

Abstract
Purpose of review
Humans eating diets low in choline develop fatty liver and liver damage. Rodents fed choline–methionine-deficient diets not only develop fatty liver, but also progress to develop fibrosis and hepatocarcinoma. This review focuses on the role of choline in liver function, with special emphasis on the epigenetic mechanisms of action.
Recent findings
Dietary intake of methyl donors like choline influences the methylation of DNA and histones, thereby altering the epigenetic regulation of gene expression. The liver is the major organ within which methylation reactions occur, and many of the hepatic genes involved in pathways for the development of fatty liver, hepatic fibrosis, and hepatocarcinomas are epigenetically regulated.
Summary
Dietary intake of choline varies over a three-fold range and many humans have genetic polymorphisms that increase their demand for choline. Choline is an important methyl donor needed for the generation of S-adenosylmethionine. Dietary choline intake is an important modifier of epigenetic marks on DNA and histones, and thereby modulates the gene expression in many of the pathways involved in liver function and dysfunction.
Keywords: choline, DNA methylation, epigenetics, fatty liver, liver carcinoma
INTRODUCTION
Humans must eat diets containing choline [1] because its metabolite phosphatidylcholine constitutes 40–50% of cellular membranes and 70–95% of phospholipids in lipoproteins, bile and surfactants [2]; it is needed to form acetylcholine, an important neurotransmitter [2]; its metabolite betaine is needed for normal kidney glomerular function, and perhaps for mitochondrial function [2]; and it provides one-carbon units, via oxidation to betaine, to the methionine cycle for methylation reactions [2].
There is a recommended adequate intake for choline (about 550 mg/day) [3], but choline intake in the diet has been estimated to vary by as much as three-fold – the lowest quartile and the highest quartile of intake were approximately 150mg and 500 mg/day choline equivalents, respectively, in the Framingham Offspring Study [4], the Atherosclerosis Risk In Communities study [5,6], and the Nurse’s Health Study [7]. Intake of choline is likely to be lower in low-income countries. Patients fed with total parenteral nutrition (TPN) solutions receive only small amounts of choline (from the lipid emulsions) and many become choline deficient [8].
CHOLINE AND LIVER FUNCTION
Much of choline metabolism occurs in the liver, and this is among the first organs to accumulate choline absorbed from the intestine [2]. When humans eat diets low in choline, fatty liver is one of the earliest adverse events, and in some people significant hepatic damage occurs (as assessed by release of hepatic enzymes into blood) [9]. People with one of several very common genetic polymorphisms (SNPs) in the genes of choline metabolism are more likely to develop hepatic dysfunction when deprived of choline [10–12], and these people have abnormal plasma metabolomic profiles even when fed a normal diet containing choline [13]. Patients fed with TPN solutions often develop liver dysfunction, and in some, this resolves when they are fed a source of choline [8,14]. We do not know whether the patients who are susceptible to TPN-associated liver damage are those who have SNPs in the genes of choline metabolism.
Rodents also develop fatty liver when fed diets low in choline and methionine [2,15], and this animal model is commonly used for the study of nonalcoholic fatty liver disease (NAFLD) which affects 20% of the global population, 50% of diabetic patients, and 90% of morbidly obese people [16]. The likely mechanism responsible for the development of fatty liver in choline deficiency is related to the synthesis of very-low-density lipoprotein (VLDL), which is the primary package within which triglycerides are secreted from the liver [17]. Phosphatidylcholine is a required component of the VLDL envelope, and when it is not available, triglycerides cannot be exported from liver and hence accumulate in the cytosol [17]. Phosphatidylcholine is formed in the liver by methylation of phosphatidylethanolamine or from incorporation of preformed choline (usually from the diet) [17]. Premenopausal women are less likely to develop fatty liver on a low-choline diet because estrogen induces the hepatic gene (PEMT) that is responsible for de novo formation of phosphatidylcholine [10]. In more than 20% of premenopausal women, a SNP in PEMT leaves them less responsive to estrogen induction of this gene, and they must eat choline to prevent development of fatty liver [10,11,18].
In people, NAFLD sometimes progresses to liver injury and hepatocarcinoma [19], and the choline–methionine-deficient rodent model may help us to understand the underlying reasons for this progression. Rats and mice fed a diet low in choline–methionine content first develop fatty liver, then the liver becomes fibrotic, followed by the development of foci of enzyme-altered hepatocytes which express γ-glutamyltranspeptidase [19] and the placental form of glutathione S-transferase [20] similar to those precancerous cells induced by chemical carcinogens [21,22▪▪]. Eventually, these animals develop adenomas and hepatocellular carcinomas [21]. Adding choline to this deficient diet completely prevents the development of cancer in experimental animals, suggesting that choline itself has an important role [21]. It is interesting that hepatocytes in cell culture, which are slowly shifted to growth media low in choline concentration, also transform into hepatocarcinoma cells [23]. This suggests that the underlying mechanisms for this response to low choline are intrinsic to the hepatocytes. Choline–methionine deficiency also sensitizes rodents to liver carcinogens such as aflatoxin B1 [24]. For example, the dose of aflatoxin B1 needed to induce hepatocarcinomas was greatly reduced in rats fed low-choline–methionine diet [24]. Thus, choline–methionine deficiency acts as an initiator and as a promoter of carcinogenesis.
Several potential mechanisms whereby diets low in choline and methionine result in hepatocarcinogenesis have been explored. These include [22▪▪] liver necrosis with consequent regeneration; induction of oxidative DNA damage and lipid peroxidation because of free radical leaks from mitochondria, with subsequent oxidation of DNA bases resulting in nicks and deletions during base repair; altered protein kinase C signaling because of accumulation of diacylglycerol; and loss of liver apoptotic responses. This review will focus on yet another proposed mechanism – the alteration in the status of labile epiloci induced by methyl devoid diets [25].
