

Abstract
Chromenes, isochromenes, and benzoxathioles react with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone to form stable aromatic cations that react with a range of nucleophiles. These oxidative fragment coupling reactions provide rapid access to structurally diverse heterocycles. Conducting the reactions in the presence of a chiral Brønsted acid results in the formation of an asymmetric ion pair that can provide enantiomerically enriched products in a rare example of a stereoselective process resulting from the generation of a chiral electrophile through oxidative carbon–hydrogen bond cleavage.
Keywords: oxidation, fragment coupling, heterocycles, chiral counterions, enantioselectivity
1. Introduction
Efficient fragment-coupling reactions expedite the preparation of structurally diverse compound collections. Cross-coupling reactions have proven to be exceedingly valuable in this regard and are now an invaluable component in the synthesis of medicinal agents and subunits that can be employed in fragment-based screening assays.1 The latter protocol generally requires the preparation of libraries of low molecular weight compounds that are rich in heteroatoms. Further diversification would be accessible through the incorporation of stereocenters into these collections.2 Access to these libraries would be facilitated by the development of new coupling reactions that are broad in scope and allow for structural variability while generating one or more stereocenters.
Fragment-coupling through oxidative carbon–hydrogen bond cleavage is emerging as a useful approach for rapidly accessing diverse structures.3 These transformations proceed through the formation of stabilized carbocations via pathways that are initiated by single electron oxidation or hydrogen atom abstraction. Tetrahydroisoquinoline derivatives have served as the substrates for a significant number of these methods due to the facility by which the intermediate iminium ion can be accessed.4 Oxygen-containing heterocycles can also engage in oxidative fragment-coupling reactions,5 though the processes often require forcing conditions or the incorporation of electron-donating groups into the structure.
We have developed a number of cyclization reactions that proceed through oxidative carbon–hydrogen bond cleavage by employing 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) as the oxidant.6 DDQ is an easily handled reagent that tolerates a wide range of solvents and can be used as a catalyst in the presence of a range of terminal oxidants.7 Our mechanistic studies for DDQ-mediated oxocarbenium ion formation from allylic and benzylic ethers8 implicate a reaction pathway that proceeds through an electron transfer to form a radical cation intermediate followed by a hydrogen atom abstraction. The rate of oxocarbenium ion formation is thus dictated by the oxidation potential, which controls the radical cation concentration, and the stability of the resulting carbocation, which controls the rate of hydrogen atom abstraction. Substrates with low oxidation potentials that can generate stable carbocations react quite quickly under these conditions, but the presence of a rapid nucleophilic addition is required to drive the reaction forward since cation formation is reversible and usually thermodynamically disfavored.
Utilizing DDQ-mediated oxidative carbon–hydrogen bond cleavage as a prelude to bimolecular coupling reactions requires the intermediacy of a highly stabilized carbocation to balance the diminished rates of addition by an untethered nucleophile. This led us to explore aromaticity as a stabilizing factor for cations. Herein we describe the use of chromenes, isochromenes, and benzoxathioles as precursors to aromatic cations that can react with a range of nucleophiles.9 The high stability of the intermediates leads to the need for a catalyst to promote the coupling. We report that the use of chiral phosphoric acids as catalysts can promote asymmetric induction in the coupling step to provide enantiomerically enriched products. This constitutes a rare example of asymmetric, oxidative fragment coupling through a chiral electrophilic intermediate.
2. Results and Discussion
2H-Chromene (1, Scheme 1) served as our initial substrate for the development of oxidative bimolecular coupling reactions. This structure has a low oxidation potential due to the electron-rich aromatic ring, thereby assuring access to radical cation intermediate 2 in the presence of a mild oxidant like DDQ. Subsequent hydrogen atom abstraction provides aromatic cation 3 that should be suitable for nucleophilic additions. Similarly, 1H-isochromene (4) should also be a suitable substrate for the oxidative formation of cation 5.
Scheme 1.
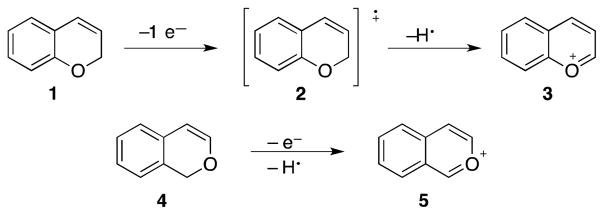
Aromatic cation formation through oxidative carbon–hydrogen bond cleavage.
The project commenced with the coupling of 1 with allyl trimethylsilane. The oxidation of 1 with DDQ proceeded rapidly (< 30 min) in a number of solvents of varying polarity, ranging from dichloromethane to nitromethane. Molecular sieves were added because the intermediate cation reacts quite rapidly with adventitious water to yield alcohol and ether products. Allyl trimethylsilane reacts with the intermediate cation, but the reaction proceeds more efficiently in the presence of LiClO4. This suggests that the intermediate that forms upon oxidation is not the oxocarbenium ion and is most likely mixed acetal 6. The addition of LiClO4 promotes the breakdown of this intermediate to form ion pair 7,10 which reacts with the nucleophile to yield 8. The addition proceeded when the nucleophile was added after the initial alkylation was complete rather than before the addition of DDQ. This “cation pool” approach11 to the coupling is useful because it allows for the addition of nucleophiles that might be oxidized in preference to the substrate. Optimal yields of 74% were observed when the reaction was conducted in CH3CN. Isochromene 4 underwent oxidation significantly more rapidly. Complete starting material disappearance was observed instantaneously at −30 °C. The intermediate electrophile, however, reacted inefficiently with allyl trimethylsilane in CH3CN. These results indicated that cation 5 is much more stable than cation 3. Therefore we conducted the reaction in CH2Cl2 to destabilize the cation and utilized the more nucleophilic12 allyl tributyltin in to form 9 in 79% yield. The rapid oxidation of 4 highlights the utility of proceeding through aromatic cation intermediates, since related dihydroisocoumarin structures require elevated temperatures and/or prolonged reaction times for complete reaction.5d
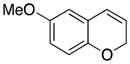
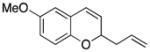
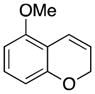
Considerable substitution was tolerated on the substrates, as shown in Table 1. The chromene derivatives were prepared through gold-mediated Friedel-Crafts cyclizations13 of aryl propargyl ethers or by ring closing metathesis reactions of allylic ethers of substituted o-hydroxystyrenes. Electron-rich chromenes 10 and 12 (entries 1 and 2) proceeded through the oxidation step smoothly but the intermediate cations were somewhat less reactive than the cation that was derived from 1. Electron poor chromene 14 (entry 3) required heating to 75 °C to effect oxidation but the intermediate cation reacted smoothly to provide 15 in 92% yield. Substitution on the pyran subunit is also tolerated. Methyl substituted chromene 16 (entry 4) reacted in a similar manner to 1 to yield 17. Silyl-substituted chromene 17 (entry 5) required 2 h to complete the oxidation step but reacted with the nucleophile smoothly to yield 19. Substituted isochromenes are not as readily accessible as substituted chromenes, but chloroisochromene 20 (entry 6) could be prepared through a known protocol.14 This compound required slightly higher temperatures than 4 for the oxidation step but the intermediate cation reacted smoothly with allyl tributyltin to yield 21.
Table 1.
Scope studies of chromenes and isochromenes.
| entry | substrate | nucleophile | product | yield (%) |
|---|---|---|---|---|
| 1 |
 10 |
|
 11 |
64 |
| 2 |
 12 |
|
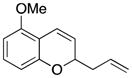 13 |
46 |
| 3 |
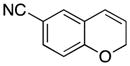 14 |
|
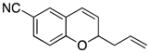 15 |
92 |
| 4 |
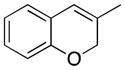 16 |
|
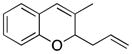 17 |
60 |
| 5 |
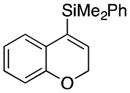 18 |
|
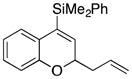 19 |
72 |
| 6 |
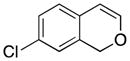 20 |
|
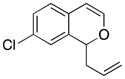 21 |
91 |
These reactions proved to be important in defining the roles of various structural features on the rates of oxidation and nucleophilic addition. Electron-donating groups facilitate carbocation formation but inhibit nucleophilic addition by stabilizing the intermediate cation. Electron-withdrawing groups slow cation formation but facilitate subsequent nucleophilic additions by destabilizing the intermediate carbocation. Forming cationic intermediates that contain electron-withdrawing groups again highlights the capacity of aromatic intermediates to facilitate oxidative carbon–hydrogen bond cleavage. The significant steric bulk of the phenyl dimethylsilyl group slows the oxidation by inhibiting the approach of DDQ,15 but the cation reacts in an expected manner. The silyl and chloro groups provide an additional handle for functionalizing these structures following the oxidative coupling reactions.
