

Abstract
The Northern Fulmar (Fulmarus glacialis) is a common tube-nosed seabird with a disjunct Holarctic range. Taxonomic divisions within the Northern Fulmar have historically been muddled by geographical variation notably including highly polymorphic plumage. Recent molecular analyses (i.e., DNA barcoding) have suggested that genetic divergence between Atlantic and Pacific populations could be on par with those typically observed between species. We employ a multigene phylogenetic analysis to better explore the level of genetic divergence between these populations and to test an old hypothesis on the origin of the modern distribution of color morphs across their range. Additionally, we test whether mutations in the melanocortin-1 receptor gene (MC1R) are associated with dark plumage in the Northern Fulmar. We confirmed that mitochondrial lineages in the Atlantic and Pacific populations are highly divergent, but nuclear markers revealed incomplete lineage sorting. Genetic divergence between these populations is consistent with that observed between many species of Procellariiformes and we recommend elevating these two forms to separate species. We also find that MC1R variation is not associated with color morph but rather is better explained by geographical divergence.
Keywords: Arctic, Atlantic, DNA barcodes, MC1R, Pacific, polymorphism
Introduction
Drawing species limits between allopatric populations is a long-standing problem in biology (Mayr 1963). However, this endeavor has been greatly facilitated in recent decades by the application of molecular methods, which yield data that are less susceptible to homoplasy and inform divergence estimates. Attempts to streamline this molecular approach to taxonomy and to generally address the “taxonomic impediment” contributed to the advent of DNA barcoding – a species identification system reliant on a short but standardized gene region (Hebert et al. 2003a,b). Although species identification is the primary role for this system, it has also been touted as a tool that can expedite the species discovery process by identifying genetically divergent populations within recognized species (Hebert et al. 2004a,b).
How much genetic divergence is necessary to distinguish species is a question that has incited controversy (Moritz and Cicero 2004; DeSalle et al. 2005; Meyer and Paulay 2005; Wiemers and Fiedler 2007). Proponents have advocated various metrics, most notoriously the “10x rule” (Hebert et al. 2004b). This and other thresholds have spurred critics who contend that a threshold of genetic divergence is not an accurate reflection of the speciation process (Hickerson et al. 2006). Kerr et al. (2009a) found that a multi-tiered approach, incorporating additional methods such as character-based measures, was the most effective in accurately portraying species limits, but this approach is heavily reliant on a priori knowledge of species boundaries and performs poorly as a species discovery tool. The current consensus is that a surveying method such as DNA barcoding is best implemented as a first pass approach and that such data should be complemented by alternative sources of evidence before taxonomic boundaries are reconsidered (DeSalle 2006; Padial and de la Riva 2007; Frezal and Leblois 2008).
DNA barcode coverage of extant birds is more comprehensive than in any other taxonomic group. This is attributable to several large-scale geographically oriented surveys (Yoo et al. 2006; Kerr et al. 2007, 2009a,b; Johnsen et al. 2010) and, more recently, to the systematic sampling of one of the world's largest avian museum collections (Schindel et al. 2011). These studies invariably highlight species harboring large intraspecific divergences. Many of these species have already been the subject of more rigorous analyses and simply await authoritative decisions (e.g., Barker et al. 2008; Areta and Pearman 2009), while divergences within other species remain poorly studied, or unknown, and require further review. The Northern Fulmar (Fulmarus glacialis) is a good example from the latter category.
The Northern Fulmar is a widespread seabird in the northern hemisphere, though Atlantic and Pacific populations occur disjunctively (Hatch and Nettleship 1998). Generally, three subspecies are recognized (sensu Clements 2007), one in the Pacific (F. g. rodgersii) and two in the Atlantic (the smaller-billed, high arctic breeder F. g. glacialis, and the larger-billed, boreal F. g. auduboni). Northern Fulmars display highly variable plumage, exhibiting light and dark morphs plus a large suite of intermediates, which historically has helped confound taxonomic boundaries. For example, pale-morph birds from the north Pacific were initially described as a separate species, F. rodgersii (American Ornithologists' Union 1910), while the remainder of Pacific birds were then known as F. g. glupischa (Bent 1964). An additional subspecies was also initially described from the Canadian arctic (F. g. minor), but was later synonymized with the nominate subspecies; some authors have prescribed the same treatment for F. g. auduboni (van Franeker and Luttik 2008). Currently, the American Ornithologists' Union (1998) recognizes F. glacialis as a single species.
Although the two color morphs occur in both the Atlantic and Pacific, their degree of contrast and their latitudinal frequencies vary substantially between these populations. Both the palest and the darkest birds occur in the Pacific, but intermediates are rare, whereas in the Atlantic light and dark morphs contrast less and intermediates are more common (Hatch and Nettleship 1998; Sibley 2000). An even more curious pattern is that light birds in the Pacific tend to be concentrated at higher latitudes, whereas the reverse trend is observed in the Atlantic (van Franeker and Wattel 1982; Warham 1990). To explain this unusual pattern, van Franeker and Wattel (1982) postulated a Pacific origin for the dark morphed birds and suggested that dark Pacific birds could have subsequently invaded the Atlantic during warm interglacial periods, and light Atlantic birds could have contemporaneously recolonized the north Pacific. Under this hypothesis, the authors predicted that additional characters should unite birds by color morph rather than by geography, but this hypothesis has not been formally tested.
While the life history and general biology of the Northern Fulmar has been studied in depth (e.g., Fisher 1952), genetic data is largely absent for this species (Hatch and Nettleship 1998). DNA barcode surveys based on cytochrome c oxidase I (COI) included only a single individual from the Pacific Ocean, but it was found to be roughly 3.2% divergent (based on a Kimura 2-parameter distance model) from the remainder of specimens analyzed, which all originated from sites distributed throughout the North Atlantic (Kerr et al. 2007; Johnsen et al. 2010). However, the color morph of the specimens was not reported in these studies.
In this study, we examine sequence data from both nuclear and mitochondrial loci to obtain a better grasp of genetic variation within the Northern Fulmar. We use multilocus phylogeographic analyses to assess genetic divergence between the Atlantic and Pacific populations and test the hypothesis that dark morph birds evolved in the Pacific. We include in our genetic survey a fragment from the melanocortin-1 receptor gene, from which mutations have been associated with melanic plumage in a host of other species including many avian examples (Mundy 2005). The melanocortin-1 receptor is a G protein-coupled receptor that affects the activity of melanocytes. It is believed that some amino acid substitutions can alter its activity resulting in constitutive activation, effectively leaving the receptor ‘on’, and resulting in an overproduction of eumelanin (Ling et al. 2003). We determine if mutations in this gene are correlated with dark plumage in the Northern Fulmar.
Methods
Taxon sampling
Tissue samples were obtained from various frozen tissue collections and were composed of pectoral muscle or liver samples (see Table S1 for complete list of specimens). One hundred and thirty-four Northern Fulmars from 17 different locations were sampled in total (Fig. 1); 81 samples represented Atlantic populations and 53 were from the Pacific. Whenever possible, tissues with associated voucher skins were used so that color morph could later be determined. Two specimens of Fulmarus glacialoides were included in the analysis, plus single representatives from four of the five remaining members of the Fulmarine clade of petrels: Macronectes giganteus, M. halli, Thalassoica antarctica, and Pagodroma nivea.
Figure 1.
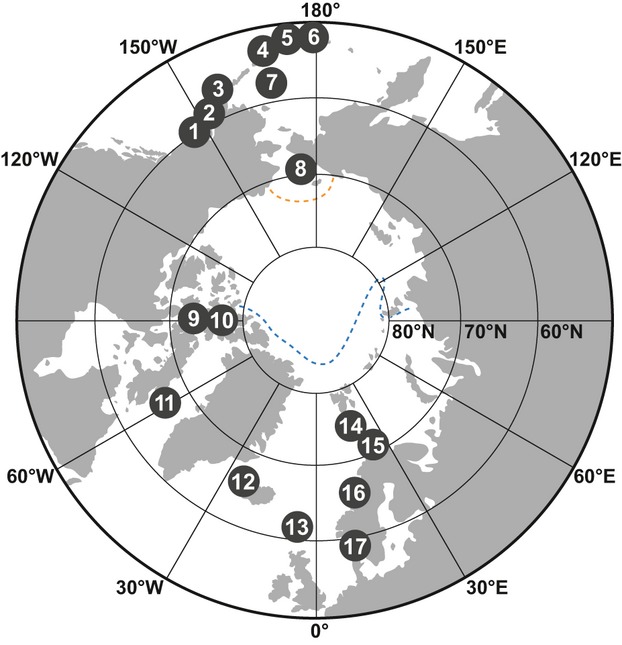
Specimens analyzed in this study originated from 17 collecting sites: 1) Kenai Peninsula, US; 2) Gulf of Alaska; 3) Chowiet I., US; 4) Chagulak I., US; 5) Adak I., US; 6) Amchitka I., US; 7) St. George I., US; 8) Chukchi Sea; 9) Prince Leopold I., CA; 10) Devon I., CA; 11) Baffin I., CA; 12) Faxaflói, IS; 13) Faeroe I., DK; 14) Bear Island, NO; 15) Nordkapp, NO; 16) Trænabanken, NO; 17) Tromlingene, NO. Dashed lines represent the northern range limits for Atlantic (blue) and Pacific (orange) populations, respectively.