ROLE OF METHYL-DONOR NUTRIENTS (METHIONINE, CHOLINE, AND FOLATE) IN SUSTAINING METHYLATION CAPACITY
After S-adenosylmethionine (SAM) is used tomethylate a substrate, S-adenosylhomocysteine and then homocysteine is formed. To regenerate methionine, homocysteine (which is toxic for cells) must be methylated, and the resulting methionine can then be converted to SAM [26]. This methionine cycle in liver utilizes methyl groups from methyl-tetrahydrofolate or from betaine to convert homocysteine to methionine. Choline is the precursor for betaine formation and, therefore, for many of the methyl groups donated to homocysteine via the enzyme betaine homocysteine methyltransferase (BHMT). BHMT processes a significant portion of cellular homocysteine as Bhmt knockout mice become hyperhomocysteinemic even under adequate supply of dietary methyl-tetrahydrofolate [22▪▪].
As discussed later, methylation of DNA and histones constitutes an important mechanism for modulating gene expression called epigenetic regulation. It is not surprising that alterations in dietary choline supply or utilization shape the epigenome.
EPIGENETICS
Epigenetic regulation of gene expression involves the chemical modification of nucleotides in DNA at specific locations. Usually, DNA is not present in cells in the linear form that we so commonly picture it as being, but rather DNA is tightly wound around proteins (histones) [27] (Fig. 1). The positively charged DNA is attracted to negatively charged histones, forming a compact spherical complex. One mechanism for modifying this tightly wound structure occurs when specific cytosine residues are methylated [approximately 70% of the cytosine residues adjacent to guanines (CpG) in genes are methylated as are the intergenic CpG islands and the CpGs in transposable elements that are so common in the human genome [27]]. This CpG methylation is achieved by the enzymatic transfer of methyl groups from SAM to cytosine. When CpGs on DNA are methylated, they attract methyl-binding proteins, which then attract histone deacetylases [27]. These enzymes remove acetyl groups on specific lysine residues in histones and thereby increase the negative charge on the protein. The tight chromatin complex formed by positively charged DNA and negatively charged histones prevents the transcription factors from reaching the DNA to activate gene expression. When DNA CpGs are not methylated, the DNA histone interaction is weaker, opening up the chromatin and creating a permissive environment for gene transcription [28,29]. Simply stated, DNA methylation usually shuts genes off.
FIGURE 1.
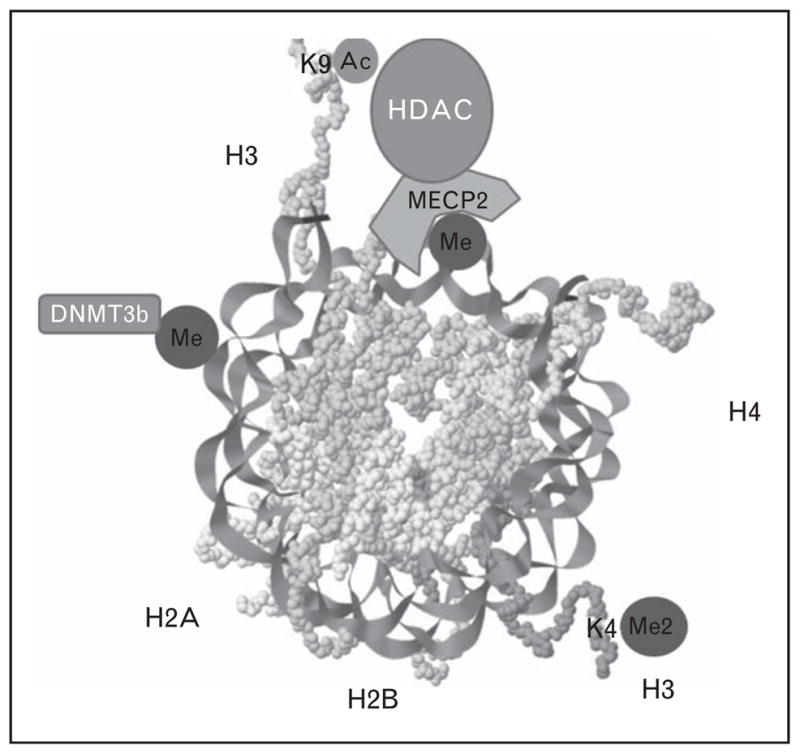
The nucleosome structure. DNA is tightly coiled in nucleosome repeating units (rendering captured with open-source java applet Jmol) composed of 147 base pairs of DNA wrapped around a histone octamer made of two copies of four core histones H2a, H2b, H3, and H4. The transcription of genes is modulated by how tightly the chromatin is packed, and epigenetic marks on histones (at specific lysines such as K9 and K4) and on DNA can modify this chromatin structure (see text). De novo methylation of the core nucleosome unit by DNA methyltransferase 3b (DNMT3b) and the mechanism of histone deacetylation by the methyl CpG-binding protein 2–histone deacetylase complex (MECP2–HDAC) are also depicted.
As discussed above, acetylation of histones is important for maintaining the structure of chromatin, but methylation of histones is also an important epigenetic signal (mono-methyl and di-methyl lysine 9 on histone H3) that represses gene transcription [30], while di-methyl and tri-methyl lysine 4 on histone H3 are enriched in areas with transcriptional active chromatin [31]. Using SAM as the methyl donor, mono-methyl and di-methyl lysine 9 on histone H3 are formed by G9a histone methylase [32], whereas tri-methyl lysine 9 on histone H3 is formed by SUV39 methylase [33]. DNA methylation and histone methylation mechanisms exhibit crosstalk, creating a reinforcing signaling system controlling gene expression.
