

Abstract
The fact that advanced NSCLC patients with wild type (wt) EGFR can benefit from erlotinib therapy makes it critical to find out biomarkers for effective selection of patients and improving the therapy effects. In present study, 3 NSCLC cell lines (U1752, Calu-6 and NCI-H292) with wt EGFR and different sensitivities to erlotinib were used for microarray analysis. The differential basal gene expression between 2 NSCLC cell lines was analyzed, about 353 genes were expression-altered with higher than 2-fold changes between Calu-6 and U1752. And Ingenuity Pathway Analysis (IPA) showed that these genes were mainly enriched in regulation of epithelial–mesenchymal transition (EMT) pathway, Wnt-β catenin signaling, Tec kinase signaling and some types of cancer-related signaling. More interestingly, RAF1 (c-raf), MAP2K1 (MEK1), SNAI and downstream signaling molecules ERK and AKT were predicted to be activated in erlotinib-resistant cell line by IPA. Subsequent immunoblotting experiments showed that the phosphorylation of ERK and AKT were exactly increased stepwise from erlotinib sensitive cell line to erlotinib resistant cell lines. Collectively, activation of RAF1-MEK1-ERK/AKT axis may determine the resistance of NSCLC cell lines bearing wt EGFR to erlotinib. Our work provides potential biomarkers and therapeutic targets for NSCLC patients harboring wt EGFR.
Keywords: Non-small cell lung cancer, NSCLC, EGFR, erlotinib, microarray, RAF1, MAP2K1, ERK, AKT
Introduction
Erlotinib, a small-molecule drug targeted to the tyrosine kinase activity of EGFR, is approved by FDA to treat advanced or metastatic non-small cell lung cancer (NSCLC) and pancreatic cancer that cannot be removed by surgery or has metastasized. Clinical trials and preclinical studies have suggested that EGFR activating mutation is a predictive marker for favorable outcome of erlotinib in NSCLC patients [1-3]. Recently, first-line erlotinib therapy in EGFR mutation-positive NSCLC patients showed profound advantage over chemotherapy in the objective response rate and progression-free survival (PFS) benefit [4,5]. However, only 10-30% of NSCLC patients harbor mutant EGFR [6-8], the majority of NSCLC patients are with wild type (wt) EGFR. There also appear to be NSCLC patients with wt EGFR who clinically benefi t from erlotinib therapy by stabilizing disease and preventing further progression [1,9,10]. However, the mechanism of this benefit remains largely unknown and the biomarkers for wt EGFR NSCLC patients who can derive benefit from erlotinib treatment need to be further uncovered.
One possible mechanism that influences the sensitivity of wt EGFR NSCLC cells to erlotinib is in the driver gene alterations other than EGFR mutation, such as gene mutation (e.g. KRAS, HER2, BRAF), gene amplification (e.g. MET, FGFR1) or gene translocation (e.g. ALK, ROS1, RET). Various studies suggest that these driver gene alterations play roles in erlotinib resistance in NSCLC cells [11-13]. For example, MET activation and amplification was recently proposed to be linked closely to erlotinib resistance [13,14]. However, most of the currently known driver mutations occur at an incidence of ≤5%. The incidences of mutations in lung cancer were as follows: KRAS 25%, BRAF 3%, HER 21%, MET amplifications 2%, and ALK rearrangements 6% [15,16]. Although KRAS mutation frequency is relative high in lung cancer, in vitro data show various degrees of sensitivity to erlotinib in KRAS-mutated NSCLC cell lines [17,18]. Moreover, clinical trial showed that KRAS mutation has no significant effect on PFS of erlotinib treatment in NSCLC patients [1]. So, driver gene alterations may confer sensitivity/resistance to erlotinib only in a small part of patients, there must be other mechanisms by which cancer cells bearing wt EGFR displayed distinct sensitivity to erlotinib.
Several reports suggested that the expression of epithelial to mesenchymal transition (EMT)-related genes mediated NSCLC and head and neck squamous cell carcinoma cells sensitivity to erlotinib or gefitinib, another small molecule drug of EGFR tyrosine kinase inhibitor (TKI) [17,19,20]. Increased expression of TGF-β, IL6 and Vimentin was observed in erlotinib resistant NSCLC cell lines, while E-cadherin was up-regulated in sensitive cell lines [19]. Furthermore, Balko et al proposed that expression of genes linked to signal transduction (NF-κB signaling cascade and PI3K/MAPK pathway) may serve as predictive markers for erlotinib sensitivity in NSCLC cell lines and patients with lung adenocarcinomas [21]. Moreover, the protein expression of EGFR [1], amphiregulin [22], HGF [13] and cyclin D3 [23] was implicated in erlotinib sensitivity in vitro or in vivo, whether the mRNA expression of these genes is related to erlotinib sensitivity is not yet well defined.
In present study, 3 NSCLC cell lines with different sensitivities to erlotinib were applied to gene expression profile analysis. The differentially expressed genes were validated by quantitative real-time PCR. The potential genes/pathways involved in erlotinib sensitivity were proposed.
Materials and methods
Cell culture
U1752, Calu6 and H292 cell lines were purchased from ATCC and maintained in DMEM or RPMI 1640 medium supplemented with 10% FBS (Hyclone), penicillin (100 IU/ml) and Streptomycin (100 μg/ml) (Life Technologies).
MTS assay for 3 NSCLC cell lines viability
Cells (4 × 103) were grown in 100 μl of DMEM or RPMI 1640 medium containing serum per well in a 96-well plate. After 24 h, the cells were treated with erlotinib (0, 0.0032, 0.01, 0.032, 0.10, 0.32, 1.00, 3.20 μmol/L, respectively) for 72 h. Every treatment was triplicate in the same experiment. Then 20 μl of MTS (CellTiter 96 AQueous One Solution Reagent; Promega) was added to each well for 2 h at 37°C. After incubation, the absorbance was read at a wavelength of 490 nm according to the manufacturer’s protocol. The IC50 calculation was performed with GraphPad Prism 5.0 software.
Microarray analysis
3 NSCLC cell lines (8 × 104) were grown in 2 ml of DMEM medium containing serum per well in a 6-well plate. Cells in the exponential growth phase were used for microarray analysis. Every treatment was duplicated in the same experiment. All the samples were homogenized with 1 ml Trizol (Invitrogen, Life Technologies) and total RNAs were extracted according to the manufacturer’s instruction.
500 ng total RNA was used to synthesize double-strand cDNA and in vitro transcribed to cRNA, purified 10 μg cRNA was used to synthesize 2nd-cycle cDNA and then hydrolyzed by RNase H and purified. Above steps were performed with Ambion WT Expression Kit. 5.5 μg 2nd-cycle cDNA was fragmented and the single-stranded cDNA was labeled with GeneChip2 WT Terminal Labeling Kit and Controls Kit (Affymetrix, PN 702880). About 700 ng fragmented and labeled single-stranded cDNA were hybridized to an Affymetrix GeneChip Human Gene 1.0 ST array, which was washed and stained with GeneChip2 Hybridization, Wash and Stain kit (Affymetrix).
