

Abstract
Sexual recombination drives genetic diversity in eukaryotic genomes and fosters adaptation to novel environmental challenges. Although strictly asexual microorganisms are often considered as evolutionary dead ends, they comprise many devastating plant pathogens. Presently, it remains unknown how such asexual pathogens generate the genetic variation that is required for quick adaptation and evolution in the arms race with their hosts. Here, we show that extensive chromosomal rearrangements in the strictly asexual plant pathogenic fungus Verticillium dahliae establish highly dynamic lineage-specific (LS) genomic regions that act as a source for genetic variation to mediate aggressiveness. We show that such LS regions are greatly enriched for in planta-expressed effector genes encoding secreted proteins that enable host colonization. The LS regions occur at the flanks of chromosomal breakpoints and are enriched for retrotransposons and other repetitive sequence elements. Our results suggest that asexual pathogens may evolve by prompting chromosomal rearrangements, enabling rapid development of novel effector genes. Likely, chromosomal reshuffling can act as a general mechanism for adaptation in asexually propagating organisms.
Sexual reproduction is thought to enable responses to environmental challenges in nature and fosters evolution of pathogen genomes (Williams 1975; Maynard Smith 1978; Colegrave 2002; Goddard et al. 2005; Heitman 2006; de Visser and Elena 2007). Consequently, pathogen populations that undergo regular sexual reproduction are thought to pose a great risk to agriculture because they can recombine alleles that contribute to virulence in the face of dynamic environmental conditions (McDonald and Linde 2002). However, sexual reproduction comes at a cost because two compatible individuals need to locate each other to generate offspring, and fitness decreases due to break up of coadapted combinations of interacting alleles may occur (Agrawal 2006; Heitman 2006; Heitman et al. 2007; de Visser and Elena 2007). Sexual compatibility between individuals depends on so-called mating types in fungi. In heterothallic fungi, mating is restricted to genetically different individuals of opposite mating types, whereas in homothallic fungi, mating and sexual reproduction can occur between all individuals, including itself (Billiard et al. 2012). Although most fungi are able to undergo asexual and sexual reproduction, ∼20% of all fungal phyla reproduce strictly asexually (Heitman et al. 2007). Such strictly asexual organisms are thought to be less flexible than sexual ones, relying solely on random mutations to adapt to changing environments. Asexual organisms are often considered as evolutionary dead ends (Burt 2000; McDonald and Linde 2002). This is mainly due to the absence of meiotic recombination resulting in increased accumulation of deleterious mutations, an effect known as Muller's ratchet (Felsenstein 1974), and a decreased ability to adapt to changing environmental conditions by generation of novel genetic combinations (Heitman 2006). Especially for pathogenic species that are continuously involved in an arms race with their hosts that try to ward off invading pathogens, quick adaptation in order to coevolve with the host immune system is essential for evolutionary success (Raffaele and Kamoun 2012). Nonetheless, many destructive plant pathogenic fungi are known as strictly asexually reproducing organisms, such as the vascular wilt fungus Verticillium dahliae (Fradin and Thomma 2006).
V. dahliae is a soil-borne broad host-range plant pathogen that invades the water-conducting xylem vessels of susceptible plant species to cause vascular wilt disease (Fradin and Thomma 2006; Klosterman et al. 2009). Hundreds of dicotyledonous plant species can be infected by V. dahliae, including many economically important crops such as lettuce, cotton, and tomato (Bolek et al. 2005; Fradin and Thomma 2006; Klosterman et al. 2009; Atallah et al. 2010). Although two mating types have been found in V. dahliae, it is predicted that propagation is solely asexual (clonal) in most if not all populations, as a sexual cycle has never been observed and the ratio between both mating types is greatly skewed toward one of the two (Usami et al. 2008, 2009; Atallah et al. 2010; Inderbitzin et al. 2011). Only in a few plant species has monogenic resistance toward Verticillium wilt been described. These include tomato, for which a Verticillium resistance locus (named Ve) has been identified that mediates resistance against race 1 strains of V. dahliae and V. albo-atrum, and that thus become avirulent (unable to cause disease). Strains that are not contained by this Ve locus remain virulent (able to cause disease) and are assigned to race 2 (Schaible et al. 1951; Kawchuk et al. 2001; Fradin et al. 2009). The resistance that is established by the Ve locus is mediated by the Ve1 gene that encodes a receptor-like protein-type cell surface receptor (Fradin et al. 2009) that activates plant immunity upon perception of a corresponding pathogen-derived ligand. Recently, through comparative genomics the race 1-specific effector Ave1 (for Avirulence on Ve1 tomato) was identified that activates Ve1-mediated resistance in tomato (de Jonge et al. 2012). Remarkably, the Ave1 effector contributes to pathogen aggressiveness on tomato plants that lack Ve1, demonstrating that Ave1 acts as a virulence factor of V. dahliae (de Jonge et al. 2012). Race 2 strains lack the Ave1 gene, and are consequently less aggressive on tomato plants that lack Ve1 when compared with race 1 strains (Amen and Shoemaker 1985; Paternotte and van Kesteren 1993; de Jonge et al. 2012).
In this study, we compared the genomes of 11 recently sequenced V. dahliae strains (Klosterman et al. 2011; de Jonge et al. 2012) that have been isolated from various geographical locations and hosts, and that are all, except for one, pathogenic on tomato. By assessing genetic diversity within this population we aimed to study the impact of asexual reproduction on the evolution of host adaptation and virulence in this plant pathogenic fungus.
Results
Identification of the core genome reveals extensive chromosomal rearrangements
With comparative genomics, we examined genetic diversity in a population of nine recently sequenced tomato pathogenic strains and one nonpathogenic strain of the asexual fungus Verticillium dahliae (Supplemental Table 1; de Jonge et al. 2012) together with the genome of V. dahliae strain VdLs17 as a reference (Klosterman et al. 2011). Read coverage mapping in 1-kb windows of all sequenced V. dahliae strains over the VdLs17 reference genome revealed a “core genome” that is shared by all strains, encompassing ∼29 Mb of sequence containing 9471 genes. The amount of single nucleotide polymorphisms (SNPs) within the core genome ranged from 5445 (JR2; ∼99.98% identity) to 163,602 (St.100; ∼99.5% identity) SNPs per strain (Table 1), collectively amounting to 236,785 nonredundant polymorphic sites. Of these, 78,342 (32.9%) occurred in protein-coding regions, of which 55% were synonymous, not affecting the protein sequence, and 45% were nonsynonymous (Table 1). To determine selection strength, the ratio of nonsynonymous (dN) substitutions per nonsynonymous site (Ka) to the number of synonymous substitutions (dS) per synonymous site (Ks) was calculated within the coding regions for each of the 9471 core genes (Stukenbrock et al. 2010). A surprisingly low number of 29 genes was found to be under positive selection (Ka/Ks > 1; P < 0.01). About half of these (13) encode proteins with a known function, including cellulose binding and regulation of transcription, whereas the other half encodes proteins of unknown function, of which only three encode secreted proteins that are candidate effectors (Supplemental Table 2; de Jonge et al. 2011).
Table 1.