DNA extraction, amplification, and sequencing
DNA was extracted from all tissue samples using the DNeasy blood and tissue kit (Qiagen, Valencia, CA), following the manufacturers’ instructions. DNA samples were eluted in 150 μL of ultrapure water. All polymerase chain reactions (PCR) were performed in total volumes of 12.5 μL (8.25 μL molecular grade H2O, 1.25 μL 5× buffer, 0.625 μL 25 mM MgCl2, 0.125 μL of each primer (10 mM), 0.0625 μL 2.5 mM each dNTPs, 0.06 μL GoTaq® Flexi polymerase (Promega, Madison, WI), and 2 μL DNA template). For difficult samples, Platinum® Taq polymerase (Invitrogen, Carlsbad, CA) was used as a substitute along with the manufacturer's reagents. All primer sequences and citations are listed in Appendix 1.
A 694-bp fragment of the mitochondrial gene cytochrome c oxidase I (COI) was amplified using the primer pair BirdF1/COIbirdR2 and the following thermal cycle: 94°C for 2 min; 30 cycles of 95°C for 30 sec, 48°C for 40 sec, 72°C for 45 sec; 72°C for 5 min. Alternative primer pairs included BirdF1/AvMiF1 and COIbirdR2/AvMiR1, which targeted smaller amplicons using internal primers, and LTyr/COI907aH2, which amplified a larger amplicon including part of the tyrosine tRNA.
A 298 bp fragment of the mitochondrial control region (CR) was amplified using the primer pair ND6*-L16406/H505 and the same thermal cycle described above. A 691 bp fragment of myoglobin intron II (MYOII) was amplified using half-nested PCR with the primers Myo2/Myo3 for the initial reaction and Myo2/Myo3F for the second. Both thermal cycles matched that described above, except with an annealing temperature of 59°C rather than 48°C. An 817 bp fragment of melanocortin-1 receptor (MC1R) was amplified using the primer pair MSHR9/MSHR72 and the thermal cycle described above, except with an annealing temperature of 61°C rather than 48°C. Additionally, two pairs of internal primers were designed to amplify sequences from more challenging samples: MC1RNoFu63/MC1RNoFu532 and MC1RNoFu362/MC1RNoFu802. A 499 bp fragment of “intron A” of the Z-linked chromodomain-helicase DNA-binding protein 1 (CHD1-Z) gene was amplified using the primer pair 2669FZ/2718R and the following touchdown thermal cycle: 94°C for 2 min; 10 cycles of 94°C for 30 sec, 60°C (decreasing by 1°C/cycle) for 30 sec, 72°C for 40 sec; 30 cycles of 94°C for 30 sec; 50°C for 30 sec, 72°C for 40 sec; 72°C for 5 min.
PCR templates were purified using ExoSAP-IT® (USB corporation, Cleveland, OH) prior to cycle sequencing. All sequencing reactions used the same primers as for PCR, except for LTyr and COI907aH2, which were sequenced using COIaRt and COI748Ht, respectively. Sequencing was performed bi-directionally (except for CR, which was sequenced with only H505) on an ABI 3730xl sequencer (Applied Biosystems, Foster City, CA). All sequences produced for this study were submitted to GenBank (accession numbers KC755410-KC755549, COI; KC755550-KC755682, CR; KC788940-KC789071, MYOII; KC755695-KC755826, MC1R; KC788823-KC788939, CHD1-Z), as well as uploaded to the data project “Fulmarine Petrels” in BOLD (http://www.boldsystems.org).
Phylogenetic analysis
Alignments were made by eye for each gene fragment using BioEdit version 7.0.5.3 (Hall 1999). Sequences were trimmed to equal lengths where necessary. For nuclear sequences containing heterozygous peaks, the allelic phase of each haplotype was resolved via one of two methods: i) simple heterozygous sequences – that is, with only one or two point mutations – were resolved using the SNP data file format in PHASE version 2.1 (Stephens et al. 2001); ii) complex heterozygous sequences were resolved by cloning. Cloning was performed using the TOPO TA Cloning kit (Invitrogen). To accommodate heterozygosity, secondary alignments were produced for nuclear gene fragments wherein each specimen was represented by two sequences, one to represent each copy (this was not done for CHD1-Z).
For each marker, we calculated haplotype diversity, nucleotide diversity, Tajima's D, and Fu & Li's D* and F* statistics using DnaSP version 5.0 (Rozas and Librado 2009), first using the entire population, and then measuring the Atlantic and Pacific populations separately. Median-joining haplotype networks were also developed separately for each marker using Network 4.6.0.0 (http://www.fluxus-engineering.com).
Phylogenetic trees were constructed using concatenated datasets. Separate trees were built for nuclear and mitochondrial markers; however, CHD1-Z was omitted from the nuclear concatenated data file due to lack of variation. For each concatenated dataset, the best-fit evolutionary model and partition scheme were selected using the Bayesian information criterion (BIC) in the program PartitionFinder version 1.0.1 (Lanfear et al. 2012). Model selection was limited to those implemented in MrBayes version 3.1.2. For each dataset, Bayesian phylogenetic trees were constructed with MrBayes version 3.1 (Huelsenbeck and Ronquist 2001; Ronquist and Huelsenbeck 2003). Data partitions are described in the results. For both datasets all parameters were unlinked, the sampling frequency was set to 5,000, and the first 25% of samples were discarded as burn-in. Each of the analyses were run sufficiently long for the average standard deviation of split frequencies to fall below 0.01 (25,000,000 generations for the nuclear dataset and 10,000,000 for the mitochondrial dataset). The initial results of the nuclear dataset yielded unusually long branch lengths, which is a well-documented ailment of partitioned datasets in MrBayes (Marshall 2010). To solve this problem, we repeated the analysis for the nuclear dataset with the branch length prior mean reduced to 1/100 as recommended by Ward et al. (2010). Pagodroma nivea was selected as the outgroup taxa for both trees (sensu Penhallurick and Wink 2004).
Estimate of divergence time
Divergence time was only estimated for the mitochondrial lineages (see Discussion) and was based on COI using the program BEAST v1.7.1 (Drummond et al. 2012). Each population of F. glacialis was represented by a single sequence. COI sequences from additional species were downloaded from BOLD to provide reasonable coverage of all Procellariiform families, plus an outgroup from the Sphenisciformes; 27 taxa were included in total (Appendix 2). Because variable molecular rates have been proposed for the Procellariiformes (e.g., Nunn and Stanley 1998; Weir and Schluter 2008) we instead used an uncorrelated lognormal relaxed molecular clock with a Yule tree prior and three fossil calibration points to date the tree: Thalassarche-Phoebetria, 5.3 Mya (Wilkinson 1969); Hydrobatidae, 10.25 Mya (Becker 1987); and Diomedeidae 30–31 Mya (Mayr and Smith 2012). All calibration dates used exponential priors with means set to 1.0 (Ho 2007). The GTR+Γ substitution model was selected based on jModeltest (Posada 2008). The gamma shape parameter and substitution parameter priors were also informed by the jModeltest results. For the clock mean, a broad gamma distribution (shape 0.001, scale 1000) was used. Default prior values were accepted for the remaining parameters. The program was run for 50 million generations and sampled every 5,000 generations. The program was rerun subsequently sampling only from the prior to observe the influence on the results. Tracer v1.5 (http://tree.bio.ed.ac.uk/software/tracer/) was used to assess convergence and assure that all effective sample sizes (ESSs) were at least greater than 200.
MC1R and color morphs
Northern Fulmar color morphs are typically divided into four categories: double light, light, dark, and double dark (van Franeker and Wattel 1982; Hatch 1991). For the purpose of this study, only three categories were necessary (two ‘pure bred’ categories, plus hybrids), so birds were only distinguished as being light (= double light), dark (= double dark), or intermediate (= light and dark categories).
Aligned MC1R sequences were translated using the standard genetic code. A previous prediction of the two-dimensional structure of MC1R in birds as collated by Mundy (2005) was used to map amino acid substitutions in the Northern Fulmar to transmembrane and loop sites. Variable amino acid positions were inspected individually for possible correlations between substituted amino acids and dark plumage (light plumage is presumed to be the ancestral condition on the basis of its absence in the more ancestral southern species), and were compared to substitutions linked to melanism in other species.
Results
Sequencing results
Not all markers could be amplified from every specimen (see Appendix 3 or Table S1). Additionally, some ambiguous base calls could not be resolved from the 3′ end of several CR and MYOII sequences, so the alignments for these two genes were trimmed to 228 bp and 650 bp, respectively, to eliminate the affected regions.
