

Abstract
Several studies have sought systematically to identify protein subcellular locations, but an even larger task is to map which of these proteins conditionally relocates in disease (the mislocalizome). Here, we report an integrative computational framework for mapping conditional location and mislocation of proteins on a proteome-wide scale, called a conditional location predictor (CoLP). Using CoLP, we mapped the locations of over 10,000 proteins in normal human brain and in glioma. The prediction showed 0.9 accuracy using 100 location tests of 20 randomly selected proteins. Of the 10,000 proteins, over 150 have a strong likelihood of mislocation under glioma, which is striking considering that few mislocation events have been identified in this disease previously. Using immunofluorescence and Western blotting in both primary cells and tissues, we successfully experimentally confirmed 15 mislocations. The most common type of mislocation occurs between the endoplasmic reticulum and the nucleus; for example, for RNF138, TLX3, and NFRKB. In particular, we found that the gene for the mislocating protein GFRA4 had a nonsynonymous point mutation in exon 2. Moreover, redirection of GFRA4 to its normal location, the plasma membrane, led to marked reductions in phospho-STAT3 and proliferation of glioma cells. This framework has the potential to track changes in protein location in many human diseases.
Protein mislocalization or mislocation, a change in the subcellular location(s) of a protein across comparable conditions, is fundamental to cell function and regulatory control in disease (Munkres et al. 1970; Reich and Liu 2006). Protein location can be governed by signal peptides, which direct the cellular transport machinery to convey proteins to the specific organelle(s) in which they are functional. It is also an important regulatory mechanism, as signal peptides can be masked or modified by carrier proteins that recognize a particular pattern of post-translational modifications. For example, STAT3 (signal transducer and activator of transcription 3) is phosphorylated by various cytokines and growth signals, resulting in its relocation to the nucleus, where it serves as a strong DNA-binding transcriptional activator (Reich and Liu 2006). Inappropriate phosphorylation and nuclear relocation of STAT3 promotes oncogenesis through abnormal cell cycle progression, angiogenesis, and invasion of tissue (Reich and Liu 2006). Changes in protein location are also associated with a host of genetic disorders. For instance, in Zellweger syndrome, mislocation of peroxisomal proteins leads to dysfunctional fatty acid oxidation (Dodt et al. 1995).
In model organisms, the location of proteins can be visualized systematically by fusion of each open reading frame to the gene encoding green fluorescent protein (GFP), either through transposon mutagenesis or polymerase chain reaction (PCR) tagging (Ross-Macdonald et al. 1999; Huh et al. 2003). In humans and other mammals, protein tagging is challenging, but immunolabeling can be used when suitable antibodies are available (Uhlen et al. 2010). Another common technique has been to fractionate the cell into different subcellular organelles and to analyze the protein content of each by tandem mass spectrometry (Gilchrist et al. 2006). Most of these approaches can be applied to multiple conditions or time points to identify protein mislocation events, although such dynamic measurements have limitations in the proteome-wide discovery of mislocation (Wickner and Schekman 2005; Reich and Liu 2006).
In addition to experimental approaches, a considerable number of methods have been developed for computationally predicting the location of proteins. Protein location prediction is a type of functional annotation and, as such, implements the principle of “guilt-by-association,” whereby the features of a target protein are matched to features of proteins whose annotations are known. The first features used for location prediction were based on protein sequences or structural characteristics (Wilson et al. 2000; Mott et al. 2002; Gardy et al. 2003; Bhasin and Raghava 2004; Scott et al. 2004; Chou and Cai 2005; Lee et al. 2006; Horton et al. 2007; Shatkay et al. 2007) and were later supplemented with gene-expression data, gene-deletion profiling, and experimental phenotypes (Chen and Xu 2004; Karaoz et al. 2004; Obozinski et al. 2008; Pena-Castillo et al. 2008). Most recently, large-scale protein–protein interaction or metabolic networks (Lee et al. 2008, 2010; Mintz-Oron et al. 2009; Jiang and Wu 2012) have been used in order to significantly increase the accuracy of location prediction, based on the principle that proteins with similar subcellular locations are likely to interact. However, these methods are limited in that they cannot identify location changes across diverse cell states or environmental conditions.
Given these experimental and bioinformatic advances, a compelling question is whether it would be feasible to mount a large-scale proteomics effort to identify all protein mislocation events across a spectrum of diseases. The essential challenges are several: (1) Identification of human protein locations is limited by the available antibodies; (2) most experimental methods fall very short of complete proteome coverage for a particular condition; and (3) a brute-force experimental survey of many conditions could be costly in reagents and time.
Here, we develop a computational framework for mapping conditional location and mislocation of proteins on a proteome-wide scale. Dynamic context is achieved through conditional network neighborhoods, in which expression profiles gathered for conditions of interest are projected onto protein–protein interaction networks. As proof-of-principle, we use this approach to develop a proteome-wide map of protein locations during the progression of glioma. From the conditional locations, we identify over 150 proteins that likely mislocate in glioma, and we observe that these events can be validated by immunofluorescent imaging and Western blot analysis with a very high rate of success. The validated mislocations lead to the hypothesis that GDNF family receptor alpha 4 (GFRA4) and persephin (PSPN), which normally interact at the plasma membrane with the product of the RET proto-oncogene (RET), are mislocated in glioma, leading to their accumulation in the endoplasmic reticulum (ER). This hypothesis is further supported by the additional finding that artificial redirection of GFRA4 to the plasma membrane results in a dramatic decrease in proliferation of glioma cells.
Results
Proteome-wide discovery of conditional location and mislocation of proteins
To predict conditional location and mislocation of proteins, we used a network-based approach that integrates proteome-wide sequences, chemical properties, gene ontology (GO) annotations, expression profiles, and well-known protein interaction databases (Fig. 1; Table 1). To generate features for location prediction, we adopted our previous approach that integrates three major types of information as indicators of protein location (S, N, and L) (Fig. 1A; Supplemental Fig. S1; Lee et al. 2008, 2010). S features capture static characteristics of a single protein including its primary amino acid sequence, its chemical properties, known structural motifs, and functional annotations. These S features have been widely used in many studies and show relatively good performance (Lee et al. 2006). N and L features describe the neighbors of the protein in a protein-interaction network: N summarizes the neighbors' static (S) features, while L represents their known locations when available. The rationale for N and L is that co-occurrence of sequence, structure, or function, including location, in a protein and its interacting partners has been shown to be useful information for location prediction (Lee et al. 2008, 2010). Instead of using all the available features, we selected a feasible combination for each distinct subcellular location using a divide-and-conquer k nearest neighbor (DC-kNN) method (Lee et al. 2008).
Figure 1.

Proteome-wide prediction of protein mislocation. (A) A protein is described by its sequence, chemical properties, motifs, and functions (single protein features) together with a description of its network neighborhood (capturing the features of its neighbors and their subcellular locations, if known). The best combination of features for each location is selected using a DC-kNN classifier. (B) Condition-dependent dynamic network features are generated by assigning different weights to each neighbor of a protein, depending on their similarity in gene expression profiles. (C) Selected features from A are combined with condition-dependent networks from B to compute a CLM for the protein, listing the quantitative possibility that the protein is in each location under each condition. (D) Mislocations are identified by calculating differences in degrees of possibility across conditions.
Table 1.