Alterations in the epigenome are more frequent during sensitive periods of development when progenitor cells are dividing and not yet differentiated [34]. During development, profound epigenomic transformations take place, including DNA methylation catalyzed by DNMT3 [35]. Once established, the epigenome maintains a relatively stable state of transcription in mature somatic cells. During mitosis, this pattern of methylation is faithfully copied to the sister DNA strands by the maintenance and chromatin maturation genes DNMT1, HDAC1, and SMARCAD1 [36]. However, selected loci on genes exhibit a degree of epigenetic plasticity and remain responsive to nutrient levels later in life.
Why is epigenetic flexibility important? As in any other living cell, the genetic information encoded in the hepatocyte genome is fixed, but an epigenetic regulatory mechanism is superim-posed to achieve flexibility in processing the genetic information. Epigenetic marks determine why cells with the same genetic code can have different differentiated phenotypes (somatic individuality): hepatocytes express different genes than do Kupffer cells, stellate cells, endothelial cells or fibroblasts in the liver [37]. Epigenetic marks can permit metabolic flexibility (adaptation of metabolic pathways in response to the environmental signals) [37]. Several genes central to hepatic metabolism are epigenetically regulated, including peroxisome proliferator- activated receptor γ (PPARγ [38]), nuclear sterol response element-binding protein 1-c (SREBP-1c) [39], alcohol dehydrogenase [39,40], glutathione S-transferase [39,40], serine dehydrase [40], CYP450 2c11 [40], glucokinase [41], pyruvate kinase [41,42], phosphoenolpyruvate carboxykinase [42], and enzymes of cholesterol metabolism via epigenetic regulation of the liver X receptor [43]. Thus, liver function is dependent, in part, on how well epigenetic regulatory mechanisms are established. At the same time, liver is probably the most important organ that controls the availability of the SAM needed to establish epigenetic marks.
THE LIVER IS AN IMPORTANT ORGAN CONTROLLING METHYLATION
Half of the methionine coming from diets is utilized by the liver for forming SAM that is needed for methylation reactions and more than 85% of methylation reactions take place in liver. Interestingly, the critical genes for controlling methyl metabolism and DNA methylation capacity are themselves regulated by methylation. For example, MAT1A (forms SAM) is underexpressed when it is hypermethylated [44], and the expression of the DNA methyltransferases DNMT1 AND DNMT3A are controlled by methylation of specific CpG sites [21,45▪,46]. The expression of G9a histone methylase is also decreased when CpGs at specific sites in the gene are methylated [47,48]. Thus, rodents fed diets low in choline and methionine undermethylate these methyltransferase genes and therefore overexpress these methyltransferases [21,45▪,46–48]. This explains why some genes are paradoxically overmethylated despite methyl-donor deficiency [46]. Interspersed elements containing repetitive DNA sequences represent 30% of the mammalian genome [49], and the methylation status of these elements is modified by the availability of dietary choline in rodents [50]. An additional potential mechanism for methyl-deficiency modulation of DNA methyltransferase activity in liver is focused on mitochondria. Abnormal membrane composition causes the release of free radicals and oxidizes the nucleotides, forming 8-hydroxydeoxyguanosine which inhibits cytosine methylation [51]. Mechanistically, the accumulation of intracellular fat, inflammation, fibrosis and eventually carcinogenesis are multi-factorial, and epigenetic mechanisms occupy central roles in this scenario (Fig. 2). We revisit here the effects of choline deficiency on the liver epigenome and on signal transduction involved in the inflammation pathways.
FIGURE 2.

Epigenetic mechanisms modify liver function. Dietary intake of methyl donors such as methionine and choline modifies hepatic inflammatory signaling pathways as well as the epigenetic marks regulating the expression of genes relevant to signaling pathways involved in hepatic steatosis, fibrosis, and carcinogenesis.
EPIGENETIC MECHANISMS INVOLVED IN NONALCOHOLIC FATTY LIVER DISEASE, NONALCOHOLIC STEATOHEPATITIS, AND PROGRESSION TOWARD LIVER TUMORIGENESIS
Some of the major signals and mechanisms involved in NAFLD, nonalcoholic steatohepatitis (NASH), and progression toward liver carcinogenesis are signaling by cytokines/chemokines (TNFα [52,53], TGFβ [54,55], IL-6 [56], and IL-10 [57]), CCL2/MCP1 targeting PPARα [58–60] (there is an increase in promoter methylation of antifibrotic PPARα receptor protein in choline-deficient livers [61]), CCL5 increased by hepatocellular lipid accumulation [62–64] and CXCL8/IL-8 [65]; increased de novo synthesis of triglycerides [66]; decreased VLDL synthesis and export [67]; and decreased long-chain fatty acid oxidation.