Summary of SNPs when compared with V. dahliae reference strain VdLs17
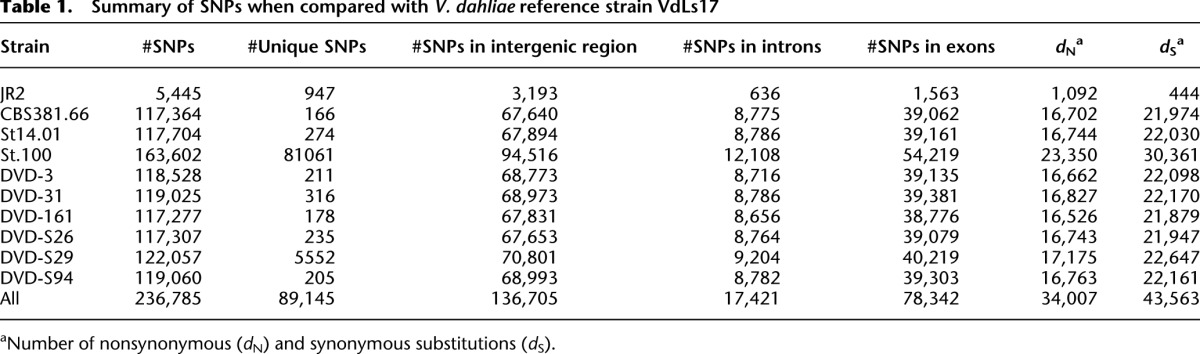
To infer evolutionary relations within the population, all 236,785 SNP positions were used to construct a phylogenetic tree (Fig. 1). Despite the high degree of conservation between the strains that were included in the population (maximum nucleotide divergence within the population is only 0.5%), strain JR2 formed a clearly separate cluster with the VdLs17 reference strain, differing only by one SNP in ∼6000 bp (0.02%). Despite the high degree of identity (99.98%), the two strains show differential aggressiveness on tomato. Strain JR2 belongs to race 1 and shows a high degree of aggressiveness on tomato lacking Ve1, while VdLs17 belongs to race 2 and displays a relatively mild aggressiveness on tomato lacking Ve1. These two strains were analyzed in further detail. To improve the de novo JR2 genome assembly, mate-pair sequencing of a 5-kb insert library was performed, leading to a drastically improved contig N50 from 59.4 kb to 1.0 Mb, and a decreased number of contigs from 4753 to 267. We subsequently used optical mapping to obtain an accurate whole-genome assembly and placed ∼30% of the contigs on eight scaffolds, covering ∼94% of the total assembly (34 Mb) (Supplemental Table 3). While the chromosome lengths of the reference strain were found to vary between 3.1 and 6.0 Mb (Klosterman et al. 2011), JR2 chromosome sizes differ and range up to 8.9 Mb (Supplemental Table 4). Thus, despite the observation that the two strains are 99.98% identical in all genomic regions that could be aligned, the karyotype of these two strains is completely different. This observation urged us to investigate collinearity between the chromosomes of the two strains.
Figure 1.

Population structure and chromosome karyotype of sequenced Verticillium dahliae isolates. An unrooted maximum likelihood phylogenetic tree based on concatenation of 236,785 SNP sites relative to reference strain VdLs17 is shown. Evolutionary distances based on the Jukes-Cantor method and bootstrap support (%) are indicated at the nodes. Race 2 strains are indicated by asterisks. Pulsed-field gel electrophoresis of sequenced V. dahliae strains are shown as well as the results of PCR analyses on structural rearrangements SR2, SR3, and SR4 using JR2-specific primer sets (J2, J3, J4) and VdLs17-specific primer sets (L3, L4), highlighting significant chromosome length and structure polymorphisms between isolates. Chromosomal DNA of Schizosaccharomyces pombe and Hansenula wingei were loaded as size makers.
Unanticipated, pairwise alignments to identify collinear (synteny) blocks between the two strains revealed repeated interruptions of syntenic regions by intra- and interchromosomal rearrangements (Fig. 2). In total, 11 intra- and 17 interchromosomal rearrangements were identified between the two strains (Fig. 2A,B). While most of these breakpoints were associated with assembly gaps due to the occurrence of repeats in the flanking genomic areas (Fig. 2B,C; Supplemental Table 6), three breakpoints could be pinpointed to the nucleotide exactly, and thus could be experimentally confirmed with PCR (Fig. 1; Supplemental Fig. 1; Supplemental Tables 5, 6). We subsequently extended the analysis of chromosomal rearrangements to all sequenced strains by screening for breakpoints within alignments of the assembled contigs for each of the strains to the reference strain. Although identification of such syntenic breakpoints in the small insert library genome assemblies is hampered by the short contig lengths, three to eight synteny breakpoints could be identified in silico for each of these strains when compared with the VdLs17 reference genome, which roughly occur in the regions of the previously identified breakpoints between VdLs17 and JR2. The occurrence of several of these breakpoints was confirmed with PCR (Fig. 1; Supplemental Fig. 1). Moreover, pulsed-field gel electrophoresis confirmed considerable chromosome length polymorphism between all strains (Fig. 1) and demonstrates that intra- and interchromosomal rearrangements frequently occur within the genomes of V. dahliae strains despite their high degree of sequence conservation.
Figure 2.
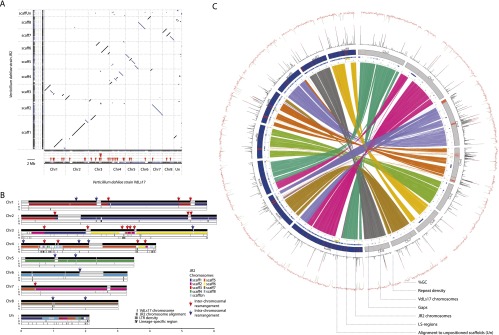
Whole-genome alignment of Verticillium dahliae strains VdLs17 and JR2 reveals extensive chromosomal rearrangements. (A) Whole-genome dot-plot comparison with forward–forward alignments (black) and inversions (blue). Red triangles mark syntenic breakpoints. (Un) Unplaced contigs during optical mapping. (B) Global view of synteny alignments between V. dahliae strains VdLs17 and JR2 and the distribution of LTR retrotransponsons and lineage-specific regions. VdLs17 chromosomes are shown as reference. For each chromosome, row I represents genomic scaffolds (black) on the chromosome separated by scaffold breaks (gray); row II displays syntenic alignment of JR2 chromosomes, highlighting 28 major intra- and interchromosomal rearrangements that are marked by red (inter) and blue (intra) arrows above each breakpoint; row III represents the density of LTR retrotransposable elements calculated in 10-kb windows; and row IV depicts lineage-specific regions that are found in only a subset of strains. (C) Circos diagram illustrating collinear blocks with alignments between VdLs17 (gray) and JR2 (blue) chromosomes, sequence gaps, sequences aligning to unpositioned scaffolds (yellow), lineage-specific sequences (red), repeat density (% coverage of 10-kb window), and GC % (per 10-kb window).
Particularly, retrotransposons have been implicated in genome rearrangements through homologous recombination between elements or by causing chromosomal breaks during excision or insertion (Mieczkowski et al. 2006; Maxwell et al. 2011). To investigate a potential involvement of retrotransposons in the genomic rearrangements of V. dahliae, we identified all repetitive elements in the genome sequences of strains VdLs17 and JR2, and found that both genomes contain only a low-repeat content of ∼4% (Klosterman et al. 2011; Supplemental Table 7). Of the different classes of repeats, we only observed significant correlation (P < 0.01) between synteny breakpoints and long terminal repeat (LTR) retrotransposons (Supplemental Tables 8–11).
The pan-genome of V. dahliae: Lineage-specific regions
In addition to the core ∼30 Mb genome, all strains carried up to ∼4 Mb of genome sequence that was unique or shared by only a subset of strains, composing a highly dynamic lineage-specific (LS) region of the genome encoding up to 1000 genes (Table 2; Klosterman et al. 2011), which is enriched for various types of transposable elements (Fig. 2B,C; Klosterman et al. 2011; Amyotte et al. 2012). These LS genomic regions are correlated (P < 0.01) with syntenic breakpoints in VdLs17 and JR2 (Supplemental Tables 6, 12). In VdLs17 as well as JR2 these regions are typically devoid of housekeeping genes that are required for the maintenance of basic cellular functions such as transcription and translation (Klosterman et al. 2011). Intriguingly, the Ave1 gene is located within the ∼4-Mb LS region of race 1 strains, suggesting that these regions contribute to niche adaptation, i.e., pathogenicity on plant hosts (de Jonge et al. 2012).
Table 2.
