

The PhyB-PIF light-inducible dimerization system is used to achieve rapid, reversible, and titratable control of protein localization in budding yeast. This system can be used to dynamically activate or inactivate proteins of interest. It is used here to probe where and when Clb2 activity is required for nuclear fission and spindle stabilization.
Abstract
Protein localization plays a central role in cell biology. Although powerful tools exist to assay the spatial and temporal dynamics of proteins in living cells, our ability to control these dynamics has been much more limited. We previously used the phytochrome B– phytochrome-interacting factor light-gated dimerization system to recruit proteins to the plasma membrane, enabling us to control the activation of intracellular signals in mammalian cells. Here we extend this approach to achieve rapid, reversible, and titratable control of protein localization for eight different organelles/positions in budding yeast. By tagging genes at the endogenous locus, we can recruit proteins to or away from their normal sites of action. This system provides a general strategy for dynamically activating or inactivating proteins of interest by controlling their localization and therefore their availability to binding partners and substrates, as we demonstrate for galactose signaling. More importantly, the temporal and spatial precision of the system make it possible to identify when and where a given protein's activity is necessary for function, as we demonstrate for the mitotic cyclin Clb2 in nuclear fission and spindle stabilization. Our light-inducible organelle-targeting system represents a powerful approach for achieving a better understanding of complex biological systems.
INTRODUCTION
Complex signaling pathways are regulated in both time and space. A comprehensive knowledge of the location of proteins is critical for understanding the mechanisms of complex biological systems. Fluorescent protein technology has allowed us to track the localization of almost every protein in established genetic systems such as budding yeast (Huh et al., 2003), and numerous genetic and pharmacological tools exist for perturbing protein localization and activity (Baudin et al., 1993; Wach et al., 1994; Bishop et al., 2000; Schuldiner et al., 2005; Sopko et al., 2006; Haruki et al., 2008).
Most perturbative tools used to understand protein localization and signaling, however, are far from ideal due to their relatively slow time scales. Constitutive genetic perturbations (deletions, mutants, fusions) and promoter-based tools operate at the level of gene expression, which is slow relative to many posttranslational physiological processes (Baudin et al., 1993; Wach et al., 1994; Sopko et al., 2006). Pharmacological tools are significantly faster, but many are not quickly reversible due to high binding affinities and the reliance on drug washout (Haruki et al., 2008). In addition, pharmacological approaches lack generality; not all proteins can be easily designed for drug targeting.
Compared to traditional genetic manipulations, the temporal and spatial precision of optogenetics allows perturbations on a time scale commensurate with rapid biological information processing, making this approach ideal for protein localization and function study (Toettcher et al., 2011c). Several classes of light-inducible protein–protein interactions have been used to study different molecular processes (Levskaya et al., 2009; Kennedy et al., 2010; Zhou et al., 2012). The phytochrome B (PhyB)–phytochrome-interacting factor (PIF) system is one of them, which takes advantage of a light-controllable binding interaction between PhyB (a fragment of Arabidopsis thaliana phytochrome B) and PIF (a fragment of phytochrome interaction factor 6; Shimizu-Sato et al., 2002; Su and Lagarias, 2007; Levskaya et al., 2009). After conjugation with the membrane-permeable small-molecule chromophore phycocyanobilin (PCB), PhyB changes conformation in response to red/infrared light. PIF binds to PhyB in the presence of red light and dissociates from PhyB in the presence of far-red light (Figure 1A). This system has been optimized for mammalian cells, where it can manipulate a multitude of signaling currencies (Levskaya et al., 2009; Toettcher et al., 2011c). Furthermore, by using computational imaging-based feedback, one can tightly control these light-gated signals in space and time (Toettcher et al., 2011a).
FIGURE 1:

PhyB anchor library construction. (A) Schematic of the PhyB-PIF interaction. (B) Schematic of PhyB anchor library construction. (C) Fluorescence images of our library of PhyB anchor strains, with dashed lines representing cell boundaries. CAAX, plasma membrane targeting sequence; NLS, nuclear localization signal; NES, nuclear export signal. For each strain, example images from all three cell cycle phases are shown. We show an extra image for cell cycle phases when there was particular interest in the dynamics. Stacks of nine images were acquired every 3 min at 30°C, and the maximum-intensity projection of the image stack is shown. (D) List of anchor proteins for which the PhyB fusions were not successful.
Here we adapt the PhyB-PIF light-gated dimerization system for unprecedented dynamical subcellular manipulation of protein localization in budding yeast in a manner that is fast, reversible, and titratable. We generated a library of PhyB constructs localizing to various subcellular addresses, allowing us to rapidly, reversibly, and quantitatively recruit PIF-tagged proteins to the PhyB-labeled subcellular compartments. This approach enables us to dynamically activate/inactivate proteins by recruiting them to or away from their normal site of action. Using this system, we can explore a wide range of biological questions that have been difficult to address in a systematic manner, such as the requirement of a given protein for establishment versus maintenance of a process or the different functions of a protein at different subcellular localizations.
RESULTS
PhyB anchors library construction
We previously used CAAX-tagged PhyB for light-gated recruitment of PIF-tagged proteins to the plasma membrane of mammalian cells. Here we sought to extend this approach for optogenetic control of protein localization to a wide variety of yeast organelles. We first constructed the PhyB anchor library by fusing PhyB (amino acids [aa] 1–908) to the N-terminus of different subcellular “anchors” (Figure 1B). Initially, 20 different “anchor” protein candidates were chosen based on their localizations, including plasma membrane, cytoplasm, nucleus, nucleolus, bud neck, myosin ring, spindle pole body (SPB), endosome, and peroxisome. Some localization signal sequences were also included (Figure 1C).
Because the efficiency of PIF-tagged protein recruitment scales with the abundance of PhyB anchors, we sought to maximize the concentration of PhyB anchors by placing them under the control of the strong constitutive promoter ADH1pr. For each anchor, we verified that PhyB-mCherry fusions displayed proper localization and that the presence of the fusion protein did not alter cell doubling time. After these assays, 9 PhyB-mCherry-anchor fusions targeting eight different locations displayed good behavior (Figure 1C), and the other 11 strains were eliminated because of growth defects or failure of the tagged anchor to localize properly (Figure 1D).
Light-gated recruitment and dissociation to and from most anchors goes to completion in seconds
Next we tested the recruitment and dissociation dynamics of PIF-tagged proteins for different anchors. To quantify performance, we tagged PIF (aa 1–100 of phytochrome-interacting factor 6) with mCitrine (yellow fluorescent protein; Figure 2A), controlled by the constitutive promoter CYC1pr. The speed, reversibility, and titratability of the system were examined by confocal microscopy (Figure 2B).