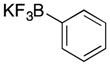
Various nucleophiles can also be used in these processes. A summary of representative transformations is shown in Table 2. (E)-Substituted allylsilane 22 (entry 1) adds with excellent diastereocontrol to form branched product 23. Enolsilane 24 (entry 2) adds efficiently, though with negligible stereocontrol, to form ketone 25. Potassium vinyltrifluoroborate 26 (entry 3) and alkynyl trifluoroborate 28 (entry 4) add to form alkenyl- and alkynyl-substituted chromenes 27 and 29, respectively. Potassium phenyltrifluoroborate (30) does not add to the parent cation (entry 5), but adds at 70 °C to the electron deficient cation derived from 14 in moderate yield (entry 6).16,17
Table 2.
Nucleophile scope.
| entry | substrate | nucleophile | product | yield (%) |
|---|---|---|---|---|
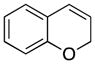
| 1 |
 1 |
22 |
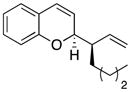 23 |
63 dr = 1:0 |
| 2 |
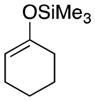
 1 |
 24 |
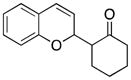 25 |
71 dr = 1.6:1 |
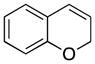
| 3 |
 1 |
26 |
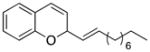 27 |
65 |
| 4 |
 1 |
28 |
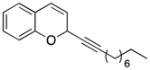 29 |
66 |
| 5 |
 1 |
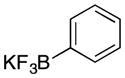 30 |
– | – |
| 6 |
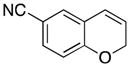 14 |
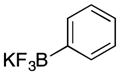 30 |
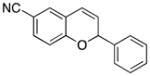 31 |
46 |
The successful completion of this phase of the project led us to consider other aromatic cation intermediates that could be accessed through carbon–hydrogen bond cleavage. Cationic aromatic five-membered rings can, in principle, be prepared from precursors that contain two heteroatoms (Scheme 3). Nucleophilic addition into these intermediates would provide heteroatom-rich products that are difficult to access through conventional protocols. For example direct condensation reactions between o-disubstituted benzene derivatives and aldehydes often fails because of the kinetically disfavored cyclization step,18 and Kwon’s elegant phosphine-mediated protocol19 requires the use of electron-deficient allenes.
Scheme 3.
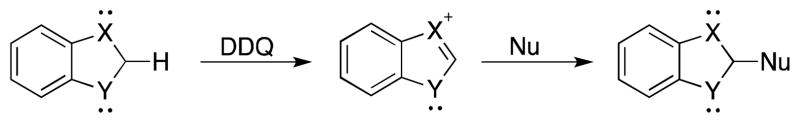
Substitution via aromatic 5-membered ring cations.
This investigation was initiated (Scheme 4) through the oxidation of benzo[d][1,3]oxathiole (32) followed by quenching with allyl tributyltin to yield 33. Extensive parameter screening revealed that non-polar solvents provided the best yields for these reactions, with 1,2-dichloroethane being optimal. The addition of LiClO4 significantly facilitated the addition and was most effective when added in excess (1.5 equiv). A 90% yield of 33 was obtained under these conditions. Benzo[d][1,3]dioxole (34) was inert toward these reaction conditions despite having the capacity to form an aromatic cation. This suggests that the intermediate radical cation intermediate is localized on the aromatic ring in the benzodioxole structure and delocalizes onto the sulfur in the benzoxathiole structure. Localizing spin density onto sulfur weakens the adjacent carbon–hydrogen bond and allows for hydrogen abstraction.
Scheme 4.

Allylation of benzoxathiole.
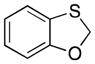
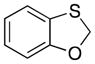
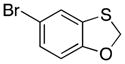
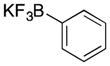
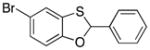
Many of the nucleophiles that successfully add to the chromene-derived cations add to the benzoxathiole-derived cations, as shown in Table 3. Allyl trimethylsilane adds with only slightly lower efficiency that allyl tributyltin (entry 1). Modest diastereocontrol is observed through the addition of substituted allylic silanes (entries 2 and 3), indicating that these reagents can marginally distinguish between oxygen and sulfur as they approach the electrophile. Alkynyl and vinyl trifluoroborates add smoothly (entries 4 and 5), but aromatic trifluoroborates are unreactive toward the parent compound (entry 6). Bromination of 32 provided 38. The incorporation of bromine in the substrate inhibited the oxidation step at room temperature. The cation could be accessed through heating to 50 °C, and the resulting species was considerably more reactive than the parent system. An improved yield was observed with allyl trimethylsilane and potassium phenyl trifluoroborate added to form 39 in 30% yield (entry 7).
Table 3.
Nucleophile scope in benzoxathiole coupling.
| entry | substrate | nucleophile | product | yield (%) |
|---|---|---|---|---|
| 1 |
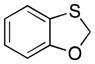 32 |
|
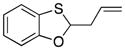 33 |
79 |
| 2a |
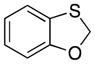 32 |
23 |
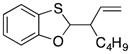 35 |
72 dr = 3.2:1 |
| 3a |
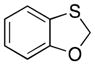 32 |
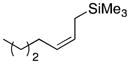 23-(cis) |
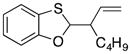 35′ |
66 dr = 1:4.6 |
| 4 |
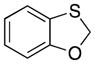 32 |
26 |
36 |
55 |
| 5 |
 32 |
 28 |
37 |
65 |
| 6 |
 32 |
 30 |
– | – |
| 7 |
 38 |
 30 |
 39 |
30 |
35 and 35′ are diastereomers. The relative stereochemical orientations have not been assigned.
The products of these coupling reactions contain a hydrogen that could also undergo oxidative cleavage, thus allowing for additional bond formation (Scheme 5). This was confirmed in the chromene series by the oxidation of 40 to form spirocycle 41. Spirocyclization was also observed through the oxidation of 42 to yield 43, which is remarkably stable despite being at the ortho ester oxidation state. The synthesis of 43 from benzoxathiole 32 and allylsilane 44 could also be achieved directly by a double oxidation sequence. This transformation represents a unique approach to spiroannulation via consecutive carbon–hydrogen bond cleavage reactions.
Scheme 5.
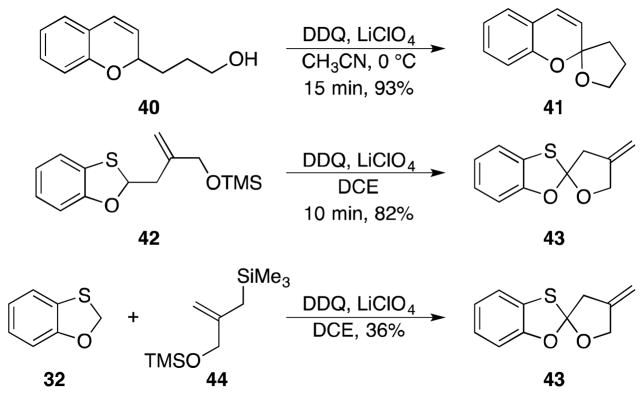
Oxidative spirocycle formation.
Asymmetric additions into oxidatively generated carbocations are quite rare, and the few examples that exist in the literature employ chiral nucleophiles to achieve enantioselectivity.5f,20 We postulated that stereoselective additions into oxocarbenium ions could be accomplished through the oxidative formation of chiral ion pairs. This could be achieved (Scheme 6) by generating acetal intermediate 6 and promoting an ion exchange with a chiral acid rather than LiClO4. The resulting chiral ion pair has the potential to engage in stereoselective nucleophilic addition reactions. Asymmetric additions through oxocarbenium ion intermediates have been promoted by chiral phosphoric acids21 and by chiral thioureas22 despite the absence of conventional hydrogen bonding sites. The association between the oxocarbenium ion and phosphoric acids has been attributed to C–H--X hydrogen bonds.23 Thus we envisioned a transformation in which a chiral phosphoric acid is added to 6 to generate ion pair 45 followed by a nucleophilic addition reaction to yield enantiomerically enriched products.
Scheme 6.