Summary of genome size and gene numbers for each sequenced strain, split by the core and lineage-specific (LS) genome
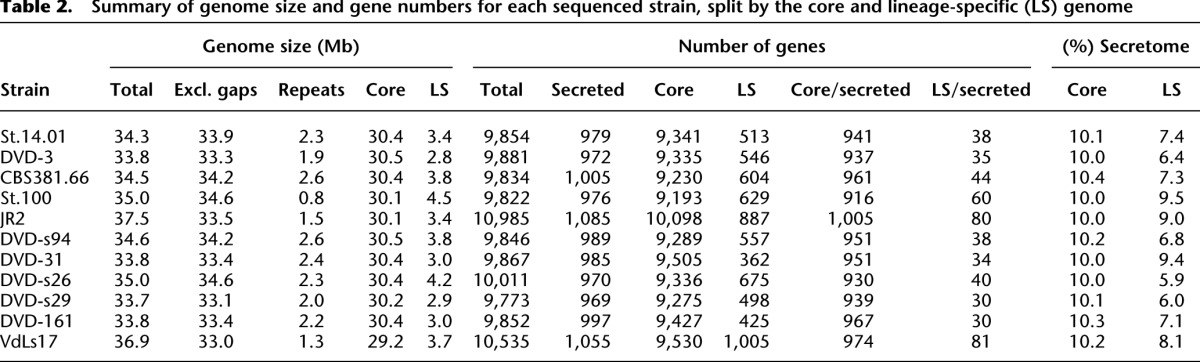
To further establish the role of LS regions in V. dahliae pathogenicity, we mined these regions, as well as the core genomic regions in VdLs17 and JR2, for candidate effectors (i.e., small, secreted proteins of unknown function). Typically, effector genes encode small proteins that are characterized by the presence of an N-terminal signal peptide that can be predicted computationally (de Jonge 2012). However, we did not observe a remarkable difference in the number of small secreted proteins between the core and LS genomic regions (Table 2). Similarly, no remarkable difference was found in the number of protein domains classified in the Pfam database (Finn et al. 2008). In a number of plant pathogenic oomycetes it was observed that effector genes are found in repeat-rich, gene-sparse regions that can be visualized by plotting the length of the 5′- and 3′-flanking intergenic regions (Raffaele et al. 2010). In the case of V. dahliae, we did not find a significant difference in gene density between core and LS genomic regions (Table 2; Supplemental Fig. 2). However, when assessing transcription (RNA-seq) data of V. dahliae-infected N. benthamiana (de Jonge et al. 2012; Faino et al. 2012), we identified 19 V. dahliae genes (P < 0.001) that were induced to the same level, or higher, as the previously characterized Ave1 effector gene [log10(FC) > 2.15; 8-d post-inoculation] when compared with expression in in vitro-cultured mycelium. Remarkably, eight of the 19 genes are located within the ∼3-Mb LS region, while the remaining 11 genes reside in the ∼30-Mb core genome, identifying a significant overrepresentation of in planta-induced genes in the LS regions (Fig. 3). Of the eight highly induced genes that reside in LS regions, five (including Ave1) encode putative secreted effectors (Fig. 3). Moreover, considering a log10(FC) > 3 or log10(FC) > 4, these ratios are even more skewed with six out of 14 and five out of six in planta-induced genes residing in LS regions, respectively.
Figure 3.

Lineage-specific genomic regions of Verticillium dahliae strain JR2 are enriched for in planta-expressed genes. Deep transcriptome sequencing of infected Nicotiana benthamiana plants harvested between 4- and 16-d post-inoculation (DPI), and fungus cultured on Czapek Dox medium (C) led to the identification of 19 highly in planta-induced genes [log10(FC) > 2.15]. Of these, eight are located in lineage-specific (LS) regions (in blue), while 11 are in the core genome (in red), demonstrating significant overrepresentation of in planta-expressed genes in LS regions. The table shows the presence (black) or absence (white) of in planta-induced JR2 genes within the set of sequenced strains and indicates hypothetical and putatively secreted proteins. Numbers in the two gray cells indicate amino acid identity and conservation, respectively, for two divergent homologs.
Lineage-specific genomic regions determine V. dahliae virulence and niche adaptation
To assess the biological significance of the significant overrepresentation of in planta-induced genes in the LS regions, we pursued targeted deletion of two randomly chosen effector genes of the four effector gene candidates in addition to Ave1 that are most highly expressed in planta; XLOC_009059 and XLOC_008951. XLOC_009059 does not occur in any of the other strains except for St.100, which contains a highly diversified copy (53% and 72% amino acid identity and conservation, respectively, as determined by BLASTp) (Fig. 3) that is unlikely to encode a functional protein due to the high number of predicted stop codons. On the other hand, XLOC_008951 was found in four of the 11 strains that were sequenced (Fig. 3). Remarkably, deletion of each of them resulted in significantly reduced disease development, confirming that they encode bona fide effectors required for virulence on tomato (Fig. 4A,B). Thus, we conclude that the LS regions of strain JR2 significantly contribute to virulence.
Figure 4.

Lineage-specific genomic regions of Verticillium dahliae harbor effectors that are required for virulence on tomato. (A) Two independent XLOC_009059 deletion strains (ΔXLOC_009059) show compromised virulence on tomato (cv. MoneyMaker), evidenced by reduced stunting when compared with inoculation with wild-type V. dahliae (WT, JR2), increased canopy area, and reduced V. dahliae biomass accumulation. (B) Two independent XLOC_008951 deletion strains (ΔXLOC_008951) show compromised virulence on tomato (cv. MoneyMaker), evidenced by reduced stunting when compared with inoculation with wild-type V. dahliae (WT, JR2), increased canopy area, and reduced V. dahliae biomass accumulation. (C) Two independent VDAG_05180 deletion strains (ΔVDAG_05180) show compromised virulence on tomato (cv. Motelle), evidenced by reduced stunting when compared with inoculation with wild-type V. dahliae (WT, VdLs17). Photographs are taken at 12 DPI. (D) Expression of VDAG_05180 in V. dahliae strain VdLs17 during infection of Nicotiana benthamiana between 4- and 16-d post-inoculation (DPI). (E) Reduced V. dahliae biomass in plants inoculated with two independent VDAG_05180 deletion strains when compared with wild-type V. dahliae (WT, VdLs17) at 8 DPI. Error bars represent standard error of three replicate experiments.
Similar to JR2, strain VdLs17 also contains a ∼4-Mb LS region (Table 2; Klosterman et al. 2011) that was assessed for unique effector genes crucial for pathogenicity. All V. dahliae strains that were sequenced contain six genes that encode LysM effectors within their core genome (VDAG_00902; VDAG_03096; VDAG_04781; VDAG_06426; VDAG_06998; VDAG_08171). LysM effectors have previously been implicated in sequestration of chitin fragments to prevent these fragments from stimulating host immunity (de Jonge and Thomma 2009; de Jonge et al. 2010; Marshall et al. 2011; Mentlak et al. 2012). VdLs17 contains an additional LysM effector gene (VDAG_05180) within a LS region, encoding a LysM effector that contains two LysM domains. Remarkably, of the seven LysM effectors in the VdLs17 genome, only this LysM effector gene is significantly expressed in planta (Fig. 4C). Furthermore, targeted deletion of this effector revealed that it is indeed required for virulence of the VdLs17 strain (Fig. 4D,E). Thus, like in JR2, the LS regions of strain VdLs17 contribute to virulence. Notably, like the race-1-specific effector Ave1 (de Jonge et al. 2012), VDAG_05180 as well as V. dahliae JR2 effector genes XLOC_009059, XLOC_008951 and the candidate effector genes XLOC_00170 and evm.model.contigUn.128 are flanked by repetitive elements such as LTR retrotransposons and FotI transposases (Fig. 5; Supplemental Figure 3), suggesting that the presence/absence polymorphism of all of these effector genes in V. dahliae is mediated by the flexibility and instability of the genomic context as a consequence of these repetitive elements (Klosterman et al. 2011; Amyotte et al. 2012).
Figure 5.

Genomic context of the lineage-specific Verticillium dahliae LysM effector VDAG_05180. (A) Genomic location of VDAG_05180 in V. dahliae strain VdLs17 revealing the presence of flanking gaps, repeats, and a predicted overlapping, but inactive LTR retrotransposon. RepeatModeler families rnd-1_family14, rnd-4_family-614, rnd-1_family-25, and rnd-4_family-1225 are classified as Ty1-Copia, Ty1-Gypsy, TcMar-Fot1, and Ty1-Copia, respectively, by RepeatMasker. (B) Repetitiveness of the genomic context of VDAG_05180 throughout the genomes of VdLs17 (top) and JR2 (bottom). The degree of conservation is indicated by a color scale, showing recent (red) and more ancient (orange to green) multiplications, especially of repeat elements. In addition, the presence of a VDAG_05181 homolog (encoding a tetrahydroxynaphthalene reductase) in the JR2 genome, a duplication of this gene in the VdLs17 genome, and the presence of multiple VDAG_05182 homologs (encoding a Kelch domain-containing protein) in both genomes is shown. The LysM effector gene VDAG_05180 is uniquely found in the VdLs17 genome.