As discussed earlier, many of the hepatic genes involved in pathways for the development of fatty liver are epigenetically regulated, including PPARγ [38], SREBP-1c [39], glucokinase [41], pyruvate kinase [41,42], phosphoenolpyruvate carboxykinase [42], and enzymes of cholesterol metabolism [43]. Fatty liver can progress to liver damage that is accompanied by fibrosis [scar tissue synthesized by activated hepatic stellate cells (aHSCs, i.e., myofibroblasts)]. Recent studies explain how liver fibrosis is increased in low methyl-donor environments [68–70]. Low levels of inflammatory signals combined with epigenetic mechanisms normally keep hepatic stellate cells (HSCs) quiescent [69]. TGF-β1 signaling mediates the activation of HSCs [71] by decreasing the expression of Phosphatase and Tensin homolog (PTEN), a repressor of phosphatidylinositol 3,4,5 triphosphate kinase/serine-threonine kinase Akt (PI3K/AKT) and extracellular signal-regulated kinase (ERK) signaling pathways [72]. A new set of epigenetic marks is acquired by aHSCs and these act to control gene expression so as to maintain the aHSC cellular phenotype [69]. Once achieved, the epigenome of aHSCs results in increased DNMT1 expression and in increased MECP2 levels with recruitment of histone modifiers [69,73,74] (Fig. 1). These changes stabilize the aHSCs’ chromatin and maintain low PTEN expression, thereby ensuring the progression of fibrosis. In summary, the fibrosis process is initiated upon HSC activation via cytokines and growth factors and a new set of epigenetic marks is acquired that maintains their new cellular phenotype [69]. The decreased availability of methyl donors thereby can initiate and sustain hepatic fibrosis (Fig. 2). Interestingly, these modified epigenetic mechanisms driving liver repair are heritable [75▪▪]. Newer generations are less responsive to fibrosis because they generate decreased numbers of HSCs, increased expression of antifibrogenic PPAR-γ, and decreased TGF-β. These adaptations were epigenetically transmitted through the male germline, via histone modifications. This may explain the presence of hypomethylated PPAR-γ in humans harboring mild forms of fibrosis in severe methyl-deficient environments.
Mechanisms responsible for tumor-initiating events or tumor progression are also, in part, epigenetic [76,77]. Many tumor suppressor genes are epigenetically regulated, including genes for cell-cycle regulation (p15 and p16), apoptosis (DAPK and APAF-1), cell adherence (CDH1 and CDH3), and DNA repair (BRCA1 and hMLH) [22▪▪]. Hepatocellular carcinomas have an epigenome that is profoundly different from normal hepatocytes, with gene-specific DNA overmethylation or undermethylation, altered histone epigenetic marks, and abnormal expression of genes for DNA methyltransferases and histone-modifying enzymes [77]. Methyl-deficient diets which caused hepatic cancers were associated with global and gene-specific epigenetic changes [25,78,79], including hypomethylation of c-myc [80], c-fos, and c-Ha-ras [81]. These changes in cytosine methylation patterns occur after short-term feeding of choline-deficient diets and before hepatocarcinomas develop, suggesting a causal rather than a consequential role. Interestingly, mouse models in which methylmetabolism has been perturbed by genetic manipulation, such as Mat1a−/− mice [in which methionine adenosyltransferase (forms SAM) is deleted [82–84]] and Bhmt−/− mice (in which BHMT, needed to transfer methyl moiety from betaine to homocysteine, is deleted [22▪▪]), develop hepatic steatosis and hepatocarcinomas.
Other products of choline metabolism influence carcinogenesis and involve epigenetic mechanisms. Lysophosphatidic acid (LPA), via G-protein-coupled transmembrane receptors, regulates cellular proliferation, differentiation, morphogenesis, and protection from apoptosis [85,86]. Phosphatidylcholine is a precursor for LPA formation [87]. The genes encoding the receptors for LPA signal are regulated by the epigenetic mechanisms [88]. Rodents fed choline–methionine-deficient diets had aberrant methylation of the gene for the LPA1 receptor in a pattern similar to the methylation abnormalities described in hepatocellular carcinomas [89].
CONCLUSION
Liver is the organ where choline, methyl folate, methionine, and SAM metabolic pathways are most active, and it is the organ where most methylation reactions occur. The liver is very sensitive to the availability of methyl donors in the diet, including choline. When deprived of these nutrients, the liver becomes fatty, hepatocytes die, fibrosis develops, and eventually foci of carcinomas appear. This progression occurs not only because these nutrients are needed to produce important structural components (membranes) and signaling molecules (e.g., LPA and acetylcholine), but also because these nutrients influence the epigenetic regulation of gene expression.
KEY POINTS.
Choline and other dietary methyl donors are important for liver function.
These nutrients are important modulators of epigenetic regulation of gene expression.
Pathways important for the development of fatty liver, hepatic fibrosis, and hepatocarcinoma are regulated via epigenetic mechanisms.
Acknowledgments
Financial support was provided by the grants from the NIH (DK05595 and DK36530).
M. G. Mehedint and S. H. Zeisel have no financial interest in relation to this article.
Footnotes
Conflicts of interest
Dr Zeisel received grant support from the Pfizer Nutrition, Balchem, and the Egg Nutrition Research Center for studies other than those described in this article. Dr Zeisel is on the Scientific Advisory Board for Solae, American Pistachio Growers, Dupont, Metabolon, and GenoVive.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
Additional references related to this topic can also be found in the Current World Literature section in this issue (p. 364).