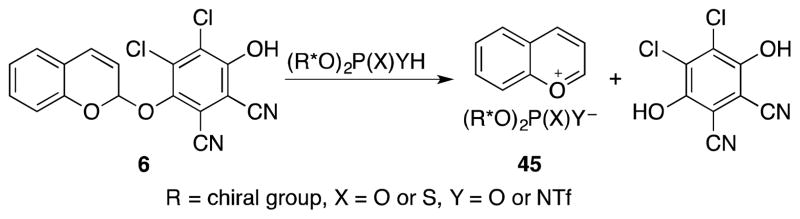
Chiral ion pair from an oxidatively generated intermediate.
The first step in this process was the identification of reaction conditions in which the non-stereoselective uncatalyzed nucleophilic addition reaction is suppressed. This was achieved by forming 6 from the reaction of 1 with DDQ, then monitoring its disappearance in the presence of allyl trimethylsilane but in the absence of LiClO4. Complete consumption of the intermediate was observed with 1.5 h in CH3CN and 2 h in CH2Cl2 at 0 °C. The reaction in toluene was considerably slower, however, requiring 12 h for consumption of the intermediate. This indicated that an uncatalyzed background reaction leading to a racemic product would be suppressed in aromatic solvents.
These reactions were subsequently run in the presence binaphthol-based phosphoric and thiophosphoric acids 46–51. The reactions were conducted by exposing chromene 1 to DDQ and the acid in toluene at 0 °C. The nucleophile was added upon consumption of the starting material (approximately 30 min). The enantiomeric ratios were determined by HPLC using a Lux Cellulose 3 column. These studies showed that triisopropylphenyl-substituted phosphoramide 4924 was the optimal catalyst for this transformation (Table 4), providing (R)-8 in 62% ee. The absolute stereochemistry was determined through independent synthesis (see below) and comparing optical rotations and HPLC retention times with the chiral stationary phase. The phosphoryl triflimide group proved to be superior to a phosphoric acid group, and thiophosphoric acids were inferior to phosphoric acids. Conducting the reaction with 49 in trifluorotoluene provided a further improvement in stereocontrol to yield (R)-8 in 77% ee.
Table 4.
Screening with one equivalent of catalyst.
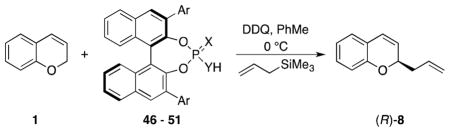
| |||||
|---|---|---|---|---|---|
| entry | catalyst | Ar | X | Y | ee (%) |
| 1 | 46 | 2,4,6-triisopropylphenyl | O | O | 26 |
| 2 | 47 | 2,4,6-triisopropylphenyl | S | O | 4 |
| 3 | 48 | 3,5-ditrifluoromethylphenyl | O | O | 4 |
| 4 | 49 | 2,4,6-triisopropylphenyl | O | NTf | 62 |
| 5 | 50 | 2,4,6-triisopropylphenyl | S | NTf | 13 |
| 6 | 51 | 2,6-diisopropyl-4-(9-anthracenyl)phenyl | O | NTf | 31 |
Additional allylation agents were screened for selectivity (Table 5). These reactions were conducted in PhCF3 with 1 equivalent of catalyst 49 at 0 °C. Allyl phenyl dimethylsilane, a less reactive nucleophile25 than allyl trimethylsilane, showed improved selectivity, providing (R)-8 in 92% ee. Allyl tributyltin proved to be less selective (48% ee), but allyl triphenyltin provided (R)-8 in 89% ee. These selectivities cannot be attributed solely to differences in nucleophilicity, since allyl triphenyltin is more nucleophilic than allyl trimethylsilane,25 indicating that electrofuge sterics or aromatic interactions could also play a role in asymmetric induction.
Table 5.
Influence of the nucleophile.

| ||
|---|---|---|
| entry | M | ee (%) |
| 1 | SiMe2Ph | 92 |
| 2 | SnBu3 | 48 |
| 3 | SnPh3 | 89 |
Allyl triphenyltin and allyl phenyl dimethylsilane were selected for subsequent studies directed toward determining the impact of lowering the catalyst loading (Table 6). Reducing the catalyst loading to 20 mol% caused a drop in selectivity, with both nucleophiles showing an ee of 62%. Lowering the temperature to −25 °C, approaching the freezing point of PhCF3, resulted in an increase in selectivity with allyl triphenyltin adding in 81% ee and 66% yield, and allyl phenyl dimethylsilane adding in 86% ee and 56% yield. Reducing the catalyst loading to 10 mol% led to the isolation of (R)-8 in 68% ee and 66% yield from allyl triphenyltin, and 79% ee and 52% yield from allyl phenyl dimethylsilane. At 5% loading the selectivity drops further for allyl triphenyltin, resulting in a 31% ee. These reactions show that the background reaction can become competitive at lower catalyst loading but useful enantioselectivities with substoichiometric quantities of the acid. To the best of our knowledge this is the first report of useful enantioselectivities through the intermediacy of chiral in pairs that originate from an oxidative carbon–hydrogen bond cleavage reaction.
Table 6.
Effects of lowering the catalyst loading and temperature.
| |||||
|---|---|---|---|---|---|
| entry | M | temperature | catalyst loading (mol%) | ee (%) | yield (%) |
| 1 | Ph3Sn | 0 °C | 20 | 62 | – |
| 2 | Ph3Sn | −25 °C | 20 | 81 | 66 |
| 3 | Ph3Sn | −25 °C | 10 | 68 | 66 |
| 4 | Ph3Sn | −25 °C | 5 | 31 | 57 |
| 5 | PhMe2Si | 0 °C | 20 | 62 | – |
| 6 | PhMe2Si | −25 °C | 20 | 86 | 56 |
| 7 | PhMe2Si | −25 °C | 10 | 79 | 52 |
| 8 | PhMe2Si | −25 °C | 5 | 17 | 53 |
Compound 8 has not previously been prepared in enantiomerically pure form. Therefore assigning the absolute stereochemistry required an independent synthesis through a vetted sequence. This was achieved as shown in Scheme 7. o-Bromohydrocinnamic acid (52) was converted to homoallylic alcohol 53 through a three step sequence that utilized a Brown allylation reaction26 to dictate a predictable stereochemical outcome. A palladium-mediated cyclization27 followed by alkene hydrogenation provided 54 (observed [α]D = +99.1).
Scheme 7.
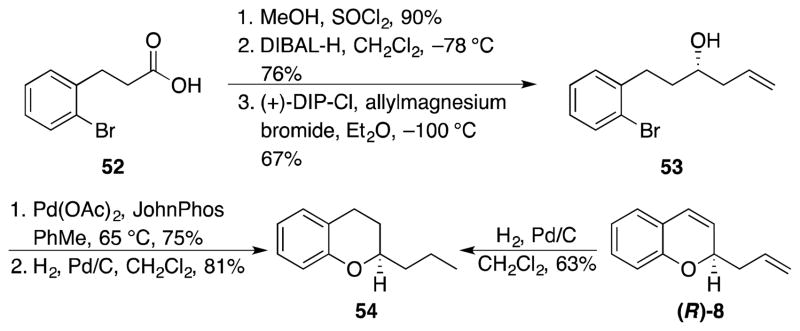
Absolute stereochemistry determination.
Attempts to expand the scope of the process met with little success (Scheme 8). Replacing allyl phenyl dimethylsilane with methallyl phenyl dimethylsilane, for example, under our optimal conditions resulted in the isolation of 55 in 33% ee. The absolute stereochemistry that is designated in Scheme 7 was based on analogy and not through independent synthesis. Conducting the reaction with methoxy chromene 10 provided 11 in 6% ee. Reactions conducted in the absence of 48, however, showed that the background reactions for both processes are quite fast. We postulate that the enhanced nucleophilicity of the methallyl nucleophile in comparison to the allyl nucleophile creates the potential for reaction with intermediates related to 6 (Scheme 2). The electron donation from the methoxy group stabilizes the intermediate cation and weakens its interaction with the counterion.
Scheme 8.
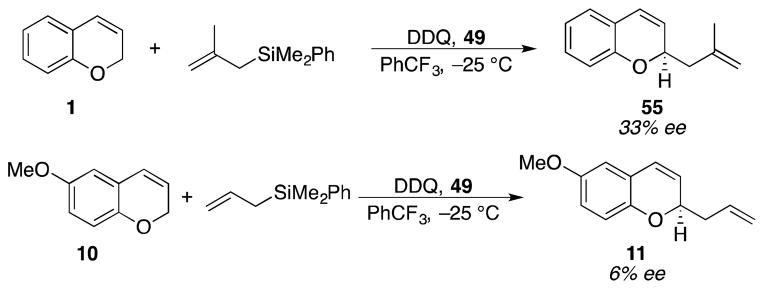
Attempts to expand the reaction scope.