FIGURE 2:
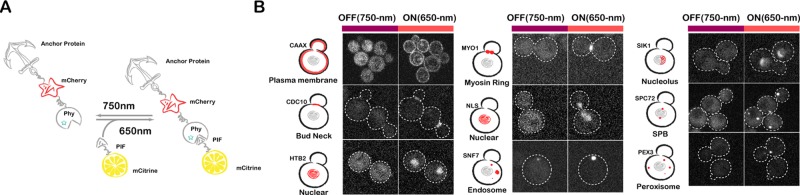
PhyB anchors recruit PIF-mCitrine. (A) Schematic of light-gated interaction of PIF-tagged protein with PhyB-tagged anchor. For fluorescence-based visualization, PhyB anchors were tagged with mCherry (red) and PIF was tagged with mCitrine (yellow). (B) Fluorescence images of light-based PIF-Citrine recruitment to PhyB anchor strains. Fluorescence images are the PIF-mCitrine channel.
Recruitment and dissociation from each anchor were rapid. For most anchors, both recruitment and dissociation go to completion in seconds (Figures 3 and 4B). The recruitment and dissociation for anchors inside the nucleus, including Htb2 (nucleus), nuclear localization signal (NLS; nucleus), and Sik1 (nucleolus), are slightly slower: ∼1 min. This is not unexpected for these anchors because recruitment/dissociation requires passing through the nuclear envelope, which is not a random diffusion process (Figure 3, A– C). Given that the nuclear pore diffusion limit is ∼40 kDa and the molecular weight of mCitrine-PIF is only 39.3 kDa, to test whether the nuclear anchor also works with high–molecular weight proteins, we further tried green fluorescent protein GFP-GFP-PIF (67.8 kDa), GFP-GFP-GFP-PIF (96.0 kDa), and Venus-Venus-Venus-PIF (95.6 kDa). The system works well with all three cases (Supplemental Figure S2). We suspect that the weak NLS of the PIF enables these large proteins to transiently visit the nucleus, thus helping them to get trapped by the PhyB-histone tag. This is consistent with the lack of nuclear exclusion of these large PIF-tagged proteins even in the presence of infrared light. These data suggest that our scheme for nuclear sequestration is likely to work even for large proteins.
FIGURE 3:

PIF-Citrine recruitment to PhyB is fast and reversible. (A) Recruitment of PIF-tagged protein (channel, PIF-mCitrine) to multiple PhyB anchors goes to completion in seconds. Top, PIF recruitment to PhyB-CAAX (plasma membrane); bottom, PIF recruitment to PhyB-HTB2 (nucleus). Cells were exposed to 750-nm light for 3 min before switching to 650-nm light. (B) Dissociation of PIF-mCitrine from PhyB anchors also goes to completion in seconds (channel, PIF-mCitrine). Top, PIF release from PhyB-CAAX (plasma membrane); bottom, PIF release from PhyB-HTB2 (nucleus). Cells were exposed to 650-nm light for 3 min before switching to 750-nm light. (C) Representative recruitment profile of PIF to the PhyB-HTB2 anchor. (D) Fluorescence images of PIF recruitment to PhyB-CAAX with the 30-s time interval (channel, PIF-mCitrine). (E, F) Single-cell recruitment (top) and dissociation (bottom) profiles of PIF to/from the PhyB-CAAX anchor. Shown are the fluorescence intensity on plasma membrane (E) and the ratio between the fluorescence intensity on plasma membrane and in cytosol (F) during PIF association to and dissociation from the PhyB-CAAX anchor, respectively.
FIGURE 4:
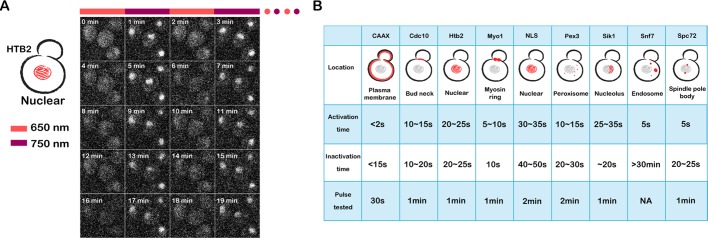
Repeated recruitment and release of PIF-tagged protein from PhyB anchors. (A) Reversibility of different anchor interactions was tested by rapid alternation between the 650- and 750-nm lights. Typically 20 cycles were tested. PIF-mCitrine images are shown. Example shown here is with a nuclear anchor (PhyB-HTB2). (B) The summary of PIF recruitment dynamics in all anchor strains (Supplemental Movie S1).
Note that the only anchor that failed to show reversibility was Snf7 (endosome), in which the PIF-tagged proteins cannot be fully dissociated after PIF recruitment. Given that Snf7 is a sorting protein, the lack of reversibility is likely due to PIF being recruited into the endosome (Babst et al., 2002). Once inside the endosome, PIF will not be released even after dissociation from PhyB-Snf7.
Although there was no obvious cell-to-cell variability in the dynamics of recruitment/dissociation (Figure 3, D and E), cell-to-cell variability in fluorescence intensity was observed when we quantified single-cell profiles over the population (Figure 3E). There are two possible sources of noise: variation in PIF expression level between cells, and variation in expression level across different cell cycle stages. To distinguish which was the major source of variation, we further quantified these two types of noise by measuring the coefficient of variation (CV) of the expression level over the population and along the cell cycle. We chose the SPB anchor (PhyB-Spc72) for this measurement because the SPBs are very dynamic along the cell cycle. Compared to the cell-to-cell gene expression variability (CV = 0.46), the variability along the cell cycle is comparably small: in the presence of red light, CV = 0.3 for the desired position SPB, and CV = 0.11 for other positions. Indeed, the cell-to-cell variability was reduced when we normalized the single-cell response to the expression level of PIF (Figure 3F).
Reversibility of recruitment
Time-varying inputs have been powerful tools for investigating the logic of signaling cascades. This approach is commonly used to dissect feedback architecture in engineering and has also been applied to a handful of extracellular inputs (Mettetal et al., 2008; Batchelor et al., 2009). Two limitations have impeded the broader application of this approach. First, many extracellular signals are difficult to control in an oscillatory manner due to the difficulties in removing bound ligand and the complexities of receptor trafficking. Second, the field has lacked general tools for producing oscillating intracellular inputs, a feat now possible with light. We sought to establish the ability of our system to generate rapid protein recruitment and dissociation from intracellular organelles by rapid alternation between the 650- and 750-nm light (Figure 4). All anchors except for Snf7 can be fully switched on and off by alternation between red and far-red light with a time interval as short as 30 s but within 2 min for all anchors tested (Supplemental Movie S1).