Discussion
So far, it remains unknown how asexually propagating organisms, including many fungal species, are able to adapt to changing environmental conditions. The lack of sexual reproduction, and consequently meiotic recombination, is thought to greatly affect the potential to generate novel genetic combinations. Especially, pathogenic microbes involved in an intimate interaction with a host need to continuously adapt their effector gene repertoire to counteract host defense responses that have evolved in the evolutionary arms race with the pathogen. Here, we examined the genetic diversity in a population of the plant pathogenic fungus V. dahliae that propagates solely through asexual reproduction. As expected for an asexually reproducing organism, we only found a low degree of genome-wide nucleotide diversity between V. dahliae strains in the population, ranging between 0.02% and 0.5%. For comparison, in a study on the sexually reproducing fungal wheat pathogen Zymoseptoria tritici (synonym of Mycosphaerella graminicola), the average genome-wide nucleotide diversity was determined to range between 2% and 4% (Stukenbrock et al. 2011). Despite the low degree of genetic diversity, we identified 29 genes that are under positive selection within the V. dahliae population (Supplemental Table 2), suggesting that these genes are under adaptive evolution. This number is comparable to the number of genes under positive selection in Z. tritici when compared with its wild sister species (Stukenbrock et al. 2011). However, only three of these V. dahliae genes encode secreted proteins that are candidate effectors, suggesting that molecular evolution at the nucleotide level does not play a major role in host adaptation of this asexual pathogen.
Various mechanisms have been described that facilitate rapid development of novel effector genes in pathogenic microbes, including diversity at genomic locations enriched for transposons, mutation, and recombination in subtelomeric regions (McDonagh et al. 2008; Chuma et al. 2011), coregulated gene clusters (Pallmer and Keller 2010; Schirawski et al. 2010), small dispensable chromosomes (Coleman et al. 2009; Ma et al. 2010; Stukenbrock et al. 2010; Goodwin et al. 2011; Raffaele and Kamoun 2012), gene sparse regions (Raffaele et al. 2010), AT-rich isochore-like regions (van der Wouw et al. 2010; Rouxel et al. 2011), genome hybridization (Stukenbrock et al. 2012), and horizontal gene transfer (Friesen et al. 2006; de Jonge et al. 2012; Gardiner et al. 2012). However, most of these mechanisms have been described in species that can reproduce sexually, and of which the genomes were shaped by repeat-driven expansion (Raffaele and Kamoun 2012).
Here we describe the observation of a novel mode of evolution of pathogenicity in the asexual plant pathogenic fungus V. dahliae, of which only 4% of the genome is repetitive (Supplemental Table 7; Klosterman et al. 2011; Amyotte et al. 2012). This is significantly less when compared with the genomes of other filamentous pathogens, such as the fungi Magnaporthe oryzae (10%), Z. tritici (17%), Fusarium oxysporum (28%), Cladosporium fulvum (47%), Blumeria graminis (64%), and the oomycetes Hyaloperonospora arabidopsidis (42%) and Phytophthora infestans (74%) (Dean et al. 2005; Haas et al. 2009; Baxter et al. 2010; Ma et al. 2010; Spanu et al. 2010; Goodwin et al. 2011; de Wit et al. 2012). Comparative analyses between highly similar V. dahliae strains revealed numerous intra- and interchromosomal rearrangements and extensive karyotype variation. This is remarkable, as it is generally assumed that genome rearrangement increases with increasing sequence divergence. Previous analyses of fungal genomes have revealed that although large syntenic regions with orthologous genes arranged in the same orientation across species (known as macrosynteny) are rare, chromosomal gene content is typically preserved among Ascomycetes, particularly in the Dothidiomycete class (Goodwin et al. 2011; Hane et al. 2011). Such preservation of chromosomal gene content without conservation of gene order or orientation is referred to as mesosynteny, and is characterized by the occurrence of numerous intrachromosomal rearrangements, presumably arising as the consequence of frequent inversions during meiosis rather than interchromosomal rearrangements (Hane et al. 2011). The extensive number of interchromosomal rearrangements between V. dahliae isolates indicates that mesosynteny is not crucial in V. dahliae.
Unlike previous observations in a number of fungal plant pathogens, including F. oxysporum, N. haematococca, and Z. tritici, which display karyotype variation as a consequence of the differential presence of small dispensable chromosomes (Coleman et al. 2009; Ma et al. 2010; Stukenbrock et al. 2010; Goodwin et al. 2011; Raffaele and Kamoun 2012), the karyotype variation that we observed between individual isolates of V. dahliae concerns chromosome length polymorphisms, and no indication for the occurrence of small dispensable chromosomes is found in V. dahliae. The observed variation in this study and previous ones among fungal karyotypes (Zolan 1995) is the result of complex chromosomal rearrangements. It has previously been suggested that karyotype variation, rather than being a mechanism of adaptation to generate novel virulence traits, occurs because nondeleterious genomic rearrangements are maintained due to the absence or rarity of a sexual cycle (Talbot et al. 1993; Zolan 1995). Our data challenge this hypothesis by showing that chromosomal plasticity, evidenced by extensive targeted chromosomal rearrangements and karyotype variability, is genetically non-neutral as it induces local sequence variation at syntenic breakpoints and increases adaptive capability, as demonstrated by the significant enrichment for novel effector genes and acquisition of the race 1-specific effector Ave1, possibly through horizontal gene transfer and subsequent loss in V. dahliae race 2 isolates (de Jonge et al. 2012). Moreover, our data reinforces the notion that V. dahliae strictly relies on asexual reproduction, as extensive karyotype variation prevents correct pairing of homologous chromosomes during meiosis (Kistler and Miao 1992).
Our phylogenetic analysis based on whole-genome sequence data (Fig. 1) has demonstrated that race 1 and race 2 isolates of V. dahliae do not form separate clades, and the fact that we did not observe any sequence variation for Ave1 within and between Verticillium species (de Jonge et al. 2012) strongly suggests that Ave1 has been acquired (in race 1) or lost (in race 2) several times. Similarly, we identified additional effector genes within the plastic genomic regions that are unique or shared by only a subset of strains and like Ave1; these are conserved and not monophyletic, suggesting that they are frequently lost and/or readily exchanged between individual strains as has been proposed for effector genes of the Avr-Pita family in M. oryzae (Chuma et al. 2011). We speculate that frequent presence/absence polymorphisms of effector genes in these plastic regions is mediated by the flexibility and instability of these regions, reflected by the enrichment for transposable elements and association with chromosomal rearrangements (Amyotte et al. 2012). Moreover, we note that particular transposable elements flank a number of the in planta highly induced effector genes located in the LS regions and speculate that, besides providing genetic flexibility, these elements might also affect the level of gene expression. An association between in planta-expressed effector genes and a particular transposable element was recently found for the tomato pathogenic vascular wilt fungus Fusarium oxysporum f. sp. lycopersici, in which a repetitive miniature Impala (mimp) element was found to be present in the promoter region of all SIX (secreted in xylem) effector genes (Schmidt et al. 2013). Similarly, members of the M. oryzae Avr-Pita effector gene family are flanked by a retrotransposon, presumably contributing to its translocation across the genome (Chuma et al. 2011). Presence/absence of polymorphism and translocation of effector genes that are associated with unstable genomic regions has also been observed in asexual lineages of M. oryzae (Yoshida et al. 2009; Chuma et al. 2011), and it was hypothesized that parasexual recombination facilitates the exchange of effector genes between these lineages (Noguchi et al. 2006; Chuma et al. 2011). Parasexual recombination involves fusion of haploid fungal hyphae, followed by the formation of diploid nuclei through hybridization that can later return to a haploid state. Mitotic recombination may result in nonsexual exchange of genetic material without meiosis. In particular cases, the hybridized nuclei are stable, resulting in a hybrid species with a doubled genome such as has been suggested for V. longisporum (Inderbitzin et al. 2011). Parasexuality has been identified in many, mostly asexually propagating fungi during in vitro culturing and was shown to contribute to adaptive capabilities of the model fungus Aspergillus nidulans in vitro (Schoustra et al. 2007). Parasexual recombination may also be involved in the exchange of effector genes between asexual lineages of V. dahliae. Laboratory studies have shown that transgenic V. dahliae strains that carry auxotrophic markers are capable of hyphal fusion and exchange of genetic material, depending on their vegetative compatibility (Hastie 1964, 1973). Likewise, transfer of pathogenicity chromosomes between asexual lineages of F. oxysporum may be mediated by parasexual cycles (Corell 1991; Ma et al. 2010). However, the significance of parasexuality in nature, and the genetic factors that control parasexual compatibility between lineages, are poorly understood, although evidence for parasexual recombination in field populations of M. oryzae has been reported (Zeigler et al. 1997; Glass et al. 2000).