Scheme 2.

Oxidative allylation reactions.
3. Conclusions
We have demonstrated that DDQ-mediated carbon–hydrogen bond cleavage reactions are useful for preparing aromatic carbocations that react with a number of nucleophile classes to generate a range of heterocyclic products. The parent chromene and isochromene structures oxidize rapidly to form the intermediate cations, and electron-deficient analogs react under slightly more forcing conditions, thereby demonstrating that the generation of aromatic intermediates significantly facilitates oxidative carbon–hydrogen bond cleavage. Several nucleophilic classes react with the cations, including allyl silanes, allylstannanes, potassium vinyl and alkynyl trifluoroborates, and enolsilanes. Cations that are derived from electron-deficient substrates are highly reactive toward nucleophiles and can react with potassium aryl trifluoroborates.
This protocol also works for the preparation of 5-membered aromatic cations that contain two heteroatoms. This process requires one of the heteroatoms to harbor sufficient spin density to weaken the adjacent carbon–hydrogen bond sufficiently for cleavage. Sulfur is particularly effective at promoting oxidation, and a number of benzoxathiole derivatives can be accessed.
Mechanistic studies implicate the formation of a mixed acetal intermediate upon cation formation that ionizes in the presence of acid to generate the reactive intermediate. Chiral Brønsted acids can be used to generate ion pairs that react with nucleophiles to yield enantiomerically enriched products. While the scope of this process is still quite limited several important observations have been noted. The background reaction to form racemic products is a significant competitive process. This reaction can be slowed by conducting the reactions in a non-polar aromatic solvent (PhCF3 is optimal) at low temperature. Selectivity is higher for weaker nucleophiles, indicating that stronger nucleophiles can react with the initially formed mixed acetal intermediate. Incorporating electron-donating groups into the substrate reduces selectivity through weakening the attraction in the ion pair. Despite these limitations we were able to conduct reactions with good enantiocontrol at reasonable catalyst loadings. These processes are, to the best of our knowledge, the first examples of enantioselective reactions with chiral electrophiles that arise from oxidative carbon–hydrogen bond cleavage.
4. Experimental section
4.1. General information
Proton (1H NMR) and carbon (13C NMR) nuclear magnetic resonance spectra were recorded at 300, 400, or 500 MHz and 75, 100, or 125 MHz, respectively. The chemical shifts are given in parts per million (ppm) on the delta (δ) scale. Tetramethylsilane (TMS) or the solvent peak was used as a reference value, for 1H NMR: TMS (in CDCl3) = 0.00 ppm, for 13C NMR: TMS (in CDCl3) = 0.00. Data are reported as follows: (s = singlet; d = doublet; t = triplet; q = quartet; dd = doublet of doublets; dt = doublet of triplets; br = broad). Samples for IR were prepared as a thin film on a NaCl plate by dissolving the compound in CH2Cl2 and then evaporating the CH2Cl2. Analytical TLC was performed on pre-coated (25 mm) silica gel 60F-254 plates. Visualization was done under UV (254 nm). Flash chromatography was done using 32–63 60 Å silica gel. Methylene chloride was distilled under N2 from CaH2. Reagent grade ethyl acetate, diethyl ether, pentane and hexanes (commercial mixture) were used as purchased for chromatography. Benzene was dried with 4Å molecular sieves. THF was distilled from sodium. Other reagents were obtained from commercial sources without further purification. All reactions were performed in oven or flame-dried glassware with magnetic stirring unless otherwise noted. Spectroscopic data and procedures for the preparation of compounds 8, 9, 11, 13, 15, 17, 19, 21, 23, 25, 27, 29, 31, and 41 have been reported elsewhere.9
4.2. Benzo[d][1,3]oxathiole (32)
Dibromomethane (5.01 mL, 59.4 mmol) and Adogen 464 (1.21 g, 2.77 mmol) were added to a flask of refluxing H2O (15 mL). A solution of 2-mercaptophenol (5.0 g, 40 mmol) and NaOH (3.96 g, 99.1 mmol) in H2O (18 mL) was added over 4.5 h. After the addition was complete the reaction mixture was stirred while refluxing for an additional 5 h. The solution cooled to room temperature, was poured into H2O, and was extracted with EtOAc (5 × 25 mL). The organic layer was dried over MgSO4, concentrated under reduced pressure, and purified via flash cromatography (SiO2, 4:1 pentane:Et2O) to yield 32 as a clear oil (2.91 g, 53% yield): 1H NMR (300 MHz, CDCl3) δ 7.18 (dd, 1H, J = 10.0, 1.6 Hz), 7.00 (td, 1H, J = 10.0, 1.6 Hz), 6.88 (td, 1H, J = 10.0, 1.6 Hz), 6.83 (dd, 1H, J = 10.0, 1.2 Hz), 5.68, (s, 1H); 13C NMR (125 MHz, CDCl3) δ 156.4, 126.2, 126.0, 122.7, 122.6, 110.6, 75.3; IR (neat) 3066, 2925, 2871, 1468, 1450, 1326, 1208, 744 cm−1; HRMS (ESI): m/z calcd for C7H6O7S [M]+ 138.0139, found 138.0136.
4.3. General procedure for oxidative coupling
To a solution of benzoxathiole (1.0 equiv) in DCE (0.1 M) were added LiClO4 (1.5 equiv) and DDQ (1.2 equiv). The reaction mixture was stirred until complete starting material consumption was indicated by TLC analysis. The nucleophile (1.2 equiv) was added and the reaction is stirred until the consumption of the intermediate is complete (monitored via TLC). The mixture was concentrated and purified directly via flash chromatography.
4.4. 2-Allylbenzo[d][1,3]oxathiole (33)
The general procedure for oxidative coupling was followed using 32 (200 mg, 1.45 mmol), LiClO4 (231 mg, 2.17 mmol), 4 Å MS (200 mg), DDQ (395 mg, 1.74 mmol), and DCE (14 mL). After 30 min the oxidation was complete and allyl tributyltin (0.67 mL, 2.17 mmol) was added. The reaction was stirred for 1 h, then was purified directly via flash chromatography (SiO2, pentane) to give 33 as a clear oil (232 mg, 90% yield): δ 1H NMR (400 MHz, CDCl3) 7.12 (dd, 1H, J = 7.6, 1.2 Hz), 6.99 (td, 1H, J = 7.6, 1.2 Hz), 6.87 (td, 1H, J = 7.6, 0.8 Hz), 6.80 (dd, 1H, J = 8.0, 0.8 Hz), 6.09 (t, 1H, J = 6.4 Hz), 5.86 (ddt, 1H, J = 24.0, 10.0, 6.8 Hz), 5.23 (dd, 1H, J = 17.2, 1.2 Hz), 5.19 (dd, 1H, J = 10.0, 1.2 Hz), 2.85–2.92 (m, 1H), 2.70–2.77 (m, 1H); 13C NMR δ (100 MHz, CDCl3) 155.9, 132.1, 126.0, 125.8, 122.5, 122.3, 119.4, 110.5, 89.7, 41.7; IR (neat) 3074, 2978, 2973, 2903, 1463, 1213, 745 cm−1; HRMS (ESI): m/z calcd for C10H10OS [M+H]+ 179.0486, found 179.0549.
4.5. 2-(Hept-1-en-3-yl)benzo[d][1,3]oxathiole (35)
The general procedure for oxidative coupling was followed using benzoxathiole 32 (100 mg, 0.723 mmol), LiClO4 (116 mg, 1.09 mmol), 4 Å MS (103 mg), DDQ (196 mg, 0.868 mmol), and DCE (7 mL). The reaction mixture was stirred for 30 min, then (E)-hept-2-en-1-yltrimethylsilane (148 mg, 0.868 mmol) was added. The solution was stirred for 1 h, then was purified directly via flash chromatography (SiO2, hexanes) to give 35 (3.1:1 dr) as a clear oil (122 mg, 72% yield). 1H NMR (400 MHz, CDCl3) δ 7.10 (dd, 1H, J = 7.6, 1.2 Hz), 6.97 (td, 1H, J = 7.6, 1.2 Hz), 6.84 (td, 1H, J = 7.6, 1.2 Hz), 6.77 (dd, 1H, J = 8.0, 0.8 Hz), 6.02 (d, 0.76H, J = 5.6 Hz), 5.99 (d, 0.26H, J = 6.8Hz), 5.61–5.71 (m, 1H), 5.14–5.23 (m, 2H), 2.54–2.64 (m, 1H), 1.58–1.68 (m, 1H), 1.30–145 (m, 5H), 0.89 (t, 3H, J = 6.9 Hz); 13C NMR (100 MHz, CDCl3) δ 156.1, 136.9, 125.6, 122.1, 121.9, 121.8, 118.4, 110.0, 93.4, 50.3, 29.6, 29.1, 22.6, 14.0; IR (neat) 3073, 2956, 2929, 2859, 1577, 1464, 1209, 1119, 920, 743 cm−1; HRMS (ESI): m/z calcd for C14H19OS [M+H]+ 235.1078, found 235.1150.