Titratability of recruitment
Both the interaction and dissociation of PhyB and PIF reaction are controllable with different frequencies of light, making precise titration of PIF recruitment possible (Toettcher et al., 2011a). We tested whether the degree of PIF protein recruitment to the anchor could be titrated by changing the ratio of 650:750-nm light intensity. Keeping 750-nm light intensity constant, we varied the 650-nm light intensity to titrate the recruitment of PIF-mCitrine to the nucleus via PhyB-Htb2. The nuclear intensity showed a linear saturating relation to 650-nm light intensity, demonstrating our ability to titrate the system to intermediate levels by changing the ratio of 650:750-nm light intensity (Figure 5, A and B). Because we automatically read out the amount of protein translocation and adjust the light inputs, this also enables us to shape the input in time, generating any desired temporal profile such as linear or sigmoidal (Toettcher et al., 2011a). In this case, only the 650-nm light voltage was varied, but, depending on the light sources used, either could be varied to generate the appropriate ratio.
FIGURE 5:

The degree of PIF recruitment is titratable and varies among PhyB anchors. (A, B) The degree of PIF protein recruitment to the anchor can be titrated by changing the ratio of 650:750-nm light intensity. The 750-nm light intensity was kept constant, and the 650-nm light intensity was increased step by step. For each step, cells were exposed to 650:750-nm light for 2 min to reach steady state before the measurement. Typical single-cell titration profiles (A) and the average over the population (B) are shown. The straight (red) line shows the linear range of the PIF recruitment in response to red light. (C) Fold increase of PIF protein recruitment to PhyB anchors for 650- vs. 750-nm light. (D) The portion of PIF protein depleted from other positions upon exposure to 650-nm light (1.0 = 100% depletion). For C and D, unlabeled wild-type cells were used to subtract cell autofluorescence, and the PhyB anchor channel was used to define the desired position. Average fluorescence intensity per pixel was used to calculate the decrease/increase. Typically, 50–100 cells were used for each anchor strain. Data were normalized by the single-cell total fluorescence intensity under 750-nm light.
The system can be used to enrich or deplete proteins of interest at specific localizations
For many signal transduction cascades, protein localization is intimately tied to function. Thus, changing the intracellular location of a protein could lead to its activation or inactivation. Previous applications of the PhyB-PIF system used a PIF-tagged protein on top of the endogenous copy, which is suitable for recruiting proteins to sites of activation. By tagging PIF with the gene of interest at the endogenous locus, our system has control over the entire pool of protein of interest in the cell, making it possible to globally mislocalize proteins for inactivation.
Toward this end, we quantified the enrichment and depletion efficiency for each anchor in the presence of red light. The enrichment/depletion efficiency varies among different anchors due to the size, shape, and diffusional accessibility of different desired position/organelles (Figure 5, C and D). Effective sequestration of cytoplasmic proteins requires anchors with a high depletion efficiency. As expected, larger organelles (CAAX, Htb2, NLS) generate more depletion than smaller organelles (Spc72). Of importance, although PhyB-Snf7 had the largest enrichment and depletion efficiency, this anchor was much less reversible than the others, making it less suitable for many applications (Figure 4B).
Optogenetic activation of yeast galactose signaling
To test our ability to control endogenous signaling through light-inducible protein localization, we first applied the system to a very well characterized pathway: galactose signaling. Gal80 is a transcriptional repressor of GAL-responsive genes; when it is removed from the nucleus, transcription of GAL-responsive genes is activated (Lohr et al., 1995; Timson et al., 2002). In the absence of galactose, Gal80 rapidly shuttles between the nucleus and cytoplasm. The presence of galactose sequesters Gal80 in the cytoplasm, removing inhibition and allowing transcription (Figure 6A, top).
FIGURE 6:

Optogenetic activation of yeast galactose signaling. (A) Schematic of the galactose signaling system. Galactose-based activation of the system: presence of galactose removes the repressor Gal80 from the nucleus, allowing transcription (top). Light-based activation of the system: we used the CAAX (plasma membrane)–anchored PhyB and Gal80 tagged with PIF at the endogenous locus; 650-nm light causes Gal80-PIF to localize to the plasma membrane, pulling it out of the nucleus (bottom), thereby activating galactose-based transcriptional targets. (B) Galactose signaling system can be turned ON by 650-nm light even in the presence of glucose and the absence of galactose. Fluorescence represents cyan fluorescent protein expressed from the Gal1 promoter.
Here we used the CAAX (plasma membrane)–anchored PhyB and the endogenous copy of Gal80 tagged with PIF at the N-terminus. Light at 650 nm causes Gal80-PIF to localize to the plasma membrane, pulling it out of the nucleus and thereby activating galactose-based transcriptional targets (Figure 6A, bottom). Our galactose-signaling system can be turned on by 650-nm light even in the presence of glucose and the absence of galactose (Figure 6B). The steady-state expression level of the GAL1 promoter is reduced in these cells, most likely because the positive feedback loops responsible for the wild-type ultrasensitive response to galactose are not operating. These feedback loops operate through galactose uptake, so they will not function in our galactose-free, light-inducible system.
Using the light-gated system to study Clb2 function at different subcellular localizations
After we demonstrated that this optogenetic system could be easily used to tune a protein's function by recruiting it to or away from the localization where it is active, we next applied it to a more complex system, cell cycle control, where the key regulatory proteins act at multiple subcellular locations. We investigated the functions of the main mitotic cyclin in budding yeast, Clb2, at different subcellular locations.
Clb2 is the primary mitotic cyclin in budding yeast, which accumulates during G2 and M phases and is degraded at the end of mitosis. Clb2 binds to Cdk1 (Cdc28) and forms a complex, which controls a wide range of substrates to coordinate the early stages of mitosis and progression from the metaphase-to-anaphase transition (Ghiara et al., 1991; Surana et al., 1991; Enserink and Kolodner, 2010). Given the importance of Clb2 in cell cycle control, it has been extensively studied in the past few decades. It is known that Clb2 contains one NLS and two nuclear export signals, and although it predominantly localizes in the nucleus (Hood et al., 2001; Bailly et al., 2003), it also localizes to a wide range of other cellular structures (Eluère et al., 2007), such as bud neck (Hood et al., 2001), cytoplasm (Bailly et al., 2003), spindle (Bailly et al., 2003), and spindle pole body (Bailly et al., 2003). Clb2 localization is highly dynamic during cell cycle progression, but due to lack of tools that can perturb protein localization on a fast and subcellular scale, the functional significance of different pools of localized Clb2 is unclear.