Sources of evidence for prediction of conditional subcellular location
A dynamic context for condition-dependent (or conditional) location is achieved through conditional network neighborhoods, in which expression profiles gathered for conditions of interest are projected onto protein–protein interaction networks (Fig. 1B). For each condition and study, each interaction was assigned a functional “coherence” score proportional to both expression level and the correlation between interacting protein pairs (see Methods). The conditional coherence scores under distinct conditions lead to conditional network features, resulting in a conditional location map (CLM) with degrees of possibility assigned to individual locations under distinct conditions (Fig. 1C). Finally, proteins that have very different degrees of possibility across different conditions indicate potential mislocation events (Fig. 1D). The use of coherence as a metric is motivated by the observation that proteins are more likely to share the same location if they are known to interact and their expression is highly correlated (Fig. 2A). This is further supported by the improved recovery of known locations when the coexpressed network is used together with protein interactions (Fig. 2B). Coexpressed protein pairs also provide insight into physical interactions, especially when coexpressed pairs common to multiple studies are considered (Fig. 2C).
Figure 2.
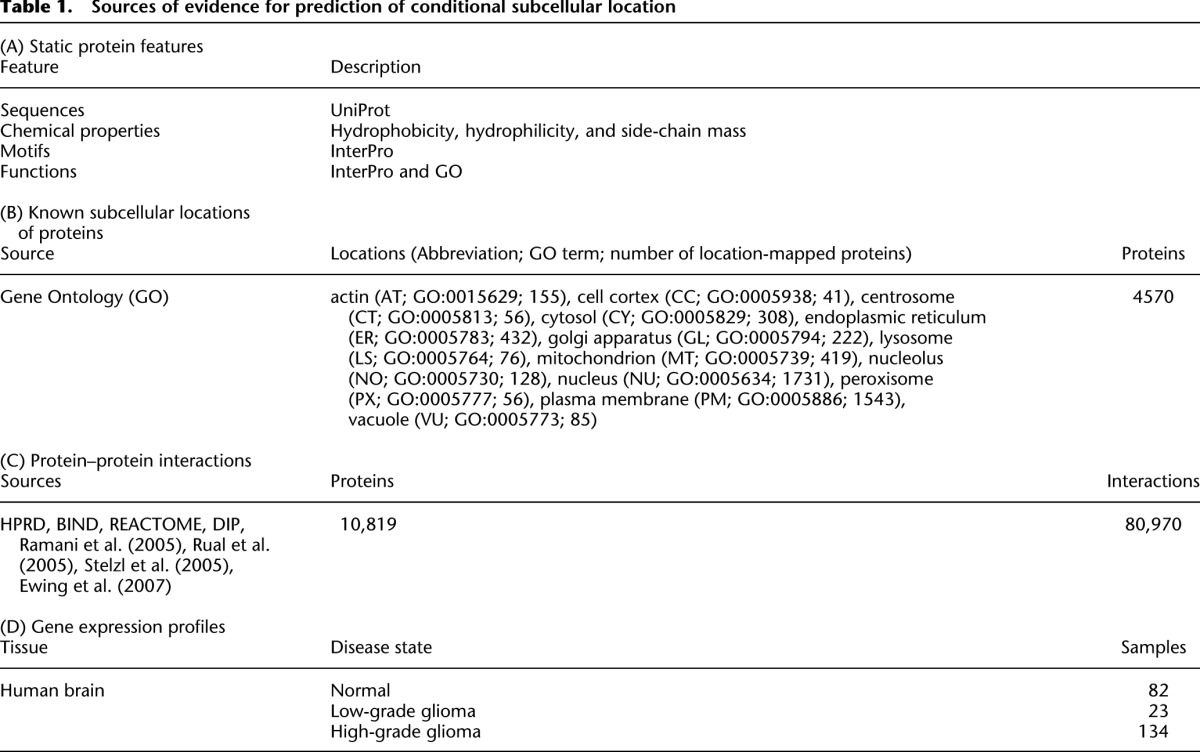
Models generated and usefulness of coherent protein interactions for location prediction. (A) The percentage of protein pairs sharing at least one location, calculated from different sets of proteins. “Random” was calculated as the average of 1000 randomly selected interaction sets with the same number of interactions as the original protein network. (B) Leave-two-out cross-validation with a DC-kNN classifier was used to assess the effect of static and network features on the accuracy of predicting known subcellular locations. (C) Fractions of protein pairs with known interactions among the top-k pairs with highest correlations in expression. “Common” indicates pairs common to normal brain (Normal) and low (Low)- and high (High)-grade gliomas. (D) Average AUC values of different feature sets, including S, ND, and LD. Here, the “TR” category means the final average AUC value of the selected models for 13 locations in the training stage. “D” indicates the distance of incorporated network neighbors. (E) Generated models with selected feature sets for individual locations using a DC-kNN classifier. Black and white squares represent selected feature sets for each location, with the white square denoting the best feature set overall. The last row indicates the AUC values for prediction of individual locations. The last column indicates the average AUC values of individual feature sets across the 13 locations considered.
Proteome-wide discovery of glioma stage-specific protein location
Before mapping conditional protein locations and mislocation events in human glioma, we first selected a feasible feature set for each location using a DC-kNN classifier. For protein, we used 4570 human proteins that have GO annotations and sequence information in the Universal Protein Resource (UniProt) database (Table 1B). We could map the 5252 locations of the proteins using GO-location mappings. For a human protein–protein interaction network, we used the pooled interactions from several well-known databases and recent studies (Table 1C). Since network features generated from network neighbors up to distance 2 were also useful in location prediction (Supplemental Fig. S2), we applied a forward selection that chose feature sets of high predictive power from the pool of features of the single protein and the network. During the feature-set selection, we used the common measure of area-under-the-receiver-operator-characteristic curve (AUC) to rank the predictive power of features and also to evaluate the performance of the resulting classifiers with a leave-two-out cross validation (LTOCV) scheme (see Methods). We observed that selecting feasible feature sets per location using the single protein and network features resulted in a dramatic increase in performance (0.93 AUC for the 13 locations) (Fig. 2D; Supplemental Fig. S3). Among the prepared features, the network features derived from the first neighbors' protein features were more useful than were any others, but many single-protein features were also selected in the final classifiers for location prediction (Fig. 2E).
Next, to predict conditional locations and mislocation events in human glioma, we downloaded gene expression profiles obtained for brain tissue under three different conditions—normal brain, low-grade glioma, and high-grade glioma (Table 1D). Because of technical noise in the microarray data, we downloaded and used multiple series of gene-expression profiles even for a single condition. These profiles included 82, 23, and 134 tissue samples, respectively (Khatua et al. 2003; Liang et al. 2007; Lockstone et al. 2007; Marucci et al. 2008; Costa et al. 2010). The expression profiles of proteins in normal brain were clearly separable from those in low- and high-grade glioma (Supplemental Fig. S4). In addition, the expression profiles were also grouped according to different studies and/or different microarray platforms. These three series of expression profiles were integrated with an accumulated human protein–protein interaction network (80,970 interactions between 10,819 proteins), which resulted in dynamic networks under the three conditions. Using the network features synthesized from the dynamic networks, we computed a conditional location map for each protein represented in the networks.