4.6. 2-(Hept-1-en-3-yl)benzo[d][1,3]oxathiole (35′)
The general procedure for oxidative coupling was followed using benzoxathiole 32 (100 mg, 0.723 mmol), LiClO4 (116 mg, 1.09 mmol), 4 Å MS (103 mg), DDQ (196 mg, 0.868 mmol), and DCE (7 mL). The reaction mixture was stirred for 30 min, then (Z)-hept-2-en-1-yltrimethylsilane (149 mg, 0.868 mmol) was added. The solution was stirred for 1 h and then purified directly via flash chromatography (SiO2, hexanes) to give 35′ (1:4.6 dr) as a clear oil (112 mg, 66% yield): 1H NMR (400 MHz, CDCl3) δ 7.10 (d, 1H, J = 7.6 Hz), 6.96 (t, 1H, J = 7.6 Hz), 6.84 (t, 1H, J = 7.6 Hz), 6.76 (d, 1H, J = 8.0 Hz), 6.01 (d, 0.18H, J = 6.8 Hz), 5.99 (d, 0.82H, J = 6.8 Hz), 5.62–5.72 (m, 1H), 5.14–5.24 (m, 2H), 2.60–2.72 (m, 1H), 1.24–1.43 (m, 6H), 0.90 (t, 3H, J = 6.4 Hz); 13C NMR (100 MHz, CDCl3) δ 156.3, 136.6, 125.8, 125.6, 122.0, 121.8, 118.8, 110.0, 93.5, 50.5, 30.2, 29.1, 22.6, 14.0; IR (neat) 3073, 2956, 2929, 2859, 1762, 1577, 1464, 1209, 1119, 920, 743 cm−1; HRMS (ESI): m/z calcd for C14H19OS [M+H]+ 235.1078, found 235.1154.
4.7. (E)-2-(Dec-1-en-1-yl)benzo[d][1,3]oxathiole (36)
The general procedure for oxidative coupling was followed using benzoxathiole 32 (100 mg, 0.723 mmol), LiClO4 (115 mg, 1.09 mmol), 4 Å MS (100 mg), DDQ (197 mg, 0.868 mmol), and DCE (7mL). The reaction mixture was stirred for 30 min, then potassium (E)-dec-1-enyltrifluoroborate (214 mg, 0.868 mmol) was added. The solution was stirred for 1 h and then purified directly via flash chromatography (SiO2, hexanes) to give 36 as a clear oil (110 mg, 55% yield). 1H NMR (400 MHz, CDCl3) δ 7.11 (dd, 1H, J = 7.6, 1.2 Hz), 6.98 (td, 1H, J = 7.6, 1.2 Hz), 6.86 (td, 1H, J = 7.6, 1.2 Hz), 6.80 (dd, 1H, J = 8.0, 0.8 Hz), 6.41 (d, 1H, J = 7.2 Hz), 5.81–5.96 (m, 2H), 2.09 (q, 2H, 6.0 Hz), 1.37–1.44 (m, 2H), 1.23–1.32 (m, 10H), 0.88 (t, 3H, J = 7.2 Hz); 13C NMR (100 MHz, CDCl3) δ 155.6, 136.6, 126.6, 126.2, 125.8, 122.3, 122.0, 110.2, 90.0, 31.9, 31.8, 29.4, 29.3, 29.2, 28.6, 22.7, 14.1; IR (neat) 3068, 2925, 2854, 1462, 1233, 1119,1150, 1020, 960, 743 cm−1; HRMS (ESI): m/z calcd for C17H25OS [M+H]+ 277.1548, found 277.1602.
4.8. 2-(dec-1-yn-1-yl)benzo[d][1,3]oxathiole (37)
The general procedure for oxidative coupling was followed using 32 (200 mg, 1.45 mmol), LiClO4 (231 mg, 2.17 mmol), 4 Å MS (200 mg), DDQ (395 mg, 1.74 mmol), and DCE (14 mL). The reaction mixture was stirred for 30 min, then the potassium alkynyl trifluoroborate (424 mg, 1.74 mmol) was added. The solution was stirred for 1 h and then was purified directly via flash chromatography (SiO2, pentane) to give 37 as a clear oil (258 mg, 65% yield). 1H NMR (400 MHz, CDCl3) δ 7.13 (d, 1H, J = 7.6 Hz), 7.02 (t, 1H, J = 7.6Hz), 6.90 (t, 1H, J = 7.6 Hz), 6.84 (d, 1H, J = 8.0 Hz), 6.54 (s, 1H), 2.27 (t, 2H, J = 7.2 Hz), 1.53 (t, 2H, J = 7.6), 1.36 (m, 2H), 1.26 (s, 10H); 13C NMR (100 MHz, CDCl3) δ 154.8, 126.3, 125.7, 122.9, 122.2, 111.0, 90.9, 76.6, 76.0, 32.0, 29.3, 29.2, 29.0, 28.3, 22.8, 19.1, 14.3; IR (neat) 3069, 2926, 2855, 2239, 1462, 1247, 962, 743 cm−1; HRMS (ESI): m/z calcd for C17H22OS [M]+ 274.1391, found 274.1374.
4.9. 5-Bromo-2-phenylbenzo[d][1,3]oxathiole (39)
The general procedure for oxidative coupling was followed using benzoxathiole 38 (100 mg, 0.461 mmol), LiClO4 (74 mg, 0.69 mmol), 4 Å MS (103 mg), DDQ (126 mg, 0.553 mmol), and DCE (5 mL). The reaction mixture was stirred for 1 h at 50 °C, then was cooled to rt and potassium trifluorophenyl borate (102 mg, 0.553 mmol) was added. The reaction was stirred for 20 min and then was purified directly via flash chromatography (SiO2, hexanes) to give 39 as a clear oil (41 mg, 30% yield): 1H NMR (400 MHz, CDCl3) δ 7.79 (s, 2H), 7.63 (s, 2H), 7.48 (d, 2H, J = 7.2 Hz), 7.33 (dd, 1H, J = 7.2, 1.2 Hz), 6.94 (m, 1H), 5.93 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 155.6, 154.9, 137.8, 129.7, 128.9, 128.8, 126.6, 124.8, 235.5, 225.4, 222.6, 91.2, 75.8; IR (neat) 3084, 2923, 2872, 1744, 1454, 1327, 1248, 1206, 1071, 999, 861, 740 cm−1; HRMS (ESI): m/z calcd for C13H9BrOS [M]+ 291.9557, found 291.9540.
4.10. 4′-Methylene-4′,5′-dihydro-3′H-spiro[benzo[d][1,3]oxathiole-2,2′-furan] (43)
To a solution of 42 (150 mg, 0.535 mmol) in DCE (5 mL) was added DDQ (292 mg, 1.28 mmol). The reaction mixture was stirred for 10 min and then was purified directly by flash chromatography (10% EtOAc in hexanes) to yield 43 as a colorless oil (91 mg, 82% yield): 1H NMR (400 MHz, CDCl3) δ 7.20 (d, 1H, J = 7.6 Hz), 7.05 (t, 1H, J = 7.6 Hz), 6.94 (t, 1H, J = 7.6 Hz), 6.90 (d, 1H, J = 8.0 Hz), 5.17 (t, 1H, J = 2.4 Hz), 5.07 (t, 1H, J = 2.4 Hz), 4.59–4.71 (m, 2H), 3.21–3.36 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 153.1, 142.7, 125.7, 124.7, 124.4, 122.5, 121.7, 110.6, 106.7, 72.1, 44.5; IR (neat) 3070, 2930, 2873, 1577, 1463, 1424, 1277, 1240, 1170, 1019, 960, 874, 745 cm−1; HRMS (ESI): m/z calcd for C11H11O2S [M+H]+ 207.0402, found 207.0472.