Heterozygous peaks occurred in 35% of MYOII sequences and 28% of MC1R sequences. In MYOII, peaks were primarily limited to single occurrences within any given sequence (two at most) and were easily resolved by phasing. In contrast, heterozygous peaks in MC1R numbered as high as eight in several sequences, often appeared at associated sites, and frequently resulted in amino acid substitutions. Twenty-one MC1R sequences could be resolved by phasing, but the remaining 14 necessitated cloning. No heterozygous peaks were observed in CHD1-Z but variation was generally low and any female specimens would only carry a single copy of this gene.
Phylogenetic results
Twelve COI haplotypes were recovered from 53 Pacific birds, and 14 COI haplotypes were recovered from 81 Atlantic birds (Fig. 2). Variation was highly conserved in the Pacific, with 79% of individuals sharing the most common haplotype. The distribution in the Atlantic was more bimodal with 56% of individuals sharing the most common haplotype and 23% sharing the second most common; however, this split did not correlate to any geographical distribution (Fig. 2). CR haplotypes were more variable, with 25 haplotypes recovered from the Pacific and 28 from the Atlantic. Although, in the Atlantic 40% of birds still shared the most common haplotype. Both COI and CR clearly differentiated the Atlantic and Pacific populations with no haplotype sharing. For both markers, the Pacific population exhibited greater divergence from the outgroup taxa.
Figure 2.
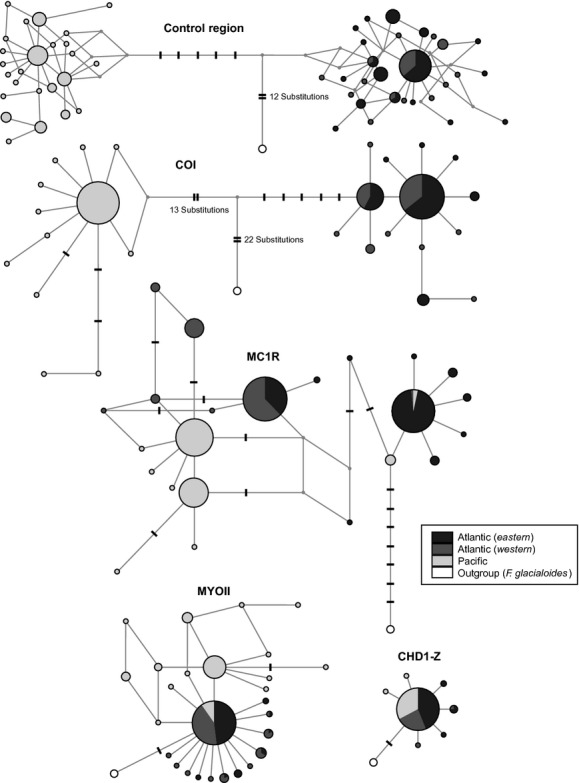
Median-joining haplotype networks for each of the five markers included in this study. The size of the circle is proportional to the number of individuals (CR, COI, and CHD1-Z) or alleles (MC1R and MYOII) with that haplotype. Adjoining circles are separated by a single nucleotide substitution. Additional substitutions are indicated by either hatch marks or, for larger values, a double hatch mark and a number. Dots indicate an unobserved median vector. Shading indicates the specimen's region of origin (the Atlantic region is divided into areas east and west of Greenland, respectively).
Haplotype sharing was observed for each of the nuclear markers, though to varying degrees (Fig. 2). The most extreme case was CHD1-Z, in which 91% of all birds shared the most common haplotype. MYOII presented a more typical case, where a subset of Pacific birds retained the most common haplotype from the Atlantic. MC1R displayed a strongly bimodal distribution within each population and also fewer rare haplotypes. In the Atlantic, one of the common haplotypes was predominantly restricted to areas east of Greenland (Fig. 2). This is also the only MC1R haplotype shared with a Pacific bird, although it was limited to one individual (this sample was reextracted and resequenced for confirmation). A very similar haplotype was recovered from birds of the Pribilof Islands (site #7 in Fig. 1), but was only ever observed in single copy. This haplotype excepted, MC1R and MYOII showed greater divergence in the Pacific as the mitochondrial markers did.
The concatenated mitochondrial gene tree was cleanly bifurcated into Atlantic and Pacific clades with high support (Fig. 3). As indicated by the network diagrams, the Atlantic clade showed no further highly supported subdivisions. The concatenated nuclear gene tree did not differentiate the Atlantic and Pacific clades, but grouped them into one highly supported cluster (Fig. 3). This is presumably due to haplotype sharing, and hence the haplotype networks offer a more informative depiction of the variation observed via nuclear markers.
Figure 3.
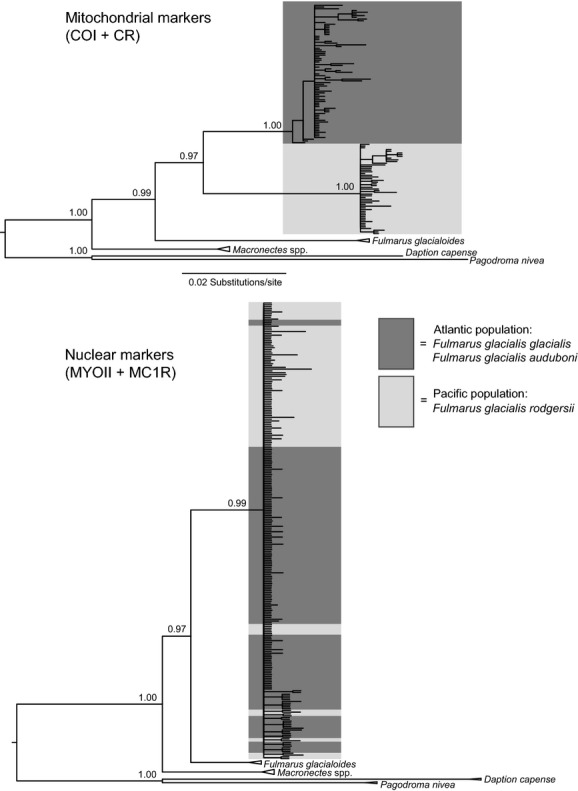
Concatenated gene trees based on mitochondrial and nuclear markers, respectively, estimated using MrBayes. The nuclear tree is based on phased haplotypes.
Divergence time
All ESS values greatly exceeded the minimum required (mean = 4054.11, range = 465.68–9001); however, the 95% highest posterior densities (HPD) for most node ages were relatively broad. The divergence time for the Atlantic and Pacific Northern Fulmar populations was estimated at 1.97 Ma (HPD = 0.72−3.37). Divergence between the Northern and Southern Fulmars was estimated at 2.95 Ma (HPD = 1.42−4.66), and between Fulmarus and Macronectes at 4.56 Ma (HPD = 2.42−6.76).
MC1R and color morphs
Of 134 specimens with MC1R sequences, 79 had associated voucher skins. The bulk of specimens lacking this information were from the Canadian arctic islands, from which no specimens included vouchers. In the MC1R fragment sequenced in this study, excluding a few low frequency mutations (i.e., those occurring in only one or two individuals) there were nine notable sites where amino acid polymorphisms were found, a couple of which were linked pairs (Fig. 4). At least one of these mutations, E92V, occurs at a site implicated in melanism in other species (Mundy 2005).
Figure 4.
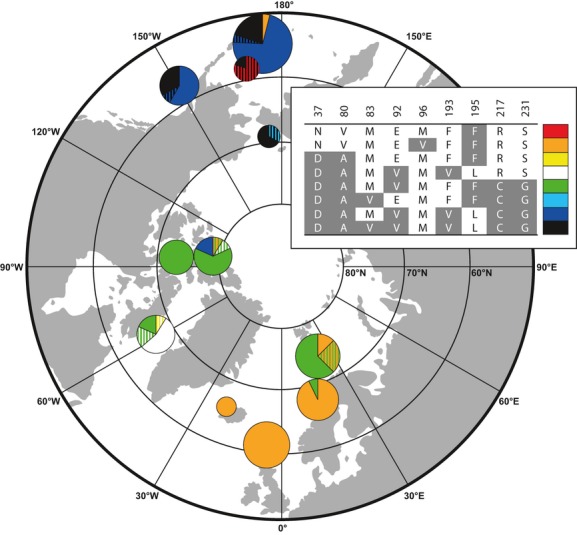
Geographical distribution of the eight most common MC1R amino acid haplotypes. The amino acid at each relevant position in the sequence is depicted in the inset for each haplotype, which are then coded by color. Circles on the map are proportional to the number of individuals represented at each site. Whole colors represent homozygous individuals and hatched colors represent the composition of heterozygous individuals.