- 1.Zeisel SH. Nutritional genomics: defining the dietary requirement and effects of choline. J Nutr. 2011;141:531–534. doi: 10.3945/jn.110.130369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zeisel SH. Choline: critical role during fetal development and dietary requirements in adults. Annu Rev Nutr. 2006;26:229–250. doi: 10.1146/annurev.nutr.26.061505.111156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeisel SH Institute of Medicine, National Academy of Sciences USA. Dietary reference intakes for folate, thiamin, riboflavin, niacin, vitamin B12, pantothenic acid, biotin, and choline. Washington, D.C: National Academy Press; 1998. Choline; pp. 390–422. [PubMed] [Google Scholar]
- 4.Cho E, Zeisel SH, Jacques P, et al. Dietary choline and betaine assessed by food-frequency questionnaire in relation to plasma total homocysteine concentration in the Framingham Offspring Study. Am J Clin Nutr. 2006;83:905–911. doi: 10.1093/ajcn/83.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bidulescu A, Chambless LE, Siega-Riz AM, et al. Usual choline and betaine dietary intake and incident coronary heart disease: the Atherosclerosis Risk in Communities (ARIC) study. BMC Cardiovasc Disord. 2007;7:20. doi: 10.1186/1471-2261-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bidulescu A, Chambless LE, Siega-Riz AM, et al. Repeatability and measurement error in the assessment of choline and betaine dietary intake: the Atherosclerosis Risk in Communities (ARIC) study. Nutr J. 2009;8:14. doi: 10.1186/1475-2891-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho E, Willett WC, Colditz GA, et al. Dietary choline and betaine and the risk of distal colorectal adenoma in women. J Natl Cancer Inst. 2007;99:1224– 1231. doi: 10.1093/jnci/djm082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sentongo TA, Kumar P, Karza K, et al. Whole-blood-free choline and choline metabolites in infants who require chronic parenteral nutrition therapy. J Pediatr Gastroenterol Nutr. 2010;50:194–199. doi: 10.1097/MPG.0b013e3181a93735. [DOI] [PubMed] [Google Scholar]
- 9.Fischer LM, daCosta K, Kwock L, et al. Sex and menopausal status influence human dietary requirements for the nutrient choline. Am J Clin Nutr. 2007;85:1275–1285. doi: 10.1093/ajcn/85.5.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Resseguie ME, da Costa KA, Galanko JA, et al. Aberrant estrogen regulation of PEMT results in choline deficiency-associated liver dysfunction. J Biol Chem. 2011;286:1649–1658. doi: 10.1074/jbc.M110.106922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Da Costa KA, Kozyreva OG, Song J, et al. Common genetic polymorphisms affect the human requirement for the nutrient choline. FASEB J. 2006;20:1336–1344. doi: 10.1096/fj.06-5734com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kohlmeier M, da Costa KA, Fischer LM, et al. Genetic variation of folate-mediated one-carbon transfer pathway predicts susceptibility to choline deficiency in humans. Proc Natl Acad Sci USA. 2005;102:16025–16030. doi: 10.1073/pnas.0504285102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sha W, da Costa KA, Fischer LM, et al. Metabolomic profiling can predict which humans will develop liver dysfunction when deprived of dietary choline. FASEB J. 2010;24:2962–2975. doi: 10.1096/fj.09-154054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buchman AL. The addition of choline to parenteral nutrition. Gastroenterology. 2009;137:S119–S128. doi: 10.1053/j.gastro.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 15.Hebbard L, George J. Animal models of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2011;8:35–44. doi: 10.1038/nrgastro.2010.191. [DOI] [PubMed] [Google Scholar]
- 16.Pagadala MR, McCullough AJ. The relevance of liver histology to predicting clinically meaningful outcomes in nonalcoholic steatohepatitis. Clin Liver Dis. 2012;16:487–504. doi: 10.1016/j.cld.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cole LK, Vance JE, Vance DE. Phosphatidylcholine biosynthesis and lipoprotein metabolism. Biochim Biophys Acta. 2012;1821:754–761. doi: 10.1016/j.bbalip.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 18.Fischer LM, da Costa KA, Kwock L, et al. Dietary choline requirements of women: effects of estrogen and genetic variation. Am J Clin Nutr. 2010;92:1113–1119. doi: 10.3945/ajcn.2010.30064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corbin KD, Abdelmalek MF, Spencer MD, et al. Genetic signatures in choline and 1-carbon metabolism are associated with the severity of hepatic steatosis. FASEB J. 2013 doi: 10.1096/fj.12-219097. [Epub ahead of print] fj.12-219097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshiji H, Yoshii J, Ikenaka Y, et al. Inhibition of renin–angiotensin system attenuates liver enzyme-altered preneoplastic lesions and fibrosis development in rats. J Hepatol. 2002;37:22–30. doi: 10.1016/s0168-8278(02)00104-6. [DOI] [PubMed] [Google Scholar]