Microarray data analysis was done using Significance Analysis of Microarrays (SAM) method, as described before [24]. Functional annotation was performed to the differential expression genes with Ingenuity Pathway Analysis (IPA) online software.
Quantitative real-time PCR (qPCR)
Total RNA above isolated was synthesized to cDNA using PrimeScript RT reagent kit with gDNA Eraser (Takara, RR074A) for RT-PCR with mixture of oligo-dT and Random Primer (9 mer). The primers used for qPCR validation were list in Supplementary Table 1. Real-time qPCR was performed on CFX-96 (Bio-lab), with endogenous control hActb. Gene expression was calculated relative to expression of hActb endogenous control and adjusted relative to expression in U1752 cells.
Protein isolation and western blotting
Cell pellets were resuspended in 1 × SDS loading buffer (1 mmol/L Na3VO4, 10 mmol/L NaF, 1 mmol/L PMSF) containing protease inhibitors. Lysates (20 μg each lane) were applied to SDS-PAGE. Immunoblotting of Abs specific for GAPDH (Abmart, 080922), EGFR (Abclonal, A0227), p-EGFR (Santa cruz, SC-12351, pY1173), p-EGFR (Epitomics, #1139-S, pY1086), AKT (Santa Cruz, sc-8312), p-AKT (Santa Cruz, SC-7985-R, pS473), ERK (Abclonal, A0228) and p-ERK (Cell signaling, #9106S, pT202/204) were detected using HRP-conjugated anti-mouse (Promega) or anti-rabbit (Promega) and visualized by chemiluminescence detection system (Millipore, WBKLS0500).
Statistical analysis
R2 values were calculated using Pearson’s correlation coefficient. The significant difference was calculated using Student’s t-test.
Results
3 NSCLC cell lines display distinct sensitivities to erlotinib
Three NSCLC cell lines harboring wt EGFR, U1752, Calu-6 and NCI-H292, were treated with 7 different concentrations of erlotinib as indicated for 72 h. Then the cell viability was determined by MTS assay (Figure 1) and IC50 s of these three cell lines were calculated. IC50 dose of U1752, Calu-6 and NCI-H292 cells to erlotinib at 72 h were 0.059 ± 0.029 (R2 = 0.99), 0.217 ± 0.134 (R2 = 0.86) and 0.503 ± 0.041 μmol/L (R2 = 0.95), respectively. According to criterion described before [25], U1752 was sensitive to erlotinib whereas Calu-6 and NCI-H292 were relatively insensitive to erlotinib.
Figure 1.
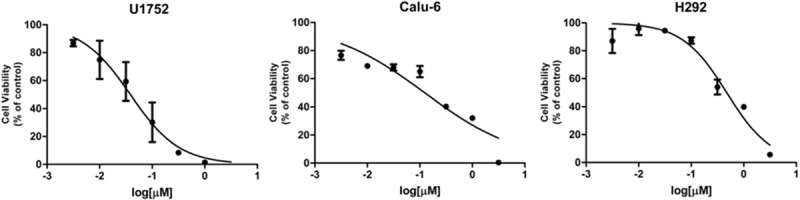
3 NSCLC cell lines display different sensitivities to erlotinib. U1752, Calu-6 and NCI-H292 cells were administrated to 7 concentrations of erlotinib (0, 0.0032, 0.01, 0.032, 0.10, 0.32, 1.00, 3.20 μmol/L, respectively) for 72 h. The cell viability was determined by MTS assay. Every treatment was triplicate in the same experiment. Bars represent the standard errors.
Differential gene expression profile analysis
To figure out the biomarkers that determine the different sensitivities to erlotinib, the 3 NSCLC cell lines were applied to microarray analysis. The raw data of microarray showed that there was dramatically difference in basal expression between U1752 and Calu-6 cells. 1163 genes were up-regulated by 2-fold and 1378 genes were down-regulated by 2-fold in Calu-6 compared to U1752 (data not shown). The most markedly expression-altered genes were TIMP3 and TFPI2, the expression of TIMP3 was increased by 191-fold and that of TEPI2 was decreased by 411-fold in Calu-6 compared to U1752, respectively. However, limited alterations were observed in basal expression between U1752 and NCI-H292 cells. Expression of only one gene (CDKN2AIPNLP1) and two genes (OR4A47 and WT1-AS) was increased and decreased by 2-fold, respectively, in NCI-H292 compared to U1752. There were no genes whose expression was increased or decreased by higher than 50% stepwise from U1752 to Calu-6 to NCI-H292, according to their sensitivities to erlotinib. These results suggested that the relative resistance of Calu-6 and NCI-H292 to erlotinib may be caused by different mechanisms; the resistance of NCI-H292 to erlotinib may be not explained by gene expression.
Then we analyzed the microarray data of Calu-6 and U1752 by SAM method. The results showed that 177 genes and 176 genes were respectively up-regulated and down-regulated by higher than 2-fold in Calu-6 compared to U1752 (Supplementary Table 2). Interestingly, EREG (epiregulin), a ligand of EGFR, was up-regulated by 2.2-fold in Calu-6 compared to U1752. The top 20 significantly expression-altered genes were list in Table 1. Within these genes, TIMP3, CDK15, TGFBI and IGFBP5 were up-regulated, while HIST1H3I, HSPA13 and CYP24A1 were down-regulated.
Table 1.