In total, 29,999 high-confidence locations were predicted for 9543 proteins, cumulative over the three conditions of normal brain and low- and high-grade glioma (P < 0.005, corresponding to a possibility degree ≥0.40 and an estimated false discovery rate [FDR] of 5%) (Supplemental Fig. S5; see Supplemental Table S1 for the predictions). One illustrative example is SMAD4, the product of a likely tumor suppressor gene, which mediates signal transduction by transforming growth factor beta and which has been previously reported to localize to either the nucleus or the cytosol (Nakao et al. 1997; Massague 1998; Sun et al. 1999). Our predictions assigned a high possibility only to the nucleus under all three conditions (Supplemental Fig. S6A); this was confirmed by immunofluorescent imaging of normal brain tissues (Supplemental Fig. S6B). The other previously reported location (cytosol) was neither computationally predicted nor experimentally observed, perhaps because of our focus on normal brain and glioma versus other tissues. In terms of the protein interaction network, SMAD4 can be seen to interact with 48 neighbors for which the expression profiles are coherent in brain and for which the location is known to be nuclear (Supplemental Fig. S6C).
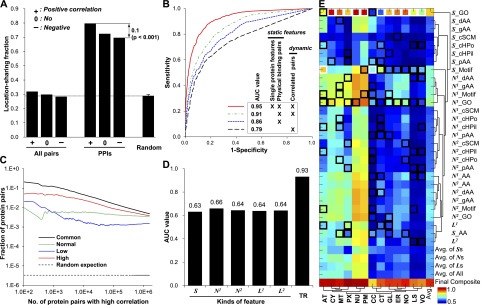
Of the predicted locations, 13,977 described a total of 5011 proteins whose locations were missed in our database (i.e., there was no mapped location based on the GO annotations and mapping relationships used here) (Supplemental Table S1). For example, NKX2-2 is a gene coding for a homeobox factor involved in central nervous system development. The subcellular location of NKX2-2 is not documented in the GO database we used, although as a transcription factor, the nucleus is one possible location (Owen et al. 2008). However, our prediction showed a very high score for NKX2-2 in the ER in all three conditions of brain tissue, and the second highest but very low score in the nucleus (Fig. 3A). Use of in vivo imaging provided confirmation that most NKX2-2 was highly associated with an ER location marker in normal brain tissue (Fig. 3B), and not with other locations, including the nucleus (Fig. 3C). We also observed that NKX2-2 was highly merged with ER markers in low- and high-grade gliomas (Supplemental Fig. S9). We also confirmed the ER location of NKX2-2 in normal brain and primary glioma cells using cellular subfractionation and Western blot assay (Fig. 3D). Signal peptide analysis using SignalP (Petersen et al. 2011) also confirmed the ER localization. As another example, the location of carboxypeptidase B1, encoded by CPB1, has not yet been clearly documented. Our prediction showed the highest signal in cytosol and the second highest, but weak, signal in ER in normal brain (Fig. 3E). We also confirmed that the major location of carboxypeptidase B1 is the cytosol and that a minor location is the ER (Fig. 3F–H).
Figure 3.

Novel protein locations in normal brain and glioma and predictive performance. (A) Conditional location map for NKX2-2. A possibility degree between 0 and 1 (blue to red gradient) was assigned to each of 13 subcellular locations (rows) across three conditions: normal brain, low- and high-grade gliomas (columns). The letter “H” in the lower left-hand corner of the panels marks the location with the highest degree of possibility among the 13 locations considered for each condition. (B) A series of three images of the same cell from normal brain tissue, showing anti-NKX2-2 (left, green), the ER (middle, red), or a merged image (right). The yellow color in the right panel indicates high overlap between NKX2-2 and the ER. (C) A second series showing anti-NKX2-2 (left, green), a nuclear marker (middle, blue), and a merged image (right). (D) Results of cellular subfractionation and Western blotting to determine the location of NKX2-2 in normal brain and primary glioma cells. (E) Conditional location map for CPB1. (F,G) Cells stained with anti-CPB1 (green) and a cytosolic (F, red) or nuclear (G, blue) marker in normal brain tissue. Scale bar, 5 μm. (H) Results of cellular subfractionation and Western blotting to determine the location of CPB1 using normal brain primary cells. (I) Heat map of immunochemistry validation results. Twenty proteins (columns) were interrogated at up to five locations (rows) using two-dimensional imaging. Predictions overlaid with experimental observations (+ present or – absent). The color of the heat map indicates the predicted score, here, the degree of possibility. Symbols are colored black if predictions are correct; otherwise, they are white. (J) Validation statistics from I summarized in tabular form. See Supplemental Figures S7 and S8 for antibody specificity tests for the locations and the proteins used in this study. (β-A) beta-actin for a cytosol marker, (CRT) calreticulin for endoplasmic reticulum, (RPII) RNA polymerase II for nucleus.
In addition to these examples, we performed confocal microscopic analysis of normal and glioma tissues to evaluate (in double-blind fashion) a total of 100 locations assigned to a set of 20 randomly selected proteins across five major locations: the cytosol, ER, Golgi apparatus, nucleus, and plasma membrane (Fig. 3I). We considered only proteins for which antibodies were commercially available. For example, the product of GSK3B, glycogen synthase kinase 3 beta, was predicted and validated to localize only to the nucleus in low-grade glioma, with one true positive and four true negatives (see Supplemental Fig. S10). Overall, 90 locations were correctly predicted among the 100 tested (Fig. 3J; Supplemental Fig. S11). This degree of accuracy of prediction would be highly unlikely by random guess (P ≈ 0) (Supplemental Fig. S12). To check the sensitivity of the predicted condition-specific location to the expression levels of proteins, we assessed the performance of 50 permutated tests using the real protein-expression data. The performance was poorer than that with the original expression data, regardless of the performance measure used (P ≈ 0) (Supplemental Fig. S13). However, the performance with permutated expression data was much better than that with totally random guesses, owing to the above-mentioned high degree of functional enrichment between interacting pairs (Fig. 2A). Moreover, the models generated with LTOCV also showed comparable performance to other types of cross-validation (Supplemental Fig. S14).
Protein mislocation during glioma progression
We next turned our attention to the 157 proteins for which predicted locations were significantly different between normal brain and glioma (P < 0.01) (Supplemental Fig. S15; Supplemental Table S2). One example was KIF13A (kinesin family member 13A). In normal brain, KIF13A showed the highest signal in the Golgi apparatus, whereas in low- and high-grade gliomas, it mapped most strongly to the nucleus (Fig. 4A). These locations were confirmed by imaging using primary cells and tissues (Fig. 4B,C; Supplemental Figs. S16, S17), indicating mislocation of KIF13A from the Golgi apparatus to the nucleus in glioma. The mislocation was frequent in a large population of primary glioma cells from multiple individuals (Fig. 4D). In terms of network dynamics, we observed that, in normal brain, KIF13A has high expression coherence with its interaction partners AP1G1 (adaptor-related protein complex 1, gamma 1 subunit) and COG2 (component of oligomeric Golgi complex 2), which are known to locate to the Golgi apparatus (Fig. 4E). However, in both low- and high-grade gliomas, KIF13A loses coherence with AP1G1 and COG2 and assumes high coherence with ATF7IP (activating transcription factor 7 interacting protein), which is known to localize to the nucleus (Fig. 4F,G).
Figure 4.
