

Abstract
This review is focused on three types of enzymes decarboxylating very different substrates: (1) Thiamin diphosphate (ThDP)-dependent enzymes reacting with 2-oxo acids; (2) Pyridoxal phosphate (PLP)-dependent enzymes reacting with α-amino acids; and (3) An enzyme with no known co-factors, orotidine 5'-monophosphate decarboxylase (OMPDC). While the first two classes have been much studied for many years, during the past decade studies of both classes have revealed novel mechanistic insight challenging accepted understanding. The enzyme OMPDC has posed a challenge to the enzymologist attempting to explain a 1017-fold rate acceleration in the absence of cofactors or even metal ions. A comparison of the available evidence on the three types of decarboxylases underlines some common features and more differences. The field of decarboxylases remains an interesting and challenging one for the mechanistic enzymologist notwithstanding the large amount of information already available.
Keywords: pyridoxal 5-phosphate, thiamin diphosphate, orotidine 5'-monophosphate, catalysis, decarboxylation, circular dichroism, 2-oxo acids, α-amino acids
A. introduction to thiamin diphosphate-dependent decarboxylases
In writing this review, the authors are keenly aware of the number of other excellent reviews available in the literature2–11 and therefore will concentrate on issues addressed for more than 30 years at Rutgers related to the coenzyme activity of ThDP, and reviewed by the senior author as well12–14. The chemistry and enzymology of ThDP is intimately dependent on three chemical moieties comprising the coenzyme: a thiazolium ring, a 4-aminopyrimidine ring and the diphosphate side chain. From the large number of high-resolution X-ray structures available for the past 20 years, starting with the structures of transketolase15 (TK), pyruvate oxidase16 (POX) from Lactobacillus plantarum and pyruvate decarboxylase from the yeast Saccharomyces cerevisae (YPDC)17,18, it has become clear that the diphosphate serves to bind the cofactor to the protein. This is achieved via electrostatic bonds of the α and β phosphoryl group negative charges with the required Mg2+ or Ca2+, the divalent metal serving as an anchor in a highly tailored environment with a conserved recognition sequence GDG-X26-NX of amino acids19. Chemically, the thiazolium ring is central to catalysis as reported in seminal studies by Breslow20, due to its ability to form a key nucleophilic center the C2-carbanion/ylide/carbene resonance forms. The 4'-aminopyrimidine moiety has gained more recognition as an important contributor to catalysis since the appearance of the X-ray structures showing its conserved proximity to the C2 thiazolium atom and the possibility for its participation in acid-base catalysis21. This issue will be a major focus of this section of the review, as it makes ThDP a truly unique and bi-functional coenzyme.
The goal of this review is to touch on themes concerned with observation and characterization of enzyme bound intermediates comprising both non-covalent and covalent complexes of ThDP. Recent studies of the mechanism have provided ever-greater insight, as detailed by recent reviews both by the authors' group and by Kluger and Tittmann22.
A.1. Detection of ThDP-related intermediates and their kinetic fates, and the information gained from such data
The presentation of thiamin-related and thiamin-bound intermediates represent pre-, or post-substrate (or substrate analogue) binding, an important distinction needed with the recent identification of several forms of ThDP on the enzymes.
A.1.a. ThDP-related intermediates prior to substrate addition
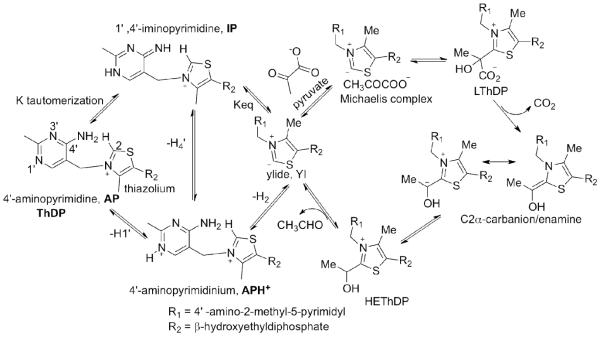
During the past decade, the authors' laboratory with collaborators (whose enzymes enable generalization of the findings) established the presence of various tautomeric and ionization states of ThDP (Schemes 1,2). The first high-resolution X-ray structures of ThDP enzymes15–18,21, clarified two issues for the first time: (i) The conformation of the bound ThDP (defining the dihedral angles formed by the thiazolium and 4'-aminopyrimidine rings with respect to the bridging methylene group was quite different (so-called V) from the conformation found for both free coenzyme (F) and coenzyme with a C2 substituent (S)23–25; (ii) This V conformation brings the N4' and C2 atoms to within 3.5Å of each other, consistent with participation of the 4'-aminopyrimidine ring in catalysis, as suggested by two groups over the years8,26–28. A need arose to identify spectral signatures for various tautomeric and ionization states of the 4'-aminopyrimidine ring of ThDP (Table 1).
Scheme 1.
Mechanism of yeast pyruvate decarboxylase YPDC.
Scheme 2.
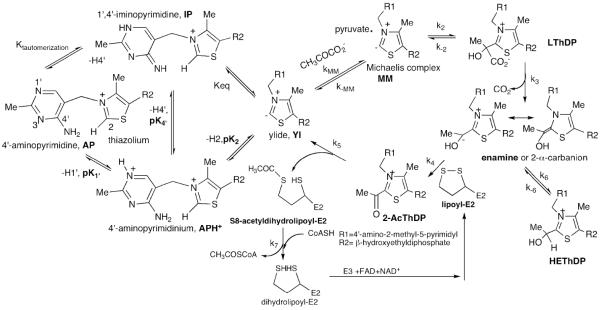
Mechanism of E. coli pyruvate dehydrogenase complex with role of ThDP on E1ec.
Table 1.
Detection of ThDP-related intermediates.
| Prior to substrate addition | With substrate present |
|---|---|
|
|
with chromophoric substrate ~400 nm.
A.1.a.i. The canonical 4'-aminopyrimidine (AP) form of ThDP
The signature for this species is a negative circular dichroism (CD) band centered near 320–330 nm as illustrated with the enzyme benzaldehyde lyase (BAL). While this CD band had been observed on the enzyme transketolase (TK)29, it was believed to be the result of a charge transfer transition between ThDP and an amino acid side chain on TK. A number of studies on YPDC and E1ec and their variants, as well as chemical model studies in the authors' laboratory suggest that this UV/CD band is due to a charge transfer transition between the 4'-aminopyrimidine ring as donor and the thiazolium ring as acceptor. The band has been observed on 10 ThDP enzymes, and its observation depends both on the pH and also on the specific enzyme environment.
A.1.a.ii. The 1',4'-iminopyrimidine (IP) form of ThDP.30–34
The notion that the 4'-aminopyrimidine could exist in its 1'.4'-iminopyrimidine tautomeric form was suggested by models attempting to mimic the reactivity of such a tautomer. The N1'-methyl analogue of both the 4-aminopyrimidine ring and of thiamin was synthesized and gave evidence of two relevant points: (i) In this N1'-methylpyrimidinium the pKa of the exocyclic amine was reduced to ca. 12–12.526,35, offering a rationale for the presence of a conserved glutamate as a catalyst for the amino⇆imino tautomerization, and (2) With the positive charge on the ring, the amino protons undergo differential exchange rates and the exchange is buffer catalyzed28. A spectroscopic signature for the 1',4'-iminopyrimidylThDP tautomer was first identified on the slow E477Q variant of YPDC30, prompting generation of chemical models by Zhang30 then Baykal35, showing that a synthetic chemical model would give rise to a UV absorption in the 300–310 nm range. It bode well for future NMR studies that the 15N chemical shifts of the three species on the left hand side of Schemes 1 and 2, the AP, IP and APH+ (two neutral and one positively charged) forms are quite distinct (consistent with early results for the AP and APH+).36
The CD bands corresponding to the AP and IP forms have different phases, enabling observation of both bands simultaneously, making the CD method useful. The signature for the IP form is a positive CD band centered near 300–314 nm, and as seen in Fig. 1, on E1h both the IP and AP tautomeric forms are seen simultaneously (Fig 1). The electronic absorption characteristics of the APH+ and the ylide (Yl) forms are yet to be established.
Figure 1.

CD spectra of E1h in the absence (apo E1h) and presence of ThDP (holo E1h). The positive CD band at 305 nm is assigned to IP and the negative at 330 nm to the AP form of ThDP.
A.1.a.iii. 1'H,4'-aminopyrimidinium (APH+) form
The presence of the protonated 4'-aminopyrimidinium ion APH+ received positive confirmation by solid state NMR measurements (T. Polenova at Univ. of Delaware) on three enzymes: YPDC, and the E1 components of both the pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase complexes from E. coli, on the basis of the characteristic 13C and 15N chemical shifts. The need for solid state NMR is due to the large molecular mass of all ThDP enzymes (at least 120 kDa).37
A.1.a.iv. Determination of the pKa for the enzyme-bound APH+ form.34
As the pH is lowered, the amplitude of the band for the AP form diminishes and titrates with an apparent pKa = 7.42 for the ([AP]+[IP])/[APH+] equilibrium on BAL. This pKa in water for ThDP is 4.8536, while on the enzymes it ranges from 5.6 to 7.5 (Table 2).34 It was concluded from data in Table 2, that the pKa for the APH+ coincides with the pH of optimum activity for each enzyme, indicating that all three forms IP, AP and APH+ must be readily accessible during the catalytic cycle. The pKa elevation on the enzymes could be rationalized by the presence of the highly conserved glutamate near the N1' position of ThDP (residue E571 on E1ec), that would tend to make the 4'-aminopyrimidine ring more basic. The tautomeric equilibrium constant Ktautomer, in conjunction with the pKas led to novel insight regarding ThDP catalysis, best viewed by the thermodynamic box for enzymes that are not substrate activated (left hand side of Schemes 1,2), such as E1h and pyruvate oxidase from Lactobacillus plantarum (POX). For these enzymes, both the IP and AP forms could be monitored over a wide pH range, providing both pKa and Ktautomer within reasonable error limits. The equilibria shown in Schemes 1,2 are valid prior to addition of substrate and lead to the following conclusions: (a) On POX and E1h, pK1', and pK4' have similar magnitudes; the enzymes shifted the pK4' from 12 in water26 to 5.6 and 7.0, respectively! (See left triangle in Schemes 1 and 2). (b). With a known forward rate constant from APH+ to the ylide (Yl) of ~50 s−1 determined for E1h38, and assuming a diffusion-controlled reverse protonation rate constant of 1010 s−1M−1 (giving a pK2 of 8.3 on E1h compared to an estimate in water of 17–1939), one could next speculate about the right triangle in Schemes 1,2. The most interesting prediction is that the proton-transfer equilibrium constant for [IP]/[Yl] is 101–102 on E1h, providing the first thermodynamic estimates for possible observation of the ylide.
Table 2.
The pKa of enzyme-bound APH+ on ThDP enzymes.
| Enzyme | pKa for the ([AP]+[IP])/(APH+) |
|---|---|
| BAL | 7.42 ± 0.02 |
| BFDC | 7.54 ± 0.11 |
| POX | 5.56 ± 0.03 |
| E1h | 7.07 ± 0.07 |
| E1o | 7.2 ± 0.01 |
| GCL V51D | 6.1 ± 0.02 |
| DXP synthase | 7.5 ± 0.09 |
A.1.a.v. The C2-carbanion/ylide/carbene
According to Breslow's findings, proton loss at the thiazolium C2 position is required to initiate the catalytic cycle. In 1997 there were two reports with significant implications regarding this issue: (i) A 1H NMR-based method was presented for measuring the rate of H/D exchange at the C2 position of bound ThDP, providing the rate constant for the dissociation of the C2H to the ylide40. ii. Arduengo and colleagues showed that the conjugate bases of imidazolium and indeed of thiazolium salts could be generated and their structure evaluated by NMR methods.41 The 13C chemical shift of the C2 resonance shifted from 157 to 253 ppm on conversion of their model thiazolium compound to its conjugate base ylide41.
A.1.b. Thiamin-bound intermediates with substrate or substrate analogue present
What makes detailed mechanistic studies of ThDP enzymes feasible is a powerful array of methods to monitor the kinetic fate of each intermediate along the catalytic cycle of all ThDP enzymes. An important method was developed for determination of the rate constants for individual steps by Tittmann and Hübner (`TH method')42. The TH method takes advantage of the known acid stability of the intermediates in Schemes 1 and 2, so that using either rapid quench or manual quench methods one can `freeze' the intermediates under acidic conditions while also precipitating the enzyme. The chemical shifts of the C6'H resonances of each intermediate are sufficiently distinct from each other and from that of the un-substituted ThDP, making 1H NMR an efficient method for evaluation of the relative concentration of the intermediates under steady state conditions, which in turn enable calculation of rate constants for individual reaction steps. The authors' group demonstrated, that substitutions of the protein distant from the active site could have a dramatic influence not only changing the rate, even changing the rate-limiting step. Further developments of this method were needed to address these issues on complex systems such as with the multienzyme complexes. The ThDP-bound intermediates have been synthesized and their chemistry established both by the authors' group and others.13,14, 42–45
A.1.b.i. The Michaelis Menten complex (MC)
The earliest detection of an MC was on addition of a substrate analogue methyl acetylphosphonate (MAP) and acetylphosphinate to several ThDP enzymes. An example is shown with acetylphosphinate added to YPDC leading to a negative CD band at ca. 325–335 nm, reminiscent of the band observed for the AP form. In this example, addition of ThDP alone did not display the AP form, the negative CD band only appeared after addition of substrate analogue, hence the band must pertain to a MC.46 Similar results were also seen when low concentrations of pyruvate were added to E1ec.32
Clear formation of the MC also resulted when adding pyruvate to the `inner loop' E1ec variants.46 Support for the claim that the MC was indeed being detected is provided by kinetic measurements: both stopped-flow photodiode array (PDA) spectra in the absorption mode, and stopped-flow CD spectra showed formation of the bands attributed to MC formation within the dead-time of the instruments (< 1ms), as expected of a non-covalent MC (see below).