Top 20 significantly expression-altered genes in Calu-6 compared to U1752
| Gene Symbol | fold change | P value | Description |
|---|---|---|---|
| FABP4 | 6.49 | 2.29E-03 | fatty acid binding protein 4, adipocyte |
| TIMP3 | 6.29 | 6.80E-04 | TIMP metallopeptidase inhibitor 3 |
| CDK15 | 5.63 | 3.55E-04 | cyclin-dependent kinase 15 |
| TGFBI | 5.60 | 4.46E-04 | transforming growth factor, beta-induced, 68 kDa |
| EMP1 | 5.23 | 6.63E-04 | epithelial membrane protein 1 |
| THBS1 | 4.91 | 3.31E-05 | thrombospondin 1 |
| NT5E | 4.67 | 7.40E-06 | 5’-nucleotidase, ecto (CD73) |
| RGS1 | 4.67 | 8.63E-04 | regulator of G-protein signaling 1 |
| IGFBP5 | 4.38 | 8.98E-04 | insulin-like growth factor binding protein 5 |
| SERPINF1 | 4.29 | 1.37E-03 | serpin peptidase inhibitor, clade F (alpha-2 antiplasmin, pigment epithelium derived factor), member 1 |
| GRAMD1B | 0.24 | 7.15E-04 | GRAM domain containing 1B |
| HIST1H3I | 0.24 | 4.36E-04 | histone cluster 1, H3i |
| FAM5C | 0.23 | 1.93E-04 | family with sequence similarity 5, member C |
| HSPA13 | 0.21 | 4.90E-04 | heat shock protein 70 kDa family, member 13 |
| OSMR | 0.21 | 1.59E-03 | oncostatin M receptor |
| NTS | 0.19 | 1.88E-04 | neurotensin |
| CYP24A1 | 0.18 | 1.88E-05 | cytochrome P450, family 24, subfamily A, polypeptide 1 |
| INA | 0.18 | 7.36E-04 | internexin neuronal intermediate filament protein, alpha |
| EDIL3 | 0.16 | 7.84E-04 | EGF-like repeats and discoidin I-like domains 3 |
| TFPI2 | 0.11 | 5.08E-06 | tissue factor pathway inhibitor 2 |
The 353 genes (177 up-regulated genes and 176 down-regulated genes) were applied to IPA. These genes were mainly enriched in regulation of epithelial–mesenchymal transition (EMT) pathway, Wnt-β catenin signaling, Tec kinase signaling and some types of cancer-related signaling (Table 2 and Figure 2). 11 genes were involved in regulation of EMT pathway (Figure 3A). Among these genes, TGFB2, TGFBR2, HMGA2, SNAI2, MMP2, TCF4, FZD7 and CDH12 were up-regulated, while Wnt2, HGF and FGF5 were down-regulated. Although expression of 3 upstream genes (Wnt2, HGF and FGF5) was decreased, the other upstream signaling molecules (TGFB2 and TGFBR2) and downstream effectors (SNAI2, MMP2 and CDH12) were up-regulated. This result suggests that EMT-regulation pathway is activated in Calu-6, cell line relatively insensitive to erlotinib, compared to U1752. Additionally, there were 7 genes linked to Tec kinase signaling (Figure 3B). Expression of 4 genes (TLR4, GNG2, PRKD1 and RND3) was elevated whereas 3 genes (TNFSF10, GNAL and GNB4) were down-regulated. Furthermore, genes associated to cytokine-cytokine receptor interaction (EGFR, CX3CL1, CXCL16 and CXCR4) were also expression-altered markedly in Calu-6 compared to U1752.
Table 2.
IPA for 353 genes expression-altered significantly in Calu-6 compared to U1752
| Ingenuity Canonical Pathways | P value | Molecules |
|---|---|---|
| Hepatic Fibrosis/Hepatic Stellate Cell Activation | 9.77E-07 | TGFBR2, TLR4, IGFBP4, FN1, CCL2, HGF, TGFB2, TGFA, LAMA1, MMP2, IGFBP5, A2M, TNFRSF11B |
| Regulation of the Epithelial-Mesenchymal Transition Pathway | 7.94E-05 | TGFBR2, TCF4, CDH12, SNAI2, HGF, TGFB2, mir-155, MMP2, WNT2, HMGA2, FZD7, FGF5 |
| Wnt/β-catenin Signaling | 8.32E-04 | TGFBR2, MYC, GJA1, TCF4, CDH12, FRZB, CD44, TGFB2, WNT2, FZD7 |
| Colorectal Cancer Metastasis Signaling | 1.00E-03 | TGFBR2, MYC, TLR4, GNB4, TCF4, RND3, TGFB2, MMP2, PTGS2, GNG2, WNT2, FZD7 |
| LPS/IL-1 Mediated Inhibition of RXR Function | 1.66E-03 | TLR4, MGST1, SULT1C2, ACSL5, FABP4, HS6ST3, CHST10, FABP3, ALDH7A1, TNFRSF11B, PPARGC1A |
| Epithelial Adherens Junction Signaling | 3.98E-03 | TGFBR2, TCF4, SNAI2, HGF, PVRL3, CTNNA1, TGFB2, JUP |
| Inhibition of Matrix Metalloproteases | 4.68E-03 | TIMP3, MMP2, A2M, TFPI2 |
| Thyroid Cancer Signaling | 5.13E-03 | PPARG, MYC, TCF4, BDNF |
| LXR/RXR Activation | 5.37E-03 | TLR4, CCL2, SERPINF1, CLU, PTGS2, PON3, TNFRSF11B |
| Polyamine Regulation in Colon Cancer | 6.31E-03 | PPARG, MYC, TCF4 |
| Role of Macrophages, Fibroblasts and Endothelial Cells in Rheumatoid Arthritis | 6.76E-03 | MYC, TLR4, TCF4, FN1, FRZB, CCL2, LTBR, IRAK3, WNT2, PRKD1, FZD7, TNFRSF11B |
| Chondroitin Sulfate Biosynthesis (Late Stages) | 7.76E-03 | SULT1C2, HS6ST3, CHST10, CSGALNACT1 |
| Ovarian Cancer Signaling | 8.51E-03 | GJA1, TCF4, CD44, MMP2, PTGS2, WNT2, FZD7 |
| RhoGDI Signaling | 1.07E-02 | GNB4, CDH12, RND3, CDH10, CD44, CDH6, GNG2, GNAL |
| Chondroitin Sulfate Biosynthesis | 1.38E-02 | SULT1C2, HS6ST3, CHST10, CSGALNACT1 |
| Dermatan Sulfate Biosynthesis | 1.55E-02 | SULT1C2, HS6ST3, CHST10, CSGALNACT1 |
| Molecular Mechanisms of Cancer | 1.62E-02 | TGFBR2, MYC, RASGRF2, TCF4, RND3, CTNNA1, CDK6, TGFB2, BIRC3, GNAL, PRKD1, FZD7 |
| Glioma Invasiveness Signaling | 1.74E-02 | TIMP3, RND3, CD44, MMP2 |
Figure 2.