Despite the relatively large number of polymorphic sites, none of these consistently correlated to color morph throughout the species range (Fig. 5). Some weak patterns did emerge. For example, all birds homozygous for glutamic acid at position 92 (the ancestral state) were light morph birds; however, in the Atlantic several birds homozygous for valine at this position had intermediate plumage, and in the Pacific some valine homozygotes were complete light morphs. The distribution of MC1R haplotypes appeared to be more heavily influenced by geography (Fig. 4). For example, some light morph birds from the Pacific differ at all of the nine polymorphic sites from light morph birds from the Atlantic.
Figure 5.
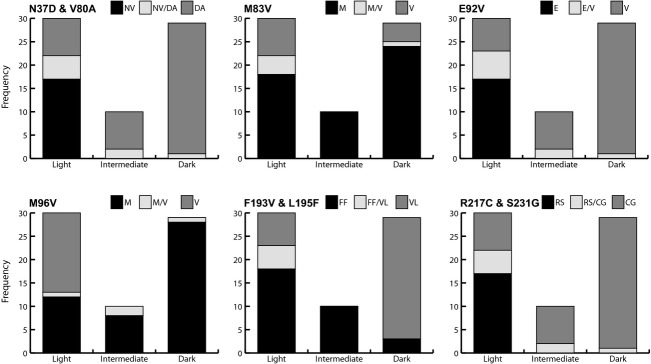
Frequency of amino acid composition at six individual or linked sites in the MC1R amino acid sequence. Shading indicates whether individuals were homozygous (black or dark gray) or heterozygous (light gray). Composition frequency is given for each plumage category: light, dark, and intermediate.
Discussion
Genetic divergence in the Northern Fulmar
Both mitochondrial markers indicated strong divergence between Atlantic and Pacific populations of Northern Fulmar, supporting the pattern initially suggested by DNA barcoding results (Kerr et al. 2007; Johnsen et al. 2010) but differing from the current taxonomic recognition (American Ornithologists' Union 1998). However, not even the fast-evolving control region indicated any clear divergence within the Atlantic population, thus failing to support the presence of two separate evolutionary significant units in that region. Results from nuclear markers were less clear. Haplotype sharing occurred in each of the three markers but to varying degrees. Sharing was most prominent for CHD1-Z, which featured almost no variation at all. Consequently, haplotype networks were much more informative than the concatenated nuclear tree. The networks demonstrate that rare haplotypes were not shared between the Atlantic and Pacific population, suggestive of early-stage divergence.
The lack of diversity at the CHD1-Z locus is slightly unusual. The Z chromosome has a smaller effective population size than its autosomal counterparts on account of its single-copy occurrence in females. Additionally, sexual selection is also thought to result in faster divergence of Z-linked genes (Borge et al. 2005). The low diversity observed within and between populations might seem to implicate male-biased gene flow and question the integrity of any postzygotic boundaries between the populations since the Z chromosome has recently been regarded as a “hotspot” for speciation genes (Sæther et al. 2007). However, CHD1-Z did not just demonstrate low variation between populations but also between other Fulmarine species. The CHD1-Z sequence in the focal taxa only differed from its congener, F. glacialoides, by two substitutions. Additionally, F. glacialoides sequences only differed from those of the genus Macronectes by a single substitution, and the two Macronectes species showed no interspecific variation at all. Further to this point, female Procellariiformes are known to disperse more than males (Burg et al. 2003). These results suggest “intron A” of the Z chromosome may simply be largely conserved in the Fulmarini clade.
In general, the discrepancy between the mitochondrial and nuclear markers is not surprising and is consistent with many similar studies (Welch et al. 2011; Gangloff et al. 2013). Time to reciprocal monophyly is generally correlated with generation time and effective population size (Avise et al. 1984; Hudson and Coyne 2002). Both of these measures are relatively large in the Northern Fulmar, with generation time generally estimated around 20 years (Koons et al. 2006; Jones et al. 2008) and census estimates suggesting both populations include millions of breeding pairs (Brooke 2004). These values collectively would predict a disproportionately long time to reciprocal monophyly relative to the divergence data, even for mitochondrial markers, though it should be borne in mind that modern population estimates might not be reflective of population trends at the time of divergence. Using values from Table 1 in Hudson and Coyne (2002) and our own divergence date estimates, it can be inferred that the long-term effective population size that would allow the observed mitochondrial divergence between Atlantic and Pacific birds would range from roughly 17,000 to 75,000 individuals/population. In turn, even the low end of this range would potentially require 2.9 million years to observe with high probability the reciprocal monophyly of a nuclear marker. This is consistent with our data, where nuclear markers appear to display reciprocal monophyly between F. glacialis and F. glacialoides, which our data suggests diverged ∼3 Mya.
The above estimates depend on the accuracy of the generation time estimates for Northern Fulmars. For example, if generation time were in fact closer to the average age at first reproduction (8–12 years, Hatch and Nettleship 1998) then the above population size estimates would double, but the values would still remain much below the current census population estimates. The settlement of the northern hemisphere is hypothesized to have followed a founder event from a southern ancestor traveling up the Pacific coastline (Voous 1949), which would likely result in a smaller initial population size in the north and subsequently a shorter predicted time to reciprocal monophyly. The division between the Atlantic and Pacific colonies on the other hand is more likely to have followed a vicariant event (i.e., the extension of Arctic ice), which theoretically would cleave the northern species into two more evenly sized populations. This too would help explain why monophyly is observed between F. glacialis and F. glacialoides, but not between F. glacialis populations. Detracting from this logic is the rapid population growth observed in the North Atlantic, where newly colonized locations such as the Faeroe Islands have seen populations surpass 500,000 breeding pairs in under 200 years (Fisher 1952). Alternatively, selective sweeps might also help explain the degree of mitochondrial divergence in F. glacialis, but evidence for such events is still largely lacking (Kerr 2011). The smaller effective population size of the mitochondrial genome appears to remain the most parsimonious explanation for the mito-nuclear discrepancy.
Northern Fulmars in the Pacific tend toward smaller body size and slimmer bills, whereas Atlantic birds are larger on average and bill size varies clinally (being smaller in the north), but measurements for both features overlap between these two regions (Brooke 2004; Pyle 2008; Howell 2012). Morphological differences are difficult to encapsulate due to the variable plumage, but Pacific birds are noted for having a darker tail that contrasts against a paler rump, whereas Atlantic birds are more uniformly colored across the tail and rump (Sibley 2000). Unfortunately, this feature is largely obscured or completely lost in Pacific dark morphs. Avian taxonomic decisions are often skewed toward characters that are diagnosable in the field, but this sometimes obscures distinct evolutionary lineages (Watson 2005).
One of the inherent challenges of studying divergent populations in allopatry is the inability to test the criteria of the biological species concept under natural conditions, i.e., assortative mating in sympatry (Mayr 1963). A common approach to taxonomic decisions for allopatric populations is to make comparisons to closely allied species pairs that do occur in sympatry (Helbig et al. 2002). Some authors have suggested that differences between Northern Fulmars from the Atlantic and Pacific are on par with that observed between other Procellariid species (Howell 2012). A notable example is Macronectes giganteus and M. halli, which differ only subtly by primarily qualitative morphological characters and exhibit far less genetic differentiation than the Northern Fulmar populations (Techow et al. 2010). Other examples, such as Pterodroma phaeopygia and P. sandwichensis echo the case of Macronectes, but these species breed on separate islands and may not be considered sympatric per se (Welch et al. 2011). It is hypothesized that high natal philopatry reduces selection for plumage divergence in Procellariids, which makes species recognition within this group very challenging in general (Brooke 2004).
The Northern Fulmar shares a common phylogeographic distribution with a few other seabird species and allospecies pairs, for example, Uria aalge (Morris-Pocock et al. 2008), U. lomvia (Birt-Friesen et al. 1992), and Fratercula arctica/corniculata (Friesen et al. 1996). Interestingly, divergence dates for these others pairs are variable, with the youngest only being 56,000–226,000 years ago (Morris-Pocock et al. 2008). Land and ice are common barriers to dispersal for all of these species (Friesen et al. 2007), so it is curious that they don't share a common divergence date. These dates are primarily predicted based on molecular clock estimates, so it could be that rate heterogeneity is underappreciated. However, the Northern Fulmar's exceptional generation time would predict a slower evolutionary rate compared to the other species, yet it yields the oldest divergence date for the group. If the other species were able to cross the Arctic passage at later dates, it would be reasonable to surmise that there would be no barrier to prevent the Northern Fulmar from completing similar movement, but the integrity of the populations remains intact.
Two historical features remain at odds with our findings: the Fulmarus fossil record and the Pacific coast origin hypothesis. Two prehistoric fossils from Kern County, California are attributed to the genus, including the proximal end of a carpometacarpus identified as F. hammeri (Howard 1969) and a complete humerus identified as F. miocaenus (Howard 1984). These fossils were said to belong to the Barstovian and Clarendonian stages, respectively, which roughly range from 10 to 16 Mya. This timeline vastly predates our estimates for the time to most recent common ancestor for even Fulmarus-Macronectes, and instead is more in line with the origin of the entire Fulmarini clade. Given that we used well-accepted fossil calibrations to obtain our date estimates, we propose that the Howard fossils likely belonged to either an ancestral lineage or to independent lineages with no modern descendents, but in either case should not be included in the genus Fulmarus. Wetmore (1926) describes a fossilized fragment of a left humerus collected in Maryland that is so similar to the modern day species that it “cannot be distinguished from [Fulmarus] glacialis”, but while he speculated that this fossil dated back to the Miocene (on account of its dark coloration and degree of fossilization), he also stated that the stratum from which it was collected is more consistent with a Pleistocene origin. The latter scenario would be consistent with our data.
