperfunctioning, and skin hyperpigmentation are caused by activating mutations in GNAS, the timing of a second mutation and the location of target stem cells may dictate the severity and tissue specificity, respectively, of disease (32). Similarly, in POH, the timing of a second hit to a stem cell (or niche cell that interacts with a stem cell) and the location of that stem or niche cell may also dictate the respective severity and tissue specificity of disease.
Potential progenitor cells involved in POH.
It is still unclear which cells give rise to heterotopic bone in POH. The lateralized dermomyotomal distribution of POH lesions suggests the involvement of an affected early mesenchymal progenitor cell, neural crest-derived precursor, or niche cell/stem cell interaction. Based on the specific location of the bone-forming lesions, we hypothesized that these cells may be derived from the somites. Our data showed that somite-derived cells, including dermomyotomal cells and sclerotomal cells, could support heterotopic bone formation. However, we cannot exclude the possibility that alternative or additional progenitor cells may contribute to HO in POH patients. Consistent with this idea, extraskeletal ossification in the subcutis and skeletal muscles was observed in conditional knockout mice in which Gsα was deleted in all tissues expressing renin, which includes renal and selected blood vessels (34); however, this model also failed to demonstrate progressive and lateralized HO formation. Due to the close association of subcutaneous fat with heterotopic bone, multipotent adipose-derived stromal cells may play an important role in this process, either serving as a progenitor cell population or providing a favorable niche to support ossification. In fact, mouse adipose-derived stromal cells with haploinsufficiency of GNAS showed a higher tendency to become osteogenic and a lower potential for adipogenesis (8, 29). Here we found that chicken adipose-derived stromal cells infected with a dominant-negative form of GNAS also showed a preference for mineralization over an adipogenic fate, consistent with prior studies.
A novel chick embryo model of POH with unilateral dermomyotomal distribution of HO.
For in vivo studies, we used the chick embryo system, which allows for gene manipulation at specific locations and developmental stages without necessarily compromising the mortality of the organism. This novel model of POH circumvented potential issues with current mouse models. For example, we were able to abrogate GNAS signaling in a specific subset of progenitors as opposed to using models with haploinsufficient GNAS expression. In this way, we essentially mimicked loss of heterozygosity of GNAS expression, or created a second hit, by using DN-GNAS. Furthermore, in addition to Gsα, several other transcripts are encoded by the GNAS locus, including an extra-long form of Gs protein, XLαs, as well as NESP55 and a noncoding sequence 1A (1, 6, 35–37). Mutations in these genes, depending on the parental origin of the mutant allele, give rise to a spectrum of bone and endocrine phenotypes (1, 37, 38). One confounding aspect of POH is that GNAS is an imprinted gene; more specifically, POH is associated with GNAS inactivation of the paternal allele (1, 2). Our DN-GNAS functioned to block the activity of endogenous GNAS, regardless of the parental origin of the endogenous GNAS transcript. In addition, DN-GNAS should inhibit not only Gsα activity, but also that of XLαs, as the same receptors that signal via XLαs would also be blocked by DN-GNAS (37).
We found that GNAS inactivity caused dramatic HO in a mosaic distribution when DN-GNAS was expressed in early somite progenitors. These progenitors, derived from either sclerotome or dermomyotome, have previously been shown give rise to the dermis, muscle, and bone of the trunk as well as limb musculature through lineage tracing studies (9, 10). Accordingly, DN-GNAS–induced HO lesions occurred only at the corresponding axial and lateral positions in these somite-derived tissues. It is important to note that in the human condition of POH, HO first originates in the dermis, then spreads to deeper tissues, such as the muscle and adjacent cartilage and bone. Because our method likely delivers DN-GNAS to progenitors of all of these cell types simultaneously, we could not ascertain whether HO progressed from a particular tissue to another. Nevertheless, these results indicate that inhibiting GNAS activity in a subset of progenitor cells can lead to mosaic distribution of heterotopic cartilage and bone formation, thus supporting the clinical findings in POH patient studies and providing functional relevance for GNAS inactivation in a population of progenitor cells relevant to the pathophysiology of the disease.
Further study of GNAS-mediated signaling.