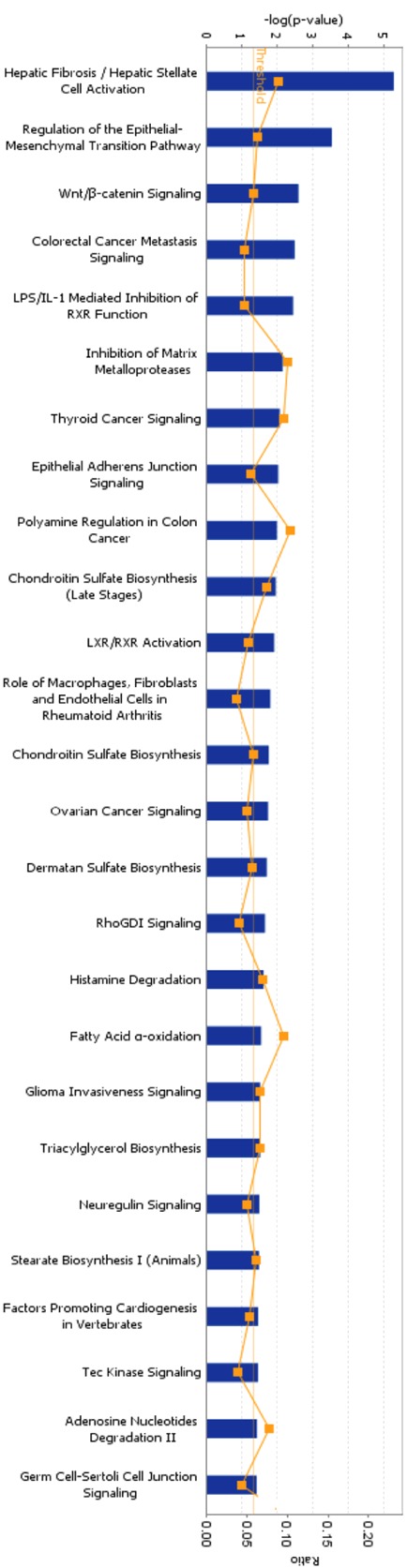
IPA for differentially expressed genes between Calu-6 and U1752. 353 genes whose expression altered by higher than 2-fold in Calu-6 and U1752 were applied to IPA. The canonical pathways (p < 0.05) were shown in this figure. The thresh line represents p = 0.05. The ratio means the proportion that the amounts of genes involved in some pathway account for the total 353 genes.
Figure 3.
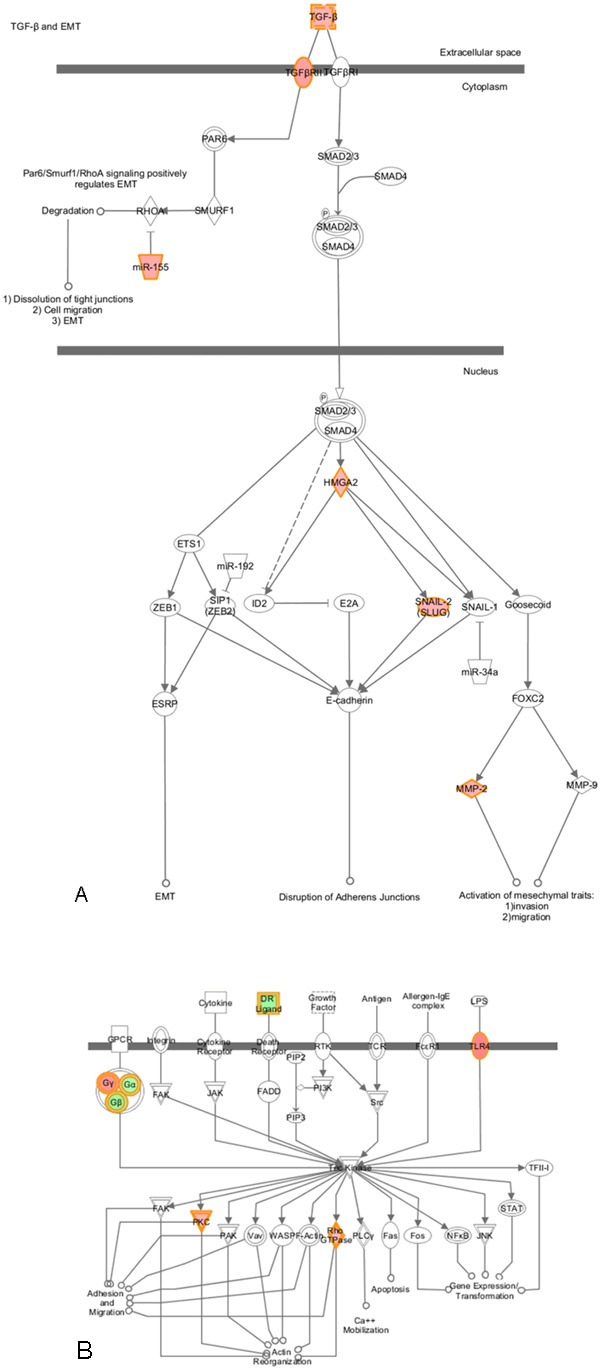
Regulation of the EMT Pathway and Tec kinase signaling in Calu-6 compared to U1752. This figure was derived from IPA. The colored molecules were genes whose expression altered significantly in Calu-6 compared to U1752. The red color represented up-regulated genes in Calu-6, while the green color represented down-regulated genes in Calu-6. A: Regulation of the EMT Pathway in Calu-6 compared to U1752. TGF-β-EMT was shown, while Wnt-EMT, Notch-EMT and Receptor tyrosine kinase-EMT were not shown. B: Tec kinase signaling in Calu-6 compared to U1752.
RAF1-MAP2K1 and downstream signaling ERK/AKT were predicted to be activated in erlotinib-resistant NSCLC cell line
In the other hand, mechanistic network analysis of IPA showed that RAF1 (C-RAF, Figure 4A), MAP2K1 (MEK1, Figure 4B) and SNAI (Figure 4C) were predicted to be activated in erlotinib-resistant NSCLC cell line compared to erlotinib-sensitive cell line. The prediction of RAF1 activation was based on that 13 genes in the 353-gene list were regulated by RAF1 and that expression directions of 9 out 13 genes were consistent with RAF1 activation, as described before [26-28] (Supplement Table 3). For example, PTGS2 [26], HMGA2 [27], and RND3 [28] were reported to be up-regulated by RAF1, while in our microarray data these 3 genes were all expression-increased in Calu-6, erlotinib-resistant cell line. This indicated that RAF1 was activated in Calu-6. Based on the same way, MAP2K1 and SNAI were predicted to be activated. Interestingly, the downstream signaling ERK and AKT were predicted to be activated following RAF1 and MAP2K1 activation.
Figure 4.