4.11. 4′-methylene-4′,5′-dihydro-3′H-spiro[benzo[d][1,3]oxathiole-2,2′-furan] (43)
The general procedure for oxidative coupling was followed using benzoxathiole 32 (100 mg, 0.723 mmol), LiClO4 (115 mg, 1.09 mmol), 4 Å MS (100 mg), DDQ (197 mg, 0.868 mmol), and DCE (7mL). The reaction mixture was stirred for 30 min, then nucleophile 4428 (188 mg, 0.868 mmol) was added. The solution was stirred for 1 h and then DDQ (199 mg, 0.876 mmol) was added. The reaction mixture was stirred for 20 min followed by a third addition of DDQ (101 mg, 0.438 mmol). The resulting solution was purified directly via flash chromatography (SiO2, 10% EtOAc in hexanes) to give 43 as a clear oil (55 mg, 36% yield). All data matched the product from the previously described stepwise protocol.
4.12. General protocol for asymmetric catalyst screening
To a solution of 1 (1.0 equiv) in a corresponding solvent (0.1 M concentration) at 0 °C were added powdered 4Å molecular sieves (60 mg/mL) and the corresponding catalyst (0.05 to 1 equiv). DDQ (1.4 equiv) was added to the reaction mixture, and it was stirred until TLC analysis showed complete consumption of the substrate. After cooling to the corresponding specified temperature, the nucleophile (2~3 equiv) was added, and the reaction was stirred until TLC analysis showed complete consumption of the oxidized intermediate. The reaction was quenched with 10% aqueous NaHCO3 solution, extracted with diethyl ether, and carefully concentrated under reduced pressure. Flash column chromatography (3% Et2O in pentane) afforded the desired product. An aliquot was taken and analyzed by chiral HPLC for enantiomeric excess.
4.13. Optimized catalytic protocol
The reaction was performed at −25 °C for 3 h, using 1 (41.5 mg, 0.32 mmol, 1.0 equiv), 29 (55.7 mg, 0.063 mmol, 20% equiv), 4Å molecular sieves powder (200 mg), DDQ (100 mg, 0.44 mmol, 1.4 equiv), allylphenyldimethylsilane (169 mg, 0.96 mmol, 3 equiv) and anhydrous á,á,á-trifluorotoluene (3 mL). Normal workup and flash column chromatography (3% Et2O in pentane) afforded the desired product (30 mg, 56%). The enantiomeric excess was determined to be 86% by chiral HPLC (1% i-PrOH:hexanes, 1.0 mL/min). Characterization data were in agreement with previously reported values.
4.14. Synthesis of 53
To a solution of 52 (2.29 g, 10.0 mmol) in methanol (20 mL) stirred at 0 °C, was added thionyl chloride (1.47 mL, 20.0 mmol) dropwise. The mixture was stirred overnight at rt. The reaction was carefully quenched with saturated aqueous NaHCO3 solution (10 mL), neutralized with powder NaHCO3, and extracted with diethyl ether (3 × 20 mL). The organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure. Flash column chromatography (10% EtOAc in hexane afforded the desired ester (2.19 g, 90%). 1H NMR (400 MHz, CDCl3) δ 7.51 (d, J = 8.0 Hz, 1H), 7.20–7.26 (m, 2H), 7.06 (dd, J = 7.2 Hz, 1H), 3.67 (s, 3H), 3.07 (t, J = 7.8 Hz, 2H), 2.65 (t, J = 7.8 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 172.7, 139.5, 132.7, 130.2, 127.9, 124.1, 51.4, 33.7, 31.2.
To the solution of the ester (1.22 g, 5.0 mmol) in dichloromethane (20 mL) at −78 °C, 1M DIBAL-H solution in hexanes (6 mL, 6.0 mmol) was added dropwise. The reaction was stirred at −78 °C for 1 hour, quenched with methanol (2 mL), and concentrated under reduced pressure. Flash column chromatography with 10% ethyl acetate in hexanes afforded the desired aldehyde (0.81 g, 76%). 1H NMR (300 MHz, CDCl3) δ 9.85 (d, J = 0.9 Hz, 1H), 7.55 (d, J = 8.1 Hz, 1H), 7.24–7.28 (m, 2H), 7.07–7.13 (m, 1H), 3.09 (t, J = 7.5 Hz, 2H), 2.82 (t, J = 7.5 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 200.9, 139.6, 132.9, 130.4, 128.0, 127.6, 124.2, 43.6, 28.6.
To a solution of (+)-IPC-Cl (1.80 g, 5.6 mmol, 1.87 equiv) in diethyl ether (3 mL) stirred at 0 °C, freshly prepared 0.5 M allylmagnesium bromide solution in diethyl ether (10.5 mmol, 5.25 mmol, 1.75 equiv) was added dropwise over 20 min. The reaction was stirred at 0 °C for an additional 30 min, decanted, extracted two times with diethyl ether (5 mL), and concentrated under reduced pressure. The residual oil was dissolved in dry pentane (10 mL), filtered through Millex syringe filter, and added dropwise into a solution of the above aldehyde (640 mg, 3.0 mmol, 1.0 equiv) in diethyl ether (5 mL) at −100 °C over 45 min. The addition was in such a pattern that the allylmagnesium bromide solution touched the inner wall of the reaction flask before entering the solution. After one additional hour at −100 °C, the reaction was quenched with methanol (2 mL) and concentrated under reduced pressure. The residual oil was dissolved in THF (3 mL), cooled to 0 °C, and treated with saturated aqueous NaHCO3 solution (7.0 mL) and 30% H2O2 (5.6 mL). After 3 hours at room temperature, the reaction mixture was extracted three time with diethyl ether (10 mL), dried over Na2SO4 and concentrated under reduced pressure. Flash column chromatography with 15% ethyl acetate in hexanes afforded the chiral alcohol (510 mg, 67%). 1H NMR (400 MHz, CDCl3) δ 7.54 (d, J = 8.1 Hz, 1H), 7.11–7.17 (m, 2H), 6.93–6.99 (m, 1H), 5.58–5.82 (m, 1H), 5.07 (d, J = 15.6 Hz, 1H), 5.06 (d, J = 11.7 Hz, 1H), 3.58–3.66 (m, 1H), 2.80–2.90 (m, 1H), 2.67–2.77 (m, 1H), 2.22–2.30 (m, 1H), 2.07–2.17 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 141.3, 134.5, 132.8, 130.4, 127.6, 127.4, 124.4, 118.3, 70.0, 41.9, 36.8, 32.4; IR (film) 3371, 3072, 2927, 1640, 1567, 1471, 1439, 1045, 1022, 994, 916, 749 cm−1; HRMS (ESI) calc. for C12H15BrONa [M+Na]+: 277.0204, found 277.0197; [α]25D = −9.7 (c = 2.56, CH2Cl2).
4.15. Synthesis of 54
A round bottom flask was charged with Pd(OAc)2 (11.2 mg, 0.05 mmol, 5 mol%), (2-biphenyl)di-tert-butylphosphine (18.6 mg, 0.063 mmol, 6.3 mol%), and Cs2CO3 (652 mg, 2.0 mmol, 2.0 equiv). The flask was evacuated and backfilled with argon, and the above alcohol (255 mg, 1.0 mmol, 1.0 equiv) in toluene (2 mL) was added via syringe. The flask was then placed into an oil bath pre-heated at 65 °C and stirred overnight. The reaction mixture was then cooled to room temperature, quenched with water (10 mL), and extracted two time with diethyl ether (10 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. Flash column chromatography with 3% Et2O/pentane afforded the chiral allyl ether (100 mg, 75%). 1H NMR (300 MHz, CDCl3) δ 7.07 (d, J = 7.4 Hz, 1H), 7.04 (d, J = 7.4 Hz, 1H), 6.80–6.86 (m, 2H), 5.86–6.00 (m, 1H), 5.10–5.20 (m, 2H), 4.02–4.09 (m, 1H), 2.71–2.85 (m, 2H), 2.54–2.61 (m, 1H), 2.37–2.45 (m, 1H), 1.98–2.06 (m, 1H), 1.70–1.79 (m, 1H).