A.1.b.ii. The covalent substrate-ThDP pre-decarboxylation complex (LThDP and analogues)
Observation of the intermediate analogues derived from substrate analogue phosphonates and phosphinates
Initial observation of the IP form (positive CD band, 300–314 nm) resulted from formation of a stable pre-decarboxylation adduct of ThDP with (a) MAP or acetylphosphinate31,32 (CH3C(C=O)P(H)O2NA)34, and (b) the aromatic 2-oxoacid analogue methyl benzoylphosphonate (MBP)47,48. With 10 enzymes tested so far, the IP form appeared on the stopped-flow time scale (either absorption or CD mode): the reaction is efficiently catalyzed by all of the enzymes. An important additional finding results from mixing YPDC and acetylphosphinate34: evidence for coexistence of the MC and the covalent pre-decarboxylation intermediate, consistent with `alternating active site reactivity' suggested by the authors' group earlier for YPDC and BFDC.49–51
The product phosphonomandelylThDP (PMThDP) formed on BFDC from MBP and ThDP was also confirmed (FT-MS) in solution,48 and of PLThDP (from MAP.ThDP) by X-ray methods on E1ec52 and on POX53. The X-ray structures of the PMThDP adduct on BAL and BFDC have also been determined to high resolution and are informative47,48, providing proof of configuration at the C2α position, not accessible with the pyruvate analogs (CH3 and OH could not be differentiated at current resolution).52,53
Additionally, all LThDP-like structures displayed some distortion where the C2-C2α bond is found non-coplanar with the thiazolium ring (unlike in the structure of PLThDP in the absence of enzyme), although the Dunathan hypothesis (according to which the C2α-P bond should be essentially perpendicular to the thiazolium plane) is adhered to.47,48,52,53
In some favorable cases, the CD band for the true pre-decarboxylation intermediate (via the IP form) could be observed from the slow substrates. This was accomplished when BAL was reacted with benzoylformate or phenylpyruvic acid,54 slow alternate substrates for this enzyme. LThDP was also observed by cryo-crystallographic methods on POX53. More recently, formation of surprisingly stable LThDP was observed on the enzyme 1-deoxy-D-xylulose 5-phosphate synthase (DXP synthase, condenses the enamine derived from pyruvate with the acceptor glyceraldehyde-3-phosphate, GAP) in the absence of GAP.60
Observation of LThDP analogues from chromophoric substrate analogs
On three enzymes, YPDC, BFDC and BAL, formation of the pre-decarboxylation adduct formed with ThDP from a chromophoric substrate analogue (E)-2-oxo-4(pyrid-3-yl)-3-butenoic acid (3-PKB) (as well as its ortho- and para isomers) was also observed.
With a stopped-flow photodiode array (SF-PDA) instrument, two transients resulted on mixing YPDC with 3-PKB: the first transient formed (T1) was assigned to the pre-decarboxylation intermediate analogous to LThDP (LThDP* with λmax near 470 nm), while the second transient (T2) was assigned to the enamine (λmax near 430 nm). The second transient T2 was formed at the same rate as the rate of depletion of T1. This compound provided outstanding information about the rates of formation of these two important intermediates on YPDC55–57, BFDC58 and BAL54, not available from any other source. On YPDC evidence was obtained whether an active center residue (a) had a role pre-, or post-decarboxylation (b) was involved in active center communication, (c) led to the alternating active site behavior,55,56 and about kinetic properties and consequences of a mobile loop near the active center.57 Where applicable, this method is complementary to the TH method, which could not differentiate the enamine from the HEThDP in Scheme 1; these species are at protolytic equilibrium, the acid quench converts the enamine to HEThDP.
On BAL, Nemeria observed simultaneous presence of both the negative CD band at 475 nm and the positive one at 312 nm on addition of PAA: the former a direct result of the tetrahedral adduct between PAA and BAL, the band at 312 nm of the IP form characteristic of C2α-tetrahedrally substituted intermediates.58
Reactivity of LThDP on and off the enzymes
With the ability to synthesize the putative intermediates LThDP and HEThDP (thanks mostly to Kluger's group), a reexamination of the reactivity of LThDP on and off the enzymes could be carried out, leading to the following results: (1) Decarboxylation of model C2α-lactylthiazolium and C2α-lactylthiamin salts could achieve first-order rate constant in aprotic solvents59 essentially identical to the enzymic turnover numbers (~60 s−1), i.e., LThDP can undergo decarboxylation at rates that are catalytically competent with the turnover numbers. A low polarity active center may be sufficient to ensure that decarboxylation of LThDP is not rate limiting. (2) When YPDC and E1ec were reconstituted with synthetic LThDP, both enzymes catalyzed its decarboxylation (unpublished observations at Rutgers). Rates at steady state are limited by the slow off-rate for ThDP, so a new molecule of LThDP can be bound. Pre-steady state experiments demonstrated that the reaction rates with LThDP reconstitution are limited by factors other than catalysis, i.e., binding and/or conformational changes. (3) Rapid acid quench of certain YPDC variants reacting with pyruvate enabled direct observation of LThDP under steady state conditions.42 It was concluded that LThDP is indeed on the pathway of both YPDC and E1ec59. As mentioned above, DXP synthase displayed the presence of a surprisingly stable LThDP. In the absence of GAP, decarboxylation of LThDP on the enzyme proceeded with k =0.07 s−1, while addition of GAP accelerated the decarboxylation rate by at least 600-fold.60
A.1.b.iii. The first post-decarboxylation intermediate: the enamine/C2α-carbanion
According to Schemes 1 and 2, the enamine is the only conjugated covalent ThDP-bound intermediate. UV-VIS observation of enzyme-bound enamine derived from aliphatic substrates is difficult due to the expected λmax near 290–295 nm, according to thiazolium-based models.61,62
The enamine intermediate derived from benzoylformate (modeled with λmax of 380 nm)63,64 has been observed directly on the enzyme BFDC at 390 nm58. While BFDC converts benzoylformate to benzaldehyde, the enzyme also catalyzes a benzoin-type condensation of benzaldehyde in the reverse reaction, similar to the reverse reaction of BAL. When reacting the benzaldehyde product with BFDC, there appeared an absorbance (and a CD band) at 390 nm, in the wavelength region predicted by models63,64, but no CD band was evident in the 300–310 nm region58. Also, when (R)-benzoin was added to BAL, there was formed the same CD band at 390 nm indicating slow release of the first benzaldehyde, and the stability of the enamine in the forward direction54. It was concluded that: (a) The `real' enamine could be observed (λmax at 390 nm) for the first time derived from benzoin or benzaldehyde; (b) The enamine was found to be in its AP or APH+, but not in its IP form; and (c) Since it gives rise to a CD signal, the enamine is chiral on the enzyme, even though it is planar and conjugated, hence all intermediates here discussed are chiral on the enzymes, both pre- and post substrate addition. The CD measurements nicely confirm this expectation for all intermediates, both covalently bound and even non-covalent ones such as the MC. With YPDC, BFDC and BAL, the enamine could be observed directly at 430 nm with 3-PKB as alternate substrate on YPDC.54–58
A.1.b.iv. The second post-decarboxylation intermediate, the product-ThDP complex (HEThDP, HBThDP)
Evidence on YPDC
Evidence for formation of this intermediate also will be reported in a later section using the TH method. Clear evidence was obtained for HEThDP-analog formation from reacting 3(pyrid-3-yl)-acrylaldehyde (PAA) (the product of decarboxylation of 3-PKB), with BAL or BFDC58. An absorbance with λmax 470 nm appeared (similar to that observed with 3-PKB), and was attributed to the HEThDP analogue. X-ray results confirmed formation of the tetrahedral post-decarboxylation intermediate on addition of PAA to BFDC. Striking confirmation of HEThDP formation resulted from mixing acetaldehyde, the product of pyruvate decarboxylation, with YPDC on the SF-PDA instrument,56 giving the characteristic absorption for IP form (λmax= 310 nm), with no alternative assignment to the IP of HEThDP (Scheme 1).
Evidence on E1ec.66,32
While HEThDP is not usually considered to be on the reaction pathway of ThDP-dependent oxidative decarboxylases (Scheme 2), groups working with POX67 and the PDHc's68 have long used HEThDP as an alternate substrate. Chemical model studies from the authors' laboratory on the enamine and related intermediates9,13,14 have unambiguously signaled that HEThDP cannot be oxidized directly by either FAD or lipoic acid as oxidizing partners. Instead, oxidation must be preceded by ionization at the C2α position to generate the enamine, which is then prone to oxidation by even molecular dioxygen from air. However, the pKa at the C2α position is very high, estimated at ca. 17–18 for HEThDP163 derived from pyruvic acid. Experiments on YPDC demonstrated substantial lowering of this pKa by the enzyme, and a low effective dielectric constant at the active center of YPDC69 was offered as explanation.
Given that HEThDP is not on the direct pathway of PDHc, reversibility of the reaction from HEThDP to enamine needed to be demonstrated on E1ec. If E1ec catalyzes the exchange, this could reflect the intervention of an enzymatic `solvent effect'. The E1ec catalyzed pre-steady state rate constant for the 2H→1H exchange from the C2α position of HEThDP-2H4 (steps k6/k−6 in Scheme 2), as an indicator of the formation of the enamine was measured. E1ec accelerates the rate of ionization of this bond by a factor of 107, corresponding to 10 kcal/mol stabilization of the enamine intermediate by the enzyme.70 This stabilization is a property of the active center per se, and, with our earlier evidence reported on YPDC, suggests that such a `solvent effect' is likely a general feature of ThDP enzymes.
A.1.b.v. 2-Acetylthiamin diphosphate, the 2-electron oxidation product of the enamine
This compound is the product of oxidation of the enamine by any one of the following oxidizing agents on enzymes: the dithiolane ring of lipoic acid covalently amidated to a lysine side chain in the 2-oxoacid dehydrogenase multienzyme complexes; less frequently by FAD in the pyruvate oxidases; finally by NAD+. The POX reaction has been studied for many years and most recent evidence suggests that the oxidation takes place via single electron transfers with the likely intermediacy of the radical cation species delocalized onto the thiazolium ring (see next section). Oxidation of the enamine by lipoic acid is usually viewed as the reductive acetylation of lipoyllysyl-E2. An important mechanistic issue of whether electron and group transfer take place in a single step via a tetrahedral intermediate (essentially `cross-linking' the ThDP-bound enamine on the E1 component with the lipoyl group on E2), or stepwise (where oxidation to the acetylThDP with concomitant reduction of lipoamide-E2 to dihydrolipoamide-E2 precedes acyl transfer to the S8 atom of the dihdyrolipoamideE2) was investigated by Frey and coworkers. Using a number of ingenious ways to generate 2-acetylThDP (for example, by reversing the reaction by addition acetylCoA), they provided evidence that the first option, redox followed by group transfer is the likely scenario.45 Pan at Rutgers demonstrated in a model system that the tetrahedral intermediate can indeed be generated from the enamine and lipoic acid analogs, although on critical examination this could be the obligatory intermediate with either scenario.71 Pan showed that the cleavage of the dithiolane ring by the enamine required electrophilic catalysis. In the model system this could be accomplished by S-methylation, while on the enzymes the reaction presumably uses an acid catalyst such as a histidinium ion, probably H407 on E1ec.72 Our group published evidence indicating that in the presence of an artificial oxidizing agent 2,6-dichlorophenoindophenol the enamine produced by E1p is converted to 2-acetylThDP, while in the presence of the E2p component, there is no apparent 2-acetylThDP on the way to reductive acetylation. This could signal that both pathways may be available to the enzymes.89
A.1.b.vi. The C2α-hydroxyethylideneThDP radical, the 1-electron oxidation product of the enamine
Early and clear evidence for a free-radical mechanism on ThDP enzymes was obtained on pyruvate-ferredoxin oxidoreductase, an enzyme that converts pyruvate to acetylCoA in anaerobes.73 In addition to ThDP, the enzyme has three Fe4S4 clusters forming a 40–50Å long electron transfer chain.74 The stability of the free radical was manifested by the fact that the crystal also displayed an electron paramagnetic resonance signal.74 A chemical model was generated for the electrochemical oxidation of the enamine leading to dimerization at the C2α atom, suggesting significant electron spin density at this atom.75 Subsequent detailed work on pyruvate-ferredoxin oxidoreductase clearly showed that the spin density is delocalized into the thiazolium ring, but there indeed is a significant fraction at the C2α atom.76 The enzyme pyruvate oxidase using both ThDP and FAD as cofactors, also uses a free radical mechanism.77
At the same time, there are ThDP enzymes with no known redox role for the bound FAD, such as glyoxylate carboligase (GCL). This enzyme carries out decarboxylative carboligation, similar to DXP synthase. Perhaps the most striking feature of glyoxylate carboligase is the replacement of the virtually universally conserved Glu residue opposite the N1' atom of ThDP by a hydrophobic residue.78 The GCL and BAL are of particular interest since on neither enzyme is there an acid-base residue within hydrogen bonding distance of the ThDP to assist with proton transfers. There is perhaps no alternative to water carrying out the proton transfers on these two enzymes. Of course, these enzymes also provide strong support for an obligatory catalytic role of the 4'-aminopyrimidine ring of the ThDP.