GPCRs mediate multiple signaling pathways that play important roles in regulating bone formation. One example is the parathyroid hormone receptor (PPR), the receptor for PTH and PTHrP (39). Indeed, Gsα-knockout mice phenocopy the PPR-null mouse with regard to regulating chondrocyte hypertropic differentiation (40). However, chondrocyte-specific Gsα knockouts also exhibit ectopic cartilage formation in the diaphysis underneath the cortical bone, which was not observed in the PPR-knockout mouse (41). This observation suggests that Gsα may signal from other receptors involved in bone formation. Smoothened and Frizzled are examples of GPCRs involved in cartilage and bone formation, serving as the receptors for hedgehog and Wnt proteins, respectively (42–46).
Our model system thus sets the stage for in vivo testing of potential roles for other candidate pathways involved in mosaicism and heterotopic bone formation in POH development and helps establish mechanistic insight into this devastating disease.
Methods
Patient criteria.
We reviewed the charts of 12 consecutive individuals with severe POH who had lost the mobility of at least 1 joint as a result of the condition. We chose subjects with at least 1 immobilized joint because less advanced cases would not have permitted evaluation of the extent in distribution and pattern of HO. All patients presented to the University of Pennsylvania Orthopaedic Surgery Outpatient Clinic and met diagnostic criteria for POH, which included the presence of superficial HO in infancy that progressed to deeper tissues during childhood, in the absence of multiple other AHO features and without hormone resistance. Patients ranged 3–43 years of age at the time of their last clinical evaluation.
Mutation analysis of the patients.
Genomic DNA was isolated from blood or lymphoblastoid cell lines (LCLs) using DNA blood-isolation reagents (QIAamp; Qiagen). If sample size was small, total genomic DNA was amplified with the Qiagen Repli-g Kit. GNAS mutation analysis was conducted by PCR amplification of genomic DNA using oligonucleotide primers (Sigma-Genosys) flanking each of the 13 exons of human GNAS, as previously described (3). Amplified samples were electrophoresed through 1% agarose gels, stained with ethidium bromide (1 μg/ml), and purified using the Qiagen Gene Clean Kit. Eluted products were sequenced by the DNA Sequencing core facilities of the University of Pennsylvania.
Evaluation of clinical features.
The mosaic distribution of HO lesions was recorded for each patient using clinical (physical examination), radiographic, and photographic documentation.
The extent and lateralization of HO lesions were scored as the number of involved regional body areas localized to the left or right of midline. The presence of HO was documented for each of the following 30 regional body areas: face/head, neck, right shoulder, left shoulder, right arm (proximal), left arm (proximal), right elbow, left elbow, right forearm, left forearm, right wrist, left wrist, right hand, left hand, chest, abdomen, back, buttocks, right hip, left hip, right thigh, left thigh, right knee, left knee, right leg, left leg, right ankle, left ankle, right foot, and left foot.
HO involvement was plotted onto body maps from information provided by direct observation, plain radiographs, CT scans, and clinical photographs. These body maps included lines depicting dermomyotomal distributions.
Generation of GNAS constructs.
A mutant construct of GNAS, GNAS-long-Q227L/D295N, was purchased from University of Missouri cDNA Resources Center. The insert of GNAS-long-Q227L/D295N was excised by HindIII and XbaI from pcDNA3.1 and cloned into Slax13 shuttle vector (47) that was also cut by HindIII and XbaI. Subsequently, the GNAS mutant form was excised from Slax13 by ClaI digestion, which was then cloned into avian retrovirus vector RCAS (23). Viruses, including the control GFP virus, were produced according to the standard protocol (23, 48) and were titered by indirect immunocytochemistry using antiviral coat protein GAG antibody. All viruses used reached a titer of 108 particles/ml.
Adipose-derived stromal cell culture.
White leghorn chicken eggs were obtained from Hy-Line International. Adipose-derived stromal cells were obtained from the pectoral fat pad based on established methods (49, 50). Briefly, pectoral fat pads of day-17 chicken embryos were excised, washed, minced, subjected to Pronase treatment (1 mg/ml in PBS) for 40 minutes at 37°C, centrifuged, and resuspended in PBS. The cell suspension was then filtered through a 40-mM cell strainer (BD Biosciences) and centrifuged, and cells were resuspended in DMEM with 10% FBS for subsequent culturing.
cAMP assay.
cAMP assays were performed using the Promega cAMP assay kit, cAMP-Glo. Briefly, 293T cells were transfected with pCDNA3 GNAS mutant constructs or GFP control (0.05 μg/well in 96-well plates) using Fugene6 transfection reagent (Roche). 24 hours after transfection, PGE1 (Sigma-Aldrich) was added to the cells at various concentrations. Cells were lysed to release cAMP, then PKA was added. The PKA reaction was detected via a luciferase reaction, where luminescence was correlated to cAMP concentrations following a standard curve.