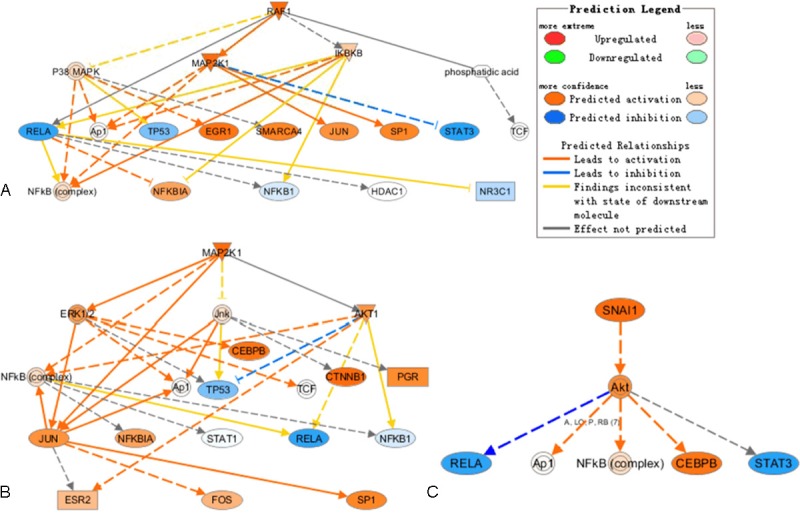
Mechanistic network analysis by IPA in Calu-6 compared to U1752. RAF1 (A), MAP2K1 (B), and SNAI (C) were predicted to be activated. The downstream molecules regulated by RAF1, MAP2K1, and SNAI and interactions between these molecules were illustrated.
qPCR validation of microarray data
To validate the microarray data, qPCR was performed for 13 genes whose basal expression altered markedly in Calu-6 compared to U1752. Among these 13 genes, CXCL16 was down-regulated by 67-fold, CXCR4 was down-regulated by 64-fold, Myc and Tp53 were down-regulated by 29-fold, CTNNA1 (α-catenin) was down-regulated by 28-fold, EGFR was up-regulated by 2.7-fold, RET up-regulated by 2.9-fold and CX3CL1 was up-regulated by 6.6-fold. Fold changes of these genes’ expression determined by qPCR and microarray were log2 transformed and shown in Figure 5. The change folds varied between qPCR and microarray data, however, the expression trends of all the 13 genes were consistent. The correlation between both data sets was examined using R2 and an R2 value of 0.93 was obtained, suggesting a strong overall concordance of expression trends between the microarray and qPCR data. The most dramatically expression-altered genes validated by qPCR were CXCL16, CXCR4, Myc, TP53, EGFR, RET and CX3CL1, genes involved in cytokine-cytokine receptor interaction, receptor tyrosine kinase and caner signaling pathways.
Figure 5.
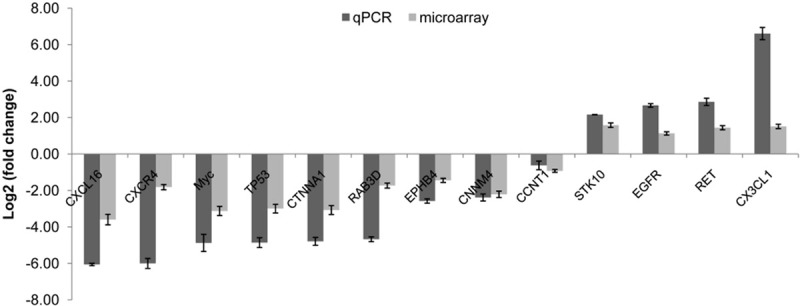
qPCR validation for microarray data. 13 genes were selected to perform qPCR. The expression was calculated relative to expression in U1752. The change folds determined by qPCR and microarray were transformed to log2. Bars represent the standard errors.
ERK and AKT were activated in erlotinib insensitive cell lines
Since expression of EGFR was up-regulated significantly and that ERK and AKT were predicted to be activated in Calu-6 compared to U1752, we next investigated the activation status of EGFR and its downstream signaling ERK and AKT in the 3 NSCLC cell lines by western blotting (Figure 6). The p-EGFR (Y1173) was up-regulated stepwise from U1752 to Calu-6 to NCI-H292, whereas the p-EGFR (Y1086) was detected only in NCI-H292 but not in Calu-6 and U1752. The p-AKT was increased gradually from U1752 to Calu-6 to NCI-H292. The p-ERK was also shown similar pattern among those three cell lines. The protein expression of p-ERK and p-AKT was basically increased from sensitive cell line to insensitive cell lines. Our data suggest that ERK and AKT are highly activated in erlotinib-resistant NSCLC cell lines.
Figure 6.
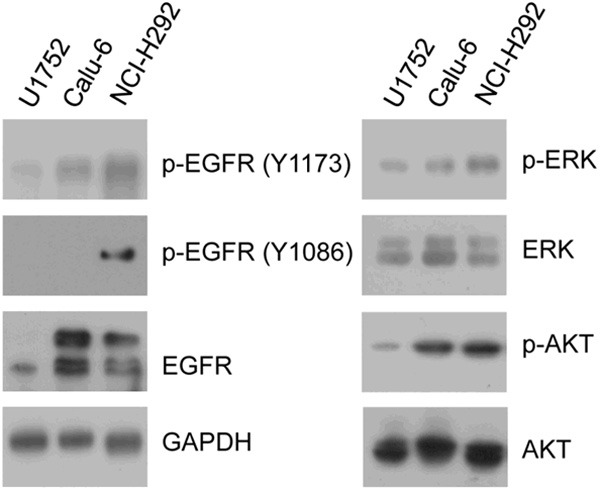
The activation status of EGFR/ERK/AKT in 3 NSCLC cell lines. U1752, Calu-6 and NCI-H292 cells were applied to western blotting for EGFR, p-EGFR (Y1173), p-EGFR (Y1086), ERK, p-ERK (T202/204), AKT, p-AKT (S473). GAPDH was used as input control.
Discussion
The fact that advanced NSCLC patients with wt EGFR can benefit from erlotinib therapy makes it essential to uncover biomarkers for effective selection of patients and improving the therapy. In present study, 3 NSCLC cell lines with wt EGFR and different sensitivities to erlotinib were employed to perform microarray analysis. The differential basal expression between NSCLC cell lines was analyzed and the expression-altered genes were applied to IPA. Expression of 13 genes was validated by qPCR and the activation status of EGFR and its downstream signal ERK/AKT were examined by western blotting.
Since erlotinib is an EGFR tyrosine kinase inhibitor (TKI), EGFR expression level is one of the most frequently mentioned signature for erlotinib sensitivity. However, in this work, the expression of EGFR was as follows: Expression in NCI-H292 was approximately equal to that in U1752 (data not shown), while expression level in Calu-6 was increased by 2.7-fold compared to U1752. This was not in line with erlotinib sensitivities of these cell lines. The subsequent immunoblotting experiments showed that p-EGFR (Y1086) was detectable only in NCI-H292 cell line but not either in U1752 and Calu-6 or in the other two erlotinib-resistant NSCLC cell lines, NCI-H1975 and NCI-H1395 (IC50 dose to erlotinib at 72 h was 0.32 ± 0.09 and 1.49 ± 0.96 μmol/L, respectively; data not shown). Although p-EGFR (Y1086) has been implicated to cancer cells invasion and proliferation [29,30], it is not likely to be a predictive marker for erlotinib sensitivity, based on our data. At the same time, p-EGFR (Y1173) was up-regulated gradually from erlotinib-sensitive cell line to erlotinib-insensitive cell lines, according to their sensitivities to erlotinib. However, previous reports have suggested that EGFR activation mutation was predictive marker for sensitivity to erlotinib. The aberrant activation of EGFR (Y1173) in cancer cells harboring wt EGFR seems likely to confer resistance to erlotinib. In summary, either mRNA expression of EGFR or p-EGFR (Y1086) may not be potential marker for erlotinib sensitivity in patients harboring wt EGFR.