A.2. Determination of rate-limiting steps and microscopic rate constants on ThDP enzymes
Starting with the 1970s, both solvent and heavy-atom kinetic isotope effects (KIE) were employed to probe the rate-limiting steps in ThDP enzymes, especially on YPDC. 13C/12C KIEs used natural abundance 13C label at the pyruvate C1 atom by measuring the mass ratio of 45CO2/44CO2 using an isotope ratio mass spectrometer.79–81 Supported by a model study in which the decarboxylation step could be isolated,82 the results of such studies could inform about the partitioning of the LThDP intermediate reverting to pyruvate and free coenzyme or going forward to decarboxylation to the enamine (Scheme 1). The method suggested that decarboxylation was not rate limiting on YPDC, the forward rate constant being 4–5-fold larger than the reversion to the Michaelis complex. YPDC is subject to substrate activation (Hill coefficient ~ 2.0) Especially, the solvent deuterium KIEs suggested that there may be a strong hydrogen bond formed during regulation and, along with heavy atom KIEs enabled construction of a free energy diagram for the reaction pathway including the regulatory mechanism.83,84 Solvent deuterium KIEs were determined on some of the cysteine variants of YPDC (Cys221 is believed to be site of substrate activation13,14), again attempting to delineate the effects of substrate activation.85,86
The earlier quoted TH method42 is based on observation of covalent ThDP-bound intermediates (Schemes 1 and 2) after their release from the enzyme by acid-quench of a reaction mixture resulting from rapid mixing of enzyme and substrate on a chemical quench instrument. Fortuitously, HEThDP, LThDP and 2-acetylThDP are all (a) stable under these conditions, and (b) have distinct 1H chemical shifts for their C6'-H resonances. Integration of these resonances provides the relative steady-state distribution of intermediates, and, with the turnover number of the enzyme, the forward rate constants could be calculated for the pathway (rate constants for formation of LThDP (k'2), its decarboxylation of (k'3) and acetaldehyde release (k4,5).42 The method has been applied to a number of ThDP enzymes.22 The power of the experiment is illustrated with the YPDC loop variants.
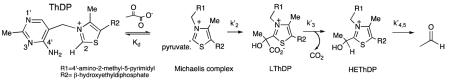 |
(1) |
It was concluded that acetaldehyde release is rate limiting for all of the loop variants, hence the loop opening/closing must be impaired.87 The method has also been applied to active center variants of YPDC,42 along with several substitutions on the E1ec.88 The method has some limitations: (a) It measures intermediate distribution once those are released from the enzyme; (b) It cannot differentiate the level of enamine and HEThDP, since under acid quench the former is converted to the latter. Therefore, the HEThDP measured in the quench corresponds to the sum of the relative concentration of enamine and HEThDP; and (c) Given that the method depends on the observation of the aromatic C6'-H resonances, many aromatic cofactors would interfere with the observations.
Balakrishnan and Chakraborty37,89 in the authors' laboratory addressed the NMR issue and extended the power of the TH method by synthesizing ThDP specifically labeled at both the C2 and C6' positions with 13C ([C2,C6'-13C2]ThDP). Using 1D-gradient HSQC NMR methods only protons attached to these two 13C-labeled atoms were detected. This enabled detection of C2H and C6'H in the aromatic region of the 1H NMR spectrum even in the presence of other cofactors with aromatic resonances. As an example, the PDHc complex, even after the protein is acid precipitated, leaves aromatic resonances pertinent to FAD, NAD+ and coenzyme A in the supernatant, in addition to the C2H and C6'H of ThDP. A comparison of the rates of formation of HEThDP in the E1ec component and in the entire PDHc-ec.89 was accomplished by using [C2,C6'-13C2]ThDP, and demonstrating that assembly to the complex accelerates the rate of HEThDP formation in a particularly slow E1ec variant by ca. 9-fold. Such information is not readily available from any other method.
The chromophoric conjugated substrates exploited by the authors' group provide complementary information to the TH method, since with the chromophoric substrates the enamine is directly observed and several microscopic rate constants could be directly assessed.
To observe directly the time-course of enzyme-bound intermediates, a method was developed which is a combination of CD methods and the TH method: the time-course of intermediate formation/depletion is assessed by stopped-flow CD, by looking at the intermediates with signatures identified as above. If there is `degeneracy' in the CD assignments (an important case in point is that all ThDP-bound intermediates with tetrahedral substitution at C2α appear to exist in their IP tautomeric forms at pH values above the pKa of the APH+ form), we rely on the TH method to identify the intermediate after acid quench under identical reaction conditions. This combined approach was recently applied to E1ec, DXP synthase and GCL.89,60,91
A major conclusion from these studies is that the rate of individual steps can be assessed directly from detailed time course studies, providing a virtually unprecedented opportunity to gain insight to ThDP-dependent reactions, including interrogation of individual enzyme residues regarding their catalytic roles. This issue has again become important in view of growing evidence that our understanding of the role of even His side chains is lacking as signaled by studies on two ThDP dependent decarboxylases: (a) Saturation mutagenesis studies on BFDC showed that a His residue did not have acid-base role,92 and (b) Similar studies on the E1 component of the E. coli 2-oxoglutarate dehydrogenase revealed that only one of the two His at the substrate's putative γ-carboxylate binding site (subjected to saturation mutagenesis) could not be replaced by hydrophobic side chains, i.e., in both enzymes studied at least one His had a role different from that assumed by biochemical intuition, such as acid-base, hydrogen bonding or nucleophilic.90
A.3. Evidence for correlation of active center loop dynamics and reactivity in E1ec
We wished to investigate the possibility that loop movement/dynamics may control catalysis (i.e., such movement may be rate limiting) rather than chemical steps.
A.3.a.i. Structural evidence pointing to mobile loops
In the X-ray structure of E1ec with ThDP, there were three regions of poorly defined electron density among the 886 amino acid residues per monomer, corresponding to residues 1–55, 401–413, and 541–557.93 A remarkable change was seen in the structure of E1ec in complex with a pre-decarboxylation intermediate phosphonolactylThDP (PLThDP- the adduct of MAP with ThDP), revealing the structure and important interactions of the two loops 401–413 (inner loop) and 541–557 (outer loop) not seen before.94 This signaled a disorder to order transition on forming the pre-decarboxylation PLThDP intermediate (Scheme 2). On E1ec, the bound PLThDP is stabilized by interactions with inner loop residues E401, H407 (via direct hydrogen bond formation) and Q408 (via a water molecule).94 The two newly ordered regions interact with each other, and propagate from the active site to the enzyme surface forming a tunnel to accept lipoamide from E2ec for the next reaction (Fig. 2). This led the authors to propose that the ordering observed in the E1ec-PLThDP complex would also be present when the true intermediate LThDP formed. Also, the crystal structure of the H407A E1ec in complex with PLThDP provided evidence that in the absence of H407, the two loops are not ordered; hence the H-bond from H407 to LThDP is the key trigger for the disorder to order transformation. The E1ec-PLThDP structure (mimicking LThDP) was among the first examples of a covalently bound pre-decarboxylation reaction intermediate analogue in any ThDP-dependent enzyme.93
Figure 2.
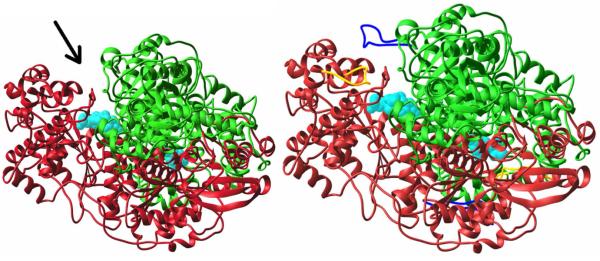
Left. E1ec.ThDP with inner, outer loops fully disordered (not seen). Arrow indicates active site entrance. Subunits in red and green, ThDP (cyan) space filling. Right. E1ec-PLThDP complex, newly ordered inner (yellow) and outer loop (blue).52
A.3.a.ii. Studies on the inner active center mobile loop of E1ec46,95,96
The above results prompted a study on the role and dynamics of the inner loop, starting with mapping of the conserved residues on the loop. Kinetic, spectroscopic and crystallographic studies on some inner loop variants led to the conclusion that charged residues flanking H407 are important for stabilization/ordering of the inner loop, thereby facilitating completion of the active site. The results further suggested that a disorder to order transition of the dynamic inner loop is essential for (a) forming LThDP, (b) sequestering active site chemistry from undesirable side carboligase side reactions, as well as (c) communication between the E1ec and E2ec components. The experiments carried out to reach these conclusions are presented briefly as they should be useful for studies of related enzymes.
Both a Michaelis complex (MC) and predecarboxylation intermediate analogue could be observed on addition of MAP: formation of PLThDP from MAP on E1ec (k1obs =3.6 ± 0.2 s−1 and k2obs = 0.35 ± 0.06 s−1) was slower than MC formation and significantly slower in E401K (k1obs = 0.37 ± 0.05 s−1and k2obs = 0.04 ± 0.01 s−1). Clearly, formation of PLThDP, hence formation of C-C covalent bond, and not formation of MC (kMC), is the rate-limiting pre-decarboxylation step. Since C-C bond formation, but not MC formation, is dramatically slowed down (10-fold compared to E1ec) in E401K, loop dynamics apparently greatly influence covalent addition of substrate to the enzyme-bound ThDP.
The mobile inner loop was found to control access to the active site, changing from the normal product distribution to more carboligase side product (Scheme 3), but the loop was even found to impact on the facial selectivity towards the electrophile.
Scheme 3.
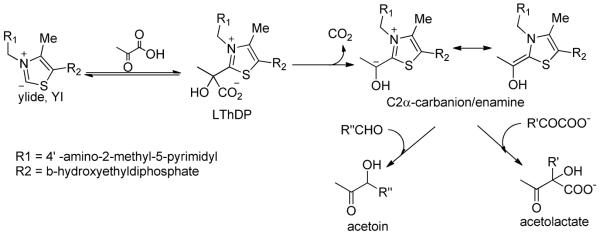
Carboligase side reactions on YPDC and other 2-oxoacid decarboxylases.
To study communication between the E1 and E2 components, MALDI-TOF MS was utilized to determine the time course of reductive acetylation of the E2 component by the E1 component and pyruvate.72,97 While the wild-type E1ec can reductively acetylate the E2ec component in less than 30 s, with the inner loop variants both unacetylated and acetylated forms are clearly visible even after 30 min, i.e., the reductive acetylation was incomplete even after this time period.95 Next, the X-ray crystal structure of a second loop variant E401K was solved, with both ThDP or PLThDP present. Again, the inner and outer loops were un-organized even with PLThDP present (as with the H407A variant).95
To investigate the rate of loop movement, a cysteine-less E1ec was constructed where five cysteines were substituted to alanine and the sixth, Cys259 to Asn47. This cysteine-less construct still possessed 22% activity for NADH production in the reconstituted PDHc-ec assay. Monitoring the kinetics of E1ec and its loop variants at various solution viscosities, it was shown that the rate of a chemical step is modulated by loop dynamics. The cysteine-less E1ec construct was site-specifically labeled on the inner loop with an electron paramagnetic resonance active nitroxide label revealing ligand-induced conformational dynamics of the loop and slow `open-close' conformational equilibrium in the un-liganded state. A CF3C(=O)CH2- (trifluoroacetonyl) 19F NMR label placed at the same residue revealed that the interconversion of the open and close forms of the inner loop takes place with a rate constant of 0.5–1.0 s−1, very similar to the turnover number from the E1-specific assay (0.38 s−1). These results suggest a quantitative correlation of E1ec catalysis and loop dynamics for the 200,000 Da protein. Thermochemical studies (isothermal titration calorimetry) revealed that these motions may promote covalent addition of substrate to the enzyme bound ThDP by reducing the free energy of activation. The results are consistent with efficient coupling of catalysis and regulation with enzyme dynamics in E1ec, and suggest the mechanism by which it is achieved, and reinforce the hypothesis ascribing catalytic and regulatory roles to enzyme dynamics.46
A.4. Do Thiamin Enzymes Provide a Paradigm for `Enzymatic Solvent Effects'?
In 1970, there appeared two publications modeling ThDP-catalyzed reactions in solvents of different dielectric solvent. Both Lienhard98,99 and coworkers and Kemp and O'Brien100 presented studies, which suggested that a solvent of low dielectric constant accelerated thiamin model catalyzed reactions. In the same year, Ullrich and Donner presented the first enzymatic evidence suggesting that the YPDC has a hydrophobic active center on the basis of studies with a fluorescent label.101 The authors' group has also probed the active center environment of ThDP enzymes using a variety of experimental approaches.