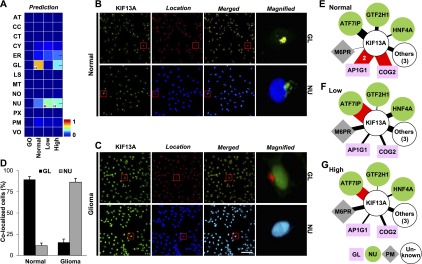
Prediction and validation of KIF13A mislocation in glioma tissues and cells. (A) Conditional location map for KIF13A, for which the highest signal under normal conditions was in the Golgi apparatus (GL), but in low- and high-grade gliomas was in the nucleus (NU). The color indicates degree of possibility and “H” indicates the location with the highest degree of possibility within each condition. (B,C) Confocal images for KIF13A (green) together with markers for GL (red, row 1) or NU (blue, row 2) in normal (B) and glioma (C) tissues reveals results consistent with predictions. Scale bar, 5 μm. (D) The fraction of colocalized cells expressing GL and NU markers using >50,000 normal brain and glioma primary cells. Samples from four normal and five glioma subjects were used. (E–G) The dynamic interaction neighborhood of KIF13A in normal brain (E), and low- (F) and high-grade (G) glioma tissues. Node color/shape indicates known protein locations. The width of each link is proportional to the expression coherence score, which is also indicated numerically for selected links. Red links indicate key interactions.
The global landscape of all 157 mislocation events in glioma is shown in Figure 5A. The most frequent mislocations were from ER to nucleus (21.66%), nucleus to ER (15.92%), or plasma membrane to ER (12.10%); thus, many mislocations involved movement into or out of the ER. For example, the conditional location map for the product of RNF138 (ring finger protein 138, E3 ubiquitin protein ligase) shows its mislocation from ER to nucleus in glioma (Fig. 5B). Confocal imaging using primary cells confirmed that, in normal brain, RNF138 overlaps with markers for the ER, but not the nucleus, while in glioma it overlaps with markers for the nucleus, but not the ER (Fig. 5D; Supplemental Figs. S18, S19). This mislocation was observed in most primary glioma cells from multiple individuals (Fig. 5E). Cellular subfractionation and Western blotting confirmed the mislocation of RNF138 (Fig. 5F). As another example, TLX3 (T-cell leukemia homeobox 3) is a member of a family of orphan homeobox genes that encode DNA-binding nuclear transcription factors; TLX3 mislocation from nucleus to ER during glioma progression was both predicted (Fig. 5C) and confirmed (Fig. 5G–I; Supplemental Figs. S20, S21).
Figure 5.

The landscape of protein mislocations in human glioma. (A) The landscape of mislocations in glioma. Each peak (z-axis) corresponds to the percentage of these mislocation candidates moving from one location (x-axis) to another (y-axis). Colors along the x and y margins represent the total percentage of proteins mislocating out of or into a location, respectively. (B,C) Conditional location maps of RNF138 and TLX3 are shown as examples of the most common mislocations from the ER to the nucleus (NU) or from the NU to ER, respectively. The color indicates degree of possibility and “H” indicates the location with the highest degree of possibility within each condition. (D–I) Validation of RNF138 (D) and TLX3 (G) using confocal images for normal brain and glioma tissues. Confirmation of RNF138 (E,F) and TLX3 (H,I) mislocations by population assay and Western blot analyses using normal brain and glioma primary cells. For the population assay, samples from four normal and five glioma subjects were used. (Green) RNF138 or TLX3, (red) ER, (blue) NU, (CRT) calreticulin for an ER marker, (RPII) RNA polymerase II for nucleus. Scale bar, 5 μm.
Next, we performed additional testing of the mislocation candidates (five positive and five negative cases). We performed a single-blind randomized controlled trial using only proteins for which antibodies were commercially available (the experimenters were blinded to the proteins being investigated). For example, ATIC (5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/IMP cyclohydrolase; predicted to mislocate from cytosol to nucleus), DIP2A (DIP2 disco-interacting protein 2 homolog A; from nucleus to ER), DLX2 (distal-less homeobox 2; from ER to nucleus), HPS5 (Hermansky–Pudlak syndrome 5; from plasma membrane to ER), and TBX19 (T-box 19; from ER to nucleus) were predicted to mislocate during glioma progression, whereas CDH2 (cadherin 2, type 1, N-cadherin; plasma membrane, regardless of glioma stage), HSF1 (heat-shock transcription factor 1; nucleus), MAGED1 (melanoma antigen family D, 1; cytosol), PAX6 (paired box 6; nucleus), and STAT3 (nucleus) were not. Among the five positive candidates, locations of four proteins, except HPS5, definitely differed between normal brain and glioma, consistent with predictions (Supplemental Figs. S22–S25 for validation using in vivo imaging, Western blot of cellular subfraction, and population assay). However, HPS5 resided at both plasma membrane and ER in glioma, as well as in normal brain (Supplemental Fig. S26). In the negatives, the locations of the four proteins, except STAT3, were consistent with predictions (Supplemental Fig. S27). STAT3, however, changed its location from plasma membrane in normal brain to nucleus in glioma (Supplemental Fig. S28). Therefore, eight out of 10 positives and negatives were observed to be correctly predicted.
In total, we successfully validated fifteen mislocation events predicted to occur during glioma progression: KIF13A (Fig. 4), RNF138 and TLX3 (Fig. 5), PSPN and GFRA4 (Fig. 6), ATIC, DIP2A, DLX2, and TBX19 (Supplemental Figs. S22–S25), AGAP1 (ArfGAP with GTPase domain, ankyrin repeat and PH domain 1; cytosol to Golgi apparatus) (Supplemental Fig. S29), carboxypeptidase B1 (cytosol to ER) (Supplemental Fig. S30), NFRKB (nuclear factor related to kappaB-binding protein; ER to nucleus) (Supplemental Fig. S31), ARHGEF15 (Rho guanine nucleotide exchange factor 15; ER to plasma membrane) (Supplemental Fig. S32), CLK2 (CDC-like kinase 2; ER to nucleus) (Supplemental Fig. S33), and SYT9 (synaptotagmin IX; Golgi apparatus to nucleus) (Supplemental Fig. S34). Interestingly, none of these mislocations had been documented in glioma previously. Moreover, these validated mislocations could not be predicted using wild-type normal or random-permutated expression sets (P ≈ 0) (Supplemental Fig. S35).
Figure 6.

Conditional location of GFRA4, PSPN, and RET in glioma. (A–C) CLMs of GFRA4 (A), PSPN (B), and RET (C), and the results of confocal images in normal brain and glioma tissues. The color of the heat maps indicates predicted degree of possibility, and “H” indicates the location with the highest degree of possibility within each condition. Scale bar, 5 μm. (D) Location fraction of GFRA4, PSPN, and RET in normal brain and glioma primary cells. (E–G) Results of cellular subfractionation and Western blotting for locations of GFRA4 (E), PSPN (F), and RET (G) in normal brain and glioma primary cells. (SPA) sodium potassium ATPase (plasma membrane marker), (CRT) calreticulin (ER marker).
Dynamic complex of GFRA4/PSPN/RET may play an essential role in glioma proliferation
Two of the confirmed proteins mislocated in glioma were PSPN and GFRA4, for which predictions suggested a strong shift from plasma membrane to ER in glioma (Fig. 6A,B). These mislocations were observed frequently (Fig. 6D; Supplemental Fig. S36). However, the common interacting partner of GFRA4 and PSPN, RET, was predicted and validated to remain in the plasma membrane (Fig. 6C,D; Supplemental Fig. S36). Western blot analysis confirmed the glioma-dependent locations of GFRA4/PSPN/RET (Fig. 6E–G). Moreover, proximity ligation assays indicated dissociation of RET from GFRA4 or PSPN in glioma (Fig. 7A; Supplemental Fig. S37). These dissociations were not caused by reduced GFRA4/PSPN/RET protein expression in glioma (Supplemental Fig. S38), but mainly by mislocation of GFRA4 and PSPN. Interestingly, the interaction between GFRA4 and PSPN was consistently observed, and the number of interacting pairs increased with progressive disease stages (Fig. 7B; Supplemental Fig. S37). However, the GFRA4/PSPN interaction was weak in glioma (Fig. 7C).