Embryonic somite injection.
Embryonic somite injection followed established protocols (23). Hamburger Hamilton Stage 17 (60 hours of incubation) chicken embryos (White Leghorn chicken eggs; Hy-Line International) were used. Briefly, an opening was made in the eggshell to reveal the living chicken embryo. Fast green dye (1 μl) was added to a 10-μl aliquot of virus, and the mixture was injected into the lumen of newly formed somites (4 caudal somites) using a micromanipulator (Parker Picospritzer II).
GFP-expressing virus was coinjected for the purpose of tracking infected cells. GFP and DN-GNAS were encoded by RCAS retroviruses of 2 different coatings (RCAS-B and RCAS-A, respectively) so that they could be expressed in the same cell. After the operation, the window of the eggshell was sealed with transparent tape, and the embryos were incubated at 37°C for 10 days before processing and fixation for histological and μCT analyses.
Histological analysis.
Embryos were fixed in 4% paraformaldehyde for histological staining of whole-mount and sectioned embryos. Frozen tissue sections (serial sections, 10 μm) were obtained using a cryostat (Microm HM560). For paraffin tissue sections, samples were dehydrated in graded ethanol solutions, cleared with xylene, embedded in paraffin, and sectioned (5 μm) for histological staining. Alcian blue and Alizarin red staining for whole-mount tissues and sections were performed using standard procedures. For immunohistochemistry, sections were incubated with primary antibodies overnight and with secondary antibodies for 4 hours. Mouse antibody against virus coat protein GAG (Developmental Studies Hybridoma Bank) and rabbit anti-pan keratin antibody (a gift from the J. Schwob laboratory, Tufts University, Boston, Massachusetts, USA) were used. Alexa 594 (Invitrogen) was the secondary antibody. ALPL activity assay was detected by a BCIP and NBT assay (Roche).
RT-PCR analysis.
Total RNA was isolated using the RNeasy Mini kit according to the manufacturer’s protocol. For RT-PCR, cDNA was mixed with gene-specific primers, quantified using BioRad’s iQ5 Real Time PCR Detection system and optical software, and normalized against the housekeeping gene GAPDH. Primer sequences were as follows: PPARG forward, 5′-TGGACAAGGATTCATGACACGGGA-3′, PPARG reverse, 5′-ACAAACCTGGGCGATCTCCACTTA-3′; ALPL forward, 5′-AACCGTGACTGGTACTCAGATGGA-3′, ALPL reverse, 5′-TCACCTCGATGTCAGGGATGTTCT-3′; GAPDH forward, 5′-CCTGCTGCCTAGGGAAGC-3′; GAPDH reverse, 5′-CAGATCAGTTTCTATCAGCCTCT-3′.
Microscopy.
Brightfield and fluorescent images from histological and immunocytochemistry analyses were obtained with an Olympus IX71 inverted microscope and a Leica dissection microscope using an Olympus DP70 digital camera and associated software (Olympus).
For histological analysis of HO, regions of interest were visualized using a Nikon Eclipse 90i microscope and ×4 and ×20 objectives for H&E staining. Image capture was performed using NIS Elements Imaging Software 3.10 Sp2 and a Photometrics Coolsnap EZ camera.
μCT analysis.
High-resolution cross-sectional images of whole chick embryos were obtained using Scanco VivaCT 40 (Bruettisellen). Embryos were scanned using a source voltage of 55 kV, source current of 142 μA, and isotropic voxel size of 10.5 μm. A 3D image of the whole embryo was then reconstructed from the cross-sectional images using Scanco μCT V6.1 software. Bone was differentiated from non-bone by an upper threshold of 105 Hounsfield units and a lower threshold of 60 Hounsfield units.
Statistics.
All data are expressed as mean ± SD. At least 3 independent experiments were performed, and at least 3 independent samples or slides were analyzed per experiment. Significance of differences were determined by 1-factor ANOVA with post-hoc Tukey test using the statistics software SYSTAT12 (Systat). Conclusions of altered gene expression (such as upregulation and downregulation) were based on statistical analysis. A P value less than 0.05 was considered significant.
Study approval.
Patient informed consent was obtained under a protocol approved by the IRB of the University of Pennsylvania.