A.4.a. Evidence from observation of the enamine on YPDC
With the understanding that YPDC has an active center which accepts 2-oxo acids with large substituents replacing the methyl group, and that the λmax of the enamine derived from HBThDP is near 380 nm, we tested whether the enzyme would convert the second post-decarboxylation intermediate HBThDP to the enamine. When racemic HBThDP added to the E91D YPDC (this variant can readily exchange its ThDP with its analogues), there resulted in a slow development of the A380, suggesting formation of the enamine, i.e., reversal of the protonation in Scheme 1.69 Given that the pKa for this ionization is 15–16 in the absence of enzyme63,64, this result strongly suggested that the YPDC could stabilize the enamine relative to an aqueous environment. To estimate the nature of the YPDC active center environment, the fluorescence emission of thiochrome diphosphate, a competitive inhibitor (competing with ThDP) of a number of ThDP enzymes, was measured on YPDC and in a series of 1-alkanols ranging from methanol to 1-hexanol.69 The correlation of solvent dielectric with emission maximum for the 1-alkanols was virtually linear, enabling us to interpolate the value on YPDC as falling between the values for 1-hexanol and 1-pentanol, i.e., a dielectric constant of 11–13. Even with the limitations of such an approach, the message is unmistakable: the active center environment of YPDC resembles that of 1-pentanol or 1-hexanol more than it resembles the aqueous milieu.
A.4.b. Evidence from solvent effects on decarboxylation rate constants in model compounds
The effect of solvent of model decarboxylation reactions carried out earlier by Lienhard98 and Kluger4 was reexamined by the authors' group in both C2α-lactylthiazolium salts and C2α-lactylthiamin.59 This model isolates the decarboxylation step of Schemes 1,2 by monitoring enamine formation via oxidative trapping with DCPIP: first-order rate constants were achieved for decarboxylation in low dielectric media that were virtually the same as the turnover number per subunit for a typical decarboxylase such as YPDC. It was concluded that acceleration by the enzymatic environment per se was so large as to need little additional catalysis.
A.4.c. Evidence from acceleration of the reaction step leading to the enamine on E1ec
While HEThDP is not directly on the pathway of the E1 component of the 2-oxo acid dehydrogenase complexes, ionization of its C2αH could be used to test the reversibility of enamine protonation, and to probe the effect of the active center on this reaction. To test whether the E1ec could accelerate the C2αH ionization step in Scheme 2, HEThDP was synthesized with 4 deuterons in the C2α-hydroxyalkyl side chain by condensing ThDP with acetaldehyde-d4. This deuterated HEThDP was then rapidly mixed with apo-E1ec (devoid of ThDP) on a KINTEK rapid quench instrument in H2O and the reaction was stopped by acid quench. The hypothesis was that the rate of D to H exchange would reflect the rate of the C2αH ionization step. The exchange was quantified using FT-MS. Compared to model systems, the rate of this reaction was accelerated on the enzyme by ca. 107. Since there is no amino acid in the active site of E1ec charged with the specific function of ionizing this C2αH bond, it was concluded that the enzyme environment was largely responsible for the observed rate acceleration, again pointing to as much as 10 kcal/mol rate acceleration due to the enzyme environment.70
A.4.d
Finally, the authors recall that the IP form of ThDP is stabilized on the enzyme, this must also be a consequence of the protein environment, since there is simply no model for partitioning of the APH+ form to the IP, rather than the AP tautomer in the absence of enzymes.
These few examples suggest that: (a) Indeed there is an important `environmental' contribution to the catalytic rate acceleration on ThDP enzymes, stabilizing high energy intermediates, including the ylide and the enamine; and (b) Such factors could account for as much as 107–109 of the total rate acceleration. This constitutes a very significant fraction of the total rate acceleration provided by the protein over and above that afforded by ThDP itself, which on YPDC was estimated to be 1012–1013-fold83,84; and finally (c) Such effects are almost certainly present on other ThDP enzymes as well, and researchers should be on the lookout for them.
B.1. Introduction to pyridoxal-5-phosphate dependent α-amino acid α-decarboxylases
Pyridoxal-5'-phosphate (vitamin B6)-dependent enzymes play an important role in amino acid metabolism of both humans and microorganisms. These enzymes catalyze a variety of reactions of α-amino acids, including transaminations, racemizations, α-decarboxylations, β-decarboxylations, etc. Several enzymes are targets for inhibitor design to treat a variety of diseases.102,103 Alignment of the amino acid sequences of PLP-dependent decarboxylases identified four distinct groups,104 among which three belong to type I fold105 (among five distinct folds106–108) which react at the α- and γ-carbon atoms of the substrate.106 The PLP-dependent α-decarboxylation mechanism is shown in Scheme 4 in which PLP is stabilizes the carbanions by negative charge delocalization. A common theme among PLP-dependent enzymes is protection of the aldehyde of PLP in an `internal aldimine' (covalent enzyme-PLP complex) where PLP is bound to a highly conserved active site Lys residue forming an imine (Schiff base). The stable enzyme-PLP complex then reacts with the α-amino group of the substrate via trans-imination forming the so-called `external aldimine'. This aldimine provides the necessary driving force for decarboxylation by placing the imine electron withdrawing group β to the departing CO2 and leading to a resonance-stabilized carbanion. Once the carbanion is protonated at Cα, the decarboxylated product is released by a reverse of the trans-imination reaction with the conserved lysine, hence reforming the internal aldimine. Unlike in the more complicated transamination mechanism, for α-decarboxylation there is no cycling required between the pyridoxal and pyridoxamine forms of the cofactor, the aldehyde form is sufficient to carry out the entire reaction sequence.
Scheme 4.

PLP-dependent α-amino acid decarboxylation.
There is an enormous literature on the mechanisms of PLP-catalyzed reactions, here only some more recent results dealing with the α-decarboxylation will be discussed. The general mechanism of PLP enzymes was proposed by David Metzler and the aldehyde carbon atom, the pyridine nitrogen and the 3-OH group were proposed to be important to catalysis.109
B.2. Catalysis of the conversion of the internal aldimine to the external aldimine and its reversal after decarboxylation
This preliminary reaction, universal to all PLP enzymes, is itself a typical nucleophilic attack leading to a tetrahedral diamine which is partitioned to one imine and an amine in each direction. Such reactions in solution are weak acid catalyzed to enhance the leaving group ability of the departing amine.
The conserved residue Cys360 in ornithine decarboxylase (ODC) was suggested to be important for the decarboxylation step, the C360A substitution decreased the enzyme's kcat by 50-fold.110–112 Recently, Eduardo et al. suggested that the conserved active site Cys360 residue has a role in the interconversion of the internal/external aldimines.113
B.3. Selection of the appropriate conformation at Cα and catalysis of CO2 loss
In an excellent review, Eliot and Kirsch114 introduce the reader to key issues in PLP catalyzed reactions. One of the unique features of the entire class of PLP enzymes is the selection of the type of reaction, i.e., decarboxylation at the Cα position, racemization or transamination (both reaction types starting with Cα-H ionization), or reactions at Cβ or Cγ. A brilliant hypothesis by Dunathan115 in 1966 presented a simple solution to the conundrum by selection of the desired conformer with respect to the Cα-N single bond as outlined in Scheme 5. For decarboxylation, the Ccarboxyl-Cα bond is lined up such that the incipient 2p orbital will be conjugated with the already conjugated external aldimine (the resulting intermediate is called the quinonoid structure and has λmax near 500 nm clearly informing of extended conjugation). By contrast, for racemization and transamination, the Cα-H must be ionized, hence that conformer must be selected where this Cα-H bond is aligned, such that its scission will provide the lone pair of electrons to participate in the π system. By this stereochemical selection, the energy of the transition state for bond scission is lowered, as it is approaching the ideal 2p-π orbital overlap. This stereochemical imperative is ensured by having tailored subsites around each of the three substituents at Cα, and it appears to be seldom violated.
Scheme 5.
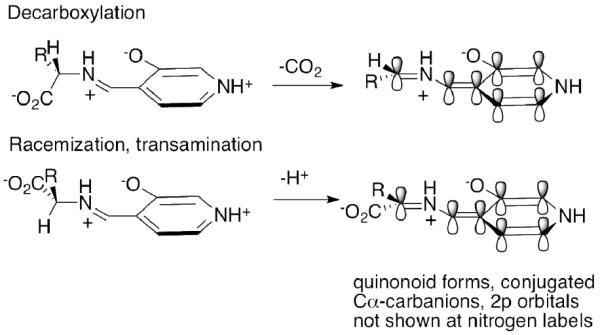
Dunathan hypothesis for conformational selection in PLP enzymes.
A fascinating example of this hypothesis is presented by the enzyme dialkylglycine decarboxylase, an enzyme which first decarboxylates the substrate, then carries out a transamination reaction. This ping-pong mechanism uses different subsites in the consecutive parts of the chemistry, first the CO2 fits into its subsite, decarboxylation ensues, next the Cα-H fits into its subsite and transamination follows, providing excellent support for the hypothesis.116
It was reported that in ornithine decarboxylase the carboxyl group of the bound substrate is buried in a region comprising hydrophobic and electron-rich residues.117 The residue Phe-397 was found to form a close contact with the carboxylate, its substitution not only reduced the rate of decarboxylation, but also affected product distribution, since there was a trans-imination side reaction producing pyridoxamine. The authors suggested that the environment enhances the decarboxylation rate to accommodate the charge-dispersed carbanion intermediate in favor of the negatively charged pre-decarboxylation carboxylate.117 This theme appears to be somewhat akin to findings on ThDP enzymes where the environment also appears to have an important impact on decarboxylation: in the case of ThDP the decarboxylation concentrates charges from the zwitterionic LThDP to the dipolar enamine.
In contrast, dialkylglycine decarboxylase has evolved an Arg group proximal to the bound carboxylate,118 and while the enzyme is an unusual PLP-dependent decarboxylase, this finding points out how difficult it is to generalize regarding themes for acceleration of decarboxylation.
B.4. The tautomeric equilibrium between the imine nitrogen and the 3-OH of PLP
There is information indicating that the 3-OH proton is sometimes transferred to the imine nitrogen of the external aldimine, increasing its electron withdrawing potential.65 In a recent report on carboxynorspermidine decarboxylase an Arg was identified water separated from the 3-OH, suggesting that this Arg residue stabilizes the ionized form of 3-OH which would be very useful to stabilize in turn the positive charge on an iminium ion derived from the α-amino group.119 Computational and kinetic isotope effect studies on L-DOPA decarboxylase also suggest that such proton transfer contributes to catalysis.120,121 Dopa decarboxylase (DDC) catalyzes the irreversible decarboxylation of aromatic L-amino acid substrates. The crystal structure of DDC in complex with its inhibitor carbidopa (PDB entry: 1JS3)122 shows that the N1 in the pyridine ring of PLP interacts with side chain of acidic amino acid Asp271 (Fig. 3).
Figure 3.
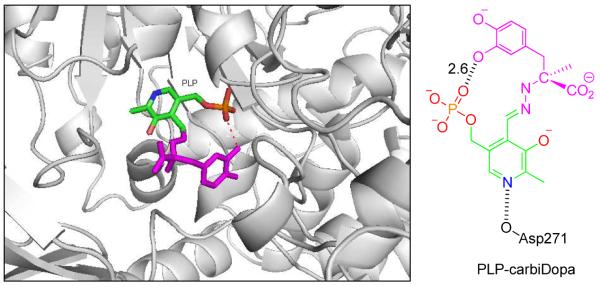
Left. View of dopa decarboxylase in complex with external PLP-carbiDopa (PDB: 1JS3)122 shown as stick, PLP in green and carbiDopa in magenta color. The image was created using the program PyMOL. Right. Schematic diagram showing interaction of N1 of PLP with Asp271. The value 2.6 Å represents the distance between connected atoms.
A very extensive study of the protonation/tautomerization states of PLP both in solution and on aspartate aminotransferase was carried out by Limbach, Toney and coworkers65 using a combination of solution and solid-state NMR methods, mostly relying on 15N labeling at the Cα-N and at the pyridinium N1 position, and supported by very extensive model studies. The methods developed enabled this group to suggest chemical shift-bond length correlations. Their results are fully consistent with an Asp residue hydrogen bonded to the pyridinium nitrogen atom, and the zwitterionic charge distribution in the external aldimine, i.e., a 3-oxy anion opposite the iminium (protonated Schiff-base) ion at a distance of perhaps 2.6 Å or less, as diagrammed in Scheme 5. The pKa of the pyridinium ion is raised compared to that in water, an increase attributed to the Asp residue within hydrogen bonding distance. These NMR studies, along with the studies reported on ThDP mechanisms, constitute some of the most precise definition of the state of ionization and tautomerization of coenzymes on such large enzymes.
B.5. Must the pyridine ring be protonated in all PLP reactions?
For many years it was assumed that the aromatic ring of PLP exists as a pyridinium ion, a protonation that would increase the electron withdrawing ability of the cofactor, and could be useful for drawing resonance structures for the intermediates, especially the so-called quinonoid form. On the basis of the above-discussed ThDP catalysis, this would be manifested by a carboxylic acid amino acid (Asp or Glu) within hydrogen bonding distance of the pyridinium ion. With the multiple structures solved for various classes of PLP enzymes, it became obvious that not all PLP enzymes use this catalytic mechanism, but, in fact, the α-decarboxylases do appear to use it. A very ingenious way to test this mechanism was developed by M. Toney and his group, via the de novo synthesis of 1-deaza-PLP, then reconstitution of the PLP enzymes with this coenzyme analogue. Indeed, some PLP enzymes could function at reduced rates with this 1-deazaPLP demonstrating that that protonation at N1 is not a universal requirement for PLP enzymes.123 At the same time, in several α-decarboxylases, there is clear evidence of a Glu side chain carboxylic acid within hydrogen-bonding distance of the pyridine nitrogen atom.119,124
B.6. Evaluation of the kinetic fates of PLP-bound intermediates
Determination of the kinetics of individual steps in PLP enzymes is facilitated by observations in the VIS spectroscopic range. Detailed analysis by multi-wavelength VIS spectroscopy both at steady state and pre-steady state time scales has taken advantage of the recognition of the different wavelength maxima of different intermediates on the pathway (see ref123 for a good summary of the spectral properties of key PLP-bound intermediate). Using a variety of sophisticated mechanistic probes to study the enzyme dialkylglycyl decarboxylase, the authors concluded that decarboxylation and C4' (aldehyde carbon) protonation were concerted.125
B.7. State of ionization of the phosphate group of PLP
The environment of the phosphate of bound PLP has been examined by 31P NMR spectroscopy.126 Of immediate interest is the state of ionization of the phosphate monoester. Application to dialkylglycine decarboxylase indicated that the pKa for second ionization is ca. 6.0–7.0, depending on the positive counterion. Equally importantly, the method in favorable cases provides information regarding the state of ionization of the iminium ion form of the external aldimine, confirming strong basicity of the imine nitrogen, a valuable piece of data for mechanistic understanding.