In our study, chromosomal rearrangements were found to be frequently associated with retrotransposons, a class of repetitive sequence elements that is known to promote genome instability through recurrent excision and insertion as well as by mediating homologous recombination during cell division (Zolan 1995; Lemoine et al. 2005; Maxwell et al. 2011). In organisms without a cycle of sexual reproduction, such as V. dahliae, homologous recombination between highly similar copies of transposable elements might help to generate genetic diversity by facilitating mitotic crossover, thereby creating new genetic combinations that are potentially beneficial. In addition, altered cosuppression, silencing of dispersed homologous genomic regions through directed DNA methylation, might act on proliferation of transposable elements to affect genome stability (Martienssen and Colot 2001; Selker et al. 2003; Muszewska et al. 2011). Although it is generally believed that asexual reproduction limits genetic variation, and consequently limits adaptive capability of the organism, we provide evidence for chromosomal plasticity as facilitator of asexual genome evolution.
Methods
SNP identification
Illumina reads (de Jonge et al. 2012) were mapped onto the Verticillium dahliae VdLs17 reference genome using GSNAP (version 2012-04-16; Wu and Nacu 2010) with default settings and processed by Picard (version 1.57; http://picard.sourceforge.net). SNPs were identified by SAMtools mpileup (version; 0.1.18; http://samtools.sourceforge.net) and filtered using vcfutils.pl varFilter (-Q20, -D100) and a minimum allele frequency of 0.8. SNP statistics were determined by VCFtools (version 0.1.10; Danecek et al. 2011) and variant annotation by SnpEff (version 2.1; http://snpeff.sourceforge.net). To specifically identify genes that are subject to positive selection, we applied the Nei Gojobori method, where rates of Ka and Ks are compared using a Z-test (Nei and Kumar 2000; Stukenbrock and Dutheil 2012). Z-values >2326 are considered significant with P < 0.01.
Coverage breadth was calculated for all genes as the percentage of nucleotides with minimum one read aligned using BEDtools (version 2.16.1; Quinlan and Hall 2010). Genes were considered absent when breadth was <0.1.
Maximum likelihood phylogenetic analysis was performed in MEGA5 (Tamura et al. 2011). For each strain, as input we extracted for all nonredundant SNP positions the respective base call, resulting in 11 sequences, each containing 236,785 bp.
Assembly, alignment, and identification of rearrangements
Draft assemblies generated previously (de Jonge et al. 2012) were used for all strains, except for JR2. Mate-pair library preparation and sequencing (50 bp, ∼5-kb insert) of JR2 was performed by the BGI (Hong Kong). For de novo assembly, Velvet (version 1.2.03; Zerbino and Birney 2008) was used with cov_cutoff=6, exp_cov=auto, k-mer=31 and shortMatePaired=yes to generate a new assembly based on the 500-bp paired-end and 5-kb mate-pair libraries. Optical mapping, i.e., the construction of ordered genome-wide, high-resolution (>150×) restriction maps, was performed by BGI using the Argus System, and contigs were placed using MapSolver (version 3.2; OpGen).
Whole-genome alignments and dot plots were generated by MUMmer 3.0 (Kurtz et al. 2004) using NUCmer with default settings (except for -l 15 and–maxmatch) and Mummerplot. We used custom scripts to identify rearrangements and associated breakpoints. Core and LS genomic regions were determined by assessing coverage breadth of the alignments on 1-kb nonoverlapping windows using BEDTools. In addition, we determined the percentage of gaps for each region. Regions were considered lineage specific with an alignment breadth of <0.2. Core genomic regions were defined based on their minimum presence in all strains except one in order to compensate for assembly and alignment inconsistencies. Similarly, genes were considered as core genes when identified minimally in all strains except one with an alignment breadth of >0.8 for each strain. Remaining genes are considered lineage specific. Collinear blocks, repetitiveness, percentage GC, and LS genomic regions in the VdLs17 and JR2 genomes were visualized using Circos (version 0.55; Krzywinski et al. 2009) and GBrowse (version 2.49; Stein et al. 2002).
Multiple chromosomal rearrangements were verified by PCR. To this end, primer pairs spanning predicted breakpoints were designed for JR2 and VdLs17 to selectively amplify breakpoint regions in either of the two genotypes and used in PCR reactions on genomic DNA. Quality of input DNA and integrity of syntenic sequences flanking the breakpoints in JR2 and VdLs17 were verified by control primer sets (Supplemental Table 5).
Repeat identification
Repetitive elements were identified and classified by RepeatModeler (version 1.0.5; http://www.repeatmasker.org/RepeatModeler.html), incorporating the two de novo repeat-finding programs RECON (version 1.0.7; Bao and Eddy 2002) and RepeatScout (version 1.0.3; Price et al. 2005), the tandem repeat finder TRF (version 3.2.1), and the annotated repeat library from RepBase (release 16.12). Repetitive sequence families were used by RepeatMasker (version open-3.3.0; http://www.repeatmasker.org) to identify and mask all repetitive sequences applying the sensitive mode. Full-length long terminal repeat (LTR) retrotransposons were identified by LTR_FINDER (version 1.0.5; Xu and Wang 2007). Statistical significant spatial correlations between repetitive sequences, breakpoints, and genomic regions were assessed by GenometriCorr (version 1.1.7; http://genometricorr.sourceforge.net).
Karyotyping
V. dahliae mycelium was prepared for protoplasting following the mycelium-based fungal biomass preparation method (Mehrabi et al. 2012). Mycelium was digested in 1 M sorbitol containing 1% (w/v) glucanex and 0.5% driselase (Sigma-Aldrich) at 32°C until protoplast concentration reached 108/mL. Protoplast plugs were prepared and stored as described (Mehrabi et al. 2012). Karyotyping was carried out using a CHEF Mapper XA pulsed field electrophoresis system (Bio-Rad) using the auto algorithm function with low- and high-molecular weight settings at 2 and 6 Mb, respectively. Chromosomes were separated in 0.8% low EEO agarose (US Biological) gels. Chromosome size markers from Hansenula wingei and Schizosaccharomyces pombe (Bio-Rad) were included as reference. After electrophoresis, gels were stained with ethidium bromide (1 μg/mL) in water for 1 h and destained in water for 2 h.
Gene prediction, expression, and annotation
Gene predictions on the genome sequences of all V. dahliae strains were performed using a combinatorial approach, applying the EVidenceModeler (version r2012-06-25; Haas et al. 2008) to combine protein-coding gene evidence from V. dahliae-trained Augustus (version 2.6; Stanke et al. 2006, 2008), GeneMarkES (version 2.3c; Borodovsky and Lomsadze 2011), V. dahliae-trained GlimmerHMM (version 3.0.1; Majoros et al. 2004), protein alignments to the latest Fusarium graminearum (release 3.2, MIPS, Munich) (Wong et al. 2011) and Magnaporthe oryzae (release 8, Broad Institute) proteins using the Analysis and Annotation Tool (AAT) package (version r03052011; Huang et al. 1997). For deep transcriptome sequencing and mapping, ∼2 Gb of reads from V. dahliae strain JR2-infected Nicotiana benthamiana plants and ∼1 Gb of reads from V. dahliae JR2 cultured in vitro on Czapek Dox medium were mapped on the V. dahliae JR2 genome using TopHat (version 2.0.8; Trapnell et al. 2009; de Jonge et al. 2012). Cufflinks (version 2.0.2; Trapnell and Salzberg 2010) was used to assemble novel transcripts and isoforms from the mapped reads that were subsequently incorporated in the final gene-set for JR2 using the EVidenceModeler after manual curation. Relative expression for each gene in each experiment was determined by assessing the number of reads mapping to each gene using HTSeq (version 0.5.4p1; www-huber.embl.de/users/anders/HTSeq/) applying the default “union” setting and subsequently reported in the number of Reads Per Kilobase of transcript per Million mapped reads (RPKM). For differential gene expression analysis we compared in planta and in vitro expression using Excel and CummeRbund (http://compbio.mit.edu/cummeRbund/). The predicted proteins were mined for candidate effectors (de Jonge 2012) by functional annotation using BLASTp analyses against the nonredundant database at NCBI (http://www.ncbi.nih.nlm.gov) and Pfam domain scanning (Finn et al. 2008) using the Blast2GO suite (Conesa et al. 2005). Secreted proteins were predicted by SignalP4 (Petersen et al. 2011). Gene density was determined as previously described (Raffaele et al. 2010). In short, 5′- and 3′-flanking intergenic regions were calculated, scored in two-dimensional bins, and plotted. Filled contour plots were generated using R (http://www.r-project.org/).