Supplementary Material
Acknowledgments
We thank Jeff Marchant for helpful discussions, James Schwob for providing the pan-keratin antibody, and Andreas Gal and Ludwig Meiss for information about their patient. L. Zeng was supported by grants from NSF (CBET-0966920) and NIH (R01AR059106). D.M. Cairns was supported by the Sackler Dean’s Fellowship from Tufts University. Work from the laboratories of R.J. Pignolo, E.M. Shore, and F.S. Kaplan was supported by the NIH (NIH R01-AR046831 and ARRA R01-AR046831-S1), the Progressive Osseous Heteroplasia Association (POHA), the Italian POH Association, the University of Pennsylvania Center for Research in FOP and Related Disorders, the Penn Center for Musculoskeletal Disorders, and the Isaac and Rose Nassau Professorship of Orthopaedic Molecular Medicine (to F.S. Kaplan).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(8):3624–3633. doi:10.1172/JCI69746.
References
- 1.Shore EM, Kaplan FS. Inherited human diseases of heterotopic bone formation. Nat Rev Rheumatol. 2010;6(9):518–527. doi: 10.1038/nrrheum.2010.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaplan FS, et al. Progressive osseous heteroplasia: a distinct developmental disorder of heterotopic ossification. Two new case reports and follow-up of three previously reported cases. J Bone Joint Surg Am. 1994;76(3):425–436. [PubMed] [Google Scholar]
- 3.Shore EM, et al. Paternally inherited inactivating mutations of the GNAS1 gene in progressive osseous heteroplasia. N Engl J Med. 2002;346(2):99–106. doi: 10.1056/NEJMoa011262. [DOI] [PubMed] [Google Scholar]
- 4.Adegbite NS, Xu M, Kaplan FS, Shore EM, Pignolo RJ. Diagnostic and mutational spectrum of progressive osseous heteroplasia (POH) and other forms of GNAS-based heterotopic ossification. Am J Med Genet A. 2008;146A(14):1788–1796. doi: 10.1002/ajmg.a.32346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaplan FS, Shore EM. Progressive osseous heteroplasia. J Bone Miner Res. 2000;15(11):2084–2094. doi: 10.1359/jbmr.2000.15.11.2084. [DOI] [PubMed] [Google Scholar]
- 6.Hayward BE, Moran V, Strain L, Bonthron DT. Bidirectional imprinting of a single gene: GNAS1 encodes maternally, paternally, and biallelically derived proteins. Proc Natl Acad Sci U S A. 1998;95(26):15475–15480. doi: 10.1073/pnas.95.26.15475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li T, et al. Tissue-specific expression of antisense and sense transcripts at the imprinted Gnas locus. Genomics. 2000;69(3):295–304. doi: 10.1006/geno.2000.6337. [DOI] [PubMed] [Google Scholar]
- 8.Liu JJ, Russell E, Zhang D, Kaplan FS, Pignolo RJ, Shore EM. Paternally inherited gsalpha mutation impairs adipogenesis and potentiates a lean phenotype in vivo. Stem Cells. 2012;30(7):1477–1485. doi: 10.1002/stem.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christ B, Huang R, Wilting J. The development of the avian vertebral column. Anat Embryol (Berl). 2000;202(3):179–194. doi: 10.1007/s004290000114. [DOI] [PubMed] [Google Scholar]
- 10.Stockdale FE, Nikovits W, Christ B. Molecular and cellular biology of avian somite development. Dev Dyn. 2000;219(3):304–321. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1057>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 11.Hunt P, Krumlauf R. Hox codes and positional specification in vertebrate embryonic axes. Annu Rev Cell Biol. 1992;8:227–256. doi: 10.1146/annurev.cb.08.110192.001303. [DOI] [PubMed] [Google Scholar]
- 12.Iimura T, Denans N, Pourquie O. Establishment of Hox vertebral identities in the embryonic spine precursors. Curr Top Dev Biol. 2009;88:201–234. doi: 10.1016/S0070-2153(09)88007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bagnall KM, Higgins SJ, Sanders EJ. The contribution made by cells from a single somite to tissues within a body segment and assessment of their integration with similar cells from adjacent segments. Development. 1989;107(4):931–943. doi: 10.1242/dev.107.4.931. [DOI] [PubMed] [Google Scholar]