A mixture of the above allyl chroman (83 mg, 0.62 mmol) and 10% Pd/C (20 mg) in dichloromethane (3 mL) was stirred under a hydrogen atmosphere for 4 h. Flash column chromatography (3% Et2O/pentane) afforded the desired ether (67 mg, 81%). 1H NMR (300 MHz, CDCl3) δ 7.03–7.10 (m, 2H), 6.78–6.84 (m, 2H), 3.94–4.04 (m, 1H), 2.72–2.90 (m, 2H), 1.95–2.03 (m, 1H), 1.72–2.02 (m, 2H), 1.45–1.62 (m, 3H), 0.98 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 155.1, 129.5, 127.1, 122.1, 119.8, 116.7, 75.6, 37.6, 34.1, 27.4, 24.8, 22.3, 18.6, 14.1, 14.0; IR (film) 2957, 2872, 1582, 1488, 1457, 1302, 1232, 1119, 984, 885, 752 cm−1; HRMS (EI) m/z calcd for C12H16O [M]+ 176.1201, found 176.1219; [α]25D = +99.1 (c = 1.96, CH2Cl2).
4.16. Hydrogenation of 8
A mixture of 8 (30 mg, 0.22 mmol, 74% ee by HPLC) and 10% Pd/C (10 mg) in dichloromethane (3 mL) was stirred under a hydrogen atmosphere for 4 h. Flash column chromatography (3% Et2O in pentane) afforded hydrogenated chiral ether 53 (19.6 mg, 64%). [α]25D = +73.0 (c = 1.96, CH2Cl2).
4.17. (R)-2-(2-methylallyl)-2H-benzopyran (55)
The reaction was performed at −25 °C for 3 h, using 1 (38 mg, 0.29 mmol, 1.0 equiv), 49 (50.9 mg, 0.058mmol, 0.2 equiv), 4Å molecular sieves (200 mg), DDQ (92 mg, 0.41 mmol, 1.4 equiv), methylallyl phenyldimethylsilane (162 mg, 0.87 mmol, 3 equiv), and anhydrous PhCF3 (3 mL). Normal workup and flash column chromatography with 3% Et2O/pentane afforded the desired product (36.4 mg, 68%). The enantiomeric excess was determined to be 33% by chiral HPLC (1% i-PrOH:hexanes, 1.0 mL/min). Characterization data were in agreement with previously reported values.9 1H NMR (300 MHz, CD2Cl2) δ 7.12 (ddd, J = 7.8, 7.8, 1.8 Hz, 1H), 6.98 (dd, J = 7.5, 1.5 Hz, 1H), 6.85 (ddd, J = 7.5, 7.5, 1.5 Hz, 1H), 6.75 (d, J = 7.8 Hz, 1H), 6.43 (d, J = 9.9 Hz, 1H), 5.74 (dd, J = 9.9, 3.6 Hz, 1H), 4.96–5.03 (m, 1H), 4.89 (aps, 1H), 4.79 (d, J = 0.9 Hz, 1H), 2.56 (dd, J = 13.8, 7.8 Hz, 1H), 2.35 (dd, J = 13.8, 6.0 Hz, 1H), 1.82 (s, 3H); 13C NMR (75 MHz, CD2Cl2) δ 153.8, 142.2, 129.7, 127.0, 126.3, 124.4, 122.7, 121.6, 116.7, 113.6, 74.2, 43.8, 23.1; IR (film) 3075, 2936, 1640, 1606, 1486, 1457, 1229, 1208, 1113, 1055, 1034, 891, 767, 753 cm−1; HRMS (ESI) m/zcalcd for C13H13O [M − H]+ 185.0966, found 185.0971; [α]25D = +55.7 (c = 2.51, CH2Cl2).
4.18. 2-Allyl-6-methoxy-2H-benzopyran (11)
The reaction was performed at −25 °C for 3 h, using 10 (48 mg, 0.30 mmol, 1.0 equiv), 49 (53 mg, 0.06 mmol, 0.2 equiv), 4Å molecular sieves (200 mg), DDQ (95 mg, 0.42 mmol, 1.4 equiv), allylphenyldimethylsilane (158 mg, 0.9 mmol, 3 equiv) and PhCF3 (3 mL). Normal workup and flash column chromatography (5% Et2O in pentane) afforded the desired product (32.4 mg, 53%). The enantiomeric excess was determined to be 9% by chiral HPLC (1% i-PrOH:hexanes, 1.0 mL/min). Characterization data were in agreement with previously reported values.9 1H NMR (400 MHz, CDCl3) δ6.72 (d, J = 8.4 Hz, 1H), 6.66 (dd, J = 8.8, 2.1 Hz, 1H), 6.54 (d, J = 2.8 Hz, 1H), 6.38 (d, J = 9.6 Hz, 1H), 5.88 (ddt, J = 18.0, 10.4, 7.2 Hz, 1H), 5.74 (dd, J = 9.6, 3.2 Hz, 1H), 5.15 (d, J = 8.8 Hz, 1H), 5.12 (s, 1H), 4.84 (brs, 1H), 3.75 (s, 3H), 2.53–2.60 (m, 1H), 2.40–2.47 (m, 1H); 13C NMR (CDCl3, 100 MHz) δ 153.9, 147.1, 133.4, 126.1, 124.3, 122.5, 117.8, 116.5, 114.2, 111.6, 74.3, 55.6, 39.4. [α]25D = −8.7 (c = 0.33, CH2Cl2).
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (R01-GM062924 and P50-GM06082) for generous support of this work.
Footnotes
Spectra for all new compounds and HPLC traces to determine enantiomeric excess can be found in the Supporting Information.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.(a) Congreve M, Chessari G, Tisi D, Woodhead AJ. J Med Chem. 2008;51:3661. doi: 10.1021/jm8000373. [DOI] [PubMed] [Google Scholar]; (b) Hajduk PJ, Greer J. Nature Rev Drug Disc. 2007;6:211. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]; (c) Congreve M, Carr R, Murray C, Jhoti H. Drug Disc Today. 2003;8:876. doi: 10.1016/s1359-6446(03)02831-9. [DOI] [PubMed] [Google Scholar]
- 2.Burke MD, Schreiber SL. Angew Chem, Int Ed. 2004;43:46. doi: 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]
- 3.For recent reviews on carbon–hydrogen bond functionalization of aliphatic molecules, see: Liu L, Floreancig PE. Curr Opin Drug Disc Devel. 2010;13:733.Gutekunst WR, Baran PS. Chem Soc Rev. 2011;40:1976. doi: 10.1039/c0cs00182a.Davies HML. Angew Chem Int Ed. 2006;45:6422. doi: 10.1002/anie.200601814.Li CJ. Acc Chem Res. 2009;42:335. doi: 10.1021/ar800164n.Robertson J, Pillai J, Lush RK. Chem Soc Rev. 2001;30:94.
- 4.For representative examples, see: Li Z, Li CJ. Org Lett. 2004;6:4997. doi: 10.1021/ol047814v.Dubs C, Hamashima Y, Sasamoto N, Seidel TM, Suzuki S, Hashizume D, Sodeoka M. J Org Chem. 2008;73:5859. doi: 10.1021/jo800800y.Condie AG, González-Gómez JC, Stephenson CRJ. J Am Chem Soc. 2010;132:1464. doi: 10.1021/ja909145y.Tsang ASK, Jensen P, Hook JM, Hashmi ASK, Todd MH. Pure Appl Chem. 2011;83:655.Zhang G, Zhang Y, Wang R. Angew Chem, Int Ed. 2011;50:10429. doi: 10.1002/anie.201105123.Alagiri K, Devadig P, Prabhu KR. Chem Eur J. 2012;18:5160. doi: 10.1002/chem.201200100.DiRocco DA, Rovis T. J Am Chem Soc. 2012;134:8094. doi: 10.1021/ja3030164.
- 5.For representative examples, see: Xu YC, Kohlman DT, Liang SX, Erikkson C. Org Lett. 1999;1:1599.Ying BP, Trogden BG, Kohlman DT, Liang SX, Xu YC. Org Lett. 2004;6:1523. doi: 10.1021/ol036314j.Zhang Y, Li CJ. J Am Chem Soc. 2006;128:4242. doi: 10.1021/ja060050p.Zhang Y, Li CJ. Angew Chem, Int Ed. 2006;45:1949. doi: 10.1002/anie.200503255.Park SJ, Price JR, Todd MH. J Org Chem. 2012;77:949. doi: 10.1021/jo2021373.Benfatti F, Capdevila MG, Zoli L, Benedetto E, Cozzi PG. Chem Commun. 2009:5919. doi: 10.1039/b910185c.Ho X-H, Mho S-i, Kang H, Jang H-Y. Eur J Org Chem. 2010:4436.Zhang B, Xiang SK, Zhang LH, Cui Y, Jiao N. Org Lett. 2011;13:5212. doi: 10.1021/ol202090a.