- 21.Shimizu K, Onishi M, Sugata E, et al. Disturbance of DNA methylation patterns in the early phase of hepatocarcinogenesis induced by a choline-deficient L-amino acid-defined diet in rats. Cancer Sci. 2007;98:1318–1322. doi: 10.1111/j.1349-7006.2007.00564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22▪▪.Teng YW, Mehedint MG, Garrow TA, et al. Deletion of betaine-homocysteine S-methyltransferase in mice perturbs choline and 1-carbon metabolism, resulting in fatty liver and hepatocellular carcinomas. J Biol Chem. 2011;286:36258–36267. doi: 10.1074/jbc.M111.265348. The rodent studies performed by Teng et al. revealed that genetic defects of choline metabolism that specifically impair the provision of methyl groups lead to steatohepatitis and progression toward adenocarcinoma and hepatocellular carcinoma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zeisel SH, Albright CD, Shin O-K, et al. Choline deficiency selects for resistance to p53-independent apoptosis and causes tumorigenic transformation of rat hepatocytes. Carcinogenesis. 1997;18:731–738. doi: 10.1093/carcin/18.4.731. [DOI] [PubMed] [Google Scholar]
- 24.Schrager TF, Newberne PM, Pikul AH, et al. Aflatoxin–DNA adduct formation in chronically dosed rats fed a choline-deficient diet. Carcinogenesis. 1990;11:177–180. doi: 10.1093/carcin/11.1.177. [DOI] [PubMed] [Google Scholar]
- 25.Pogribny IP, Shpyleva SI, Muskhelishvili L, et al. Role of DNA damage and alterations in cytosine DNA methylation in rat liver carcinogenesis induced by a methyl-deficient diet. Mutat Res. 2009;669:56–62. doi: 10.1016/j.mrfmmm.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 26.Baric I. Inherited disorders in the conversion of methionine to homocysteine. J Inherit Metab Dis. 2009;32:459–471. doi: 10.1007/s10545-009-1146-4. [DOI] [PubMed] [Google Scholar]
- 27.Rodenhiser D, Mann M. Epigenetics and human disease: translating basic biology into clinical applications. CMAJ. 2006;174:341–348. doi: 10.1503/cmaj.050774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li G, Reinberg D. Chromatin higher-order structures and gene regulation. Curr Opin Genet Dev. 2011;21:175–186. doi: 10.1016/j.gde.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Steensel B. Chromatin: constructing the big picture. EMBO J. 2011;30:1885–1895. doi: 10.1038/emboj.2011.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fierz B, Muir TW. Chromatin as an expansive canvas for chemical biology. Nat Chem Biol. 2012;8:417–427. doi: 10.1038/nchembio.938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bassols J, Prats-Puig A, Vazquez-Ruiz M, et al. Placental FTO expression relates to fetal growth. Int J Obes (Lond) 2010;34:1365–1370. doi: 10.1038/ijo.2010.62. [DOI] [PubMed] [Google Scholar]
- 32.Kubicek S, O’Sullivan RJ, August EM, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. 2007;25:473–481. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 33.Loyola A, Tagami H, Bonaldi T, et al. The HP1alpha–CAF1–SetDB1-containing complex provides H3K9me1 for Suv39-mediated K9me3 in pericentric heterochromatin. EMBO Rep. 2009;10:769–775. doi: 10.1038/embor.2009.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Niculescu MD, Craciunescu CN, Zeisel SH. Dietary choline deficiency alters global and gene-specific DNA methylation in the developing hippocampus of mouse fetal brains. FASEB J. 2006;20:43–49. doi: 10.1096/fj.05-4707com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chedin F. The DNMT3 family of mammalian de novo DNA methyltransferases. Prog Mol Biol Transl Sci. 2011;101:255–285. doi: 10.1016/B978-0-12-387685-0.00007-X. [DOI] [PubMed] [Google Scholar]
- 36.Blusztajn JK, Mellott TJ. Choline nutrition programs brain development via DNA and histone methylation. Cent Nerv Syst Agents Med Chem. 2012;12:82–94. doi: 10.2174/187152412800792706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fujiki K, Kano F, Shiota K, et al. Expression of the peroxisome proliferator activated receptor gamma gene is repressed by DNA methylation in visceral adipose tissue of mouse models of diabetes. BMC Biol. 2009;7:38. doi: 10.1186/1741-7007-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Esfandiari F, Medici V, Wong DH, et al. Epigenetic regulation of hepatic endoplasmic reticulum stress pathways in the ethanol-fed cystathionine beta synthase-deficient mouse. Hepatology. 2010;51:932–941. doi: 10.1002/hep.23382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pal-Bhadra M, Bhadra U, Jackson DE, et al. Distinct methylation patterns in histone H3 at Lys-4 and Lys-9 correlate with up- & down-regulation of genes by ethanol in hepatocytes. Life Sci. 2007;81:979–987. doi: 10.1016/j.lfs.2007.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lomba A, Milagro FI, Garcia-Diaz DF, et al. Obesity induced by a pair-fed high fat sucrose diet: methylation and expression pattern of genes related to energy homeostasis. Lipids Health Dis. 2010;9:60. doi: 10.1186/1476-511X-9-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiong Y, Lei QY, Zhao S, et al. Regulation of glycolysis and gluconeogenesis by acetylation of PKM and PEPCK. Cold Spring Harb Symp Quant Biol. 2011;76:285–289. doi: 10.1101/sqb.2011.76.010942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Plosch T, Gellhaus A, van Straten EM, et al. The liver X receptor (LXR) and its target gene ABCA1 are regulated upon low oxygen in human trophoblast cells: a reason for alterations in preeclampsia? Placenta. 2010;31:910– 918. doi: 10.1016/j.placenta.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 44.Zhang J, Gong C, Bing Y, et al. Hypermethylation-repressed methionine adenosyltransferase 1A as a potential biomarker for hepatocellular carcinoma. Hepatol Res. 2012 doi: 10.1111/j.1872-034X.2012.01099.x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 45▪.Pogribny IP, Tryndyak VP, Bagnyukova TV, et al. Hepatic epigenetic phenotype predetermines individual susceptibility to hepatic steatosis in mice fed a lipogenic methyl-deficient diet. J Hepatol. 2009;51:176–186. doi: 10.1016/j.jhep.2009.03.021. Pogribny et al. clearly indicated a causal rather than a consequential role of epigenetics in the development of NASH. New disease prevention strategies and treatments targeting unstable epiloci can be developed based on these findings. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kovacheva VP, Mellott TJ, Davison JM, et al. Gestational choline deficiency causes global and Igf2 gene DNA hypermethylation by up-regulation of Dnmt1 expression. J Biol Chem. 2007;282:31777–31788. doi: 10.1074/jbc.M705539200. [DOI] [PubMed] [Google Scholar]