Functional analysis
Knockouts were generated by amplifying the sequences flanking coding sequences using the primer sets KO-XLOC_009059-F1/KO-XLOC_009059-R1 and KO-XLOC_009059-F2/KO-XLOC_009059-R2 for XLOC_009059, KO-XLOC_008951-F1/KO-XLOC_008951-R1 and KO-XLOC_008951-F2/KO-XLOC_008951-R2 for XLOC_008951, and KO-VDAG_05180-F1/KO-VDAG_05180-R1 and KO-VDAG_05180-F2/KO-VDAG_05180-R2 for VDAG_05180 (Supplemental Table 4). Likewise, knockouts of XLOC_009059 and XLOC_008951 were generated by amplification of the flanking sequences (Supplemental Table 4). PCR products were subsequently cloned into pRF-HU2 (Frandsen et al. 2008). V. dahliae transformation and subsequent inoculations on tomato (cv. Motelle and MoneyMaker) plants to assess the impact on virulence were performed as described (Fradin et al. 2009). Plants were regularly inspected during a 2-wk interval and photographed at 8- and 12-d post -inoculation (DPI). For biomass quantification, the roots and stem below cotyledons of three plants per V. dahliae genotype were flash-frozen in liquid nitrogen. To determine canopy surface areas, we measured the leaf surface areas of multiple plants from photographs taken from the top using ImageJ. The samples were ground to powder, of which an aliquot was used for DNA isolation (Fulton et al. 1995). Real-time PCR was conducted with primer sets SlRub-F1/SlRub-F2 for tomato RuBisCo and VdGAPDH-F/VdGAPDH-R for V. dahliae GAPDH (Supplemental Table 4). For expression analyses, 3-wk-old Nicotiana benthamiana plants were inoculated with strain VdLs17 as previously described (Fradin et al. 2009), harvested at 4, 8, 12, and 16 DPI, and flash-frozen in liquid nitrogen. Total RNA was extracted using the RNeasy Kit (Qiagen) and cDNA was synthesized by SuperScript III (Invitrogen). Real-time PCR was conducted with primer sets VdGAPDH-F/VdGAPDH-R for V. dahliae GAPDH and qVDAG_05180-F1/qVDAG_05180-R1 for V. dahliae VDAG_05180 (Supplemental Table 4).
Data access
All genome and transcriptome raw read data have been deposited at the NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) under BioProject PRJNA169154 (http://www.ncbi.nlm.nih.gov/bioproject). The V. dahliae strain JR2 draft genome sequence is deposited at the NCBI WGA archive database (http://www.ncbi.nlm.nih.gov/genome) under accession number APFD00000000 and described under BioProject PRJNA175765.
Acknowledgments
We thank X. Wang for technical assistance and T. Friesen, H. de Jong, F. Debets, P. de Wit, M. Joosten, J. van Kan, Eva Stukenbrock, and Julien Dutheil for helpful discussions. B.P.H.J.T. is supported by the Netherlands Organization for Scientific Research (NWO-ALW), the Centre for BioSystems Genomics (CBSG), Technology Foundation STW, and ERA-NET Plant Genomics. M.D.B. is supported by USDA-ARS CRIS project 5442-22000-042-00D.
Author contributions: R.d.J. and B.P.H.J.T. conceived the project. R.d.J., M.D.B., A.K., G.C.M.v.d.B. and B.P.H.J.T. designed the experiments. B.P.H.J.T. supervised the project. K.A.Y. analyzed pathogenicity of Verticillium strains. R.d.J., G.C.M.v.d.B. and M.D.B. prepared the samples for genome sequencing and optical mapping. M.D.B. performed karyotype experiments. A.K. performed functional analysis of VDAG_05180. G.C.M.v.d.B performed PCR reactions and functional analyses of XLOC_009059 and XLOC_008951. R.d.J. performed the bioinformatic analyses. R.d.J. and B.P.H.J.T. wrote the paper. All authors discussed results and commented on the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.152660.112.
Freely available online through the Genome Research Open Access option.
References
- Agrawal AF 2006. Evolution of sex: Why do organisms shuffle their genotypes? Curr Biol 16: R696–R704 [DOI] [PubMed] [Google Scholar]
- Amen J, Shoemaker PB 1985. Histopathology of resistant and susceptible tomato cultivars inoculated with Verticillium dahliae races 1 and 2. Phytopathology 75: 1361–1362 [Google Scholar]
- Amyotte SG, Tan X, Pennermann K, del Mar Jimenez-Gasco M, Klosterman SJ, Ma L-J, Dobinson KF, Veronese P 2012. Transposable elements in phytopathogenic Verticillium spp.: Insights into genome evolution and inter- and intra-specific diversification. BMC Genomics 12: 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atallah ZK, Maruthachalam K, du Toit L, Koike ST, Michael Davis R, Klosterman SJ, Hayes RJ, Subbarao KV 2010. Population analyses of the vascular plant pathogen Verticillium dahliae detect recombination and transcontinental gene flow. Fungal Genet Biol 47: 416–422 [DOI] [PubMed] [Google Scholar]
- Bao Z, Eddy SR 2002. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res 12: 1269–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter L, Tripathy S, Ishaque N, Boot N, Cabral A, Kemen E, Thines M, Ah-Fong A, Anderson R, Badejoko W, et al. 2010. Signatures of adaptation to obligate biotrophy in the Hyaloperonospora arabidopsidis genome. Science 330: 1549–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billiard S, López-Villavicencio M, Hood ME, Giraud T 2012. Sex, outcrossing and mating types: Unsolved questions in fungi and beyond. J Evol Biol 25: 1020–1038 [DOI] [PubMed] [Google Scholar]
- Bolek Y, El-Zik KM, Pepper AE, Bell AA, Magill CW, Thaxton PM, Reddy OUK 2005. Mapping of Verticillium wilt resistance genes in cotton. Plant Sci 168: 1581–1590 [Google Scholar]
- Borodovsky M, Lomsadze A 2011. Eukaryotic gene prediction using GeneMark.hmm-E and GeneMark-ES. Curr Protoc Bioinformatics 35: 4.6.1.–4.6.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt A 2000. Perspective: Sex, recombination, and the efficacy of selection—was Weismann right? Evolution 54: 337–351 [DOI] [PubMed] [Google Scholar]
- Chuma I, Isobe C, Hotta Y, Ibaragi K, Futamata N, Kusuba M, Yoshida K, Terauchi R, Fujita Y, Nakayaskiki H, et al. 2011. Multiple translocation of the AVR-Pita effector gene among chromosomes of the rice blast fungus Magnaporthe oryzae and related species. PLoS Pathog 7: e1002147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegrave N 2002. Sex releases the speed limit on evolution. Nature 420: 664–666 [DOI] [PubMed] [Google Scholar]
- Coleman JJ, Rounsley SD, Rodriguez-Carres M, Kuo A, Wasmann CC, Grimwood J, Schmutz J, Taga M, White GJ, Zhou S 2009. The genome of Nectria haematococca: Contribution of supernumerary chromosomes to gene expansion. PLoS Genet 5: e1000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M 2005. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21: 3674–3676 [DOI] [PubMed] [Google Scholar]
- Corell JC 1991. The relationship between formae speciales, races and vegetative compatibility groups in Fusarium oxysporum. Phytopathology 81: 1061–1064 [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. 2011. The variant call format and VCFtools. Bioinform 27: 2156–2158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jonge R 2012. In silico identification and characterization of effector catalogs. Methods Mol Biol 835: 415–425 [DOI] [PubMed] [Google Scholar]
- de Jonge R, Thomma BPHJ 2009. Fungal LysM effectors: Extinguishers of host immunity? Trends Microbiol 17: 151–157 [DOI] [PubMed] [Google Scholar]
- de Jonge R, van Esse HP, Kombrink A, Shinya T, Desaki Y, Bours R, van der Krol S, Shibuya N, Joosten MHAJ, Thomma BPHJ 2010. Conserved fungal LysM effector Ecp6 prevents chitin-triggered immunity in plants. Science 329: 953–955 [DOI] [PubMed] [Google Scholar]
- de Jonge R, Bolton MD, Thomma BPHJ 2011. How filamentous pathogens co-opt plants: The ins and outs of fungal effectors. Curr Opin Plant Biol 14: 400–406 [DOI] [PubMed] [Google Scholar]
- de Jonge R, van Esse HP, Maruthachalam K, Bolton MD, Santhanam P, Keykha Saber M, Zhang Z, Usami T, Lievens B, Subbarao KV, et al. 2012. Tomato immune receptor Ve1 recognizes effector of multiple fungal pathogens uncovered by genome and RNA sequencing. Proc Natl Acad Sci 109: 5110–5115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Visser JAGM, Elena SF 2007. The evolution of sex: Empirical insights into the roles of epistasis and drift. Nat Rev Genet 8: 139–149 [DOI] [PubMed] [Google Scholar]