- 6.(a) Tu W, Liu L, Floreancig PE. Angew Chem, Int Ed. 2008;47:4184. doi: 10.1002/anie.200706002. [DOI] [PubMed] [Google Scholar]; (b) Tu W, Floreancig PE. Angew Chem, Int Ed. 2009;48:4567. doi: 10.1002/anie.200901489. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu L, Floreancig PE. Org Lett. 2009;11:3152. doi: 10.1021/ol901188q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liu L, Floreancig PE. Angew Chem, Int Ed. 2010;49:3069. doi: 10.1002/anie.201000033. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Liu L, Floreancig PE. Angew Chem, Int Ed. 2010;49:5894. doi: 10.1002/anie.201002281. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Brizgys GJ, Jung HH, Floreancig PE. Chem Sci. 2012;2:438. [Google Scholar]; (g) Cui Y, Floreancig PE. Org Lett. 2012;14:1720. doi: 10.1021/ol3002877. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Han X, Floreancig PE. Org Lett. 2012;14:3808. doi: 10.1021/ol301720u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Peh GR, Floreancig PE. Org Lett. 2012;14:5614. doi: 10.1021/ol302744t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Chandrasekhar S, Sumithra G, Yadav JS. Tetrahedron Lett. 1996;37:1645. [Google Scholar]; (b) Sharma GVM, Lavanya B, Mahalingam AK, Krishna PR. Tetrahedron Lett. 2000;41:10323. [Google Scholar]; (c) Liu L, Floreancig PE. Org Lett. 2010;12:4686. doi: 10.1021/ol102078v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cosner CC, Cabrera PJ, Byrd KM, Thomas AMA, Helquist P. Org Lett. 2011;13:2071. doi: 10.1021/ol200441g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Shen Z, Dai J, Xiong J, He X, Mo W, Hu B, Sun N, Hu X. Adv Synth Catal. 2011;353:3031. [Google Scholar]; (f) Alagiri K, Devadig P, Prabhu KR. Chem Eur J. 2012;18:5160. doi: 10.1002/chem.201200100. [DOI] [PubMed] [Google Scholar]; (g) Ghosh AK, Xu C. Tetrahedron Lett. 2012;53:2568. doi: 10.1016/j.tetlet.2012.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Cheng D, Ye X, Cui W, Sun R, Yan J. J Chem Res. 2012:729. [Google Scholar]
- 8.Jung HH, Floreancig PE. Tetrahedron. 2009;65:10830. doi: 10.1016/j.tet.2009.10.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.A portion of this work was previously published. See: Clausen DJ, Floreancig PE. J Org Chem. 2012;77:6574. doi: 10.1021/jo301185h.
- 10.Hayashi Y, Mukaiyama T. Chem Lett. 1987:1811. [Google Scholar]
- 11.Yoshida J-i, Suga S. Chem Eur J. 2002;8:2651. doi: 10.1002/1521-3765(20020617)8:12<2650::aid-chem2650>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 12.Mayr H, Kempf B, Ofial AR. Acc Chem Res. 2003;36:66. doi: 10.1021/ar020094c. [DOI] [PubMed] [Google Scholar]
- 13.Lykakis IN, Efe C, Gryparis C, Stratakis M. Eur J Org Chem. 2011:2334. [Google Scholar]
- 14.Varela-Fernández A, González-Rodríguez C, Verela JA, Castedo L, Saá C. Org Lett. 2009;11:5350. doi: 10.1021/ol902212h. [DOI] [PubMed] [Google Scholar]
- 15.Hubig SM, Rathore R, Kochi JK. J Am Chem Soc. 1999;121:617. [Google Scholar]
- 16.For reviews on the chemistry of organotrifluoroborate salts, see: Darses S, Genet JP. Chem Rev. 2008;108:288. doi: 10.1021/cr0509758.Stefani HA, Cella R, Vieira A. Tetrahedron. 2007;63:3623.Molander GA, Ellis N. Acc Chem Res. 2007;40:275. doi: 10.1021/ar050199q.
- 17.For recent examples of organoborates adding into carbocations, see: Vieira AS, Fiorante PF, Hough TLS, Ferreira FP, Lüdtke DS, Stefani HA. Org Lett. 2008;10:5215. doi: 10.1021/ol8022177.Mitchell TA, Bode JW. J Am Chem Soc. 2009;131:18057. doi: 10.1021/ja906514s.Moquist PN, Kodama T, Schaus SE. Angew Chem, Int Ed. 2010;49:7096. doi: 10.1002/anie.201003469.Vo CVT, Mitchell TA, Bode JWJ. Am Chem Soc. 2011;133:14082. doi: 10.1021/ja205174c.Graham TJA, Doyle AG. Org Lett. 2012;14:1616. doi: 10.1021/ol300364s.
- 18.(a) Prakash GKS, Mathew T, Panja C, Vaghoo H, Venkataraman K, Olah GA. Org Lett. 2007;9:179. doi: 10.1021/ol062562e. [DOI] [PubMed] [Google Scholar]; (b) Iwagami H, Yatagai M, Nakazawa M, Orita H, Honda Y, Ohnuki T, Yukawa T. Bull Chem Soc Jpn. 1991;64:175. [Google Scholar]; (c) Chan TH, Brook MA, Chaly T. Synthesis. 1983:203. [Google Scholar]
- 19.Szeto J, Sriramurthy V, Kwon O. Org Lett. 2011;13:5420. doi: 10.1021/ol201730q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Terada M, Tanaka H, Sorimachi K. J Am Chem Soc. 2009;131:3430. doi: 10.1021/ja8090643. [DOI] [PubMed] [Google Scholar]; (b) Zhang QW, Fan CA, Zhang HJ, Tu YQ, Zhao YM, Gu P, Chen ZM. Angew Chem, Int Ed. 2009;48:8572. doi: 10.1002/anie.200904565. [DOI] [PubMed] [Google Scholar]; (c) Coric I, Vellalath S, List B. J Am Chem Soc. 2010;132:8536. doi: 10.1021/ja102753d. [DOI] [PubMed] [Google Scholar]; (d) Coric I, List B. Nature. 2012;483:315. doi: 10.1038/nature10932. [DOI] [PubMed] [Google Scholar]; (e) Sun Z, Winschel GA, Borovika A, Nagorny P. J Am Chem Soc. 2012;134:8074. doi: 10.1021/ja302704m. [DOI] [PubMed] [Google Scholar]
- 21.(a) Dubs C, Hashiyama Y, Samamoto N, Seidel TM, Suzuki S, Hashizume D, Sodeoka M. J Org Chem. 2008;73:5859. doi: 10.1021/jo800800y. [DOI] [PubMed] [Google Scholar]; (b) Li Z, Li CJ. Org Lett. 2004;6:4997. doi: 10.1021/ol047814v. [DOI] [PubMed] [Google Scholar]
- 22.(a) Reisman SE, Doyle AG, Jacobsen EN. J Am Chem Soc. 2008;130:7198. doi: 10.1021/ja801514m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Burns NZ, Witten MR, Jacobsen EN. J Am Chem Soc. 2011;133:14578. doi: 10.1021/ja206997e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.For recent reviews of chiral ion pairs, see: Mahlau M, List B. Angew Chem, Int Ed. 2013;52:518. doi: 10.1002/anie.201205343.Brak K, Jacobsen EN. Angew Chem, Int Ed. 2013;52:534. doi: 10.1002/anie.201205449.Phipps RJ, Hamilton GL, Toste FD. Nature Chem. 2012;4:603. doi: 10.1038/nchem.1405.Terada M. Synthesis. 2010:1929.
- 24.(a) Nakashima D, Yamamoto H. J Am Chem Soc. 2006;128:9626. doi: 10.1021/ja062508t. [DOI] [PubMed] [Google Scholar]; (b) Cheon CH, Yamamoto H. J Am Chem Soc. 2008;130:9246. doi: 10.1021/ja8041542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hagen G, Mayr H. J Am Chem Soc. 1991;113:4954. [Google Scholar]
- 26.Racherla US, Brown HC. J Org Chem. 1991;56:401. [Google Scholar]
- 27.Kuwabe S, Torraca KE, Buchwald SL. J Am Chem Soc. 2001;123:12202. doi: 10.1021/ja012046d. [DOI] [PubMed] [Google Scholar]
- 28.Redpath P, Macdonald S, Migaud ME. Org Lett. 2008;10:3323. doi: 10.1021/ol801183e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.