C.1. Introduction to orotidine 5'-monophosphate decarboxylase
The cofactor independent enzyme orotidine 5'-monophosphate decarboxylase (OMPDC) catalyzes the decarboxylation of orotidine 5'-monophosphate to uridine 5'-monophosphate during pyrimidine nucleotide biosynthesis (Scheme 6).127,128 The finding that this enzyme catalyzes the enzymatic reaction by a factor of 1017 over the non-enzymic rate makes this enzyme perhaps the most proficient one known to date,129,130 and also carries out a reaction in the apparent absence of any obvious electron sink in the system, akin to those created in ThDP and PLP enzymes. Wolfenden's discovery spawned enormous interest, producing many structures,131,132 many novel experimental tests and even computational tests. For this brief review, the authors had to be selective and apologize to all research groups whose work is not referred to. There are a number of reviews on the subject including refs. 133,134. The authors will present what they perceive to be the key current theories to explain the catalytic power of OMPDC. Experiments indicating that this is a single substrate enzyme with no known cofactor or metal ion requirement make such studies more tractable.
Scheme 6.
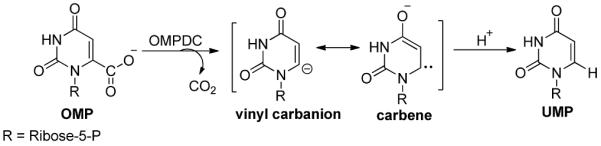
OMPDC-catalyzed reaction mechanism.
C.2. Early chemical model studies135
led to a mechanism, according to which an initial protonation at the pyrimidine O2 or O4 atoms creates a potential electron sink via an ylide involving the N1 atom, and this drives the subsequent decarboxylation reaction. Solvent kinetic isotope effects suggest that there is an isotope-sensitive rate-determining step, perhaps consistent with the protonation idea.136 Arguments against protonation include: (a) the absence of any suitably positioned amino acid that could donate the proton to O2 or O4, albeit hydrogen bonding is a possibility via amides according to the x-ray structures of OMPDC in the presence of substrate, and (b) 15N kinetic isotope effects appear to rule out ylide formation in the rate-determining step.137
C.3. Mechanistic hypotheses based on active center amino acid side chains
The structure of Saccharomyces cerevisiae (ScOMPDC) in a complex with 6-hydroxyuridine-5'-phosphate (BMP) suggests that Lys 93 (Lys 72 in Methanothermobacter thermautotrophicus, MtOMPDC,138 as in Fig. 4) is positioned to stabilize the negative charge at C-6 and also to protonate the intermediate, which then forms the product UMP.139 Saturation mutagenesis experiments were carried out on 24 residues of the E. coli OMPDC140 in and around the active site. All positions could be substituted to some extent when the library mutants were expressed from a multi-copy plasmid. For the conserved quartet of charged residues Lys44, Asp71, Lys73 and Asp76 (Lys42, Asp70, Lys72 and Asp75 inMtOMPDC), a cysteine substitution was found to provide function at positions 71 and 76. The authors suggested that a lower pKa for both cysteine variants supports a mechanism whereby the thiolate group of cysteine substitutes for the negatively charged aspartate side chain. The partial function variants such as D71C and D76C exhibit reduced catalytic efficiency relative to wild type OMPDC, but still reacted at 1015 times as fast as the un-catalyzed rate, indicating the catalytic proficiency of the enzyme is tolerant to amino acid substitutions.
Figure 4.
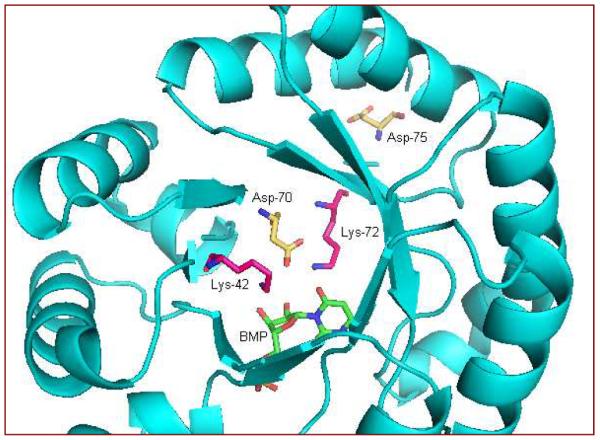
Partial view of Methanothermobacter thermautotrophicus OMPDC in complex with BMP (PDB: 1LOR)138 showing four charged side chains Lys-42, Asp-70, Lys-72 and Asp-75.
A different proposal invokes `Electrostatic Stress' by nearby negative charges,141,142 where the carboxylate group of substrate to be removed is forced into the immediate proximity of the Asp-91 (yeast) by the bound phosphate of the substrate. Assuming both carboxylates are indeed negatively charged, this electrostatic repulsion would raise the energy of the ground state, thereby lowering the energy of activation. Results of computational analyses predict that electrostatic stress furnishes the majority of the catalytic driving force for OMP decarboxylation. Several independent experimental and computational studies, along with careful thermodynamic considerations, have questioned the significance of electrostatic stress as a catalytic device for OMPDC.143,144
Based on studies of H to D exchange rates at the C6 product position, as well as in the 5-F substituted product, the authors concluded that both carbanion stabilization and substrate destabilization by the proximal Asp contribute to the overall catalytic efficiency.145 On the basis of experiments with a 6-phosphonic acid derivative of uridine-5'-phosphate, as an analogue of the substrate that cannot be decarboxylated, the authors claimed to also have support for the substrate destabilization idea,146 as did a recent X-ray study at 1.0 Å resolution with product bound.147
When the hydrophobic environment around the substrate was investigated with site-directed mutagenesis, some evidence was obtained for the importance of a hydrogen bond between a backbone NH of the enzyme and the O4 atom,148 along with as much as four orders of magnitude reduction in kcat. The authors suggested that their results supported both the electrostatic repulsion idea via the conserved Asp and the presence of a transient C6 carbanion/carbene intermediate. Iiams et al reported that the substitution S127P has a great impact on kcat/KM,148 supporting the explanation by Lee and Houk149 that the hydrogen bond between O4 of the substrate and NH backbone is important in reducing the energy barrier.
C.4. A concerted protonation-CO2 loss at C6 to avoid an unstable carbanion
The notion that there is a distinct negative charge at C6 on CO2 loss was challenged by a different model proposed by Begley and Ealick on the basis of their structure of OMPDC. This model proposes concerted protonation at C6 and C6-C-carboxyl scission, abrogating the need for stabilization of a distinct carbanion and preceding transition state.128,139,150,151
C.5 Role of conserved lysine in stabilizing the carbanion
There is also a suggestion on the basis of a highly conserved Lys, presumably positively charged, that part of the catalytic power of OMPDC resides in simple electrostatic attraction. Here, formation of the putative C6 carbanion creates favorable dipole interactions with a cationic, active site lysine residue.152 Depending on the distance of these charges (2.7–3.0 Å) and the local dielectric constant, such interactions could indeed change pKa's significantly (the authors refer the reader to a 3–5 pKa unit elevation of the active center His of serine proteases on juxtaposing tetrahedral oxyanionic transition state analogues on the Ser residue).153–156 A prediction made by proponents of the electrostatic attraction theory is that the lysine residue thought to pair with the carbanion will have a perturbed pKa value from its value in solution, so as to optimize its potential as an H-bond donor. It is also argued that the solvation behaviour of the OMPDC active site differs considerably from that of bulk water.157 Indeed it has been estimated that the pKa for C6-H is suppressed by at least 10 units on the enzyme158 compared to the value estimated by Wolfenden's group for the aqueous value,159–161 leading to as much as 14 kcal/mol transition state stabilization.
The H to D exchange at the product C6 position was studied over a range of pH values and the results were interpreted as being consistent with proton transfer from the product (UMP) C6 atom to a neutral conserved lysine nearby thus producing an enzyme-bound vinyl-carbanion (or carbane in resonance with it) and concluding that the enzyme stabilizes the carbanionic intermediate by 1010-fold, consistent with the earlier conclusion above.
C.6. Test of the conversion of intrinsic binding energy to transition state stabilization
In a series of papers Amyes and Richard and coworkers tested the idea by the late WP Jencks162 that the intrinsic binding energy could be converted into transition state stabilization. This notion appears to have been first tested on OMPDC by Sievers and Wolfenden who reported that a substrate analogue lacking the phosphate group underwent decarboxylation at a much-impaired rate.164 In the first communication by Amyes and Richard, 1-(α–D-erythrofuranosyl)orotic acid (EO) was complemented with exogenous phosphite dianion, and it was found that binding of the phosphite away from the reactive center caused a significant lowering of the transition state energy for decarboxylation, consistent with the Jencks theory.165 Further research by the group has explored both altered substrates (such as the use of an electron withdrawing 5-F substituent on the pyrimidine ring) in conjunction with added phosphite dianion, and concluded that phosphodianion binding interactions are utilized to stabilize a rare closed enzyme form that exhibits a high catalytic activity for decarboxylation.166–169 This continuing work not only sheds light on the OMPDC mechanism but also provides a very important protocol for others wishing to study the effects of various parts of sugar phosphates in catalysis.
The continuing fascination with this enzyme (as evidenced by the yearly publication output on it) is justified as it provides additional issues on decarboxylation mechanisms, which extensive studies of the ThDP and PLP mechanisms have not yet provided.
D. Comparison of the catalytic strategies employed by the three enzymes
There is an important parallel in the catalytic strategies employed by ThDP and PLP, while some parallels also appear to exist with all three reactions.
1. Simply forming a covalent linkage with the substrate thus juxtaposing a β-electron sink, the electrophilicity assisting CO2 loss is enhanced by many orders of magnitude. According to computational studies, formation of the external aldimine with substrate per se provides as much as 16 kcal/mol lowering of the barrier to decarboxylation of L-DOPA, while the protein lowers it further by an additional 8 kcal/mol, producing perhaps 5–6 orders of magnitude additional catalysis by the protein.170 At the same time, it had been estimated that YPDC, the prototypical ThDP enzyme lowers the rate of enzymatic decarboxylation of pyruvate by 1012-1013 -fold compared to ThDP in solution.171 Even with ThDP present, benzoin condensations are the prototypical model reactions for ThDP, rather than decarboxylations, and the classical benzoin condensation is also catalyzed by cyanide anion. For decarboxylation of 2-oxo acids we appear to have no reliable experimental models with which to assess the catalytic effects of ThDP itself on the reaction. This factor is obviously absent in the case of OMPDC.
2. The possibility of contribution to catalysis by substrate distortion has been raised especially in the case of OMPDC, where a carboxylate group is juxtaposed very near the departing CO2 of the substrate. This juxtaposition would impart severe electrostatic distortion, were both the substrate carboxylate and the protein Asp carboxylate negatively charged, not a simple question to resolve experimentally. It is to be noted, that while there are a number of ThDP-dependent decarboxylases with Asp and or Glu at their active sites, there are also some with no potentially ionizable or protonated side chains of any kind, such as glyoxylate carboligase, certainly ruling out this mechanism as a universal property of ThDP enzymes. At the same time, as quoted in the section on ThDP, there is evidence of distortion in the pre-decarboxylation covalent intermediate, specifically of the thiazolium ring, perhaps also accelerating the CO2 loss. Perhaps, the fact that ThDP is found in the `V' conformation when enzyme-bound, already strained compared to what is observed for free ThDP and even LThDP, could also be construed as substrate distortion, this by itself then enables intramolecular acid-base catalysis.
3. All three enzymes appear to share an electrostatic stabilization by a positive charge near the departing CO2 group. In some reported structures of PLP-dependent α-amino acid α-decarboxylases and in OMPDC this is provided by a Lys group, while the authors suggest a similar role for the APH+ form of the coenzyme in the pre-decarboxylation intermediate. The observation of the APH+ form on all three enzymes examined so far by solid state NMR methods, along with the elevated pKa's observed for this form on all enzymes where such data are available (seven cases as of writing), suggest such a possibility with ThDP as well. Such a positive charge would provide significant electrostatic stabilization for any negative charge nearby, including the transition state preceding the carbanion product of CO2 loss.
4. Apparently, both ThDP enzymes and OMPDC have evolved to significantly suppress pKa's of weak carbon acids as discussed in the review. Measurement of pKa's on the enzymes themselves provides a very powerful tool to evaluate the effect of the enzyme environment on reactivity. Some of the acidification observed and deduced indirectly is indeed very impressive, perhaps as many as 9–10 orders of magnitude. Such pKa suppressions are almost certainly due to the hydrophobic environment of the protein, and provide the opportunity for quantitative assessment of catalytic contributions.