Figure 7.

Dynamics of the GFRA4/PSPN/RET complex in glioma. (A,B) A proximity ligation assay was used to measure groups of close physical interactions between RET and PSPN, RET and GFRA4, and PSPN and GFRA4 in normal brain and glioma tissues. Red spots indicate physical proximity of the corresponding protein pair. Insets: 4× magnification. Scale bar, 20 μm. (C) Two fluorescence autocorrelation functions, G(τ), of GFP-PSPN (blue), TagRFP-GFRA4 (red), and one cross-correlation function (black), calculated from time traces of fluorescent fluctuations with high-grade glioma primary cells, as a function of correlation lag time τ (μs). (D) Reverse transcription-PCR of GFRA4 with axon 2 mutation. (E) Cell proliferation assay using thymidine incorporation in high-grade glioma cells with (“siGFRA4”) or without GFRA4 silencing (“glioma”), and using rapamycin for GFRA4 redirection to the plasma membrane (“FKBP-FBP”) in high-grade primary glioma cells. Bars indicate radioactivity in counts per minute (CPM; average ± standard deviation). (F) Immunoblot results using primary high-grade glioma (Glioma, FKBP-FRB, siGFRA4) and normal brain (Normal) cells. (G) Snapshots of GFRA4 redirection to the plasma membrane using the rapamycin technique in high-grade glioma cells. (Cyan) GFRA4. Scale bar, 100 μm.
We observed that GFRA4 had a point mutation in exon 2 in glioma cells (Fig. 7D), which altered its encoded protein sequence from Cys to Ser at a position common to the three known isoforms of GFRA4. Cys residues drive protein folding and are therefore closely related to subcellular protein trafficking (Herrmann and Riemer 2010; Vascotto et al. 2011). We also observed a marked decrease in proliferation of glioma cells after GFRA4 silencing (Fig. 7E). GFRA4 silencing also reduced the levels of phosphorylated STAT3 (pSTAT3), which was widely found in the nucleus in glioma cells with no marked change in total pSTAT3 expression level (Fig. 7F; Supplemental Fig. S39). Surprisingly, artificial redirection of GFRA4 to the plasma membrane in live glioma cells (Fig. 7G; Supplemental Movie 1) markedly decreased cell proliferation (Fig. 7E) and the pSTAT3 expression level (Fig. 7F). Inappropriate location and complex formation of GFRA4 potentiated cell cycle progression and tumor growth.
Discussion
To the best of our knowledge, this paper describes the first computational approach for predicting conditional changes in the subcellular location of proteins in a genome-wide manner. The core concept behind this approach is that functional coherence in the gene expression profiles of protein pairs that are known to interact physically is an indicator of their subcellular location. As shown in Figure 2, A and B, interacting protein pairs with similar expression patterns at the mRNA level showed the highest degree of location sharing among different kinds of protein pairs, including pairs with inversely correlated expression patterns. Thus, to make inferences with regard to dynamic properties, such as protein location, one need only look at neighbors showing high functional coherence scores under a specific condition. However, high functional coherence scores for the expression profiles do not necessary imply physical interactions between proteins, even though they give some indication as to physical interactions (Fig. 2C). This principle may not cover every mislocation event, as one can imagine that interacting proteins might colocalize even if their mRNA expression patterns are not coherent. However, the conditional location maps are not based on any single interacting protein but on trends integrated over the entire network neighborhood. To address issues of coverage, the definition of the network ‘neighborhood’ can be extended to include not only a protein's direct interactors but all proteins reachable within a certain network distance (here, we used a network distance of two). In prediction of static protein locations, network-based methods have been shown to be surprisingly robust to missing interaction data (Lee et al. 2008).
In a sense, because we categorized our samples as normal brain and low- and high-grade glioma, the current study has a limited ability to discover different location changes across various normal conditions at different time points or in different glioma subtypes such as IDH mutant and IDH wild type. However, if the expression data could be recategorized according to the subconditions of the various normal tissues or glioma subtypes, the conditional location mapping framework presented here could easily be used to discover time-dependent or glioma type-specific location changes. Even though our current focus is location change in a specific disease (glioma), this framework can be used to map location changes under any type of condition for which gene expression profiles are available, such as diseases, stem cell differentiation, and responses to drugs and external stresses.
The landscape of cancer-related mislocations (Fig. 5) suggests that many instances involve the ER. Misfolding of some proteins is known to occur in the ER under oxidative stress conditions or in diverse diseases (Uehara et al. 2006). Because of such misfolding, proteins might not be transported to their target locations from the ER. This might result in malfunction of the corresponding proteins, leading to disease. Interestingly, among the mislocation events that we observed were striking changes in locations and interactions between RET, GFRA4, and PSPN, suggesting that these might play a key role in glioma progression (Figs. 6, 7). These findings are supported in part by previous evidence. RET is an established proto-oncogene associated with various cancers, including multiple endocrine neoplasia type 2A and 2B (Mulligan et al. 1994; Rossel et al. 1997; Hansford and Mulligan 2000). It encodes a receptor tyrosine kinase involved in control of cell survival, differentiation, proliferation, migration, chemotaxis, branching morphogenesis, neurite outgrowth, and synaptic plasticity. RET itself is activated by members of the GDNF family of ligands that includes PSPN (Lin et al. 1993; Milbrandt et al. 1998). Such ligands bind to RET through the GDNF receptors GFRA1–4, a family of glycosyl phosphatidylinositol-anchored proteins. GFRA4 is specific for PSPN, and its role in familial medullary thyroid carcinoma has been documented (Lindahl et al. 2001). Using our conditional location-mapping framework, we could identify that mislocation of GFRA4 and PSPN might be one of the key events in glioma progression. However, the precise mechanisms by which GFRA4 and PSPN mislocate to the ER rather than the plasma membrane need to be investigated further. Nonetheless, it appears likely that inappropriate location and complex formation of GFRA4 potentiates cell cycle progression and tumor growth.
In this study, we predicted 157 mislocation candidates and successfully validated 15. As we tested only a small portion of the predicted candidates, the other candidates also need to be validated further.
Methods
Protein locations, interactions, and expression profiles
For known static locations of human proteins, we obtained GO Cellular Component annotations using the AmiGO tool (http://amigo.geneontology.org/cgi-bin/amigo/go.cgi) and mapped these to 13 high-level subcellular locations using only experimental evidence codes (including EXP, IDA, IPI, IMP, IGI, and IEP) for the GO annotations (Table 1B). A total of 4570 proteins had GO annotations and were represented with sequence information in the UniProt database. For the protein–protein interaction network, we downloaded interactions from the HPRD (Keshava Prasad et al. 2009), BIND (Bader et al. 2001), REACTOME (Joshi-Tope et al. 2005), and DIP (Salwinski et al. 2004) databases (Table 1C). We also included two recent Y2H results (Rual et al. 2005; Stelzl et al. 2005), the results of immunoprecipitation followed by LC-ESI-MS/MS (Ewing et al. 2007), and interactions mined from prior literature (Ramani et al. 2005). Expression profiles of normal brain and low- and high-grade gliomas were downloaded from the NCBI Gene Expression Omnibus (GEO) database (Table 1D). For studies in GEO that used two channel microarray platforms, total RNA abundance values were extracted for a specific channel of interest. When multiple probes per gene were available, we computed the average value. Expression profiles were quantile normalized. For missing expression values within a single experiment set, we applied a kNN imputation method if the percentage of missing values for a gene was <30%; otherwise, the expressions were discarded.