CLB1 and CLB2 genes encode a closely related pair of M-phase cyclins in budding yeast. Clb1 is the major cyclin regulating meiosis, and Clb2 is the most important cyclin in regulating mitosis (Grandin and Reed, 1993). Neither CLB1 nor CLB2 is essential; however, disruption of both is lethal and causes a mitotic defect (Surana et al., 1991; Fitch et al., 1992; Richardson et al., 1992). To facilitate optogenetic control of cyclin function, we deleted CLB1 and directly tagged endogenous CLB2 with PIF at its C-terminus. By tagging cyclin at the endogenous locus, we gain the ability of optical control of the spatial and temporal dynamics of the sole M-phase cyclin. This enables us to control when and where cyclin is active in the cell by recruiting cyclins either toward or away from various intracellular sites of action.
Recruiting Clb2 to the nucleus results in nuclear fission failure
We first recruited Clb2 to the nucleus using PhyB-Htb2 (Figure 7A). In the presence of far-red light, these cells always split the two nuclei, which are then accurately distributed to the two daughter cells (Supplemental Movie S2). When exposed to red light, Clb2 is sequestered in the nucleus, and 83% of cells fail to split the two nuclei (Figure 7, B and C, and Supplemental Movie S3). The nucleus does split eventually, presumably when cytokinesis divides the two cells, but the split is significantly delayed with respect to WT cells. Some cells completely fail to split the nuclei, resulting in two SPBs in the same daughter cell. None of these cells progresses through the next cell cycle successfully (Supplemental Movie S4).
FIGURE 7:

Tethering Clb2 inside the nucleus results in nuclear fission failure. (A) Schematic of optogenetic nuclear recruitment of Clb2, and Clb2 localization under 750- and 650-nm light (inset). (B) Sequestering Clb2 to nucleus (PhyB-HTB2) results in nuclear fission defect. Combined phase and fluorescence time-course images (3-min interval) when Clb2 is not recruited to the nucleus (750-nm light; top and Supplemental Movie S2) vs. when Clb2 is recruited to the nucleus (with 650-nm light; bottom and Supplemental Movies S3 and S4). Red channel represents the nucleus (PhyB-mCherry-HTB2), and the green channel represents the spindle pole body (Spc42-GFP). (C) Kymograph of spindle pole dynamics when Clb2 is unrecruited (system is OFF, left) vs. when Clb2 is recruited to the nucleus (system is ON, right) with a time interval of 3 min. (D) The nuclear fission failure is rescued by adding an extra untagged copy of CLB2, suggesting that nuclear fission failure is likely due to cytoplasmic Clb2 depletion. The average cell size in different strains is shown as the inset.
Tethering Clb2 to the nucleus has two consequences: cytoplasmic Clb2 is depleted, and nuclear Clb2 concentration is increased. To distinguish whether the nuclear fission failure was due to gain of function in the nucleus or loss of function in the cytoplasm, we introduced an additional untagged copy of CLB2. If the phenotype is due to loss of function because of cytoplasmic depletion, adding an extra copy should rescue the phenotype. In contrast, if the phenotype is due to gain of function in the nucleus, adding an extra copy should not rescue the phenotype. Consistent with the defect being due to cytoplasmic depletion, the percentage of cells with nuclear fission failure was rescued significantly, decreasing from 83 to 29.5%, after addition of an untagged copy of CLB2 (Figure 7D). These results suggest that nuclear fission failure is likely due to Clb2 depletion from the cytoplasm rather than enrichment in the nucleus.
The defect was not fully rescued by adding one copy of CLB2, presumably due to the expression level of the additional copy of CLB2, which was not inserted at the endogenous CLB2 genome location and may not have brought CLB2 levels back to endogenous levels. We cannot rule out the possibility that some portion of the phenotype is a result of recruiting Clb2 to histone, which may change the chromatin structure and impede chromatin separation. To further clarify the relevant site of Clb2 for nuclear fission, we tethered Clb2 to the plasma membrane. Under these conditions, we observed no nuclear fission defect (Supplemental Figure S3). This could be either because the cytosolic substrates are still accessible for a plasma membrane recruited Clb2 or because the relevant site of action (such as the bud neck) overlaps the plasma membrane recruited Clb2.
Recruiting Clb2 to the spindle pole body stabilizes the spindle at the end of mitosis
We next recruited Clb2 to the spindle pole body by using PhyB-Spc72 (Figure 8A). When Clb2 was recruited to the SPBs, the fully elongated spindle persisted for significantly longer than for wild-type cells. This resulted in cells with a significantly longer duration of maximum spindle length (τ650 nm = 40 min in the presence of red light vs. τ0 750 nm = 8 min in the presence of far-red light), suggesting that the mitotic spindle was stabilized at the end of mitosis (Figure 8B).
FIGURE 8:
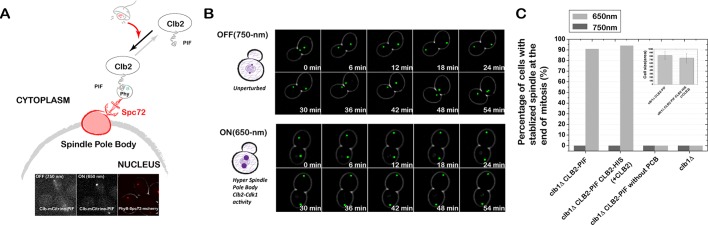
Tethering Clb2 to the spindle pole body stabilizes the spindle at the end of mitosis. (A) Schematic of optogenetic spindle pole body recruitment of Clb2, and Clb2 localization under 750- and 650-nm light (inset). (B) Combined phase and fluorescence time-course images (6-min interval) when Clb2 is not recruited to the spindle pole body (750-nm light; top) vs. when Clb2 is recruited to the spindle pole body (650-nm light; bottom). Green channel represents the spindle pole body (Spc42-GFP). (C) Clb2 recruitment to the spindle pole body results in significant spindle stabilization. This phenotype is not rescued by adding an extra untagged copy of Clb2, suggesting that the defect is mainly due to the gain of Clb2–CDK function at the SPB. The average cell size in different strains is shown as the inset.