- 47.Davison JM, Mellott TJ, Kovacheva VP, et al. Gestational choline supply regulates methylation of histone H3, expression of histone methyltransferases G9a (Kmt1c) and Suv39h1 (Kmt1a), and DNA methylation of their genes in rat fetal liver and brain. J Biol Chem. 2009;284:1982–1989. doi: 10.1074/jbc.M807651200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mehedint MG, Niculescu MD, Craciunescu CN, et al. Choline deficiency alters global histone methylation and epigenetic marking at the Re1 site of the calbindin 1 gene. FASEB J. 2010;24:184–195. doi: 10.1096/fj.09-140145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Estecio MR, Gallegos J, Dekmezian M, et al. SINE retrotransposons cause epigenetic reprogramming of adjacent gene promoters. Mol Cancer Res. 2012;10:1332–1342. doi: 10.1158/1541-7786.MCR-12-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Asada K, Kotake Y, Asada R, et al. LINE-1 hypomethylation in a choline-deficiency-induced liver cancer in rats: dependence on feeding period. J Biomed Biotechnol. 2006;2006:17142. doi: 10.1155/JBB/2006/17142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hitchler MJ, Domann FE. Metabolic defects provide a spark for the epigenetic switch in cancer. Free Radic Biol Med. 2009;47:115–127. doi: 10.1016/j.freeradbiomed.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hui JM, Hodge A, Farrell GC, et al. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology. 2004;40:46–54. doi: 10.1002/hep.20280. [DOI] [PubMed] [Google Scholar]
- 53.Zhou YJ, Li YY, Nie YQ, et al. Influence of polygenetic polymorphisms on the susceptibility to nonalcoholic fatty liver disease of Chinese people. J Gastroenterol Hepatol. 2010;25:772–777. doi: 10.1111/j.1440-1746.2009.06144.x. [DOI] [PubMed] [Google Scholar]
- 54.Annoni G, Weiner FR, Zern MA. Increased transforming growth factor-beta 1 gene expression in human liver disease. J Hepatol. 1992;14:259–264. doi: 10.1016/0168-8278(92)90168-o. [DOI] [PubMed] [Google Scholar]
- 55.Castilla A, Prieto J, Fausto N. Transforming growth factors beta 1 and alpha in chronic liver disease. Effects of interferon alfa therapy. N Engl J Med. 1991;324:933–940. doi: 10.1056/NEJM199104043241401. [DOI] [PubMed] [Google Scholar]
- 56.Haukeland JW, Damas JK, Konopski Z, et al. Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J Hepatol. 2006;44:1167–1174. doi: 10.1016/j.jhep.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 57.Esposito K, Pontillo A, Giugliano F, et al. Association of low interleukin-10 levels with the metabolic syndrome in obese women. J Clin Endocrinol Metab. 2003;88:1055–1058. doi: 10.1210/jc.2002-021437. [DOI] [PubMed] [Google Scholar]
- 58.Kanda H, Tateya S, Tamori Y, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ito M, Suzuki J, Tsujioka S, et al. Longitudinal analysis of murine steatohepatitis model induced by chronic exposure to high-fat diet. Hepatol Res. 2007;37:50–57. doi: 10.1111/j.1872-034X.2007.00008.x. [DOI] [PubMed] [Google Scholar]
- 60.Rull A, Rodriguez F, Aragones G, et al. Hepatic monocyte chemoattractant protein-1 is upregulated by dietary cholesterol and contributes to liver steatosis. Cytokine. 2009;48:273–279. doi: 10.1016/j.cyto.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 61.Mikael LG, Pancer J, Wu Q, et al. Disturbed one-carbon metabolism causing adverse reproductive outcomes in mice is associated with altered expression of apolipoprotein AI and inflammatory mediators PPARalpha, interferon-gamma, and interleukin-10. J Nutr. 2012;142:411–418. doi: 10.3945/jn.111.151753. [DOI] [PubMed] [Google Scholar]
- 62.Berres ML, Koenen RR, Rueland A, et al. Antagonism of the chemokine Ccl5 ameliorates experimental liver fibrosis in mice. J Clin Invest. 2010;120:4129–4140. doi: 10.1172/JCI41732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Desai MS, Mariscalco MM, Tawil A, et al. Atherogenic diet-induced hepatitis is partially dependent on murine TLR4. J Leukoc Biol. 2008;83:1336–1344. doi: 10.1189/jlb.0607390. [DOI] [PubMed] [Google Scholar]
- 64.Wu H, Ghosh S, Perrard XD, et al. T-cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation. 2007;115:1029–1038. doi: 10.1161/CIRCULATIONAHA.106.638379. [DOI] [PubMed] [Google Scholar]
- 65.Jarrar MH, Baranova A, Collantes R, et al. Adipokines and cytokines in nonalcoholic fatty liver disease. Aliment Pharmacol Ther. 2008;27:412–421. doi: 10.1111/j.1365-2036.2007.03586.x. [DOI] [PubMed] [Google Scholar]
- 66.Bradbury MW, Berk PD. Lipid metabolism in hepatic steatosis. Clin Liver Dis. 2004;8:639–671. xi. doi: 10.1016/j.cld.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 67.Zeisel SH, Blusztajn JK. Choline and human nutrition. Annu Rev Nutr. 1994;14:269–296. doi: 10.1146/annurev.nu.14.070194.001413. [DOI] [PubMed] [Google Scholar]