- de Wit PJ, van der Burgt A, Ökmen B, Stergiopoulos I, Abd-Elsalam KA, Aerts AL, Bahkali AH, Beenen HG, Chettri P, Cox MP, et al. 2012. The genomes of the fungal plant pathogens Cladosporium fulvum and Dothistroma septosporum reveal adaptation to different hosts and lifestyles but also signatures of common ancestry. PLoS Genet 8: e1003088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean RA, Talbot NJ, Ebbole DJ, Farman ML, Mitchell TK, Orbach MJ, Thon M, Kulkarni R, Xu JR, Pan H, et al. 2005. The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 434: 980–986 [DOI] [PubMed] [Google Scholar]
- Faino L, de Jonge R, Thomma BPHJ 2012. The transcriptome of Verticillium dahliae-infected Nicotiana benthamiana determined by deep RNA sequencing. Plant Signal Behav 7: 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J 1974. The evolutionary advantage of recombination. Genet 78: 737–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer ELL, et al. 2008. The Pfam protein families database. Nucleic Acids Res 36: D281–D288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradin EF, Thomma BPHJ 2006. Physiology and molecular aspects of Verticillium wilt diseases caused by V. dahliae and V. albo-atrum. Mol Plant Pathol 12: 71–86 [DOI] [PubMed] [Google Scholar]
- Fradin EF, Zhang Z, Juarez Ayala JC, Castroverde CDM, Nazar RN, Robb J, Liu C-M, Thomma BPHJ 2009. Genetic dissection of Verticillium wilt resistance mediated by tomato Ve1. Plant Physiol 150: 320–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frandsen RJ, Andersson JA, Kristensen MB, Giese H 2008. Efficient four fragment cloning for the construction of vectors for targeted gene replacement in filamentous fungi. BMC Mol Biol 9: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen TL, Stukenbrock EH, Liu Z, Meinhardt S, Ling H, Faris JD, Rasmussen JB, Solomon PS, McDonald BA, Oliver RP 2006. Emergence of a new disease as a result of interspecific virulence gene transfer. Nat Genet 38: 953–956 [DOI] [PubMed] [Google Scholar]
- Fulton TM, Chunwongse J, Tanksley SD 1995. Microprep protocol for extraction of DNA from tomato and other herbaceous plants. Plant Mol Biol Rep 13: 207–209 [Google Scholar]
- Gardiner DM, McDonald MC, Covarelli L, Solomon PS, Rusu AG, Marshall M, Kazan K, Chakraborty S, McDonald BA, Manners JM 2012. Comparative pathogenomics reveals horizontally acquired novel virulence genes in fungi infecting cereal hosts. PLoS Pathog 8: e1002952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass NL, Jacobsen DJ, Shiu PK 2000. The genetics of hyphal fusion and vegetative incompatibility in filamentous ascomycete fungi. Ann Rev Genet 34: 165–168 [DOI] [PubMed] [Google Scholar]
- Goddard MR, Godfray HCJ, Burt A 2005. Sex increases the efficacy of natural selection in experimental yeast populations. Nature 434: 636–640 [DOI] [PubMed] [Google Scholar]
- Goodwin SB, Ben M'Barek S, Dhillon B, Wittenberg AHJ, Crane CF, Hane JK, Foster AJ, Van der Lee TAJ, Grimwood J, Aerts A, et al. 2011. Finished genome of the fungal wheat pathogen Mycosphaerella graminicola reveals dispensome structure, chromosome plasticity, and stealth pathogenesis. PLoS Genet 7: e1002070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Salzberg SJ, Zhu W, Pertea M, Allen JE, Orvis J, White O, Buell CR, Wortman JR 2008. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9: R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Kamoun S, Zody MC, Jiang RHY, Handsaker RE, Cano LM, Grabherr M, Kodira CD, Raffaele S, Torto-Alalibo T, et al. 2009. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 461: 393–398 [DOI] [PubMed] [Google Scholar]
- Hane JK, Rouxel T, Howlett BJ, Kema GHJ, Goodwin SB, Oliver RP 2011. A novel mode of chromosomal evolution peculiar to filamentous Ascomycete fungi. Genome Biol 12: R45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie AC 1964. The parasexual cycle in Verticillium albo-atrum. Genetical Research 5: 305–315 [Google Scholar]
- Hastie AC 1973. Hybridization of Verticillium albo-atrum and Verticillium dahliae. Trans Br Mycol Soc 60: 511–523 [Google Scholar]
- Heitman J 2006. Sexual reproduction and the evolution of microbial pathogens. Curr Biol 16: R711–R725 [DOI] [PubMed] [Google Scholar]
- Heitman J, Kronstad JW, Taylor JW, Casselton LA, ed. 2007. Sex in fungi: Molecular determination and evolutionary implications. ASM Press, Washington DC. [Google Scholar]
- Huang X, Adams MD, Zhou H, Kerlavage AR 1997. A tool for analyzing and annotating genomic sequences. Genomics 46: 37–45 [DOI] [PubMed] [Google Scholar]
- Inderbitzin P, Davis RM, Bostock RM, Subbarao KV 2011. The ascomycete Verticillium longisporum is a hybrid and a plant pathogen with an expanded host range. PLoS ONE 6: e18260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawchuk LM, Hachey J, Lynch DR, Kulcsar F, van Rooijen G, Waterer DR, Robertson A, Kokko E, Byers R, Howard RJ, et al. 2001. Tomato Ve disease resistance genes encode cell surface-like receptors. Proc Natl Acad Sci 98: 6511–6515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kistler HC, Miao VP 1992. New modes of genetic change in filamentous fungi. Annu Rev Phytopathol 30: 131–153 [DOI] [PubMed] [Google Scholar]
- Klosterman SJ, Atallah ZK, Vallad GE, Subbarao KV 2009. Diversity, pathogenicity, and management of Verticillium species. Annu Rev Phytopathol 47: 39–62 [DOI] [PubMed] [Google Scholar]
- Klosterman SJ, Subbarao KV, Kang S, Veronese P, Gold SE, Thomma BPHJ, Chen Z, Henrissat B, Lee Y-H, Park J, et al. 2011. Comparative genomics yields insights into niche adaptation of plant vascular wilt pathogens. PLoS Pathog 7: e1002137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA 2009. Circos: An information aesthetic for comparative genomics. Genome Res 19: 1639–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL 2004. Versatile and open software for comparing large genomes. Genome Biol 5: R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoine FJ, Degtyareva NP, Lobachev K, Petes TD 2005. Chromosomal translocations in yeast induced by low levels of DNA polymerase: A model for chromosome fragile sites. Cell 120: 587–598 [DOI] [PubMed] [Google Scholar]
- Ma L-J, van der Does HC, Borkovich KA, Coleman JJ, Daboussi MJ, Di Pietro A, Dufresne M, Freitag M, Grabherr M, Henrissat B, et al. 2010. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464: 367–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoros WH, Pertea M, Salzberg SL 2004. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 20: 2878–2879 [DOI] [PubMed] [Google Scholar]
- Marshall R, Kombrink A, Motteram J, Loza-Reyes E, Lucas J, Hammond-Kosack KE, Thomma BPHJ, Rudd JJ 2011. Analysis of two in planta expressed LysM effector homologs from the fungus Mycosphaerella graminicola reveals novel functional properties and varying contributions to virulence on wheat. Plant Physiol 156: 756–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martienssen RA, Colot V 2001. DNA methylation and epigenetic inheritance in plants and filamentous fungi. Science 293: 1070–1074 [DOI] [PubMed] [Google Scholar]
- Maxwell PH, Burhans WC, Curcio MJ 2011. Retrotransposition is associated with genome instability during chronological aging. Proc Natl Acad Sci 108: 20376–20381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Smith J. 1978. The evolution of sex. Cambridge University Press, Cambridge, UK. [Google Scholar]
- McDonagh A, Fedorova ND, Crabtree J, Yu Y, Kim S, Chen D, Loss O, Cairns T, Goldman G, Armstrong-James D, et al. 2008. Sub-telomere directed gene expression during initiation of invasive Aspergillosis. PLoS Pathog 4: e1000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald BA, Linde C 2002. Pathogen population genetics, evolutionary potential, and durable resistance. Annu Rev Phytopathol 40: 349–379 [DOI] [PubMed] [Google Scholar]
- Mehrabi R, Taga M, Aghaee M, de Wit PJGM, Kema GHJ 2012. Karyotyping methods for fungi. Methods Mol Biol 835: 591–602 [DOI] [PubMed] [Google Scholar]