To distinguish whether the defect was due to gain of function at the SPB or loss of function due to depletion at other positions, we introduced an additional untagged CLB2 copy. Because adding extra copies of CLBs also changes the doubling time, this makes it difficult to compare the absolute spindle duration times between different strains. To do a fair comparison, we used the maximum duration time in the presence of far-red light as the threshold for each strain. This threshold varies between strains due to the altered doubling time. Then we determined the percentage of cells in red light whose duration exceeded the corresponding threshold. The percentage of cells with significantly stabilized spindles did not decrease when an additional copy of CLB2 was introduced (i.e., before adding CLB2, 90%; after adding CLB2, 94%), suggesting that the phenotype is likely due to gain of function at the SPB (Figure 8C). Thresholds using duration time of 95 and 99% cells were tested, with the same conclusion.
Using our optogenetic system to identify when Clb–Cdk1 activity is necessary for nuclear fission
The ability to control protein activity by light is enormously powerful for studying protein function within a physiological context. The speed of the light-gated anchoring system offers the possibility of probing biological systems in real time, allowing us to identify when a given protein is required for a process. In this case, we wanted to determine when Clb2–Cdk1 activity is necessary for nuclear fission. Because we determined that the nuclear fission phenotype was likely due to loss of function via cytoplasmic depletion, we can use far-red light to release Clb2 at various points during the cell cycle and measure whether the phenotype is rescued.
We grew PhyB-Htb2 Clb2-PIF cells in the presence of red light and then applied short far-red pulses at different stages of the cell cycle to transiently release Clb2 from the nucleus. By monitoring these cells for nuclear fission failure, we could determine what pulse timing was required to rescue the nuclear fission failure. We first used a far-red light pulse with duration of 12 min. The nuclear fission failure was rescued by applying the far-red light pulses at the end of mitosis, whereas no rescue occurred when pulses were applied at the earlier stages of the cell cycle (G1, S, and early M phase; Figure 9, A and B). We further tested shorter pulses of 9 min and observed the same result: the nuclear fission failure could be rescued only by applying far-red pulses at the end of mitosis (Figure 9C), suggesting that Clb2–Cdk1’s function in nuclear fission is at the end of mitosis.
FIGURE 9:
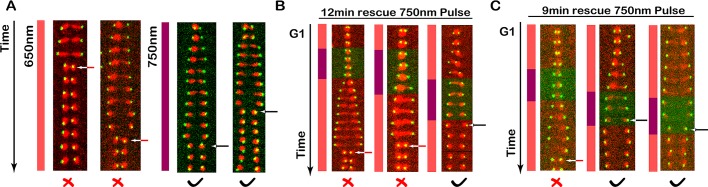
Use of our optogenetic system to identify when Clb2–Cdk1 activity is necessary for nuclear fission. Nuclear fission failure can be rescued by applying a short far-red pulse at the end of mitosis to transiently release Clb2 from the nucleus. (A) Tethering Clb2 to nucleus (PhyB-mCherry-HTB2) results in nuclear fission failure. Kymograph of the nucleus (red channel) and the SPB (green channel) when Clb2 is sequestered in the nucleus (650-nm light) vs. unsequestered Clb2 (750-nm light). The time interval is 3 min. Kymographs of two cells are shown for each condition. (B) We applied 12-min, 750-nm pulses (purple) at the different stages of the cell cycle (time interval is 3 min) to release Clb2 from the nucleus, with 650-nm light (red, Clb2 sequestered in nucleus) delivered at all other times. Red X's and red arrows indicate failed nuclear fission, and black check marks and black arrows indicate successful nuclear fission. Clb2 function is required at the end of mitosis for nuclear fission. (C) We applied 9-min, 750-nm pulses at the different stages of the cell cycle (time interval is 3 min). Red X's and red arrows indicate failed nuclear fission, and black check marks and black arrows indicate successful nuclear fission. These experiments indicate that cytoplasmic Clb2–Cdk1 activity is required at the end of mitosis for nuclear fission.
Using our optogenetic system to establish the spatial requirements of Clb2–Cdk1 activity in budding yeast
Although optical control is capable of precisely illuminating small regions of the cell, in practice, it is extremely challenging to pattern light on specific organelles, since both the size and shape of organelles are very dynamic. By combining optical control with an organelle-anchoring strategy, our optogenetic system provides the opportunity to manipulate the experimental system with user-defined complex spatial patterns. Whereas the PhyB anchors ensure perfect organelle specificity, the optical patterning control allows us to apply the perturbation on only one or several cells in a microscopy field of view and leave the rest as controls or even to direct protein recruitment to a small portion of a single organelle.
Here we tested the spatial control of the system on PhyB-Spc72 Clb2-PIF cells. Because we could not visualize Clb2-PIF recruitment on the epifluorescence scope used for spatial control, we first verified that we had adequate spatial control by using the PhyB-Spc72 PIF-mCitrine strain to demonstrate recruitment to just one SPB. Patterning red light only on the daughter cell, with far-red light on all other cells, allowed us to recruit PIF-mCitrine to the single SPB in the daughter cell only (Figure 10A). We repeated the same experiment on PhyB-Spc72 Clb2-PIF cells. Shining red light either on the daughter cell alone or on the mother cell alone was not sufficient to stabilize the spindle at the end of mitosis. These data suggest that spindle stabilization requires a threshold amount of total Clb2 recruitment or Clb2 must act at both SPBs (Figure 10B).
FIGURE 10:

Use of our optogenetic system to establish the spatial requirements of Clb2–Cdk1 activity in budding yeast. (A) The system is capable of recruiting PIF-mCitrine to only one spindle pole body by restricting the activating light to the daughter. (B) Whereas illuminating both mother and daughter with 650-nm light stabilizes the spindle at the end of mitosis, restricting Clb2 recruitment to either the mother SPB or daughter SPB is not sufficient to stabilize the spindle. Green channel represents the SPB (Spc42-GFP). To control for day-to-day variations in doubling time due to ambient temperature in the microscope room and timing of movie since yeast dilution, experimental (650-nm illuminated) cells were compared with control cells (no 650-nm illumination) in the same microscope field of view.
DISCUSSION
Cell signal transduction pathways are highly regulated in time and space. The activities of many proteins are controlled by changes in localization, and much of this regulation takes place on relatively short time scales, ranging from seconds to hours. To understand these dynamic processes, we need tools that can perturb protein localization and function on similar spatiotemporal scales. The powerful set of genetic tools available in yeast makes it simple to generate constitutive genetic changes and induce or deplete a given protein, but most of these tools operate through transcription and translation and are therefore slow relative to many cell signaling pathways. Over the time needed to change protein activity with these tools, compensatory changes can accumulate, up- or down-regulating other proteins to compensate, and masking the true effect of depleting the protein of interest. Approaches that act on faster time scales, such as pharmacological approaches, have limitations in their generality and are often irreversible or difficult to titrate.