- 14.O’Rahilly R, Muller F. The development of the neural crest in the human. J Anat. 2007;211(3):335–351. doi: 10.1111/j.1469-7580.2007.00773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drews U. Color Atlas Of Embryology . New York, New York, USA: Stuttgart Thieme Medical Publishers; 1995. [Google Scholar]
- 16.Murtaugh LC, Zeng L, Chyung JH, Lassar AB. The chick transcriptional repressor Nkx3.2 acts downstream of Shh to promote BMP-dependent axial chondrogenesis. Dev Cell. 2001;1(3):411–422. doi: 10.1016/S1534-5807(01)00039-9. [DOI] [PubMed] [Google Scholar]
- 17.Mattera R, Codina J, Crozat A, Kidd V, Woo SL, Birnbaumer L. Identification by molecular cloning of two forms of the alpha-subunit of the human liver stimulatory (GS) regulatory component of adenylyl cyclase. FEBS Lett. 1986;206(1):36–42. doi: 10.1016/0014-5793(86)81336-9. [DOI] [PubMed] [Google Scholar]
- 18.Gille A, Wenzel-Seifert K, Doughty MB, Seifert R. GDP affinity and order state of the catalytic site are critical for function of xanthine nucleotide-selective Galphas proteins. J Biol Chem. 2003;278(10):7822–7828. doi: 10.1074/jbc.M210162200. [DOI] [PubMed] [Google Scholar]
- 19.Yu B, Simon MI. Interaction of the xanthine nucleotide binding Goalpha mutant with G protein-coupled receptors. J Biol Chem. 1998;273(46):30183–30188. doi: 10.1074/jbc.273.46.30183. [DOI] [PubMed] [Google Scholar]
- 20.Xu X, et al. Promiscuous coupling of receptors to Gq class alpha subunits and effector proteins in pancreatic and submandibular gland cells. J Biol Chem. 1998;273(42):27275–27279. doi: 10.1074/jbc.273.42.27275. [DOI] [PubMed] [Google Scholar]
- 21.Terrin A, et al. PGE(1) stimulation of HEK293 cells generates multiple contiguous domains with different [cAMP]: role of compartmentalized phosphodiesterases. J Cell Biol. 2006;175(3):441–451. doi: 10.1083/jcb.200605050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo J, Chung UI, Kondo H, Bringhurst FR, Kronenberg HM. The PTH/PTHrP receptor can delay chondrocyte hypertrophy in vivo without activating phospholipase C. Dev Cell. 2002;3(2):183–194. doi: 10.1016/S1534-5807(02)00218-6. [DOI] [PubMed] [Google Scholar]
- 23.Zeng L, Kempf H, Murtaugh LC, Sato ME, Lassar AB. Shh establishes an Nkx3.2/Sox9 autoregulatory loop that is maintained by BMP signals to induce somitic chondrogenesis. Genes Dev. 2002;16(15):1990–2005. doi: 10.1101/gad.1008002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gown AM, Vogel AM. Monoclonal antibodies to intermediate filament proteins of human cells: unique and cross-reacting antibodies. J Cell Biol. 1982;95(2 pt 1):414–424. doi: 10.1083/jcb.95.2.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frank J, Happle R. Cutaneous mosaicism: right before our eyes. J Clin Invest. 2007;117(5):1216–1219. doi: 10.1172/JCI32111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Happle R. An early drawing of Blaschko’s lines. Br J Dermatol. 1993;128(4):464. doi: 10.1111/j.1365-2133.1993.tb00212.x. [DOI] [PubMed] [Google Scholar]
- 27.Paller AS. Piecing together the puzzle of cutaneous mosaicism. J Clin Invest. 2004;114(10):1407–1409. doi: 10.1172/JCI23580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murray RO, McCredie J. Melorheostosis and the sclerotomes: a radiological correlation. Skeletal Radiol. 1979;4(2):57–71. doi: 10.1007/BF00349329. [DOI] [PubMed] [Google Scholar]
- 29.Pignolo RJ, et al. Heterozygous inactivation of Gnas in adipose-derived mesenchymal progenitor cells enhances osteoblast differentiation and promotes heterotopic ossification. J Bone Miner Res. 2011;26(11):2647–2655. doi: 10.1002/jbmr.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Happle R. Epidermal nevus syndromes. Semin Dermatol. 1995;14(2):111–121. doi: 10.1016/S1085-5629(05)80006-9. [DOI] [PubMed] [Google Scholar]
- 31.Gimelbrant A, Hutchinson JN, Thompson BR, Chess A. Widespread monoallelic expression on human autosomes. Science. 2007;318(5853):1136–1140. doi: 10.1126/science.1148910. [DOI] [PubMed] [Google Scholar]