Expression coherence score
An expression coherence score is computed between proteins a and b under a specific condition:
where 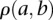 (–1 to 1, inclusive) is the Pearson correlation coefficient between the gene expression levels of
(–1 to 1, inclusive) is the Pearson correlation coefficient between the gene expression levels of 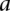 and
and 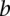 ,
, 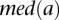 (or
(or 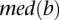 ) is the median expression level of
) is the median expression level of 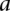 (or
(or 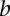 ),
), 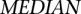 is the median value of all median expression levels of genes used, and
is the median value of all median expression levels of genes used, and 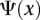 is the P-value of
is the P-value of 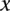 versus the distribution of the correlation coefficients of all interacting protein pairs. If
versus the distribution of the correlation coefficients of all interacting protein pairs. If 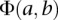 cannot be directly calculated owing to missing or insufficient expression values, it is assigned the median coherence value of all interactions involving
cannot be directly calculated owing to missing or insufficient expression values, it is assigned the median coherence value of all interactions involving 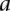 or
or 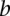 and otherwise the median of all interactions in the network. If
and otherwise the median of all interactions in the network. If 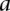 and
and 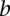 is not a direct interaction (e.g.,
is not a direct interaction (e.g., 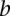 is in the second neighborhood of
is in the second neighborhood of 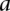 ), we multiplied the coherence scores along all the shortest paths between
), we multiplied the coherence scores along all the shortest paths between 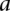 and
and 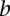 and chose the product with the maximum value.
and chose the product with the maximum value.
Generation of protein features and prediction model generation
In total, 29 types of single protein and network feature sets were generated for each protein as described previously (Lee et al. 2008). Among the prepared feature sets, we selected a combination of feature sets using a DC-kNN classifier for each location for model generation. Individual models produce a confidence score for each location of a protein (see Supplemental Methods for details).
Conditional location map and scoring the degree of possibility
A conditional location map for a protein consists of degrees of possibility for each location under individual conditions. For each protein, a DC-kNN classifier was used to assign a confidence c to each location l with a selected feature set. This confidence score was further expressed as a degree of possibility P(0∼1). Using guidelines proposed by Dubois and colleagues (Dubois et al. 2004), we converted the confidence score to a degree of possibility according to the following formula:
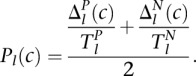 |
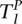 and
and 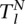 represent the total areas under the confidence score distributions of Positive and Negative gold-standard examples for subcellular location l. These areas are computed from
represent the total areas under the confidence score distributions of Positive and Negative gold-standard examples for subcellular location l. These areas are computed from 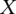 , the minimum value of the positive distribution, to
, the minimum value of the positive distribution, to 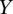 , the maximum value of the negative distribution.
, the maximum value of the negative distribution. 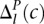 and
and 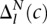 represent the areas under these Positive and Negative distributions from
represent the areas under these Positive and Negative distributions from 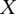 to
to  . For the gold-standard data set, we used the assigned locations of 4570 proteins based on GO Cellular Component terms (Table 1B).
. For the gold-standard data set, we used the assigned locations of 4570 proteins based on GO Cellular Component terms (Table 1B). 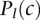 is ‘0’ if
is ‘0’ if  <
< 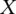 and ‘1’ if
and ‘1’ if  >
> 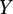 . A higher value of
. A higher value of 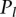 means a higher degree of possibility that a protein has location l.
means a higher degree of possibility that a protein has location l.
Brain tissue preparation and primary cell culture
Human normal brain and glioma tissues were acquired from the Brain Bank of Seoul National University Hospital in Korea. Brain tissues were fixed in 4% paraformaldehyde in 0.1 M phosphate buffer, followed by cryoprotection in 30% sucrose overnight (see the Supplemental Methods for details).
Immunohistochemistry
Tissue sections were incubated overnight with protein-specific antibodies at 4°C (Supplemental Table S3A). Tissues were rinsed with phosphate-buffered saline (PBS) and incubated for 1 h at room temperature with secondary antibodies (Supplemental Table S3B). For counterstaining of the nucleus, cells were incubated with DAPI (4′,6-diamidino-2-phenylindole; 1 μg/mL; Sigma Aldrich) for 40 sec. After washing with PBS, coverslips were mounted on glass slides using VectaShield mounting media (Vector Laboratories), and analyzed using an LSM 710 confocal microscope (Carl Zeiss).
Proximity ligation assay
A proximity ligation assay (PLA) was performed in both primary cells and tissues to visualize the population of protein–protein interactions. Tissues were washed with chilled PBS and incubated overnight with protein-specific antibodies at 4°C (Supplemental Table S3A). Proximity ligation was performed according to the manufacturer's protocol using the Duolink detection kit (Olink Bioscience). Hoechst stain was included in the Duolink detection kit during the detection reaction. Specimens were mounted with VECTASHIELD mounting media (Vector Laboratories) and analyzed using an LSM 710 confocal microscope (Carl Zeiss). The number of in situ PLA signals per cell was counted by semiautomated image analysis using BlobFinder V3.0 (see Supplemental Methods for more information about PLA).
Transfection and short-interfering RNA synthesis
Human glioma primary cells were transiently transfected using the OneDrop Microporator MP kit (NanoEnTek) after short-interfering RNA (siRNA) synthesis according to the manufacturer's instructions (Supplemental Table S3C).
Subcellular fractionation
Nuclear, cytoplasmic, ER, and plasma membrane fractions from human normal primary and glioma cells were purified using the Subcellular Protein Fractionation Kit (78840; Thermo Scientific), Endoplasmic Reticulum Isolation Kit (ER0100; Sigma-Aldrich), and Plasma Membrane Protein Extraction Kit (ab65400; Abcam), according to the manufacturers' instructions. Briefly, 1 × 106 cells were cultured on 10-cm2 dishes (Nunc) and rinsed with PBS. The cells were homogenized in extraction buffer containing a protease inhibitor cocktail. Extracts were centrifuged at 4°C, and the supernatants were saved as the nuclear, cytoplasmic, ER, and plasma membrane protein fractions. Validation of successful nuclear, cytoplasmic, ER, and plasma membrane separation was confirmed by Western blotting of each fraction for RNA polymerase II, beta-actin, calreticulin, and sodium potassium ATPase (Supplemental Table S3A).
Immunoblot
Cell lysates were prepared with lysis buffer containing 7 M urea, 2 M thiourea, and 4% CHAPS. Equal amounts (25 μg) of protein from each group were separated in 4%–12% polyacrylamide gels (Invitrogen) and transferred to a nitrocellulose membrane (Millipore). Proteins were detected with protein-specific antibodies (Supplemental Table S3A).
GFRA4 gene mutation
Mutation analysis of GFRA4 was performed by reverse transcription-PCR as described previously (Zhou et al. 2001).
FCS and FCCS measurements
Both LSM510/ConfoCor2 and LSM710/ConfoCor3 (Carl Zeiss) were used for two-dimensional imaging, FCS, and FCCS measurements to analyze colocalization and dynamics of protein–protein interactions in live primary cells. FCS and FCCS measure the binding strength of protein–protein interactions, in addition to dynamic colocalization (Bacia et al. 2006). Each pair of proteins (persephin, GFRA4, and RET) was transfected into primary normal or glioma cells at a very low expression level (0.005 μg/μl plasmid transfection concentration) in order to minimize perturbation to the system. Data were analyzed with the ConfoCor2/ConfoCor3 software as described in our previous study (Pack et al. 2006; Noda et al. 2008).