Voous (1949) proposed that the Northern Hemisphere was colonized by ancestral fulmars traveling up the cooler water of the west coast of the Americas. He used bill shape to support this hypothesis, indicating that the slender bill of F. glacialoides was the ancestral condition and the broader bill of the North Atlantic F. glacialis was the most derived (the relatively slim bill of Pacific birds would thus reflect an intermediate state). This hypothesis has perpetuated unchallenged (Hatch and Nettleship 1998), though before Voous (1949) it was thought that the Atlantic was colonized prior to the Pacific (Fisher and Waterston 1941). Contrary to Voous' hypothesis, our molecular data reveal more derived characters in the sequences of Pacific birds for four out of five markers (CHD1-Z was the exception because the two populations predominantly shared a single haplotype). This could detract from the Pacific coast origin hypothesis, though it is possible that the increased divergence in the Pacific is reflective of that region's smaller population size, which can have a positive effect on the molecular evolutionary rate (Ohta 1973).
MC1R and color morphs in the Northern Fulmar
The association between MC1R mutants and melanism was first deduced in model laboratory species such as mouse and subsequently chicken, but a common role was later confirmed in such disparate avian taxa as skuas, geese, and passerines (see Mundy 2005 for review). MC1R has since been regularly employed as a candidate gene in avian plumage studies, though often arguably in vain. Although MC1R-melanism correlations have been found in additional avian taxa (Baiao et al. 2007; Pointer and Mundy 2008; Uy et al. 2009; Vidal et al. 2010), a near equal number of studies have unsurprisingly yielded negative results (MacDougall-Shackleton et al. 2003; Cheviron et al. 2006; Haas et al. 2009; Hull et al. 2010; Cadena et al. 2011; Dobson et al. 2012). Several of the latter studies examined species that exhibited finely scaled patterns, not melanism per se (MacDougall-Shackleton et al. 2003; Cadena et al. 2011; Dobson et al. 2012). Others examined species where melanic plumage was limited to a single sex and age class (Cheviron et al. 2006).
In the absence of a functional analysis, there is an intrinsic risk of falsely attributing cause to MC1R mutations when it could simply correlate with plumage variation because of population divergence. Doucet et al. (2004) described an association between MC1R and melanism in the island subspecies of Malurus leucopterus, which features melanic plumage in males, but Mundy (2005) correctly speculated that this association might be an artifact of population divergence, as was later demonstrated by Driskell et al. (2010). In his review, Mundy (2005) offers strong evidence for how demographic history can be ruled out as an explanation for the association in at least three iconic species, which bolsters the role of MC1R in at least some cases. Interestingly, shared amino acid substitutions are often recorded in unrelated species (Mundy 2005), which seemingly gives further credence to causation, but these substitutions are also found in nonmelanic species (Pointer and Mundy 2008). If MC1R mutations do in fact play a role in determining color morph in the Northern Fulmar then a strenuous explanation would be required to explain the mechanism.
Conclusions
The distribution and degree of variation of color morphs across the range of F. glacialis has long been of interest. Determining the genetic cause of this variation will prove to be equally interesting, but might remain a challenge for some time. The gene implicated in melanic plumage in several other avian examples, MC1R, shows a surprising amount of nonsynonymous variation within F. glacialis, yet this variation does not correlate with the plumage color of specimens.
The current taxonomic status of F. glacialis is inconsistent with the treatment of other species within the Procellariiformes. The Atlantic and Pacific populations display genetic (and morphological) divergence comparable to that observed between other Procellariid sister species, and appear to be on independent evolutionary trajectories. Divergence date estimates indicate that the Atlantic and Pacific populations may have persisted independently for nearly 2 million years, spanning over warm interglacial periods that may have drastically reduced ice cover (Anderson et al. 2006). Consequently, taxonomic boundaries within this genus warrant reconsideration and we would recommend elevating the Pacific population to a separate species. Despite the significant genetic divergence between these two populations, not even fast-evolving regions such as the mitochondrial control region distinguish the two named Atlantic subspecies and thus we support the decision of some authors not to recognize F. g. auduboni as a valid subspecies.
The association of genetic markers and geographical distribution but not color morph fails to support an old hypothesis on the origin of the Northern Fulmar's modern day color morph distribution (van Franeker and Wattel 1982). The Northern Fulmar is just one of several seabirds with ranges bisected by Arctic ice. Some current climate predictions indicate that summer arctic sea ice is likely to recede within the lifetime of some contemporary biologists (Kerr 2009), which may present a unique opportunity for future biographical study if long separated populations are reunited.
Acknowledgments
Funding was provided through a Smithsonian post-doctoral fellowship to KCRK. Specimen access was provided by Jim Dean, Chris Huddleston, and Chris Milensky from the Smithsonian Institution, National Museum of Natural History; Sharon Birks from the Burke Museum of Natural History and Culture; Paul Sweet from the American Museum of Natural History; Kevin Winker from the University of Alaska; Jan Lifjeld and Arild Johnsen from Oslo Museum of Natural History; Maria Dam of the Faeroe Islands Environmental Agency; and Guy Savard and Charles Francis via the National Wildlife Specimen Bank, part of the Science and Technology branch of Environment Canada. Pablo Tubaro from the Museo Argentino de Ciencias Naturales “Bernardino Rivadavia” supplied additional tissues for outgroup taxa and access to unpublished COI sequences. John Gerwin (North Carolina Museum of Natural Sciences), Roar Solheim (Agder Nature Museum), Arild Johnsen, and Kevin Winker provided specimen details, collection information, and/or specimen photographs. Nor Faridah Dahlan (Smithsonian Institution) aided in the lab and Gabriel Johnson (Smithsonian Institution) provided assistance with cloning. We thank Oliver Haddrath, Ben Wielstra, and one anonymous reviewer for beneficial comments on earlier drafts of this manuscript.
Appendix 1
Sequences and citations for primers used in this study.
| Primer | Sequence (5′ to 3′) | Gene | Dir. | Citation |
|---|---|---|---|---|
| BirdF1 | TTCTCCAACCACAAAGACATTGGCAC | COI | F | (Hebert et al. 2004b) |
| COIbirdR2 | ACGTGGGAGATAATTCCAAATCCTGG | COI | R | (Hebert et al. 2004b) |
| AvMiF1 | CCCCCGACATAGCATTCC | COI | F | (Kerr et al. 2009b) |
| AvMiR1 | ACTGAAGCTCCGGCATGGGC | COI | R | (Kerr et al. 2009b) |
| LTYR | TGTAAAAAGGWCTACAGCCTAACGC | COI | F | (Tavares and Baker 2008) |
| COI907aH2 | GTRGCNGAYGTRAARTATGCTCG | COI | R | (Tavares and Baker 2008) |
| COIaRt | AACAAACCACAAAGATATCGG | COI | F | (Tavares and Baker 2008) |
| COI748Ht | CCAGGCTTYGGAATTATYTCCCA | COI | R | (Tavares and Baker 2008) |
| ND6*-L16406 | CCACCCCATAATACGGCGAAGG | CR | F | (Burg et al. 2003) |
| H505 | GAAAGAATGGTCCTGAAGC | CR | R | (Burg et al. 2003) |
| Myo2 | GCCACCAAGCACAAGATCCC | MYOII | F | (Slade et al. 1993) |
| Myo3 | CGGAAGAGCTCCAGGGCCTT | MYOII | R | (Slade et al. 1993) |
| Myo3F | TTCAGCAAGGACCTTGATAATGACTT | MYOII | R | (Heslewood et al. 1998) |
| MSHR72 | ATGCCAGTGAGGGCAACCA | MC1R | F | (Mundy et al. 2004) |
| MSHR9 | CTGGCTCCGGAAGGCATAGAT | MC1R | R | (Mundy et al. 2004) |
| MC1RNoFu63 | GCAACCAGAGCAACACCAC | MC1R | F | This study |
| MC1RNoFu802 | GAAGAAGCAGGTGCAGAAGG | MC1R | R | This study |
| MC1RNoFu362 | GGACAACGTCATCAACATGC | MC1R | F | This study |
| MC1RNoFu532 | GCGTTGTTGTGGTAGTAGGTGA | MC1R | R | This study |
| 2669FZ | CTCAGAT GGTGAGGATGCTG | CHD1-Z | F | (Berlin et al. 2006) |
| 2718R | ATTGAAATGATCCAGTGCTTG | CHD1-Z | R | (Fridolfsson and Ellegren 1999) |
Run direction is indicated by F (forward) or R (reverse).