- 68.Koca SS, Bahcecioglu IH, Poyrazoglu OK, et al. The treatment with antibody of TNF-alpha reduces the inflammation, necrosis and fibrosis in the nonalcoholic steatohepatitis induced by methionine- and choline-deficient diet. Inflammation. 2008;31:91–98. doi: 10.1007/s10753-007-9053-z. [DOI] [PubMed] [Google Scholar]
- 69.Bian EB, Huang C, Ma TT, et al. DNMT1-mediated PTEN hypermethylation confers hepatic stellate cell activation and liver fibrogenesis in rats. Toxicol Appl Pharmacol. 2012;264:13–22. doi: 10.1016/j.taap.2012.06.022. [DOI] [PubMed] [Google Scholar]
- 70.Tomita K, Teratani T, Suzuki T, et al. p53/p66Shc-mediated signaling contributes to the progression of nonalcoholic steatohepatitis in humans and mice. J Hepatol. 2012;57:837–843. doi: 10.1016/j.jhep.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 71.Gressner AM, Weiskirchen R, Breitkopf K, et al. Roles of TGF-beta in hepatic fibrosis. Front Biosci. 2002;7:d793–d807. doi: 10.2741/A812. [DOI] [PubMed] [Google Scholar]
- 72.Liu Y, Wen XM, Lui EL, et al. Therapeutic targeting of the PDGF and TGF-beta-signaling pathways in hepatic stellate cells by PTK787/ZK22258. Lab Invest. 2009;89:1152–1160. doi: 10.1038/labinvest.2009.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mann J, Chu DC, Maxwell A, et al. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology. 2010;138:705–714. 714.e701–714.e704. doi: 10.1053/j.gastro.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tao H, Huang C, Yang JJ, et al. MeCP2 controls the expression of RASAL1 in the hepatic fibrosis in rats. Toxicology. 2011;290:327–333. doi: 10.1016/j.tox.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 75▪▪.Zeybel M, Hardy T, Wong YK, et al. Multigenerational epigenetic adaptation of the hepatic wound-healing response. Nat Med. 2012;18:1369–1377. doi: 10.1038/nm.2893. Short-term adaptations in liver repair mechanisms are crucial for choline-induced liver injury. This is a first evidence of how these rapid epigenetic modifications escape the resetting process during early life and manifest themselves as a new phenotype in the next generation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Herceg Z, Paliwal A. Epigenetic mechanisms in hepatocellular carcinoma: how environmental factors influence the epigenome. Mutat Res. 2011;727:55–61. doi: 10.1016/j.mrrev.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 77.Pogribny IP, Rusyn I. Role of epigenetic aberrations in the development and progression of human hepatocellular carcinoma. Cancer Lett. 2012 doi: 10.1016/j.canlet.2012.01.038. [Epub ahead of print]. S0304-3835(12)00082-1 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Poirier LA. Methyl group deficiency in hepatocarcinogenesis. Drug Metab Rev. 1994;26:185–199. doi: 10.3109/03602539409029790. [DOI] [PubMed] [Google Scholar]
- 79.Christman JK. Lipotrope deficiency and persistent changes in DNA methylation. Lipotrope deficiency and DNA methylation. Adv Exp Med Biol. 1995;375:97–106. doi: 10.1007/978-1-4899-0949-7_9. [DOI] [PubMed] [Google Scholar]
- 80.Tsujiuchi T, Tsutsumi M, Sasaki Y, et al. Hypomethylation of CpG sites and c-myc gene overexpression in hepatocellular carcinomas, but not hyperplastic nodules, induced by a choline-deficient L-amino acid-defined diet in rats. Jpn J Cancer Res. 1999;90:909–913. doi: 10.1111/j.1349-7006.1999.tb00834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Christman JK, Sheikhnejad G, Dizik M, et al. Reversibility of changes in nucleic acid methylation and gene expression induced in rat liver by severe dietary methyl deficiency. Carcinogenesis. 1993;14:551–557. doi: 10.1093/carcin/14.4.551. [DOI] [PubMed] [Google Scholar]
- 82.Lu SC, Alvarez L, Huang ZZ, et al. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proc Natl Acad Sci USA. 2001;98:5560–5565. doi: 10.1073/pnas.091016398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tomasi ML, Iglesias-Ara A, Yang H, et al. S-adenosylmethionine regulates apurinic/apyrimidinic endonuclease 1 stability: implication in hepatocarcinogenesis. Gastroenterology. 2009;136:1025–1036. doi: 10.1053/j.gastro.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ko KS, Tomasi ML, Iglesias-Ara A, et al. Liver-specific deletion of prohibitin 1 results in spontaneous liver injury, fibrosis, and hepatocellular carcinoma in mice. Hepatology. 2010;52:2096–2108. doi: 10.1002/hep.23919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Contos JJ, Ishii I, Chun J. Lysophosphatidic acid receptors. Mol Pharmacol. 2000;58:1188–1196. doi: 10.1124/mol.58.6.1188. [DOI] [PubMed] [Google Scholar]
- 86.Fang X, Schummer M, Mao M, et al. Lysophosphatidic acid is a bioactive mediator in ovarian cancer. Biochim Biophys Acta. 2002;1582:257–264. doi: 10.1016/s1388-1981(02)00179-8. [DOI] [PubMed] [Google Scholar]
- 87.Selvy PE, Lavieri RR, Lindsley CW, et al. Phospholipase D: enzymology, functionality, and chemical modulation. Chem Rev. 2011;111:6064–6119. doi: 10.1021/cr200296t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Okabe K, Hayashi M, Yoshida I, et al. Distinct DNA methylation patterns of lysophosphatidic acid receptor genes during rat hepatocarcinogenesis induced by a choline-deficient L-amino acid-defined diet. Arch Toxicol. 2011;85:1303–1310. doi: 10.1007/s00204-011-0656-7. [DOI] [PubMed] [Google Scholar]
- 89.Obo Y, Yamada T, Furukawa M, et al. Frequent mutations of lysophosphatidic acid receptor-1 gene in rat liver tumors. Mutat Res. 2009;660:47–50. doi: 10.1016/j.mrfmmm.2008.10.005. [DOI] [PubMed] [Google Scholar]