Activation of bypass pathway or downstream signaling molecules is one of the crucial mechanisms by which cancer cells confer resistance to targeted therapy. For example, constitutive phosphorylation and activation of HER2 and EGFR caused by HER2 kinase domain mutation [11], activating ERBB3 signaling resulted from MET amplification [14], high IκB in NSCLC patients [31] and downstream BRAF activation [12] all confer resistance of cancer cells to EGFR TKI. Recently, a number of studies suggest that the PI3K/Akt signaling pathway is central to NSCLC growth and survival [32-34]. In this work, mechanistic network analysis by IPA predicted that RAF1, MAR2K1, SNAI and downstream signaling ERK/AKT were activated. Although the mRNA expression levels of these 3 genes (RAF1, MAR2K1, SNAI) were not altered significantly (data not shown), the up-regulation of downstream molecules positively regulated by these 3 genes indicated that protein expression levels of these 3 genes were increased. Subsequent immunoblotting experiments validated that p-ERK and p-AKT protein expression increased stepwise from erlotinib sensitive cell line to insensitive cell lines, proposing that p-ERK and p-AKT expression is potential biomarkers for erlotinib resistance and p-ERK and p-AKT may serve as a therapeutic target for combination therapy with EGFR TKI in NSCLC patients bearing wt EGFR. Furthermore, PRKD1, one member of PKC family, was up-regulated in Calu-6 compared to U1752. PRKD1 was proposed to mediate anchorage-dependent and -independent growth of tumor cells via the zinc finger transcription factor Snail1 [35]. The activation of PRKD1 may be another reason that NSCLC cells confer resistance to erlotinib. The possible mechanism of erlotinib resistance in NSCLC cell lines with wt EGFR was illustrated in Figure 7.
Figure 7.
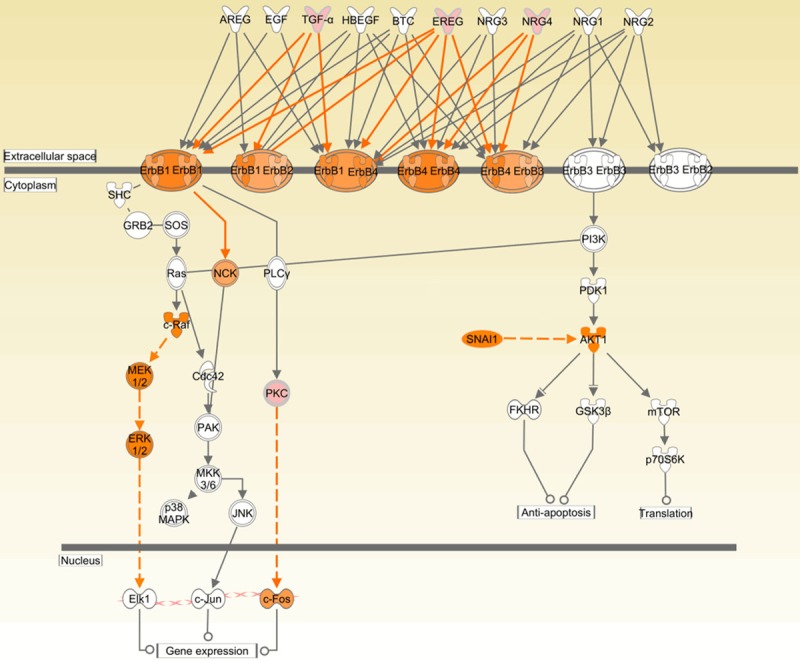
Potential mechanism by which NSCLC cells with wt EGFR display resistance to erlotinib. The up-regulation of ErbB ligands (TGF-α, EREG and NRG4) activates EGFR. Activation of RAF1-MEK1-ERK and PKC/SNAI-AKT up-regulates the downstream signaling and hence confers NSCLC cells harboring wt EGFR resistance to erlotinib. The pink molecules represent that its mRNA expression is up-regulated. The orange molecules mean that it is activated.
As for the increased mRNA level of EGFR in Calu-6, it is probable to be illuminated by the up-regulation of ErbB ligands (TGF-α, EREG and NRG4, see in Figure 7) and cytokine (CX3CL1). In our data, TGF-α, EREG, NRG4 and CX3CL1 were up-regulated by 2.2-, 2.2-, 2.1- and 6.6-fold, respectively, in Calu-6 compared to U1752 (Supplementary Table 2 and Figure 5). It was reported that CX3CL1 has anti-apoptotic and proliferative effects on human vascular smooth muscle cells via epiregulin-induced EGFR signaling [36], CX3CL1 promotes breast cancer via transactivation of the EGFR pathway [37] and that CX3CL1 induced the phosphorylation and activation of PI3K, AKT and ERK in rheumatoid arthritis fibroblast-like synoviocyte [38] and chronic lymphocytic leukemia cells [39]. So, in some NSCLC cell lines, CX3CL1 may be potential marker for EGFR TKI resistance.
Furthermore, our data also showed that EMT-related pathways were elevated in erlotinib insensitive cell line, which is exactly consistent with previous report [17,19,20] and suggests that our microarray data were reliable. Moreover, genes associated to Tec kinase signaling were expression-altered significantly in erlotinib insensitive cell line, indicating that tec kinases may be novel targets for cancer therapy as described before [40,41].
Taken together, our results present a serial of potential biomarker candidates for further validation in erlotinib sensitivity prediction. And the AKT and EKT hyperphosphorylation contributes to erlotinib resistance might provide the indication of combination with AKT/EKT inhibitor in clinical erlotinib therapy. Moreover, our finding might also suggest a mechanism of secondary drug tolerance in patients treated with erlotinib, although it needs to be addressed more.
Acknowledgments
This work is support by the grant from Health Bureau Research Fund of Shanghai, China (No. 20114291).