Appendix 2
List of all COI sequences included in the BEAST divergence dating analysis.
| BOLD Process ID | GenBank no. | Species | Family |
|---|---|---|---|
| FULMR104-10 | KC755410 | Fulmarus glacialis (Atlantic) | Procellariidae |
| FULMR116-11 | KC755530 | F. glacialis (Pacific) | Procellariidae |
| KPARG309-08 | KC755545 | F. glacialoides | Procellariidae |
| FULMR121-11 | KC755546 | Macronectes giganteus | Procellariidae |
| FULMR122-11 | KC755547 | M. halli | Procellariidae |
| BROMB704-07 | n/a | Daption capense | Procellariidae |
| FULMR016-10 | KC755549 | Thalassoica antarctica | Procellariidae |
| FULMR120-11 | KC755548 | Pagodroma nivea | Procellariidae |
| GBIR1243-09 | AY567883 | Calonectris diomedea | Procellariidae |
| BROMB706-07 | n/a | Procellaria westlandica | Procellariidae |
| ROMC325-07 | n/a | Pterodroma cookii | Procellariidae |
| SIBHI087-11 | JF498895 | Puffinus pacificus | Procellariidae |
| KAARG101-07 | FJ027984 | Pelecanoides georgicus | Pelecanoididae |
| KAARG032-07 | FJ027985 | P. magellani | Pelecanoididae |
| ROMC300-07 | n/a | P. urinatrix | Pelecanoididae |
| NZCOI449-09 | n/a | Phoebetria palpebrata | Diomedeidae |
| NZCOI234-08 | JW1033 | Diomedea epomophora | Diomedeidae |
| BROMB337-06 | n/a | D. exulans | Diomedeidae |
| SIBHI060-11 | JF498886 | Phoebastria immutabilis | Diomedeidae |
| KKBNA002-04 | DQ433935 | P. nigripes | Diomedeidae |
| KBARG059-07 | FJ028397 | Thalassarche chlororhynchos | Diomedeidae |
| KPARG374-08 | n/a | T. melanophris | Diomedeidae |
| KPARG321-08 | n/a | Oceanites oceanicus | Hydrobatidae |
| TZBNA330-03 | AY666400 | Oceanodroma castro | Hydrobatidae |
| TZBNA229-03 | AY666306 | O. furcata | Hydrobatidae |
| BON034-06 | GU571506 | O. leucorhoa | Hydrobatidae |
| KPARG109-08 | n/a | Spheniscus magellanicus | Spheniscidae |
Appendix 3
Characterization of nucleotide polymorphism within and between populations, including number of segregating sites (S), number of haplotypes (H), nucleotide diversity (π), Watterson's estimator (θW), and several neutrality tests calculated using DnaSP version 5.0 (Rozas and Librado 2009).
| Population | n | S | H | π·10−3 | θW·10−3 | Tajima's D | Fu & Li's D* | Fu & Li's F* |
|---|---|---|---|---|---|---|---|---|
| COI (694 bp) | 134 | 35 | 24 | 0.51 | 9.52 | 1.6688 | −1.4569 | −0.21859 |
| Atlantic | 81 | 13 | 14 | 1.39 | 3.77 | −1.7565 | −3.0538* | −3.0844** |
| Pacific | 53 | 9 | 10 | 0.56 | 2.95 | −2.2444*** | −3.9924** | −4.0287** |
| CR (228 bp) | 131 | 42 | 62 | 40.52 | 33.81 | 0.1958 | −2.3238 | −1.5312 |
| Atlantic | 78 | 25 | 30 | 8.49 | 22.06 | −1.9949* | −3.5059** | −3.5106** |
| Pacific | 53 | 22 | 32 | 13.14 | 21.26 | −1.2214 | −1.4206 | −1.6044 |
| MYOII (694 bp) | 252 (2·126) | 21 | 27 | 1.37 | 5.74 | −2.0075* | −4.3904** | −4.1363** |
| Atlantic | 164 (2·82) | 12 | 13 | 0.60 | 3.25 | −2.0281* | −2.6307* | −2.8806* |
| Pacific | 88 (2·44) | 12 | 15 | 1.86 | 3.97 | −1.4359 | −2.2006 | −2.2943 |
| CHD1-Z (694 bp) | 112 | 6 | 7 | 0.44 | 2.81 | −1.8639* | −2.7728* | −2.9186* |
| Atlantic | 75 | 4 | 5 | 0.52 | 2.03 | −1.5573 | −1.3637 | −1.6707 |
| Pacific | 37 | 2 | 3 | 0.22 | 0.96 | −1.4933 | −2.3888 | −2.4664 |
| MC1R (694 bp) | 252 (2·126) | 21 | 23 | 6.09 | 4.73 | 0.7600 | −1.8218 | −0.9439 |
| Atlantic | 162 (2·81) | 16 | 15 | 5.61 | 3.88 | 1.1732 | −0.0888 | 0.4677 |
| Pacific | 90 (2·45) | 15 | 8 | 2.17 | 4.06 | −1.3083 | −1.5951 | −1.7762 |
*P < 0.05; **P < 0.02; ***P < 0.01.
Data Archive
DNA sequences have been deposited in GenBank (accession numbers: KC755410-KC755682, KC755695-KC755826, KC788823-KC789071) and BOLD (in the project FULMR Fulmarine Petrels). Alignments of phased DNA sequences for MYOII and MC1R have been deposited in the Dryad Repository (doi:10.5061/dryad.0520f).
Conflict of Interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1. Complete list of all the specimens included in the analysis, their respective BOLD Process IDs, their respective GenBank accession numbers for each gene sequenced, their tissue composition, and the donor institution.
References
- American Ornithologists' Union. Check-list of North American Birds. 3rd ed. New York: American Ornithologists' Union; 1910. [Google Scholar]
- American Ornithologists' Union. Check-list of North American birds. 7th ed. Washington, D.C: American Ornithologists' Union; 1998. [Google Scholar]
- Anderson P, Bermike O, Bigelow N, Brigham-Grette J, Duval M, Edwards M, et al. Last Interglacial Arctic warmth confirms polar amplification of climate change. Quatern. Sci. Rev. 2006;25:1383–1400. [Google Scholar]
- Areta JI, Pearman M. Natural history, morphology, evolution, and taxonomic status of the earthcreeper Upucerthia saturatior (Furnariidae) from the Patagonian forests of South America. Condor. 2009;111:135–149. [Google Scholar]
- Avise JC, Neigel JE, Arnold J. Demographic influences on mitochondrial DNA lineage survivorship in animal populations. J. Mol. Evol. 1984;20:99–105. doi: 10.1007/BF02257369. [DOI] [PubMed] [Google Scholar]
- Baiao PC, Schreiber FA, Parker PG. The genetic basis of the plumage polymorphism in red-footed boobies (Sula sula): a melanocortin-1 receptor (MC1R) analysis. J. Hered. 2007;98:287–292. doi: 10.1093/jhered/esm030. [DOI] [PubMed] [Google Scholar]
- Barker FK, Vandergon AJ, Lanyon SM. Assessment of species limits among yellow-breasted meadowlarks (Sturnella spp.) using mitochondrial and sex-linked markers. Auk. 2008;125:869–879. [Google Scholar]
- Becker JJ. Neogene avian localities of North America Smithsonian. Washington, D.C: Institution Press; 1987. [Google Scholar]
- Bent AC. Life histories of North American petrels and pelicans and their allies. New York: Dover Publications; 1964. [Google Scholar]
- Berlin S, Brandström M, Backström N, Axelsson E, Smith NG, Ellegren H. Substitution rate heterogeneity and the male mutation bias. J. Mol. Evol. 2006;62:226–U217. doi: 10.1007/s00239-005-0103-6. [DOI] [PubMed] [Google Scholar]
- Birt-Friesen VL, Montevecchi WA, Gaston AJ, Davidson WS. Genetic structure of thick-billed Murre (Uria lomvia) populations examined using direct sequence analysis of amplified DNA. Evolution. 1992;46:267–272. doi: 10.1111/j.1558-5646.1992.tb02001.x. [DOI] [PubMed] [Google Scholar]
- Borge T, Webster MT, Andersson G, Sætre GP. Contrasting patterns of polymorphism and divergence on the Z chromosome and autosomes in two Ficedula flycatcher species. Genetics. 2005;171:1861–1873. doi: 10.1534/genetics.105.045120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooke M. Albatrosses and petrels across the world. New York: Oxford University Press; 2004. [Google Scholar]
- Burg TM, Lomax J, Almond R, Brooke MD, Amos W. Unravelling dispersal patterns in an expanding population of a highly mobile seabird, the northern fulmar (Fulmarus glacialis. Proceedings of the Royal Society of London Series B-Biological Sciences. 2003;270:979–984. doi: 10.1098/rspb.2002.2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadena CD, Cheviron ZA, Funk WC. Testing the molecular and evolutionary causes of a ‘leapfrog’ pattern of geographical variation in coloration. J. Evol. Biol. 2011;24:402–414. doi: 10.1111/j.1420-9101.2010.02175.x. [DOI] [PubMed] [Google Scholar]
- Cheviron ZA, Hackett SJ, Brumfield RT. Sequence variation in the coding region of the melanocortin-1 receptor gene (MC1R) is not associated with plumage variation in the blue-crowned manakin (Lepidothrix coronata. Proceedings of the Royal Society B-Biological Sciences. 2006;273:1613–1618. doi: 10.1098/rspb.2006.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JF. The Clements checklist of the birds of the world. 6th ed. New York: Cornell University Press; 2007. [Google Scholar]