Here we report a generalizable light-inducible organelle targeting system that is fast, reversible, and titratable. We can inactivate or activate proteins by inducibly changing their localization. This system allows us to identify when and where a given protein's activity is necessary for cellular function. Given that increasing evidence suggests that the spatial context of a protein is a major mechanism for regulating its function, methods to perturb localization are becoming necessary. Here we systematically tested our system with anchors located at different organelles, including cell components lacking a membrane, such as SPB and bud neck. We quantified dynamics (both on and off) and the depletion/hyperactivation efficiency for each of them, which should greatly facilitate its adoption by others in the yeast community.
Dissecting the many roles of multifunctional proteins has been very challenging. Clb2 is the primary mitotic cyclin in budding yeast and acts with Cdk1 (Cdc28) to coordinate the early stages of mitosis and progression from the metaphase-to-anaphase transition. Clb2 localization is highly dynamic during cell cycle progression. Unfortunately, due to the lack of general tools for precision subcellular control, the functions of different pools of localized Clb2 have been unclear. Here, by employing our optogenetic system, we uncover different functions of Clb2 at different subcellular locations. It is known that CDK activity is down-regulated during mitotic exit and that Clb2 overexpression will delay or even block the mitotic exit (Cross et al., 2005). Of interest, we show that the cytoplasmic Clb2–Cdk1 activity at the end of mitosis is necessary for nuclear fission, suggesting that a certain level of cytoplasmic Clb2–Cdk1 activity at the end of mitosis is required for some mitotic exit events, such as nuclear fission. On the other hand, we also show that enrichment of Clb2 at the spindle pole body stabilizes the spindle at the end of the mitosis, suggesting that spindle disassembly at the end of mitosis requires the down-regulation of CDK activity. Spindle stabilization could be one of the reasons that Clb2 overexpression blocks the mitotic exit. Recruiting to the mother spindle pole body alone or daughter spindle pole body alone was not sufficient to stabilize the spindle, suggesting that the threshold of Clb2–Cdk1 for spindle stabilization is quite high, consistent with the idea that the mitotic block requires two copies of Gal1 promoter–controlled Clb2 (Cross et al., 2005).
These data demonstrate that our approach is likely to be a powerful tool for studying multifunctional proteins. Because this system acts on similar spatiotemporal scales to cell signaling pathways, it can lead to a better understanding of complex biological networks. In addition, this system can be applied to any protein of interest that can tolerate a PIF tag and samples the portion of the cell containing the PhyB anchor. Depending on the PhyB anchor, this is likely to be true for most cytoplasmic proteins. It should also be possible to control the subcellular distribution of lipid-modified membrane proteins that transiently sample the cytosol (Silvius et al., 2006).
In principle, any inducible protein dimerization system can be used to control protein localization. For example, the “anchor-away” system uses a chemically inducible protein dimerization system in which two binding partners bind with high-affinity upon addition of the small-molecule drug rapamycin (Haruki et al., 2008). By targeting one of the binding partners to a subcellular location and fusing the other binding partner to a protein of interest, this system has been used to induce the localization of proteins (Komatsu et al., 2010). Our light-gated dimerization system operates on the same concept, but it offers unique advantages over the rapamycin-gated system. First, our system can be turned off and on with the same time scale, a few seconds, whereas the rapamycin system is effectively irreversible over the time scale of many biological signaling events. Second, because we can turn the interaction on and off with different frequencies of light, we can achieve a wide dynamic range and titrate protein concentrations to intermediate levels. Finally, our system allows perturbations to be made on a single cell or subpopulation of cells when performing experiments on a microscope in real time as opposed to adding a drug to an entire well at a time.
Although several different light-inducible protein–protein interactions have been adapted for studying cellular signaling pathways, some unique properties of the PhyB-PIF system make it ideal in practice for cell culture–based experiments. First, both directions of the PhyB and PIF reaction are light inducible (Ni et al., 1999), which has a number of benefits: the system can be turned on and off with the same time scale and can be titrated to intermediate levels by varying the ratio of red to far-red light. This allows precise shaping of the input, generating any desired curve, including linear, sigmoidal, and so on (Toettcher et al., 2011a). Second, the PhyB-PIF system uses low-energy lights, red and far-red, which are much less toxic than the blue light used in other light-inducible system and are compatible with a wider rage of fluorescently encoded proteins than other, blue-activated optogenetic systems (Kennedy et al., 2010; Zhou et al., 2012). This is especially important in budding yeast, which are sensitive to imaging in the blue/ultraviolet (Carlton et al., 2010). Unlike some of the other optogenetic approaches, the PhyB-PIF system requires the addition of a small-molecule chromophore PCB for light control in nonphotosynthetic organisms (Levskaya et al., 2009). Although this is likely to be a disadvantage for experiments in multicellular organismal contexts in which chromophore delivery may be difficult, it can be an advantage in single-cell applications, for which the chromophore can be delivered easily and gives another degree of control over the system. Addition of PCB allows the user to determine exactly when cells become light-responsive. For many applications, it is convenient to be able to grow the cells in room light without activating the system. Once PCB is added, cells must be maintained either in the dark or under far-red light to keep the system off. Furthermore, this facilitates controls establishing that the expression of the optogenetic components do not effect the cells (i.e., no phenotype should be observed in the absence of PCB).
This system also holds the potential to be a powerful tool for high-throughput studies. For example, by tagging each component of a signaling pathway with PIF and crossing the resulting strains with the PhyB library, one can easily identify the importance of each component's temporal and spatial regulation in signal processing. Given the relative ease of tagging proteins at the endogenous locus in yeast, even larger-scale studies are possible. This system is only easily applicable, however, to cytoplasmic proteins or lipid-modified proteins that transiently visit the cytosol (Silvius et al., 2006).
In summary, we report a light-inducible organelle-targeting system that is fast, reversible, and titratable. This system allows us to dynamically perturb biological systems on a subcellular scale, where we can inducibly activate/inactivate proteins by recruiting them to or away from their normal sites of action. The toolkit developed here could enable a new generation of spatiotemporal perturbations in budding yeast.
MATERIALS AND METHODS
Yeast strain construction
Standard methods were used throughout. All strains used in this study were congenic w303 (MATa his3-11,15 trp1-1 leu2-3 ura3-1 ade2-1). All anchor genes were cloned from the genome directly and tagged with PhyB (aa 1–908) and mCherry at the N-terminus with a 15-aa linker (EFDSAGSAGSAGGSS) between the PhyB and mCherry and a 10-aa linker (SAGSAGKASG) between mCherry and anchor gene. Endogenous GAL80 and CLB2 were tagged with mCitrine and PIF at the C-terminus with an 11-aa linker (AAAGDGAGLIN) between GAL80/CLB2 and mCitrine. CLB1 was deleted by using the KanMX2 fragment. Endogenous SPC42 was tagged with GFP at the C-terminus with an 11-aa linker (AAAGDGAGLIN). All strains were characterized by sequencing PCR products.