Supporting Information
References
- 1.Brugger W, Triller N, Blasinska-Morawiec M, Curescu S, Sakalauskas R, Manikhas GM, Mazieres J, Whittom R, Ward C, Mayne K, Trunzer K, Cappuzzo F. Prospective molecular marker analyses of EGFR and KRAS from a randomized, placebo-controlled study of erlotinib maintenance therapy in advanced non-small-cell lung cancer. J. Clin. Oncol. 2011;29:4113–4120. doi: 10.1200/JCO.2010.31.8162. [DOI] [PubMed] [Google Scholar]
- 2.Janne PA, Engelman JA, Johnson BE. Epidermal growth factor receptor mutations in non-small-cell lung cancer: implications for treatment and tumor biology. J. Clin. Oncol. 2005;23:3227–3234. doi: 10.1200/JCO.2005.09.985. [DOI] [PubMed] [Google Scholar]
- 3.Ji H, Li D, Chen L, Shimamura T, Kobayashi S, McNamara K, Mahmood U, Mitchell A, Sun Y, Al-Hashem R, Chirieac LR, Padera R, Bronson RT, Kim W, Janne PA, Shapiro GI, Tenen D, Johnson BE, Weissleder R, Sharpless NE, Wong KK. The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer Cell. 2006;9:485–495. doi: 10.1016/j.ccr.2006.04.022. [DOI] [PubMed] [Google Scholar]
- 4.Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, Zhang S, Wang J, Zhou S, Ren S, Lu S, Zhang L, Hu C, Hu C, Luo Y, Chen L, Ye M, Huang J, Zhi X, Zhang Y, Xiu Q, Ma J, Zhang L, You C. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12:735–742. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 5.Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R, Pallares C, Sanchez JM, Porta R, Cobo M, Garrido P, Longo F, Moran T, Insa A, De Marinis F, Corre R, Bover I, Illiano A, Dansin E, De Castro J, Milella M, Reguart N, Altavilla G, Jimenez U, Provencio M, Moreno MA, Terrasa J, Munoz-Langa J, Valdivia J, Isla D, Domine M, Molinier O, Mazieres J, Baize N, Garcia-Campelo R, Robinet G, Rodriguez-Abreu D, Lopez-Vivanco G, Gebbia V, Ferrera-Delgado L, Bombaron P, Bernabe R, Bearz A, Artal A, Cortesi E, Rolfo C, Sanchez-Ronco M, Drozdowskyj A, Queralt C, De Aguirre I, Ramirez JL, Sanchez JJ, Molina MA, Taron M, Paz-Ares L Spanish Lung Cancer Group in collaboration with Groupe Francais De Pneumo-Cancérologie and Associazione Italiana Oncologia T. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 6.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba II, Fong KM, Lee H, Toyooka S, Shimizu N, Fujisawa T, Feng Z, Roth JA, Herz J, Minna JD, Gazdar AF. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97:339–346. doi: 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- 8.Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 9.Engelman JA, Janne PA. Factors predicting response to EGFR tyrosine kinase inhibitors. Semin Respir Crit Care Med. 2005;26:314–322. doi: 10.1055/s-2005-871990. [DOI] [PubMed] [Google Scholar]
- 10.Giaccone G. Epidermal growth factor receptor inhibitors in the treatment of non-small-cell lung cancer. J. Clin. Oncol. 2005;23:3235–3242. doi: 10.1200/JCO.2005.08.409. [DOI] [PubMed] [Google Scholar]
- 11.Wang SE, Narasanna A, Perez-Torres M, Xiang B, Wu FY, Yang S, Carpenter G, Gazdar AF, Muthuswamy SK, Arteaga CL. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell. 2006;10:25–38. doi: 10.1016/j.ccr.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 12.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 13.Gusenbauer S, Vlaicu P, Ullrich A. HGF induces novel EGFR functions involved in resistance formation to tyrosine kinase inhibitors. Oncogene. 2012 doi: 10.1038/onc.2012.396. doi: 10.1038/onc.2012.396. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 14.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Janne PA. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 15.Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011;12:175–180. doi: 10.1016/S1470-2045(10)70087-5. [DOI] [PubMed] [Google Scholar]
- 16.Cheng L, Alexander RE, Maclennan GT, Cummings OW, Montironi R, Lopez-Beltran A, Cramer HM, Davidson DD, Zhang S. Molecular pathology of lung cancer: key to personalized medicine. Mod Pathol. 2012;25:347–369. doi: 10.1038/modpathol.2011.215. [DOI] [PubMed] [Google Scholar]
- 17.Yauch RL, Januario T, Eberhard DA, Cavet G, Zhu W, Fu L, Pham TQ, Soriano R, Stinson J, Seshagiri S, Modrusan Z, Lin CY, O’Neill V, Amler LC. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res. 2005;11:8686–8698. doi: 10.1158/1078-0432.CCR-05-1492. [DOI] [PubMed] [Google Scholar]
- 18.Sos ML, Michel K, Zander T, Weiss J, Frommolt P, Peifer M, Li D, Ullrich R, Koker M, Fischer F, Shimamura T, Rauh D, Mermel C, Fischer S, Stuckrath I, Heynck S, Beroukhim R, Lin W, Winckler W, Shah K, LaFramboise T, Moriarty WF, Hanna M, Tolosi L, Rahnenfuhrer J, Verhaak R, Chiang D, Getz G, Hellmich M, Wolf J, Girard L, Peyton M, Weir BA, Chen TH, Greulich H, Barretina J, Shapiro GI, Garraway LA, Gazdar AF, Minna JD, Meyerson M, Wong KK, Thomas RK. Predicting drug susceptibility of non-small cell lung cancers based on genetic lesions. J Clin Invest. 2009;119:1727–1740. doi: 10.1172/JCI37127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao Z, Fenoglio S, Gao DC, Camiolo M, Stiles B, Lindsted T, Schlederer M, Johns C, Altorki N, Mittal V, Kenner L, Sordella R. TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc Natl Acad Sci USA. 2010;107:15535–15540. doi: 10.1073/pnas.1009472107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frederick BA, Helfrich BA, Coldren CD, Zheng D, Chan D, Bunn PA Jr, Raben D. Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non-small cell lung carcinoma. Mol Cancer Ther. 2007;6:1683–1691. doi: 10.1158/1535-7163.MCT-07-0138. [DOI] [PubMed] [Google Scholar]