- 32.Michienzi S, et al. GNAS transcripts in skeletal progenitors: evidence for random asymmetric allelic expression of Gs alpha. Hum Mol Genet. 2007;16(16):1921–1930. doi: 10.1093/hmg/ddm139. [DOI] [PubMed] [Google Scholar]
- 33.Gilbert-Barness E. Teratogenic causes of malformations. Ann Clin Lab Sci. 2010;40(2):99–114. [PubMed] [Google Scholar]
- 34.Castrop H, et al. Skeletal abnormalities and extra-skeletal ossification in mice with restricted Gsalpha deletion caused by a renin promoter-Cre transgene. Cell Tissue Res. 2007;330(3):487–501. doi: 10.1007/s00441-007-0491-6. [DOI] [PubMed] [Google Scholar]
- 35.Weinstein LS, Chen M, Liu J. Gs(alpha) mutations and imprinting defects in human disease. Ann N Y Acad Sci. 2002;968:173–197. doi: 10.1111/j.1749-6632.2002.tb04335.x. [DOI] [PubMed] [Google Scholar]
- 36.Weinstein LS, Yu S, Liu J. Analysis of genomic imprinting of Gs alpha gene. Methods Enzymol. 2002;344:369–383. doi: 10.1016/s0076-6879(02)44728-3. [DOI] [PubMed] [Google Scholar]
- 37.Bastepe M. Relative functions of Gαs and its extra-large variant XLαs in the endocrine system. Horm Metab Res. 2012;44(10):732–740. doi: 10.1055/s-0032-1316331. [DOI] [PubMed] [Google Scholar]
- 38.Huso DL, et al. Heterotopic ossifications in a mouse model of albright hereditary osteodystrophy. PLoS One. 2011;6(6):e21755. doi: 10.1371/journal.pone.0021755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lanske B, et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science. 1996;273(5275):663–666. doi: 10.1126/science.273.5275.663. [DOI] [PubMed] [Google Scholar]
- 40.Bastepe M, et al. Stimulatory G protein directly regulates hypertrophic differentiation of growth plate cartilage in vivo. Proc Natl Acad Sc U S A. 2004;101(41):14794–14799. doi: 10.1073/pnas.0405091101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakamoto A, Chen M, Kobayashi T, Kronenberg HM, Weinstein LS. Chondrocyte-specific knockout of the G protein G(s)alpha leads to epiphyseal and growth plate abnormalities and ectopic chondrocyte formation. J Bone Miner Res. 2005;20(4):663–671. doi: 10.1359/JBMR.041210. [DOI] [PubMed] [Google Scholar]
- 42.Long F, Zhang XM, Karp S, Yang Y, McMahon AP. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development. 2001;128(24):5099–5108. doi: 10.1242/dev.128.24.5099. [DOI] [PubMed] [Google Scholar]
- 43.Zhang XM, Ramalho-Santos M, McMahon AP. Smoothened mutants reveal redundant roles for Shh and Ihh signaling including regulation of L/R symmetry by the mouse node. Cell. 2001;106(2):781–792. [PubMed] [Google Scholar]
- 44.Liu T, et al. G protein signaling from activated rat frizzled-1 to the β-catenin-Lef-Tcf pathway. Science. 2001;292(5522):1718–1722. doi: 10.1126/science.1060100. [DOI] [PubMed] [Google Scholar]
- 45.Malbon CC. Frizzleds: new members of the superfamily of G-protein-coupled receptors. Front Biosci. 2004;9:1048–1058. doi: 10.2741/1308. [DOI] [PubMed] [Google Scholar]
- 46.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 47.Riddle RD, Johnson RL, Laufer E, Tabin C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell. 1993;75(7):1401–1416. doi: 10.1016/0092-8674(93)90626-2. [DOI] [PubMed] [Google Scholar]
- 48.Johnson RL, Laufer E, Riddle RD, Tabin C. Ectopic expression of Sonic hedgehog alters dorsal-ventral patterning of somites. Cell. 1994;79(7):1165–1173. doi: 10.1016/0092-8674(94)90008-6. [DOI] [PubMed] [Google Scholar]
- 49.Cryer J, Woodhead BG, Cryer A. The isolation and characterisation of a putative adipocyte precursor cell type from the white adipose tissue of the chicken (Gallus domesticus). Comp Biochem Physiol A Comp Physiol. 1987;86(3):515–521. doi: 10.1016/0300-9629(87)90535-4. [DOI] [PubMed] [Google Scholar]
- 50.Wang XG, Yu JF, Zhang Y, Gong DQ, Gu ZL. Identification and characterization of microRNA from chicken adipose tissue and skeletal muscle. Poult Sci. 2012;91(1):139–149. doi: 10.3382/ps.2011-01656. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.