Time-lapse microscopy
Cells growing exponentially in synthetic liquid medium were seeded onto thin 1.5–2% agarose slabs of the same medium. For titration and spatial control experiments, cells were seeded onto concanavalin A–coated well plates with 0.17-µm coverglass bottoms. Multiple different positions were followed simultaneously. For most experiments, stacks of nine images were acquired every 3 min at 30°C, with 30-ms exposure for green channel and 50 ms for red channel. For reversibility experiments, only one image was acquired for each time point. For light control experiments, PCB was added to cells 2 h before imaging with a final concentration of 27 μM (stock, 5.4 mM in dimethyl sulfoxide). PCB was purified according to Toettcher et al. (2011b) or purchased from ChemPep (Wellington, FL). Because room light activates the system, cells were kept in the dark once PCB was added.
For most experiments, fluorescence and phase microscopies were performed in the University of California, San Francisco (UCSF), Nikon Imaging Center using a TE2000U inverted microscope (Nikon, Melville, NY) with Yokogawa CSU22 spinning-disk confocal illumination (Solamere Technology Group, Salt Lake City, UT) and a Cascade II CCD Camera (Photometrics, Tucson, AX). Images were acquired using Micromanager software (http://micromanager.org/) and analyzed using ImageJ (National Institutes of Health, Bethesda, MD) with the SpotTracker2D plug-in (http://bigwww.epfl.ch/sage/soft/spottracker/gasser.html). Potential toxicity of PCB and fluorescence illumination was evaluated in control experiments (Supplemental Figure S1).
Titration and spatial control experiments were performed on a Nikon Eclipse Ti inverted microscope using a 100× PlanApo total internal reflection fluorescence, 1.49 numerical aperture objective, a xenon arc-lamp (Sutter Instrument, Novato, CA), and an Evolve electron-multiplying charge-coupled device camera (Photometrics). For these experiments, the microscope, dichroic positions, filters, shutters, and camera were controlled using the open-source Micromanager software package with additional custom Matlab code (Toettcher et al., 2011a). Epifluorescence images were computationally denoised in collaboration with John Sedat (UCSF), using an algorithm built into the Priism image analysis package (Kervrann and Boulanger, 2006).
For fully activating and inactivating light control experiments, we used one 650- and one 750-nm light-emitting diode (LED; Lightspeed Technologies, Campbell, CA), which are directly attached on the microscope condenser. For the titration and spatial control experiments, we used one 650-nm LED and two 750-nm LEDs (Lightspeed Technologies). Light intensity was controlled by changing the applied voltage (0–5 V). Voltage was controlled using custom Matlab code by connecting the LEDs to the analogue outputs of a DT9812 board (Data Translation, Marlboro, MA). For spatial control, user-defined patterns of LED light were projected on the sample using a custom dual-input digital micromirror device (DMD; Andor, Belfast, United Kingdom). Pixels on this device can be in two states, ON or OFF; ON pixels are illuminated with both 650- and 750-nm light, and OFF pixels are illuminated by the second 750-nm light source with a constant voltage. Sample exposure to DMD light was controlled using a 620-nm short-pass filter (Chroma Technology, Brattleboro, VT). In addition, we used a 625-nm sputtered short-pass emission filter (Chroma) to block DMD light from reaching the camera during imaging, allowing us to keep the 620-nm short-pass filter in place (and thus continue exposing the sample to 650- and 750-nm light) while images were collected.
Image analyses
Image segmentation and fluorescence quantification were performed using custom Matlab software and ImageJ with Image5D plug-in. Maximum-intensity projections of z-stacks were reported for most experiments, except for plasma membrane images. For the plasma membrane, the middle plane was used.
For assaying the concentration increase/decrease measurements as the system is turned ON and OFF (Figure 5, C and D), PhyB library strains were mixed wild-type cells that do not contain fluorescence labeling. Nonfluorescent cells were used to subtract cell autofluorescence, and the anchor images were used to define the desired position. Average fluorescence intensity per pixel was used to calculate the decrease/increase. Typically, 50–100 cells were used for each anchor strain.
Supplementary Material
Acknowledgments
We thank Jared Toettcher for constructs and strains and John Sedat for image denoising collaboration. We also thank Delquin Gong, Dan Lu, Xili Liu, and Volkan Sevim for a critical reading of the manuscript. This work was supported by National Institutes of Health Grants GM097115 (X.Y. and C.T.), P50 GM081879 (X.Y. and C.T.), GM096164 (O.D.W. and A.P.J.), and GM084040 (O.D.W. and A.P.J.). A.P.J. is supported by an American Heart Association Predoctoral Fellowship, and X.Y. is supported by a Lui Fellowship and the Li Foundation.
Abbreviations used:
- PCB
phycocyanobilin
- PhyB
phytochrome B
- PIF
phytochrome-interacting factor
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-03-0126) on June 12, 2013.