FRBP-rapamycin-FKB dimerization
Human glioma primary cells were transfected with siGFRA4 and then singly transfected with fusion vectors (RET-FRB and FKBP-GFRA4). Cells were incubated at room temperature for 20 min with 1 nmol/L of rapamycin and evaluated in a growth/proliferation assay.
Densitometry and statistical analysis
The densitometric intensity of each immunoreactive band was determined using gel digitizing Image-Pro software. All data in this report represent the results from at least three independent experiments, unless stated otherwise. The cells positive for corresponding antigens in immunohistochemical staining of normal human brain and glioma tissues were analyzed by confocal microscope (710; Carl Zeiss) using Zen software for unbiased counting. Statistical analyses were performed using the Student's t-test, and P < 0.05 was considered statistically significant.
Data access
The results of the predictions are available in the Supplemental Material and at http://nbm.ajou.ac.kr/colp/SampleResult.jsp. The web server to allow the prediction of condition locations and mislocations is available at http://nbm.ajou.ac.kr/colp/.
Acknowledgments
We thank Drs. Y. Sako and M. Hatakeyama for kindly providing facilities for live-cell imaging, and Mr. Seungleal Paek for assistance with prediction validation. This research was supported by the programs through the National Research Foundation of Korea grant funded by the Korean government (MSIP) (2010-0022887 and 2010-0028631) to K.L., and a grant from the U.S. National Institute of General Medical Sciences to T.I. (GM070743). Funding in part was also provided by a grant from the National R&D Program for Cancer Control, Ministry for Health and Welfare, Republic of Korea (1020380) (B.L.).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.155499.113.
References
- Bacia K, Kim SA, Schwille P 2006. Fluorescence cross-correlation spectroscopy in living cells. Nat Methods 3: 83–89 [DOI] [PubMed] [Google Scholar]
- Bader GD, Donaldson I, Wolting C, Ouellette BF, Pawson T, Hogue CW 2001. BIND—the Biomolecular Interaction Network Database. Nucleic Acids Res 29: 242–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhasin M, Raghava GP 2004. ESLpred: SVM-based method for subcellular localization of eukaryotic proteins using dipeptide composition and PSI-BLAST. Nucleic Acids Res 32: W414–W419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Xu D 2004. Global protein function annotation through mining genome-scale data in yeast Saccharomyces cerevisiae. Nucleic Acids Res 32: 6414–6424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou KC, Cai YD 2005. Predicting protein localization in budding yeast. Bioinformatics 21: 944–950 [DOI] [PubMed] [Google Scholar]
- Costa BM, Smith JS, Chen Y, Chen J, Phillips HS, Aldape KD, Zardo G, Nigro J, James CD, Fridlyand J, et al. 2010. Reversing HOXA9 oncogene activation by PI3K inhibition: Epigenetic mechanism and prognostic significance in human glioblastoma. Cancer Res 70: 453–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodt G, Braverman N, Wong C, Moser A, Moser HW, Watkins P, Valle D, Gould SJ 1995. Mutations in the PTS1 receptor gene, PXR1, define complementation group 2 of the peroxisome biogenesis disorders. Nat Genet 9: 115–125 [DOI] [PubMed] [Google Scholar]
- Dubois D, Foulloy L, Mauris G, Prade H 2004. Probability-possibility transformations, triangular fuzzy sets, and probabilistic inequalities. Reliable Computing 10: 273–297 [Google Scholar]
- Ewing RM, Chu P, Elisma F, Li H, Taylor P, Climie S, McBroom-Cerajewski L, Robinson MD, O'Connor L, Li M, et al. 2007. Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol Syst Biol 3: 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardy JL, Spencer C, Wang K, Ester M, Tusnady GE, Simon I, Hua S, deFays K, Lambert C, Nakai K, et al. 2003. PSORT-B: Improving protein subcellular localization prediction for Gram-negative bacteria. Nucleic Acids Res 31: 3613–3617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist A, Au CE, Hiding J, Bell AW, Fernandez-Rodriguez J, Lesimple S, Nagaya H, Roy L, Gosline SJ, Hallett M, et al. 2006. Quantitative proteomics analysis of the secretory pathway. Cell 127: 1265–1281 [DOI] [PubMed] [Google Scholar]
- Hansford JR, Mulligan LM 2000. Multiple endocrine neoplasia type 2 and RET: From neoplasia to neurogenesis. J Med Genet 37: 817–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann JM, Riemer J 2010. Oxidation and reduction of cysteines in the intermembrane space of mitochondria: Multiple facets of redox control. Antioxid Redox Signal 13: 1323–1326 [DOI] [PubMed] [Google Scholar]
- Horton P, Park KJ, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, Nakai K 2007. WoLF PSORT: Protein localization predictor. Nucleic Acids Res 35: W585–W587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK 2003. Global analysis of protein localization in budding yeast. Nature 425: 686–691 [DOI] [PubMed] [Google Scholar]
- Jiang JQ, Wu M 2012. Predicting multiplex subcellular localization of proteins using protein-protein interaction network: A comparative study. BMC Bioinformatics (Suppl 10) 13: S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi-Tope G, Gillespie M, Vastrik I, D'Eustachio P, Schmidt E, de Bono B, Jassal B, Gopinath GR, Wu GR, Matthews L, et al. 2005. Reactome: A knowledgebase of biological pathways. Nucleic Acids Res 33: D428– D432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaoz U, Murali TM, Letovsky S, Zheng Y, Ding C, Cantor CR, Kasif S 2004. Whole-genome annotation by using evidence integration in functional-linkage networks. Proc Natl Acad Sci 101: 2888–2893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- doi: 10.1093/nar/gkn892. Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R, Shafreen B, Venugopal A, et al. 2009. Human Protein Reference Database—2009 update. Nucleic Acids Res37: D767–D772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatua S, Peterson KM, Brown KM, Lawlor C, Santi MR, LaFleur B, Dressman D, Stephan DA, MacDonald TJ. 2003. Overexpression of the EGFR/FKBP12/HIF-2α pathway identified in childhood astrocytomas by angiogenesis gene profiling. Cancer Res63: 1865–1870. [PubMed] [Google Scholar]
- Lee K, Kim DW, Na D, Lee KH, Lee D 2006. PLPD: Reliable protein localization prediction from imbalanced and overlapped datasets. Nucleic Acids Res 34: 4655–4666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Chuang HY, Beyer A, Sung MK, Huh WK, Lee B, Ideker T 2008. Protein networks markedly improve prediction of subcellular localization in multiple eukaryotic species. Nucleic Acids Res 36: e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Thorneycroft D, Achuthan P, Hermjakob H, Ideker T 2010. Mapping plant interactomes using literature curated and predicted protein-protein interaction data sets. Plant Cell 22: 997–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, Walker DG, Caselli RJ, Kukull WA, McKeel D, Morris JC, et al. 2007. Gene expression profiles in anatomically and functionally distinct regions of the normal aged human brain. Physiol Genomics 28: 311–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F 1993. GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 260: 1130–1132 [DOI] [PubMed] [Google Scholar]