- 21.Balko JM, Potti A, Saunders C, Stromberg A, Haura EB, Black EP. Gene expression patterns that predict sensitivity to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer cell lines and human lung tumors. BMC Genomics. 2006;7:289. doi: 10.1186/1471-2164-7-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yonesaka K, Zejnullahu K, Lindeman N, Homes AJ, Jackman DM, Zhao F, Rogers AM, Johnson BE, Janne PA. Autocrine production of amphiregulin predicts sensitivity to both gefitinib and cetuximab in EGFR wild-type cancers. Clin Cancer Res. 2008;14:6963–6973. doi: 10.1158/1078-0432.CCR-08-0957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petty WJ, Voelzke WR, Urbanic JJ, Varela VA, Waller LL, Swift CB, Graham RM, Memoli VA, Dragnev KH. High cyclin D3 expression confers erlotinib resistance in aerodigestive tract cancer. Lung Cancer. 2011;74:384–391. doi: 10.1016/j.lungcan.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 24.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McDermott U, Sharma SV, Dowell L, Greninger P, Montagut C, Lamb J, Archibald H, Raudales R, Tam A, Lee D, Rothenberg SM, Supko JG, Sordella R, Ulkus LE, Iafrate AJ, Maheswaran S, Njauw CN, Tsao H, Drew L, Hanke JH, Ma XJ, Erlander MG, Gray NS, Haber DA, Settleman J. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc Natl Acad Sci USA. 2007;104:19936–19941. doi: 10.1073/pnas.0707498104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xuan YT, Guo Y, Zhu Y, Wang OL, Rokosh G, Messing RO, Bolli R. Role of the protein kinase C-epsilon-Raf-1-MEK-1/2-p44/42 MAPK signaling cascade in the activation of signal transducers and activators of transcription 1 and 3 and induction of cyclooxygenase-2 after ischemic preconditioning. Circulation. 2005;112:1971–1978. doi: 10.1161/CIRCULATIONAHA.105.561522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li D, Lin HH, McMahon M, Ma H, Ann DK. Oncogenic raf-1 induces the expression of non-histone chromosomal architectural protein HMGI-C via a p44/p42 mitogen-activated protein kinase-dependent pathway in salivary epithelial cells. J Biol Chem. 1997;272:25062–25070. doi: 10.1074/jbc.272.40.25062. [DOI] [PubMed] [Google Scholar]
- 28.Goh LL, Manser E. The RhoA GEF Syx is a target of Rnd3 and regulated via a Raf1-like ubiquitin-related domain. PLoS One. 2010;5:e12409. doi: 10.1371/journal.pone.0012409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cardoso AP, Pinto ML, Pinto AT, Oliveira MI, Pinto MT, Goncalves R, Relvas JB, Figueiredo C, Seruca R, Mantovani A, Mareel M, Barbosa MA, Oliveira MJ. Macrophages stimulate gastric and colorectal cancer invasion through EGFY Y, c-Src, Erk1/2 and Akt phosphorylation and smallGTPase activity. Oncogene. 2013 doi: 10.1038/onc.2013.154. doi:10.1038/onc.2013.154. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 30.Humtsoe JO, Kramer RH. Differential epidermal growth factor receptor signaling regulates anchorage-independent growth by modulation of the PI3K/AKT pathway. Oncogene. 2010;29:1214–1226. doi: 10.1038/onc.2009.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bivona TG, Hieronymus H, Parker J, Chang K, Taron M, Rosell R, Moonsamy P, Dahlman K, Miller VA, Costa C, Hannon G, Sawyers CL. FAS and NF-kappaB signalling modulate dependence of lung cancers on mutant EGFR. Nature. 2011;471:523–526. doi: 10.1038/nature09870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Faber AC, Li D, Song Y, Liang MC, Yeap BY, Bronson RT, Lifshits E, Chen Z, Maira SM, Garcia-Echeverria C, Wong KK, Engelman JA. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc Natl Acad Sci USA. 2009;106:19503–19508. doi: 10.1073/pnas.0905056106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sos ML, Koker M, Weir BA, Heynck S, Rabinovsky R, Zander T, Seeger JM, Weiss J, Fischer F, Frommolt P, Michel K, Peifer M, Mermel C, Girard L, Peyton M, Gazdar AF, Minna JD, Garraway LA, Kashkar H, Pao W, Meyerson M, Thomas RK. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 2009;69:3256–3261. doi: 10.1158/0008-5472.CAN-08-4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang YC, Kulp SK, Wang D, Yang CC, Sargeant AM, Hung JH, Kashida Y, Yamaguchi M, Chang GD, Chen CS. Targeting endoplasmic reticulum stress and Akt with OSU-03012 and gefitinib or erlotinib to overcome resistance to epidermal growth factor receptor inhibitors. Cancer Res. 2008;68:2820–2830. doi: 10.1158/0008-5472.CAN-07-1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eiseler T, Kohler C, Nimmagadda SC, Jamali A, Funk N, Joodi G, Storz P, Seufferlein T. Protein kinase D1 mediates anchorage-dependent and -independent growth of tumor cells via the zinc finger transcription factor Snail1. J Biol Chem. 2012;287:32367–32380. doi: 10.1074/jbc.M112.370999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.White GE, Tan TC, John AE, Whatling C, McPheat WL, Greaves DR. Fractalkine has anti-apoptotic and proliferative effects on human vascular smooth muscle cells via epidermal growth factor receptor signalling. Cardiovasc Res. 2010;85:825–835. doi: 10.1093/cvr/cvp341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tardaguila M, Mira E, Garcia-Cabezas MA, Feijoo AM, Quintela-Fandino M, Azcoitia I, Lira SA, Manes S. CX3CL1 promotes breast cancer via transactivation of the EGF pathway. Cancer Res. 2013 doi: 10.1158/0008-5472.CAN-12-3828. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Volin MV, Huynh N, Klosowska K, Chong KK, Woods JM. Fractalkine is a novel chemoattractant for rheumatoid arthritis fibroblast-like synoviocyte signaling through MAP kinases and Akt. Arthritis Rheum. 2007;56:2512–2522. doi: 10.1002/art.22806. [DOI] [PubMed] [Google Scholar]
- 39.Ferretti E, Bertolotto M, Deaglio S, Tripodo C, Ribatti D, Audrito V, Blengio F, Matis S, Zupo S, Rossi D, Ottonello L, Gaidano G, Malavasi F, Pistoia V, Corcione A. A novel role of the CX3CR1/CX3CL1 system in the cross-talk between chronic lymphocytic leukemia cells and tumor microenvironment. Leukemia. 2011;25:1268–1277. doi: 10.1038/leu.2011.88. [DOI] [PubMed] [Google Scholar]
- 40.Jarboe JS, Dutta S, Velu SE, Willey CD. Mini-Review: Bmx Kinase Inhibitors for Cancer Therapy. Recent Pat Anticancer Drug Discov. 2012;8:228–38. doi: 10.2174/15748928113089990043. [DOI] [PubMed] [Google Scholar]
- 41.Hur W, Velentza A, Kim S, Flatauer L, Jiang X, Valente D, Mason DE, Suzuki M, Larson B, Zhang J, Zagorska A, Didonato M, Nagle A, Warmuth M, Balk SP, Peters EC, Gray NS. Clinical stage EGFR inhibitors irreversibly alkylate Bmx kinase. Bioorg Med Chem Lett. 2008;18:5916–5919. doi: 10.1016/j.bmcl.2008.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.