5. As mentioned in Section A.4, there is significant evidence both from models and on enzymes that ThDP-dependent decarboxylases would accelerate the decarboxylation step, in which the zwitterionic LThDP and analogues would experience reduced charge separation on decarboxylation to the enamine, by creating an apolar (lower effective dielectric constant) environment. Richard and Amyes had pointed out the advantages of such zwitterionic decarboxylation transition states172, including for PLP-dependent decarboxylases and OMPDC, in addition to ThDP-dependent enzymes. While ThDP- and PLP-dependent decarboxylases offer clear examples, charge concentration experienced for rate reduction appears to be more modest in the case of OMPDC according to current understanding.
6. Regarding the issue of the form of CO2 released from the active site, whether as carbon dioxide or bicarbonate, Kluger and associates recently proposed a mechanism according to which the pre-decarboxylation intermediate of the enzyme benzoylformate decarboxylase (analogous to LThDP with a phenyl ring replacing the methyl group in YPDC) undergoes hydration of the carboxylate group, followed by expulsion of bicarbonate concerted with C-C scission.173 The hypothesis is based on decarboxylation model studies for BFDC. While an intriguing idea, the mechanism cannot be universal for all ThDP decarboxylases, given that some enzymes such as GCL have no serine residues at their active centers, and the 4'-imino nitrogen appears to be the only conserved nucleophile across all ThDP-dependent decarboxylases. At the same time, there were early studies by Vennesland and coworkers, who in the 1960's identified CO2 to be released by GCL.174 For further test of the Kluger hypothesis, more research is required addressing whether CO2 or HCO3 leaves the active center on the ThDP decarboxylases themselves, and indeed on other decarboxylases.
In conclusion, notwithstanding the large amount of excellent research on all three types of enzyme-catalyzed decarboxylation mechanisms, much remains to be learnt about each, and each class offers its own uniqueness and challenges: ThDP its dual catalytic mode, PLP its need for conformational selection, and OMPDC its deceptively (for a simple looking reaction) difficult mechanism.
OVERVIEW OF THE REVIEW.
Enzymatic decarboxylation mechanisms have been studied for six decades. The more classical studies dealt with decarboxylation of α-amino acids, 2-oxo acids and 3-oxo acids, elucidating pyridoxal phosphate (PLP), thiamin diphosphate (ThDP), and either Schiff-base (lysine-dependent) or divalent metal-ion dependent mechanisms, respectively. There have been numerous reviews of all such decarboxylations, most recently a general review in Bioorganic Chemistry in 2012.1 In view of this, the authors have selected only three such enzyme families, for which much mechanistic detail has been obtained during the past decade, focusing on the mechanistic aspects of two coenzyme-dependent and one coenzyme-independent reaction: The ThDP-dependent decarboxylation of 2-oxo acids, the PLP-dependent α-decarboxylation of α-amino acids (both of them examples of electrophilic covalent catalysis), and orotidine monophosphate decarboxylase (OMPDC) an enzyme carrying out its reaction via non-covalent complexes that offers a continuing mechanistic challenge. ThDP is of special and long-standing interest to the authors and is the only one of the three enzyme types discussed, which appears to have dual catalytic functions, electrophilic covalent and acid-base, the latter only recently elucidated. The plethora of methods used in these studies also demonstrates the maturity of the field.
Acknowledgements
The authors are grateful to Jordan group members whose names appear in this review, with special thanks to Dr. Natalia Nemeria, a long-term pivotal presence in the Jordan laboratory. Fruitful collaborations over the years are gratefully acknowledged with: William Furey on the structures of YPDC and E1ec; John Guest for invaluable advice on PDHc from E. coli; Kai Tittmann, Gerhard Hübner for NMR studies of intermediates; Mike McLeish, George Kenyon and (the late) Mimi Hasson, Gabriel Brandt, Dagmar Ringe and Gregory Petsko for structural work on BAL and BFDC; David Chipman on GCL; Caren Freel Meyers on DXP synthase, and Mulchand Patel and Lioubov Korotchkina on human PDHc. Finally, financial support of research on thiamin by the NIH (currently GM050380) is gratefully acknowledged.
Supported by NIH grant GM050380.
Glossary of abbreviations used
- ThDP
thiamin diphosphate
- YPDC
yeast pyruvate decarboxylase from Saccharomyces cerevisiae
- PDHc
pyruvate dehydrogenase complex
- TK
transketolase
- POX
pyruvate oxidase from Lactobacillus plantarum
- BFDC
benzoylformate decarboxylase
- BAL
benzaldehyde lyase;
- E1ec
the first component of the E. coli PDHc
- E1h
the first component of the human PDHc
- PDA
photodiode array
- Sf
stopped-flow
- CD
circular dichroism
- IP
the 1',4'-iminopyrimidine tautomer of ThDP or its C2-substituted derivatives
- AP
the canonical 4-aminopyrimidine tautomer of ThDP or its C2-substituted derivatives
- APH+
the N1-protonated 4-aminopyridinium form of ThDP or its C2-substituted derivatives
- Yl
the C2-carbanion/ylide/carbene form conjugate base of ThDP
- HEThDP
C2α-hydroxyethylThDP, the adduct of acetaldehyde and ThDP
- HBThDP
C2α-hydroxybenzylThDP, the adduct of benzaldehyde and ThDP
- LThDP
C2α-lactylThDP, the adduct of pyruvic acid and ThDP
- MThDP
C2α-mandelylThDP, the adduct of benzoylformic acid and ThDP
- MAP
methyl acetylphosphonate
- MBP
benzoylphosphonic acid monomethyl ester
- PMThDP
C2α-phosphonomandelylThDP, the adduct of MBP and ThDP
- PLThDP
C2α-phosphonolactylThDP, the adduct of MAP and ThDP
- 2-AcThDP
2-acetylThDP
- 3-PKB
(E)-4-(pyrid-3-yl)-2-oxo-3-butenoic acid
- GCL
glyoxylate carboligase
- DXP synthase
1-deoxy-D-xylulose-5-phosphate synthase
- PLP
pyridoxal -5-phosphate
- DDC
dopa decarboxylase
- OMPDC
orotidine 5' -monophosphate decarboxylase
- UMP
uridine 5' –monophosphate
- EO
1-(α-D-erythrofuranosyl)orotic acid)
- BMP
6-hydroxyuridine-5'-phosphate
- PDB
protein data bank
References
- (1).Li T, Huo L, Pulley C, Liu A. Bioorg. Chem. 2012;43:2–14. doi: 10.1016/j.bioorg.2012.03.001. [DOI] [PubMed] [Google Scholar]
- (2).Krampitz LO. Thiamin Diphosphate and its Catalytic Functions. Marcel Dekker; N.Y.: 1970. [Google Scholar]
- (3).Sable HZ, Gubler CJ, editors. Thiamin, 20 Years of Progress,Ann. N.Y. Acad. Sci. 1982;378:7–122. [Google Scholar]
- (4).Kluger R. Chem. Rev. 1987;87:863–876. [Google Scholar]
- (5).Schellenberger A, Schowen RL, editors. Thiamin Pyrophosphate Biochemistry. Vol. 1. CRC Press; Boca Raton, Fl: 1988. [Google Scholar]
- (6).Bisswanger H, Ullrich J, editors. Biochemistry and Physiology of Thiamin Diphosphate Enzymes. VCH Publishers, Weinheim; Germany: 1991. [Google Scholar]
- (7).Bisswanger H, Schellenberger A, editors. Biochemistry and Physiology of Thiamin Diphosphate Enzymes. A.u.C. Intemann, Wissenschaftlicher Verlag, Prien; Germany: 1996. [Google Scholar]
- (8).Schellenberger A. Biochim. Biophys. Acta. 1998;1385:177–186. doi: 10.1016/s0167-4838(98)00067-3. [DOI] [PubMed] [Google Scholar]
- (9).Frank RA, Leeper FJ, Luisi BF. Cell Mol Life Sci. 2007;64:892–905. doi: 10.1007/s00018-007-6423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Pohl M, Sprenger GA, Müller M. Curr Opin Biotechnol. 2004;15:335–42. doi: 10.1016/j.copbio.2004.06.002. [DOI] [PubMed] [Google Scholar]
- (11).Jordan F, Patel MS, editors. Thiamine. Catalytic Mechanisms in Normal and Disease States. Marcel Dekker, Inc.; New York, NY: 2004. [Google Scholar]
- (12).Jordan F. FEBS Lett. 1999;457:298–301. doi: 10.1016/s0014-5793(99)01061-3. [DOI] [PubMed] [Google Scholar]
- (13).Jordan F. Nat. Prod. Rep. 2003;20:184–201. doi: 10.1039/b111348h. [DOI] [PubMed] [Google Scholar]
- (14).Jordan F, Nemeria NS. Bioorg. Chem. 2005;33:190–215. doi: 10.1016/j.bioorg.2005.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lindqvist Y, Schneider G, Ermler V, Sundstrom M. EMBO J. 1992;11:2373–2379. doi: 10.1002/j.1460-2075.1992.tb05301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Muller Y, Schulz G. Science. 1993;259:965–967. doi: 10.1126/science.8438155. [DOI] [PubMed] [Google Scholar]
- (17).Dyda F, Furey W, Swaminathan S, Sax M, Farrenkopf B, Jordan F. Biochemistry. 1993;32:6165–6170. doi: 10.1021/bi00075a008. [DOI] [PubMed] [Google Scholar]
- (18).Arjunan P, Umland T, Dyda F, Swaminathan S, Furey W, Sax M, Farrenkopf B, Gao Y, Zhang D, Jordan F. J. Mol. Biol. 1996;256:590–600. doi: 10.1006/jmbi.1996.0111. [DOI] [PubMed] [Google Scholar]
- (19).Hawkins CF, Borges A, Perham RN. FEBS Lett. 1989;255:77–82. doi: 10.1016/0014-5793(89)81064-6. [DOI] [PubMed] [Google Scholar]
- (20).Breslow R. J.Am.Chem.Soc. 1957;79:1762–1763. [Google Scholar]
- (21).Muller YA, Lindqvist Y, Furey W, Schulz GE, Jordan F, Schneider G. Structure. 1993;1:95–103. doi: 10.1016/0969-2126(93)90025-c. [DOI] [PubMed] [Google Scholar]
- (22).Kluger R, Tittmann K. Chem. Rev. 2008;108:1797–1833. doi: 10.1021/cr068444m. [DOI] [PubMed] [Google Scholar]
- (23).Sax M, Pulsinelli P, Pletcher J. J. Am. Chem. Soc. 1974;96:155–165. doi: 10.1021/ja00808a026. [DOI] [PubMed] [Google Scholar]
- (24).Pletcher J, Sax M, Blank G. J Am Chem Soc. 1977;99:1396–403. doi: 10.1021/ja00447a019. [DOI] [PubMed] [Google Scholar]
- (25).Jordan F. J. Am.Chem.Soc. 1976;98:808–813. doi: 10.1021/ja00419a030. [DOI] [PubMed] [Google Scholar]
- (26).Jordan F, Mariam YH. J. Am.Chem.Soc. 1978;100:2534–2541. [Google Scholar]
- (27).Jordan F, Chen G, Nishikawa S, Sundoro-Wu B. Ann. New York Acad. Sci. 1982;378:14–21. doi: 10.1111/j.1749-6632.1982.tb31183.x. [DOI] [PubMed] [Google Scholar]
- (28).Jordan F. J. Org. Chem. 1982;47:2748–2753. [Google Scholar]
- (29).Kochetov GA, Usmanov RA. Biochem Biophys Res Commun. 1970;41:1134–40. doi: 10.1016/0006-291x(70)90203-2. [DOI] [PubMed] [Google Scholar]
- (30).Jordan F, Zhang Z, Sergienko E. Bioorg. Chem. 2002;30:188–198. doi: 10.1006/bioo.2002.1249. [DOI] [PubMed] [Google Scholar]
- (31).Jordan F, Nemeria N, Zhang S, Yan Y, Arjunan P, Furey W. J. Am. Chem. Soc. 2003;125:12732–12738. doi: 10.1021/ja0346126. [DOI] [PubMed] [Google Scholar]
- (32).Nemeria N, Baykal A, Joseph E, Zhang S, Yan Y, Furey W, Jordan F. Biochemistry. 2004;43:6565–6575. doi: 10.1021/bi049549r. [DOI] [PubMed] [Google Scholar]