- Lindahl M, Poteryaev D, Yu L, Arumae U, Timmusk T, Bongarzone I, Aiello A, Pierotti MA, Airaksinen MS, Saarma M 2001. Human glial cell line-derived neurotrophic factor receptor α 4 is the receptor for persephin and is predominantly expressed in normal and malignant thyroid medullary cells. J Biol Chem 276: 9344–9351 [DOI] [PubMed] [Google Scholar]
- Lockstone HE, Harris LW, Swatton JE, Wayland MT, Holland AJ, Bahn S 2007. Gene expression profiling in the adult Down syndrome brain. Genomics 90: 647–660 [DOI] [PubMed] [Google Scholar]
- Marucci G, Morandi L, Magrini E, Farnedi A, Franceschi E, Miglio R, Calo D, Pession A, Foschini MP, Eusebi V 2008. Gene expression profiling in glioblastoma and immunohistochemical evaluation of IGFBP-2 and CDC20. Virchows Arch 453: 599–609 [DOI] [PubMed] [Google Scholar]
- Massague J 1998. TGF-β signal transduction. Annu Rev Biochem 67: 753–791 [DOI] [PubMed] [Google Scholar]
- Milbrandt J, de Sauvage FJ, Fahrner TJ, Baloh RH, Leitner ML, Tansey MG, Lampe PA, Heuckeroth RO, Kotzbauer PT, Simburger KS, et al. 1998. Persephin, a novel neurotrophic factor related to GDNF and neurturin. Neuron 20: 245–253 [DOI] [PubMed] [Google Scholar]
- Mintz-Oron S, Aharoni A, Ruppin E, Shlomi T 2009. Network-based prediction of metabolic enzymes' subcellular localization. Bioinformatics 25: i247–i252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott R, Schultz J, Bork P, Ponting CP 2002. Predicting protein cellular localization using a domain projection method. Genome Res 12: 1168–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan LM, Eng C, Healey CS, Clayton D, Kwok JB, Gardner E, Ponder MA, Frilling A, Jackson CE, Lehnert H, et al. 1994. Specific mutations of the RET proto-oncogene are related to disease phenotype in MEN 2A and FMTC. Nat Genet 6: 70–74 [DOI] [PubMed] [Google Scholar]
- Munkres KD, Benveniste K, Gorski J, Zuiches CA 1970. Genetically induced subcellular mislocation of Neurospora mitochondrial malate dehydrogenase. Proc Natl Acad Sci 67: 263–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao A, Imamura T, Souchelnytskyi S, Kawabata M, Ishisaki A, Oeda E, Tamaki K, Hanai J, Heldin CH, Miyazono K, et al. 1997. TGF-β receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J 16: 5353–5362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda Y, Horikawa S, Kanda E, Yamashita M, Meng H, Eto K, Li Y, Kuwahara M, Hirai K, Pack C, et al. 2008. Reciprocal interaction with G-actin and tropomyosin is essential for aquaporin-2 trafficking. J Cell Biol 182: 587–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obozinski G, Lanckriet G, Grant C, Jordan MI, Noble WS 2008. Consistent probabilistic outputs for protein function prediction. Genome Biol (Suppl 1) 9: S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen LA, Kowalewski AA, Lessnick SL 2008. EWS/FLI mediates transcriptional repression via NKX2.2 during oncogenic transformation in Ewing's sarcoma. PloS one 3: e1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pack C, Saito K, Tamura M, Kinjo M 2006. Microenvironment and effect of energy depletion in the nucleus analyzed by mobility of multiple oligomeric EGFPs. Biophys J 91: 3921–3936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena-Castillo L, Tasan M, Myers CL, Lee H, Joshi T, Zhang C, Guan Y, Leone M, Pagnani A, Kim WK, et al. 2008. A critical assessment of Mus musculus gene function prediction using integrated genomic evidence. Genome Biol (Suppl 1) 9: S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H 2011. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat Methods 8: 785–786 [DOI] [PubMed] [Google Scholar]
- Ramani AK, Bunescu RC, Mooney RJ, Marcotte EM 2005. Consolidating the set of known human protein-protein interactions in preparation for large-scale mapping of the human interactome. Genome Biol 6: R40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich NC, Liu L 2006. Tracking STAT nuclear traffic. Nat Rev Immunol 6: 602–612 [DOI] [PubMed] [Google Scholar]
- Ross-Macdonald P, Coelho PS, Roemer T, Agarwal S, Kumar A, Jansen R, Cheung KH, Sheehan A, Symoniatis D, Umansky L, et al. 1999. Large-scale analysis of the yeast genome by transposon tagging and gene disruption. Nature 402: 413–418 [DOI] [PubMed] [Google Scholar]
- Rossel M, Pasini A, Chappuis S, Geneste O, Fournier L, Schuffenecker I, Takahashi M, van Grunsven LA, Urdiales JL, Rudkin BB, et al. 1997. Distinct biological properties of two RET isoforms activated by MEN 2A and MEN 2B mutations. Oncogene 14: 265–275 [DOI] [PubMed] [Google Scholar]
- Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, et al. 2005. Towards a proteome-scale map of the human protein-protein interaction network. Nature 437: 1173–1178 [DOI] [PubMed] [Google Scholar]
- Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, Eisenberg D. 2004. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res 32: D449–D451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott MS, Thomas DY, Hallett MT 2004. Predicting subcellular localization via protein motif co-occurrence. Genome Res 14: 1957–1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatkay H, Hoglund A, Brady S, Blum T, Donnes P, Kohlbacher O 2007. SherLoc: High-accuracy prediction of protein subcellular localization by integrating text and protein sequence data. Bioinformatics 23: 1410–1417 [DOI] [PubMed] [Google Scholar]
- Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S, et al. 2005. A human protein-protein interaction network: A resource for annotating the proteome. Cell 122: 957–968 [DOI] [PubMed] [Google Scholar]
- Sun Y, Liu X, Eaton EN, Lane WS, Lodish HF, Weinberg RA 1999. Interaction of the Ski oncoprotein with Smad3 regulates TGF-β signaling. Mol Cell 4: 499–509 [DOI] [PubMed] [Google Scholar]
- Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA 2006. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 441: 513–517 [DOI] [PubMed] [Google Scholar]
- Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, Zwahlen M, Kampf C, Wester K, Hober S, et al. 2010. Towards a knowledge-based Human Protein Atlas. Nat Biotechnol 28: 1248–1250 [DOI] [PubMed] [Google Scholar]
- Vascotto C, Bisetto E, Li M, Zeef LA, D'Ambrosio C, Domenis R, Comelli M, Delneri D, Scaloni A, Altieri F, et al. 2011. Knock-in reconstitution studies reveal an unexpected role of Cys-65 in regulating APE1/Ref-1 subcellular trafficking and function. Mol Biol Cell 22: 3887–3901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner W, Schekman R 2005. Protein translocation across biological membranes. Science 310: 1452–1456 [DOI] [PubMed] [Google Scholar]
- Wilson CA, Kreychman J, Gerstein M 2000. Assessing annotation transfer for genomics: Quantifying the relations between protein sequence, structure and function through traditional and probabilistic scores. J Mol Biol 297: 233–249 [DOI] [PubMed] [Google Scholar]
- Zhou B, Bae SK, Malone AC, Levinson BB, Kuo YM, Cilio MR, Bertini E, Hayflick SJ, Gitschier JM 2001. hGFRα-4: A new member of the GDNF receptor family and a candidate for NBIA. Pediatr Neurol 25: 156–161 [DOI] [PubMed] [Google Scholar]