REFERENCES
- Babst M, Katzmann DJ, Estepa-Sabal EJ, Meerloo T, Emr SD. Escrt-III: an endosome-associated heterooligomeric protein complex required for mvb sorting. Dev Cell. 2002;3:271–282. doi: 10.1016/s1534-5807(02)00220-4. [DOI] [PubMed] [Google Scholar]
- Bailly E, Cabantous S, Sondaz D, Bernadac A, Simon M-N. Differential cellular localization among mitotic cyclins from Saccharomyces cerevisiae: a new role for the axial budding protein Bud3 in targeting Clb2 to the mother-bud neck. J Cell Sci. 2003;116:4119–4130. doi: 10.1242/jcs.00706. [DOI] [PubMed] [Google Scholar]
- Batchelor E, Loewer A, Lahav G. The ups and downs of p53: understanding protein dynamics in single cells. Nat Rev Cancer. 2009;9:371–377. doi: 10.1038/nrc2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudin A, Ozier-Kalogeropoulos O, Denouel A, Lacroute F, Cullin C. A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res. 1993;21:3329–3330. doi: 10.1093/nar/21.14.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop, et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- Carlton PM, et al. Fast live simultaneous multiwavelength four-dimensional optical microscopy. Proc Natl Acad Sci USA. 2010;107:16016–16022. doi: 10.1073/pnas.1004037107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross FR, Schroeder L, Kruse M, Chen KC. Quantitative characterization of a mitotic cyclin threshold regulating exit from mitosis. Mol Biol Cell. 2005;16:2129–2138. doi: 10.1091/mbc.E04-10-0897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eluère R, Offner N, Varlet I, Motteux O, Signon L, Picard A, Bailly E, Simon M-N. Compartmentalization of the functions and regulation of the mitotic cyclin Clb2 in S. cerevisiae. J Cell Sci. 2007;120:702–711. doi: 10.1242/jcs.03380. [DOI] [PubMed] [Google Scholar]
- Enserink JM, Kolodner RD. An overview of Cdk1-controlled targets and processes. Cell Div. 2010;5:11. doi: 10.1186/1747-1028-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch I, Dahmann C, Surana U, Amon A, Nasmyth K, Goetsch L, Byers B, Futcher B. Characterization of four B-type cyclin genes of the budding yeast Saccharomyces cerevisiae. Mol Biol Cell. 1992;3:805–818. doi: 10.1091/mbc.3.7.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiara JB, Richardson HE, Sugimoto K, Henze M, Lew DJ, Wittenberg C, Reed SI. A cyclin B homolog in S. cerevisiae: chronic activation of the Cdc28 protein kinase by cyclin prevents exit from mitosis. Cell. 1991;65:163–174. doi: 10.1016/0092-8674(91)90417-w. [DOI] [PubMed] [Google Scholar]
- Grandin N, Reed SI. Differential function and expression of Saccharomyces cerevisiae B-type cyclins in mitosis and meiosis. Mol Cell Biol. 1993;13:2113–2125. doi: 10.1128/mcb.13.4.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J, Laemmli UK. The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol Cell. 2008;31:925–932. doi: 10.1016/j.molcel.2008.07.020. [DOI] [PubMed] [Google Scholar]
- Hood JK, Hwang WW, Silver PA. The Saccharomyces cerevisiae cyclin Clb2p is targeted to multiple subcellular locations by cis- and trans-acting determinants. J Cell Sci. 2001;114:589–597. doi: 10.1242/jcs.114.3.589. [DOI] [PubMed] [Google Scholar]
- Huh W-K, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Kennedy MJ, Hughes RM, Peteya LA, Schwartz JW, Ehlers MD, Tucker CL. Rapid blue-light-mediated induction of protein interactions in living cells. Nat Methods. 2010;7:973–975. doi: 10.1038/nmeth.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kervrann C, Boulanger J. Unsupervised patch-based image regularization and representation. In: A Leonardis, H Bischof, A Pinz., editors. Computer Vision–ECCV. Berlin: Springer; 2006. pp. 555–567. [Google Scholar]
- Komatsu T, Kukelyansky I, McCaffery JM, Ueno T, Varela LC, Inoue T. Organelle-specific, rapid induction of molecular activities and membrane tethering. Nat Methods. 2010;7:206–208. doi: 10.1038/nmeth.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levskaya A, Weiner OD, Lim WA, Voigt CA. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature. 2009;461:997–1001. doi: 10.1038/nature08446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr D, Venkov P, Zlatanova J. Transcriptional regulation in the yeast GAL gene family: a complex genetic network. FASEB J. 1995;9:777–787. doi: 10.1096/fasebj.9.9.7601342. [DOI] [PubMed] [Google Scholar]
- Mettetal JT, Muzzey D, Gómez-Uribe C, Van Oudenaarden A. The frequency dependence of osmo-adaptation in Saccharomyces cerevisiae. Science. 2008;319:482–484. doi: 10.1126/science.1151582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni M, Tepperman JM, Quail PH. Binding of phytochrome B to its nuclear signalling partner PIF3 is reversibly induced by light. Nature. 1999;400:781–784. doi: 10.1038/23500. [DOI] [PubMed] [Google Scholar]
- Richardson H, Lew DJ, Henze M, Sugimoto K, Reed SI. Cyclin-B homologs in Saccharomyces cerevisiae function in S phase and in G2. Genes Dev. 1992;6:2021–2034. doi: 10.1101/gad.6.11.2021. [DOI] [PubMed] [Google Scholar]
- Schuldiner, et al. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell. 2005;123:507–519. doi: 10.1016/j.cell.2005.08.031. [DOI] [PubMed] [Google Scholar]
- Shimizu-Sato S, Huq E, Tepperman JM, Quail PH. A light-switchable gene promoter system. Nat Biotechnol. 2002;20:1041–1044. doi: 10.1038/nbt734. [DOI] [PubMed] [Google Scholar]
- Silvius JR, Bhagatji P, Leventis R, Terrone D. K-ras4B and prenylated proteins lacking “second signals” associate dynamically with cellular membranes. Mol Biol Cell. 2006;17:192202. [Google Scholar]
- Sopko R, et al. Mapping pathways and phenotypes by systematic gene overexpression. Mol Cell. 2006;21:319–330. doi: 10.1016/j.molcel.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Su Y, Lagarias JC. Light-independent phytochrome signaling mediated by dominant GAF domain tyrosine mutants of Arabidopsis phytochromes in transgenic plants. Plant Cell. 2007;19:2124-39. doi: 10.1105/tpc.107.051516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surana U, Robitsch H, Price C, Schuster T, Fitch I, Futcher AB, Nasmyth K. The role of CDC28 and cyclins during mitosis in the budding yeast S. cerevisiae. Cell. 1991;65:145–161. doi: 10.1016/0092-8674(91)90416-v. [DOI] [PubMed] [Google Scholar]
- Timson DJ, Ross HC, Reece RJ. Gal3p and Gal1p interact with the transcriptional repressor Gal80p to form a complex of 1:1 stoichiometry. Biochem J. 2002;363:515–520. doi: 10.1042/0264-6021:3630515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toettcher JE, Gong D, Lim WA, Weiner OD. Light-based feedback for controlling intracellular signaling dynamics. Nat Methods. 2011a;8:837–839. doi: 10.1038/nmeth.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toettcher JE, Gong D, Lim WA, Weiner OD. Light control of plasma membrane recruitment using the Phy-PIF system. Methods Enzymol. 2011b;497:409-23. doi: 10.1016/B978-0-12-385075-1.00017-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toettcher JE, Voigt CA, Weiner OD, Lim WA. The promise of optogenetics in cell biology: interrogating molecular circuits in space and time. Nat Methods. 2011c;8:35–38. doi: 10.1038/nmeth.f.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach A, Brachat A, Pöhlmann R, Philippsen P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 1994;10:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- Zhou XX, Chung HK, Lam AJ, Lin MZ. Optical control of protein activity by fluorescent protein domains. Science. 2012;338:810–814. doi: 10.1126/science.1226854. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.