- (33).Nemeria N, Chakraborty S, Baykal A, Korotchkina LG, Patel MS, Jordan F. Proc.Natl.Acad.Sci. USA. 2007;104:78–82. doi: 10.1073/pnas.0609973104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Nemeria N, Korotchkina LG, McLeish MJ, Kenyon GL, Patel MS, Jordan F. Biochemistry. 2007;46:10739–10744. doi: 10.1021/bi700838q. [DOI] [PubMed] [Google Scholar]
- (35).Baykal A, Kakalis L, Jordan F. Biochemistry. 2006;45:7522–7528. doi: 10.1021/bi060395k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Cain AH, Sullivan GR, Roberts JD. J. Am.Chem.Soc. 1977;99:6423–6425. doi: 10.1021/ja00461a040. [DOI] [PubMed] [Google Scholar]
- (37).Balakrishnan A, Paramasivam S, Chakraborty S, Polenova T, Jordan F. J. Am.Chem.Soc. 2012;134:665–672. doi: 10.1021/ja209856x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Seifert F, Golbik R, Brauer J, Lilie H, Schröder-Tittmann K, Hinze E, Korotchkina LG, Patel MS, Tittmann K. Biochemistry. 2006;45:12775–12785. doi: 10.1021/bi061582l. [DOI] [PubMed] [Google Scholar]
- (39).Washabaugh MW, Jencks WP. Biochemistry. 1988;27:5044–5053. doi: 10.1021/bi00414a015. [DOI] [PubMed] [Google Scholar]
- (40).Kern D, Kern G, Neef H, Tittmann K, Killenberg-Jabs M, Wikner C, Schneider G, Hübner G. Science. 1997;275:67–70. doi: 10.1126/science.275.5296.67. [DOI] [PubMed] [Google Scholar]
- (41).Arduengo AJ, Goerlich JR, Marshall WJ. J. Liebigs Ann. Chem. 1997:365–374. [Google Scholar]
- (42).Tittmann K, Golbik R, Uhlemann K, Khailova L, Schneider G, Patel M, Jordan F, Chipman DM, Duggleby RG, Hübner G. Biochemistry. 2003;42:7885–7891. doi: 10.1021/bi034465o. [DOI] [PubMed] [Google Scholar]
- (43).Kluger R, Pike DC. J. Am. Chem. Soc. 1977;99:4504–4506. doi: 10.1021/ja00455a052. [DOI] [PubMed] [Google Scholar]
- (44).Kluger R, Pike DC. J. Am. Chem. Soc. 1979;101:6425–6428. [Google Scholar]
- (45).Frey PA. In: Thiamine. Catalytic Mechanisms in Normal and Desease States. Jordan F, Patel MS, editors. Marcel Dekker, Inc.; New York, NY: 2004. [Google Scholar]
- (46).Kale S, Ulas G, Song J, Brudvig GW, Jordan F. Proc. Natl. Acad. Sci. USA. 2008;105:1158–1163. doi: 10.1073/pnas.0709328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Brandt GS, Nemeria N, Chakraborty S, McLeish MJ, Yep , Kenyon GL, Petsko GA, Jordan F, Ringe D. Biochemistry. 2008;47:7734–7743. doi: 10.1021/bi8004413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Brandt GS, Kneen M, Chakraborty S, Baykal A, Nemeria N, Yep A, Ruby D, Petsko GA, Kenyon GL, McLeish MJ, Jordan F, Ringe D. Biochemistry. 2009;48:3247–3257. doi: 10.1021/bi801950k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Sergienko EA, Jordan F. Biochemistry. 2001;40:7382–7403. doi: 10.1021/bi002857e. [DOI] [PubMed] [Google Scholar]
- (50).Sergienko EA, Jordan F. Biochemistry. 2002;41:3952–3967. doi: 10.1021/bi011860a. [DOI] [PubMed] [Google Scholar]
- (51).Sergienko EA, Wang J, Polovnikova L, Hasson MS, McLeish MJ, Kenyon GL, Jordan F. Biochemistry. 2000;39:13862–13869. doi: 10.1021/bi001214w. [DOI] [PubMed] [Google Scholar]
- (52).Arjunan P, Sax M, Brunskill A, Chandrasekhar K, Nemeria N, Zhang S, Jordan F, Furey W. J. Biol. Chem. 2006;281:15296–15303. doi: 10.1074/jbc.M600656200. [DOI] [PubMed] [Google Scholar]
- (53).Wille G, Meyer D, Steinmetz A, Hinze E, Golbik R, Tittmann K. Nat. Chem. Biol. 2006;2:324–328. doi: 10.1038/nchembio788. [DOI] [PubMed] [Google Scholar]
- (54).Chakraborty S, Nemeria N, Yep A, McLeish MJ, Kenyon GL, Jordan F. Biochemistry. 2008;47:3800–3809. doi: 10.1021/bi702302u. [DOI] [PubMed] [Google Scholar]
- (55).Zhang S. Ph.D. Dissertation. Rutgers University, Campus at Newark; NJ: 2003. [Google Scholar]
- (56).Chakraborty S. Ph.D. Dissertation. Rutgers University, Campus at Newark; NJ: 2007. [Google Scholar]
- (57).Joseph E. Ph.D. Dissertation. Rutgers University, Campus at Newark; NJ: 2005. [Google Scholar]
- (58).Chakraborty S, Nemeria NS, Balakrishnan A, Brandt GS, Kneen MM, Yep A, McLeish MJ, Kenyon GL, Petsko GA, Ringe D, Jordan F. Biochemistry. 2009;48:981–994. doi: 10.1021/bi801810h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Zhang S, Liu M, Yan Y, Zhang Z, Jordan F. J. Biol. Chem. 2004;279:54312–54318. doi: 10.1074/jbc.M409278200. [DOI] [PubMed] [Google Scholar]
- (60).Patel H, Nemeria NS, Brammer LA, Freel Meyers CL, Jordan F. J. Am. Chem. Soc. 2012;134:18374–18279. doi: 10.1021/ja307315u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Jordan F, Kudzin ZH, Rios CB. J. Am. Chem. Soc. 1987;109:4415–4417. [Google Scholar]
- (62).Rios CB. PhD thesis. Rutgers University; 1988. [Google Scholar]
- (63).Barletta GL, Huskey WP, Jordan F. J. Am. Chem. Soc. 1992;114:7607–7608. [Google Scholar]
- (64).Barletta GL, Huskey WP, Jordan F. J. Am. Chem. Soc. 1997;119:2356–2362. [Google Scholar]
- (65).Limbach HH, Chan-Huot M, Sharif S, Tolstoy PM, Shenderovich IG, Denisov GS, Toney MD. Biochim Biophys Acta. 2011;1814:1426–1437. doi: 10.1016/j.bbapap.2011.06.004. [DOI] [PubMed] [Google Scholar]
- (66).Gruys KJ, Halkides CJ, Frey PA. Biochemistry. 1987;26:7575–7585. doi: 10.1021/bi00398a007. [DOI] [PubMed] [Google Scholar]
- (67).Bertagnolli BL, Hager LP. J. Biol. Chem. 1991;266:10168–10173. [PubMed] [Google Scholar]
- (68).Khailova LS, Korochkina LG, Severin SE. Ann. N.Y.Acad.Sci. 1989;573:37–54. doi: 10.1111/j.1749-6632.1989.tb14985.x. [DOI] [PubMed] [Google Scholar]
- (69).Jordan F, Li H, Brown AM. Biochemistry. 1999;38:6369–6373. doi: 10.1021/bi990373g. [DOI] [PubMed] [Google Scholar]
- (70).Zhang S, Zhou L, Nemeria N, Yan Y, Zhang Z, Zou Y, Jordan F. Biochemistry. 2005;44:2237–2243. doi: 10.1021/bi047696j. [DOI] [PubMed] [Google Scholar]
- (71).Pan K, Jordan F. Biochemistry. 1998;37:1357–1364. doi: 10.1021/bi971835y. [DOI] [PubMed] [Google Scholar]
- (72).Nemeria N, Arjunan P, Brunskill A, Sheibani A, Wei W, Yan Y, Zhang S, Jordan F, Furey W. Biochemistry. 2002;41:15459–15467. doi: 10.1021/bi0205909. [DOI] [PubMed] [Google Scholar]
- (73).Kerscher L, Oesterhelt D. Eur J Biochem. 1981;116:595–600. doi: 10.1111/j.1432-1033.1981.tb05377.x. [DOI] [PubMed] [Google Scholar]
- (74).Chabriere E, Charon M, Volbeda A, Pieulle L, Hatchikian E, Fontecilla-Camps J. Nature Struct. Biol. 1999;6:182–190. doi: 10.1038/5870. [DOI] [PubMed] [Google Scholar]
- (75).Barletta G, Chung AC, Rios CB, Jordan F, Schlegel JM. J. Am. Chem. Soc. 1990;112:8144–8149. [Google Scholar]
- (76).Ragsdale SW. Chem. Rev. 2003;103:2333–2346. doi: 10.1021/cr020423e. [DOI] [PubMed] [Google Scholar]
- (77).Tittmann K, Wille G, Golbik R, Weidner A, Ghisla S, Hübner G. Biochemistry. 2005;44:13291–13303. doi: 10.1021/bi051058z. [DOI] [PubMed] [Google Scholar]
- (78).Kaplun A, Binshtein E, Vyazmensky M, Steinmetz A, Barak Z, Chipman DM, Tittmann K, Shaanan B. Nat Chem Biol. 2008;4:113–118. doi: 10.1038/nchembio.62. [DOI] [PubMed] [Google Scholar]
- (79).Jordan F, Kuo DJ, Monse EU. J. Am. Chem. Soc. 1978;100:2872–2878. [Google Scholar]
- (80).DeNiro MJ, Epstein S. Science. 1977;197:261–263. doi: 10.1126/science.327543. [DOI] [PubMed] [Google Scholar]
- (81).O'Leary MH. Biochem Biophys Res Commun. 1976;73:614–618. doi: 10.1016/0006-291x(76)90854-8. [DOI] [PubMed] [Google Scholar]
- (82).Jordan F, Kuo DJ, Monse EU. J. Org. Chem. 1978;43:2828–2830. [Google Scholar]
- (83).Huhta DW, Heckenthaler T, Alvarez FJ, Ermer J, Hübner G, Schellenberger A, Schowen RL. Acta Chem Scand. 1992;46:778–788. doi: 10.3891/acta.chem.scand.46-0778. [DOI] [PubMed] [Google Scholar]
- (84).Alvarez F. j., Ermer J, Huebner G, Schellenberger A, Schowen RL. J. Am. Chem. Soc. 1995;117:1678–1683. [Google Scholar]
- (85).Wang J, Golbik R, Seliger B, Spinka M, Tittmann K, Hübner G, Jordan F. Biochemistry. 2001;40:1755–1763. doi: 10.1021/bi001003r. [DOI] [PubMed] [Google Scholar]
- (86).Wei W, Liu M, Jordan F. Biochemistry. 2002;41:451–461. doi: 10.1021/bi0112964. [DOI] [PubMed] [Google Scholar]
- (87).Joseph E, Wei W, Tittmann K, Jordan F. Biochemistry. 2006;45:13517–13527. doi: 10.1021/bi0615588. [DOI] [PubMed] [Google Scholar]
- (88).Nemeria N, Tittmann K, Joseph E, Zhou L, Vazquez-Coll MB, Arjunan P, Hübner G, Furey W, Jordan F. J. Biol. Chem. 2005;280:21473–21482. doi: 10.1074/jbc.M502691200. [DOI] [PubMed] [Google Scholar]
- (89).Balakrishnan A, Nemeria NS, Chakraborty S, Kakalis L, Jordan F. J. Am. Chem. Soc. 2012;134:18644–18655. doi: 10.1021/ja3062375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Shim DJ, Nemeria NS, Balakrishnan A, Patel H, Song J, Wang J, Jordan F, Farinas ET. Biochemistry. 2011;50:7705–7709. doi: 10.1021/bi200936n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Nemeria N, Binshtein E, Patel H, Balakrishnan A, Vered I, Shaanan B, Barak Z, Chipman D, Jordan F. Biochemistry. 2012;51:7940–7952. doi: 10.1021/bi300893v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Yep A, Kenyon GL, McLeish MJ. Proc Natl Acad Sci USA. 2008;105:5733–5738. doi: 10.1073/pnas.0709657105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Arjunan P, Nemeria N, Brunskill A, Chandrasekhar K, Sax M, Yan Y, Jordan F, Guest JR, Furey W. Biochemistry. 2002;41:5213–5221. doi: 10.1021/bi0118557. [DOI] [PubMed] [Google Scholar]
- (94).Arjunan P, Sax M, Brunskill A, Chandrasekhar K, Nemeria N, Zhang S, Jordan F, Furey W. J. Biol. Chem. 2006;281:15296–15303. doi: 10.1074/jbc.M600656200. [DOI] [PubMed] [Google Scholar]
- (95).Kale S, Arjunan P, Furey W, Jordan F. J.Biol.Chem. 2007;282:28106–28116. doi: 10.1074/jbc.M704326200. [DOI] [PubMed] [Google Scholar]
- (96).Kale S. PhD Dissertation. Rutgers University Graduate Faculty; Newark: 2008. Characterization of the dynamics in the E1 component of Escherichia coli Pyruvate Dehydrogenase. [Google Scholar]
- (97).Wei W, Li H, Nemeria N, Jordan F. Protein Expression & Purification. 2003;28:140–150. doi: 10.1016/s1046-5928(02)00674-5. [DOI] [PubMed] [Google Scholar]
- (98).Crosby J, Lienhard G. e. J. Am. Chem. Soc. 1970;92:5707–5716. doi: 10.1021/ja00722a027. [DOI] [PubMed] [Google Scholar]
- (99).Crosby J, Stone R, Lienhard G. e. J. Am. Chem. Soc. 1970;92:2891–2900. doi: 10.1021/ja00712a048. [DOI] [PubMed] [Google Scholar]
- (100).Kemp DS, O'Brien JT. J. Am. Chem. Soc. 1970;92:2554–2555. doi: 10.1021/ja00711a062. [DOI] [PubMed] [Google Scholar]
- (101).Ullrich J, Donner I. Hoppe Seylers Z. Physiol Chem. 1970;351:1030–1034. doi: 10.1515/bchm2.1970.351.2.1030. [DOI] [PubMed] [Google Scholar]