- DeSalle R. Species discovery versus species identification in DNA barcoding efforts: response to Rubinoff. Conserv. Biol. 2006;20:1545–1547. doi: 10.1111/j.1523-1739.2006.00543.x. [DOI] [PubMed] [Google Scholar]
- DeSalle R, Egan MG, Siddall M. The unholy trinity: taxonomy, species delimitation and DNA barcoding. Philosophical Transactions of the Royal Society B-Biological Sciences. 2005;360:1905–1916. doi: 10.1098/rstb.2005.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson AE, Schmidt DJ, Hughes JM. Sequence variation in the Melanocortin-1 Receptor (MC1R) does not explain continent-wide plumage color differences in the Australian magpie (Cracticus tibicen. J. Hered. 2012;103:769–780. doi: 10.1093/jhered/ess053. [DOI] [PubMed] [Google Scholar]
- Doucet SM, Shawkey MD, Rathburn MK, Mays HL, Montgomerie R. Concordant evolution of plumage colour, feather microstructure and a melanocortin receptor gene between mainland and island populations of a fairy-wren. Proceedings of the Royal Society B-Biological Sciences. 2004;271:1663–1670. doi: 10.1098/rspb.2004.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driskell AC, Prum RO, Pruett-Jones S. The evolution of black plumage from blue in Australian fairy-wrens (Maluridae): genetic and structural evidence. J. Avian Biol. 2010;41:505–514. [Google Scholar]
- Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher J. The fulmar. London: Collins; 1952. [Google Scholar]
- Fisher J, Waterston G. The breeding distribution, history and population of the fulmar (Fulmarus glacialis) in the British Isles. J. Anim. Ecol. 1941;10:204–272. [Google Scholar]
- van Franeker JA, Luttik R. Colour and size variation in the Northern Fulmar Fulmarus glacialis on Bear Island, Svalbard. Circumpolar Studies. 2008;4:39–58. [Google Scholar]
- van Franeker JA, Wattel J. Geographical variation of the fulmar Fulmarus glacialis in the North-Atlantic. Ardea. 1982;70:31–44. [Google Scholar]
- Frezal L, Leblois R. Four years of DNA barcoding: current advances and prospects. Infection Genetics and Evolution. 2008;8:727–736. doi: 10.1016/j.meegid.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Fridolfsson AK, Ellegren H. A simple and universal method for molecular sexing of non-ratite birds. J. Avian Biol. 1999;30:116–121. [Google Scholar]
- Friesen VL, Baker AJ, Piatt JF. Phylogenetic relationships within the Alcidae (Charadriiformes: Aves) inferred from total molecular evidence. Mol. Biol. Evol. 1996;13:359–367. doi: 10.1093/oxfordjournals.molbev.a025595. [DOI] [PubMed] [Google Scholar]
- Friesen VL, Burg TM, McCoy KD. Mechanisms of population differentiation in seabirds. Mol. Ecol. 2007;16:1765–1785. doi: 10.1111/j.1365-294X.2006.03197.x. [DOI] [PubMed] [Google Scholar]
- Gangloff B, Zino F, Shirihai H, González-Solís J, Couloux A, Pasquet E, et al. The evolution of north-east Atlantic gadfly petrels using statistical phylogeography. Mol. Ecol. 2013;22:495–507. doi: 10.1111/mec.12119. [DOI] [PubMed] [Google Scholar]
- Haas F, Pointer MA, Saino N, Brodin A, Mundy NI, Hansson B. An analysis of population genetic differentiation and genotype-phenotype association across the hybrid zone of carrion and hooded crows using microsatellites and MC1R. Mol. Ecol. 2009;18:294–305. doi: 10.1111/j.1365-294X.2008.04017.x. [DOI] [PubMed] [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999;41:95–98. [Google Scholar]
- Hatch SA. Evidence for color phase effects on the breeding and life history of northern fulmars. Condor. 1991;93:409–417. [Google Scholar]
- Hatch SA, Nettleship DN. Northern fulmar (Fulmarus glacialis. In: Poole A, editor. Birds of North America online. Ithica, NY: Cornell Lab of Ornithology; 1998. Available at http://bna.bird.cornell.edu/bna/species/361. [Google Scholar]
- Hebert PDN, Cywinska A, Ball SL, DeWaard JR. Biological identifications through DNA barcodes. Proceedings of the Royal Society of London Series B-Biological Sciences. 2003a;270:313–321. doi: 10.1098/rspb.2002.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert PDN, Ratnasingham S, deWaard JR. Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proceedings of the Royal Society of London Series B-Biological Sciences. 2003b;270:S96–S99. doi: 10.1098/rsbl.2003.0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert PDN, Penton EH, Burns JM, Janzen DH, Hallwachs W. Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc. Natl. Acad. Sci. USA. 2004a;101:14812–14817. doi: 10.1073/pnas.0406166101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert PDN, Stoeckle MY, Zemlak TS, Francis CM. Identification of birds through DNA barcodes. PLoS Biol. 2004b;2:1657–1663. doi: 10.1371/journal.pbio.0020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbig AJ, Knox AG, Parkin DT, Sangster G, Collinson M. Guidelines for assigning species rank. Ibis. 2002;144:518–525. [Google Scholar]
- Heslewood MM, Elphinstone MS, Tidemann SC, Baverstock PR. Myoglobin intron variation in the Gouldian Finch Erythrura gouldiae assessed by temperature gradient gel electrophoresis. Electrophoresis. 1998;19:142–151. doi: 10.1002/elps.1150190203. [DOI] [PubMed] [Google Scholar]
- Hickerson MJ, Meyer CP, Moritz C. DNA barcoding will often fail to discover new animal species over broad parameter space. Syst. Biol. 2006;55:729–739. doi: 10.1080/10635150600969898. [DOI] [PubMed] [Google Scholar]
- Ho SYW. Calibrating molecular estimates of substitution rates and divergence times in birds. J. Avian Biol. 2007;38:409–414. [Google Scholar]
- Howard H. A new avian fossil from Kern County, California. Condor. 1969;71:68–69. [Google Scholar]
- Howard H. Additional records from the Miocene of Kern County, California with the description of a new species of fulmar (Aves: Procellariidae) Bulletin of the Southern California Academy of Sciences. 1984;83:84–89. [Google Scholar]
- Howell SNG. Petrels, albatrosses, and storm-petrels of North America: a photographic guide. Princeton, NJ: Princeton University Press; 2012. [Google Scholar]
- Hudson RR, Coyne JA. Mathematical consequences of the genealogical species concept. Evolution. 2002;56:1557–1565. doi: 10.1111/j.0014-3820.2002.tb01467.x. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- Hull JM, Mindell DP, Talbot SL, Kay EH, Hoekstra HE, Ernest HB. Population structure and plumage polymorphism: the intraspecific evolutionary relationships of a polymorphic raptor, Buteo jamaicensis harlani. BMC Evol. Biol. 2010;10:224. doi: 10.1186/1471-2148-10-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsen A, Rindal E, Ericson PGP, Zuccon D, Kerr KCR, Stoeckle MY, et al. DNA barcoding of Scandinavian birds reveals divergent lineages in trans-Atlantic species. J. Ornithol. 2010;151:565–578. [Google Scholar]
- Jones OR, Gaillard J-M, Tuljapurkar S, Alho JS, Armitage KB, Becker PH, et al. Senescence rates are determined by ranking on the fast-slow life-history continuum. Ecol. Lett. 2008;11:664–673. doi: 10.1111/j.1461-0248.2008.01187.x. [DOI] [PubMed] [Google Scholar]
- Kerr RA. Arctic summer sea ice could vanish soon but not suddenly. Science. 2009;323:1655. doi: 10.1126/science.323.5922.1655. [DOI] [PubMed] [Google Scholar]
- Kerr KCR. Searching for evidence of selection in avian DNA barcodes. Molecular Ecology Resources. 2011;11:1045–1055. doi: 10.1111/j.1755-0998.2011.03049.x. [DOI] [PubMed] [Google Scholar]
- Kerr KCR, Stoeckle MY, Dove CJ, Weigt LA, Francis CM, Hebert PD. Comprehensive DNA barcode coverage of North American birds. Mol. Ecol. Notes. 2007;7:535–543. doi: 10.1111/j.1471-8286.2007.01670.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr KCR, Birks SM, Kalyakin MV, Red'kin YA, Koblik EA, Hebert PD. Filling the gap – COI barcode resolution in eastern Palearctic birds. Frontiers in Zoology. 2009a;6:29. doi: 10.1186/1742-9994-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr KCR, Lijtmaer DA, Barreira AS, Hebert PDN, Tubaro PL. Probing evolutionary patterns in Neotropical birds through DNA barcodes. PLoS ONE. 2009b;4:6. doi: 10.1371/journal.pone.0004379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koons DN, Grand JB, Arnold JM. Population momentum across vertebrate life histories. Ecol. Model. 2006;197:418–430. [Google Scholar]