- Mentlak TA, Kombrink A, Shinya T, Ryder LS, Otomo I, Saitoh H, Terauchi R, Nishizawa Y, Shibuya N, Thomma BPHJ, et al. 2012. Effector-mediated suppression of chitin-triggered immunity by Magnaporthe oryzae is necessary for rice blast disease. Plant Cell 24: 322–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieczkowski PA, Lemoine FJ, Petes TD 2006. Recombination between retrotransposons as a source of chromosome rearrangements in the yeast Saccharomyces cerevisiae. DNA Repair (Amst) 5: 1010–1020 [DOI] [PubMed] [Google Scholar]
- Muszewska A, Hoffmann-Sommer M, Grynberg M 2011. LTR retrotransposons in fungi. PLoS ONE 6: e29425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Kumar S 2000. Molecular evolution and phylogenetics. Oxford University Press, Oxford, UK [Google Scholar]
- Noguchi MT, Yasuda N, Fujita Y 2006. Evidence of genetic exchange by parasexual recombination and genetic analysis of pathogenicity and mating type of parasexual recombinants in rice blast fungus, Magnaporthe oryzae. Phytopathology 96: 746–750 [DOI] [PubMed] [Google Scholar]
- Pallmer JM, Keller NP 2010. Secondary metabolism in fungi: Does chromosomal location matter? Curr Opin Microbiol 13: 431–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paternotte SJ, van Kesteren HA 1993. A new aggressive strain of Verticillium albo-atrum in Verticillium-resistant cultivars of tomato in the Netherlands. Neth J Plant Pathol 99: 169–172 [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H 2011. SignalP4.0: Discriminating signal peptides from transmembrane regions. Nat Methods 8: 785. [DOI] [PubMed] [Google Scholar]
- Price AL, Jones NC, Pevzner PA 2005. De novo identification of repeat families in large genomes. Bioinformatics 21: i351–i358 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM 2010. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinform 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaele S, Kamoun S 2012. Genome evolution in filamentous plant pathogens: Why bigger can be better. Nat Rev Microbiol 10: 417–430 [DOI] [PubMed] [Google Scholar]
- Raffaele S, Farrer RA, Cano LM, Studholme DJ, MacLean D, Thines M, Jiang RHY, Zody MC, Kunjeti SG, Donofrio NM, et al. 2010. Genome evolution following host jumps in the Irish potato famine pathogen lineage. Science 330: 1540–1543 [DOI] [PubMed] [Google Scholar]
- Rouxel T, Grandaubert J, Hane JK, Hoede C, van de Wouw AP, Couloux A, Dominguez V, Anthouard V, Bally P, Bourras S, et al. 2011. Effector diversification within compartments of the Leptosphaeria maculans genome affected by repeat-induced point mutations. Nature Commun 2: 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaible L, Cannon OS, Waddoups V 1951. Inheritance of resistance to Verticillium wilt in a tomato cross. Phytopathology 41: 986–990 [Google Scholar]
- Schirawski J, Mannhaupt G, Münch K, Brefort T, Schipper K, Doehlemann G, Stasio MD, Rössel N, Mendoza-Mendoza A, Pester D, et al. 2010. Pathogenicity determinants in smut fungi revealed by genome comparison. Science 330: 1546–1548 [DOI] [PubMed] [Google Scholar]
- Schmidt SM, Houterman PM, Schreiver I, Ma L, Amyotte S, Chellappan B, Boeren S, Takken FL, Rep M 2013. MITEs in the promoters of effector genes allow prediction of novel virulence genes in Fusarium oxysporum. BMC Genomics 14: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoustra SE, Debets AJ, Slakhorst M, Hoekstra RF 2007. Mitotic recombination accelerates adaptation in the fungus Aspergillus nidulans. PLoS Genet 3: e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selker EU, Tountas NA, Cross SH, Margolin BS, Murphy JG, Bird AP, Freitag M 2003. The methylated component of the Neurospora crassa genome. Nature 422: 893–897 [DOI] [PubMed] [Google Scholar]
- Spanu PD, Abbot JC, Amselem J, Burgis TA, Soanes DM, Stuber K, Ver Loren van Themaat E, Brown JKM, Butcher SA, Gurr SJ, et al. 2010. Genome expansion and gene loss in powdery mildew fungi reveal tradeoffs in extreme parasitism. Science 330: 1543–1546 [DOI] [PubMed] [Google Scholar]
- Stanke M, Tzvetkova A, Morgenstern B 2006. AUGUSTUS at EGASP: Using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biol 7: S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Diekhans M, Baertsch R, Haussler D 2008. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinform 24: 637–644 [DOI] [PubMed] [Google Scholar]
- Stein LD, Mungall C, Shu S, Caudy M, Mangone M, Day A, Nickerson E, Stajich JE, Harris TW, Arva A, et al. 2002. The generic genome browser: A building block for a model organism system database. Genome Res 12: 1599–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stukenbrock EH, Dutheil JY 2012. Comparing fungal genomes: Insight into functional and evolutionary processes. Methods Mol Biol 835: 531–548 [DOI] [PubMed] [Google Scholar]
- Stukenbrock EH, Jørgensen FG, Zala M, Hansen TT, McDonald BA, Schierup MH 2010. Whole-genome and chromosome evolution associated with host adaptation and speciation of the wheat pathogen Mycosphaerella graminicola. PLoS Genet 6: e1001189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stukenbrock EH, Bataillon T, Dutheil JY, Hansen TT, Li R, Zala M, McDonald BA, Wang J, Schierup MH 2011. The making of a new pathogen: Insights from comparative population genomics of the domesticated wheat pathogen Mycosphaerella graminicola and its wild sister species. Genome Res 21: 2157–2166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stukenbrock EH, Christiansen FB, Hansen TT, Dutheil JY, Schierup MH 2012. Fusion of two divergent fungal individuals led to the recent emergence of a unique widespread pathogen species. Proc Natl Acad Sci 109: 10954–10959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot NJ, Yangkyo PS, Ma M, Hamer JE 1993. Karyotypic variation within clonal lineages of the rice blast fungus, Magnaporthe grisea. Appl Environ Microbiol 59: 585–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S 2011. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Salzberg SL 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoforms switching during cell differentiation. Nat Biotechnol 28: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL 2009. TopHat: Discovering splice junctions with RNA-Seq. Bioinform 25: 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usami T, Itoh M, Amemiya Y 2008. Mating type gene MAT 1-2-1 is common among Japanese isolates of Verticillium dahliae. Physiol Mol Plant Pathol 73: 133–137 [Google Scholar]
- Usami T, Itoh M, Amemiya Y 2009. Asexual fungus Verticillium dahliae is potentially heterothallic. J Gen Plant Pathol 75: 422–427 [Google Scholar]
- van de Wouw AP, Cozijnsen AJ, Hane JK, Brunner PC, McDonald BA, Oliver RP, Howlett BJ 2010. Evolution of linked avirulence effectors in Leptosphaeria maculans is affected by genomic environment and exposure to resistance genes in host plants. PLoS Pathog 6: e1001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GC. 1975. Sex and evolution. Princeton University Press, Princeton, NJ. [Google Scholar]
- Wong P, Walter M, Lee W, Mannhaupt G, Münsterkötter M, Mewes HW, Adam G, Güldener U 2011. FGDB: Revisiting the genome annotation of the plant pathogen Fusarium graminearum. Nucleic Acids Res 39: D637–D639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, Nacu S 2010. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinform 26: 873–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Wang H 2007. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 35: W265–W268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Saitoh H, Fujisawa S, Kanzaki H, Matsumura H, Yoshida K, Tosa Y, Chuma I, Takano Y, Win J, et al. 2009. Association genetics reveals three novel avirulence genes from the rice blast fungal pathogen Magnaporthe oryzae. Plant Cell 21: 1573–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeigler RS, Scott RP, Leung H, Bordeos A 1997. Evidence of parasexual exchange of DNA in the rice blast fungus challenges its exclusive clonality. J Phytopathol 87: 284–294 [DOI] [PubMed] [Google Scholar]
- Zerbino DR, Birney E 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolan ME 1995. Chromosome-length polymorphism in fungi. Microbiol Rev 59: 686–698 [DOI] [PMC free article] [PubMed] [Google Scholar]