- (102).Amadasi A, Bertoldi M, Contestabile R, Bettati S, Cellini B, di Salvo ML, Borri-Voltattorni C, Bossa F, Mozzarelli A. Curr med chem. 2007;14:1291–1324. doi: 10.2174/092986707780597899. [DOI] [PubMed] [Google Scholar]
- (103).Silverman RB. Angew Chem Int Ed Engl. 2008;47:3500–3504. doi: 10.1002/anie.200704280. [DOI] [PubMed] [Google Scholar]
- (104).Sandmeier E, Hale TI, Christen P. Eur J Biochem. 1994;221:997–1002. doi: 10.1111/j.1432-1033.1994.tb18816.x. [DOI] [PubMed] [Google Scholar]
- (105).Grishin NV, Phillips MA, Goldsmith EJ. Protein Sci. 1995;4:1291–1304. doi: 10.1002/pro.5560040705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Alexander FW, Sandmeier E, Mehta PK, Christen P. Eur J Biochem. 1994;219:953–960. doi: 10.1111/j.1432-1033.1994.tb18577.x. [DOI] [PubMed] [Google Scholar]
- (107).John RA. Biochim Biophys Acta. 1995;1248:81–96. doi: 10.1016/0167-4838(95)00025-p. [DOI] [PubMed] [Google Scholar]
- (108).Jansonius JN. Curr Opin Struct Biol. 1998;8:759–769. doi: 10.1016/s0959-440x(98)80096-1. [DOI] [PubMed] [Google Scholar]
- (109).Metzler DE, Ikawa M, Snell EE. J. Am. Chem. Soc. 1954;76:648–652. [Google Scholar]
- (110).Jackson LK, Baldwin J, Akella R, Goldsmith EJ, Phillips MA. Biochemistry. 2004;43:12990–12999. doi: 10.1021/bi048933l. [DOI] [PubMed] [Google Scholar]
- (111).Grishin NV, Osterman AL, Brooks HB, Phillips MA, Goldsmith EJ. Biochemistry. 1999;38:15174–15184. doi: 10.1021/bi9915115. [DOI] [PubMed] [Google Scholar]
- (112).Swanson T, Brooks HB, Osterman AL, O'Leary MH, Phillips MA. Biochemistry. 1998;37:14943–14937. doi: 10.1021/bi981154i. [DOI] [PubMed] [Google Scholar]
- (113).Oliveira EF, Cerqueira NMFSA, Fernandes PA, Ramos MJ. J Ame Chem Soc. 2011;133:15496–15505. doi: 10.1021/ja204229m. [DOI] [PubMed] [Google Scholar]
- (114).Eliot AC, Kirsch JF. Annu Rev Biochem. 2004;73:383–415. doi: 10.1146/annurev.biochem.73.011303.074021. [DOI] [PubMed] [Google Scholar]
- (115).Dunathan HC. Proc. Natl. Acad. Sci. USA. 1966;55:712–716. doi: 10.1073/pnas.55.4.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Toney MD. Biochim. Biophys. Acta. 2011;1814:1407–1418. doi: 10.1016/j.bbapap.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Jackson LK, Brooks HB, Myers DP, Phillips MA. Biochemistry. 2003;42:2933–2940. doi: 10.1021/bi026795z. [DOI] [PubMed] [Google Scholar]
- (118).Fogle EJ, Toney MD. Biochemistry. 2010;49:6485–6493. doi: 10.1021/bi100648w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Deng X, Lee J, Michael AJ, Tomchick DR, Goldsmith EJ, Phillips MA. J Biol Chem. 2010;285:25708–25719. doi: 10.1074/jbc.M110.121137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Lin YL, Gao J. J Am Chem Soc. 2011;133:4398–4403. doi: 10.1021/ja108209w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Lin YL, Gao J, Rubinstein A, Major DT. Biochim Biophys Acta. 2011;1814:1438–1446. doi: 10.1016/j.bbapap.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Burkhard P, Dominici P, Borri-Voltattorni C, Jansonius JN, Malashkevich VN. Nat Struct Biol. 2001;8:963–967. doi: 10.1038/nsb1101-963. [DOI] [PubMed] [Google Scholar]
- (123).Griswold WR, Toney MD. J Am Chem Soc. 2011;133:14823–14830. doi: 10.1021/ja2061006. [DOI] [PubMed] [Google Scholar]
- (124).Gokulan K, Rupp B, Pavelka MS, Jr., Jacobs WR, Jr., Sacchettini JC. J Biol Chem. 2003;278:18588–18596. doi: 10.1074/jbc.M301549200. [DOI] [PubMed] [Google Scholar]
- (125).Zhou X, Jin X, Medhekar R, Chen X, Dieckmann T, Toney MD. Biochemistry. 2001;40:1367–1377. doi: 10.1021/bi001237a. [DOI] [PubMed] [Google Scholar]
- (126).Schnackerz KD, Andi B, Cook PF. Biochim Biophys Acta. 2011;1814:1447–1458. doi: 10.1016/j.bbapap.2011.02.001. [DOI] [PubMed] [Google Scholar]
- (127).Lieberman I, Kornberg A, Simms ES. J Biol Chem. 1955;215:403–451. [PubMed] [Google Scholar]
- (128).Appleby TC, Kinsland C, Begley TP, Ealick SE. Proc Natl Acad Sci U S A. 2000;97:2005–2010. doi: 10.1073/pnas.259441296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Radzicka A, Wolfenden R. Science. 1995;267:90–93. doi: 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- (130).Miller BG, Wolfenden R. Annu Rev Biochem. 2002;71:847–885. doi: 10.1146/annurev.biochem.71.110601.135446. [DOI] [PubMed] [Google Scholar]
- (131).Krungkrai SR, Tokuoka K, Kusakari Y, Inoue T, Adachi H, Matsumura H, Takano K, Murakami S, Mori Y, Kai Y, Krungkrai J, Horii T. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006;62:542–545. doi: 10.1107/S1744309106015594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Tokuoka K, Kusakari Y, Krungkrai SR, Matsumura H, Kai Y, Krungkrai J, Horii T, Inoue T. J. Biochem. 2008;143:69–78. doi: 10.1093/jb/mvm193. [DOI] [PubMed] [Google Scholar]
- (133).Smailey JA. Top Curr Chem. 2004;238:63–78. [Google Scholar]
- (134).Callahan BP, Miller BG. Bioorg Chem. 2007;35:465–469. doi: 10.1016/j.bioorg.2007.07.004. [DOI] [PubMed] [Google Scholar]
- (135).Beak P, Siegel B. J. Am. Chem. Soc. 1976;98:3601–3606. doi: 10.1021/ja00428a035. [DOI] [PubMed] [Google Scholar]
- (136).Ehrlich JI, Hwang CC, Cook PF, Blanchard JS. J. Am. Chem. Soc. 1999;121:6966. [Google Scholar]
- (137).Rishavy MA, Cleland WW. Biochemistry. 2000;39:4569. doi: 10.1021/bi000376p. [DOI] [PubMed] [Google Scholar]
- (138).Wu N, Pai EF. J. Biol. Chem. 2002;277:28080–28087. doi: 10.1074/jbc.M202362200. [DOI] [PubMed] [Google Scholar]
- (139).Miller BG, Hassell AM, Wolfenden R, Milburn MV, Short SA. Proc Natl Acad Sci U S A. 2000;97:2011–2016. doi: 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Yuan J, Cardenas AM, Gilbert HF, Palzkill T. Protein Sci. 2011;20:1891–906. doi: 10.1002/pro.728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Wu N, Gillon W, Pai EF. Biochemistry. 2002;41:4002–4011. doi: 10.1021/bi015758p. [DOI] [PubMed] [Google Scholar]
- (142).Tran NL, Colvin ME, Gronert S, Wu W. Bioorg. Chem. 2003;31:271–277. doi: 10.1016/s0045-2068(03)00028-2. [DOI] [PubMed] [Google Scholar]
- (143).Warshel A, Strajbl M, Villa J, Florian J. Biochemistry. 2000;39:14728–14738. doi: 10.1021/bi000987h. [DOI] [PubMed] [Google Scholar]
- (144).Callahan BP, Wolfenden R. J. Am. Chem. Soc. 2004;126:14698–14699. doi: 10.1021/ja0450049. [DOI] [PubMed] [Google Scholar]
- (145).Chan KK, Wood BM, Fedorov AA, Fedorov EV, Imker HJ, Amyes TL, Richard JP, Almo SC, Gerlt JA. Biochemistry. 2009;48:5518–5531. doi: 10.1021/bi900623r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Thirumalairajan S, Mahaney B, Bearne SL. Chem Commun (Camb) 2010;46:3158–3160. doi: 10.1039/b926894d. [DOI] [PubMed] [Google Scholar]
- (147).Fujihashi M, Mito K, Pai EF, Miki K. J Biol Chem. 2013;288:9011–9016. doi: 10.1074/jbc.M112.427252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Iiams V, Desai BJ, Fedorov AA, Fedorov EV, Almo SC, Gerlt JA. Biochemistry. 2011;50:8497–8507. doi: 10.1021/bi2012355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Lee JK, Houk KN. Science. 1997;276:942–945. doi: 10.1126/science.276.5314.942. [DOI] [PubMed] [Google Scholar]
- (150).Wu N, Mo Y, Gao J, Pai EF. Proc. Natl. Acad. Sci. USA. 2000;97:2017–2022. doi: 10.1073/pnas.050417797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Harris P, Navarro Poulsen JC, Jensen KF, Larsen S. Biochemistry. 2000;39:4217–4224. doi: 10.1021/bi992952r. [DOI] [PubMed] [Google Scholar]
- (152).Warshel A, Sharma PK, Kato M, Xiang Y, Liu H, Olsson MHM. Chem. Rev. 2006;106:3210–3235. doi: 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- (153).Adebodun F, Jordan F. J. Cell. Biochem. 1989;40:249–260. doi: 10.1002/jcb.240400213. [DOI] [PubMed] [Google Scholar]
- (154).Zhong S, Haghjoo K, Kettner C, Jordan F. J. Am. Chem. Soc. 1995;117:7047–7055. [Google Scholar]
- (155).Bao D, Cheng J-T, Kettner C, Jordan F. J. Am. Chem. Soc. 1998;120:3485–3489. [Google Scholar]
- (156).Kahyaoglu A, Jordan F. Protein Science. 2002;11:965–973. doi: 10.1110/ps.3890102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Callahan BP, Bell AF, Tonge PJ, Wolfenden R. Bioorg. Chem. 2006;34:59–65. doi: 10.1016/j.bioorg.2005.12.001. [DOI] [PubMed] [Google Scholar]
- (158).Richard JP. Pure Appl Chem. 2011;83:1555–1565. doi: 10.1351/PAC-CON-11-02-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Sievers A, Wolfenden R. J. Am. Chem. Soc. 2002;124:13986–13987. doi: 10.1021/ja021073g. [DOI] [PubMed] [Google Scholar]
- (160).Yeoh FY, Cuasito RR, Capule CC, Wong FM, Wu W. Bioorg. Chem. 2007;35:338–343. doi: 10.1016/j.bioorg.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Tsang WY, Wood BM, Wong FM, Wu W, Gerlt JA, Amyes TL, Richard JP. J Am Chem Soc. 2012;134:14580–14594. doi: 10.1021/ja3058474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Jencks WP. Proc. Natl. Acad. Sci. U.S.A. 1981;78:4046–4050. doi: 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Zou Y. Ph.D Dissertation. Rutgers University Graduate faculty; Newark, NJ: 1999. [Google Scholar]
- (164).Sievers A, Wolfenden R. Bioorg. Chem. 2005;33:45–52. doi: 10.1016/j.bioorg.2004.08.005. [DOI] [PubMed] [Google Scholar]
- (165).Amyes TL, Richard JP, Tait JJ. J Am Chem Soc. 2005;127:15708–15709. doi: 10.1021/ja055493s. [DOI] [PubMed] [Google Scholar]
- (166).Goryanova B, Spong K, Amyes TL, Richard JP. Biochemistry. 2013;52:537–546. doi: 10.1021/bi301650d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Wood BM, Amyes TL, Fedorov AA, Fedorov EV, Shabila A, Almo SC, Richard JP, Gerlt JA. Biochemistry. 2010;49:3514–3516. doi: 10.1021/bi100443a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Goryanova B, Amyes TL, Gerlt JA, Richard JP. J Am Chem Soc. 2011;133:6545–6548. doi: 10.1021/ja201734z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Amyes TL, Ming SA, Goldman LM, Wood BM, Desai BJ, Gerlt JA, Richard JP. Biochemistry. 2012;51:4630–4632. doi: 10.1021/bi300585e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Giardina G, Montioli R, Gianni S, Cellini B, Paiardini A, Voltattorni CB, Cutruzzolà F. Proc Natl Acad Sci U S A. 2011;108:20514–20519. doi: 10.1073/pnas.1111456108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Alvarez FJ, Ermer J, Hübner G, Schellenberger A, Schowen RL. J. Am. Chem. Soc. 1991;113:8402–8409. [Google Scholar]
- (172).Richard JP, Amyes TL. Bioorg. Chem. 2004;32:354–366. doi: 10.1016/j.bioorg.2004.05.002. [DOI] [PubMed] [Google Scholar]
- (173).Howe GW, Bielecki M, Kluger R. J Am Chem Soc. 2012;134:20621–20623. doi: 10.1021/ja310952a. [DOI] [PubMed] [Google Scholar]
- (174).Hall RL, Vennesland B, Kézdy FJ. J Biol Chem. 1969;244:3991–3998. [PubMed] [Google Scholar]