- Lanfear R, Calcott B, Ho SYW, Guindon S. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012;29:1695–1701. doi: 10.1093/molbev/mss020. [DOI] [PubMed] [Google Scholar]
- Ling MK, Lagerström MC, Fredriksson R, Okimoto R, Mundy NI, Takeuchi S, et al. Association of feather colour with constitutively active melanocortin 1 receptors in chicken. Eur. J. Biochem. 2003;270:1441–1449. doi: 10.1046/j.1432-1033.2003.03506.x. [DOI] [PubMed] [Google Scholar]
- MacDougall-Shackleton EA, Blanchard L, Gibbs HL. Unmelanized plumage patterns in old world leaf warblers do not correspond to sequence variation at the melanocortin-1 receptor locus (MC1R) Mol. Biol. Evol. 2003;20:1675–1681. doi: 10.1093/molbev/msg186. [DOI] [PubMed] [Google Scholar]
- Marshall DC. Cryptic failure of partitioned Bayesian phylogenetic analyses: lost in the land of long trees. Syst. Biol. 2010;59:108–117. doi: 10.1093/sysbio/syp080. [DOI] [PubMed] [Google Scholar]
- Mayr E. Animal species and evolution. Cambridge: Harvard University Press; 1963. [Google Scholar]
- Mayr G, Smith T. A fossil albatross from the early Oligocene of the North Sea Basin. Auk. 2012;129:87–95. [Google Scholar]
- Meyer CP, Paulay G. DNA barcoding: error rates based on comprehensive sampling. PLoS Biol. 2005;3:2229–2238. doi: 10.1371/journal.pbio.0030422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moritz C, Cicero C. DNA barcoding: promise and pitfalls. PLoS Biol. 2004;2:1529–1531. doi: 10.1371/journal.pbio.0020354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris-Pocock JA, Taylor SA, Birt TP, Damus M, Piatt JF, Warheit KI, et al. Population genetic structure in Atlantic and Pacific Ocean common murres (Uria aalge): natural replicate tests of post-Pleistocene evolution. Mol. Ecol. 2008;17:4859–4873. doi: 10.1111/j.1365-294X.2008.03977.x. [DOI] [PubMed] [Google Scholar]
- Mundy NI. A window on the genetics of evolution: MC1R and plumage colouration in birds. Proceedings of the Royal Society B-Biological Sciences. 2005;272:1633–1640. doi: 10.1098/rspb.2005.3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundy NI, Badcock NS, Hart T, Scribner K, Janssen K, Nadeau NJ. Conserved genetic basis of a quantitative plumage trait involved in mate choice. Science. 2004;303:1870–1873. doi: 10.1126/science.1093834. [DOI] [PubMed] [Google Scholar]
- Nunn GB, Stanley SE. Body size effects and rates of cytochrome b evolution in tube-nosed seabirds. Mol. Biol. Evol. 1998;15:1360–1371. doi: 10.1093/oxfordjournals.molbev.a025864. [DOI] [PubMed] [Google Scholar]
- Ohta T. Slightly deleterious mutant substitutions in evolution. Nature. 1973;246:96–98. doi: 10.1038/246096a0. [DOI] [PubMed] [Google Scholar]
- Padial JM, de la Riva I. Integrative taxonomists should use and produce DNA barcodes. Zootaxa. 2007;1586:67–68. [Google Scholar]
- Penhallurick J, Wink M. Analysis of the taxonomy and nomenclature of the Procellariiformes based on complete nucleotide sequences of the mitochondrial cytochrome b gene. Emu. 2004;104:125–147. [Google Scholar]
- Pointer MA, Mundy NI. Testing whether macroevolution follows microevolution: are colour differences among swans (Cygnus) attributable to variation at the MC1R locus? BMC Evol. Biol. 2008;8:249. doi: 10.1186/1471-2148-8-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada D. jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 2008;25:1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- Pyle P. Identification guide to North American birds. Part II. Point Reyes Station, CA: Slate Creek Press; 2008. [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Rozas J, Librado P. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Sæther SA, Sætre GP, Borge T, Wiley C, Svedin N, Andersson G, et al. Sex chromosome-linked species recognition and evolution of reproductive isolation in flycatchers. Science. 2007;318:95–97. doi: 10.1126/science.1141506. [DOI] [PubMed] [Google Scholar]
- Schindel DE, Stoeckle MY, Milensky CM, Trizna M, Schmidt BK, Gebhard CA, et al. Project description: DNA barcodes of bird species in the National Museum of Natural History, Smithsonian Institution, USA. Zookeys. 2011;152:87–92. doi: 10.3897/zookeys.152.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley DA. The Sibley guide to birds. New York: Knopf; 2000. [Google Scholar]
- Slade RW, Moritz C, Heideman A, Hale PT. Rapid assessment of single-copy nuclear DNA variation in diverse species. Mol. Ecol. 1993;2:359–373. doi: 10.1111/j.1365-294x.1993.tb00029.x. [DOI] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares ES, Baker AJ. Single mitochondrial gene barcodes reliably identify sister-species in diverse clades of birds. BMC Evol. Biol. 2008;8:81. doi: 10.1186/1471-2148-8-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Techow N, O'Ryan C, Phillips RA, Gales R, Marin M, Patterson-Fraser D, et al. Speciation and phylogeography of giant petrels Macronectes. Mol. Phylogenet. Evol. 2010;54:472–487. doi: 10.1016/j.ympev.2009.09.005. [DOI] [PubMed] [Google Scholar]
- Uy JAC, Moyle RG, Filardi CE, Cheviron ZA. Difference in plumage color used in species recognition between incipient species is linked to a single amino acid substitution in the melanocortin-1 receptor. American Naturalist. 2009;174:244–254. doi: 10.1086/600084. [DOI] [PubMed] [Google Scholar]
- Vidal O, Araguas RM, Fernandez R, Heras S, Sanz N, Pla C. Melanism in guinea fowl (Numida meleagris) is associated with a deletion of Phenylalanine-256 in the MC1R gene. Anim. Genet. 2010;41:656–658. doi: 10.1111/j.1365-2052.2010.02056.x. [DOI] [PubMed] [Google Scholar]
- Voous KH. The morphological, anatomical, and distributional relationship of the Arctic and Antarctic Fulmars (Aves, Procellariidae) Ardea. 1949;37:113–122. [Google Scholar]
- Ward PS, Brady SG, Fisher BL, Schultz TR. Phylogeny and biogeography of dolichoderine ants: effects of data partitioning and relict taxa on historical inference. Syst. Biol. 2010;59:342–362. doi: 10.1093/sysbio/syq012. [DOI] [PubMed] [Google Scholar]
- Warham J. The petrels: their ecology and breeding systems. London: Academic Press; 1990. [Google Scholar]
- Watson DM. Diagnosable versus distinct: evaluating species limits in birds. Bioscience. 2005;55:60–68. [Google Scholar]
- Weir JT, Schluter D. Calibrating the avian molecular clock. Mol. Ecol. 2008;17:2321–2328. doi: 10.1111/j.1365-294X.2008.03742.x. [DOI] [PubMed] [Google Scholar]
- Welch AJ, Yoshida AA, Fleischer RC. Mitochondrial and nuclear DNA sequences reveal recent divergence in morphologically indistinguishable petrels. Mol. Ecol. 2011;20:1364–1377. doi: 10.1111/j.1365-294X.2011.05008.x. [DOI] [PubMed] [Google Scholar]
- Wetmore A. Observations on fossil birds described from the Miocene of Maryland. Auk. 1926;43:462–468. [Google Scholar]
- Wiemers M, Fiedler K. Does the DNA barcoding gap exist? – a case study in blue butterflies (Lepidoptera: Lycaenidae) Frontiers in Zoology. 2007;4:8. doi: 10.1186/1742-9994-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson HE. Description of an Upper Miocene albatross from Beaumaris, Victoria, Australia, and a review of the fossil Diomedeidae. Memoirs of the National Museum of Victoria. 1969;29:41–51. [Google Scholar]
- Yoo HS, Eah JY, Kim JS, Kim YJ, Min MS, Paek WK, et al. DNA barcoding Korean birds. Mol. Cells. 2006;22:323–327. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.