

Abstract
Blood-sucking lice of humans have extensively fragmented mitochondrial (mt) genomes. Human head louse and body louse have their 37 mt genes on 20 minichromosomes. In human pubic louse, the 34 mt genes known are on 14 minichromosomes. To understand the process of mt genome fragmentation in the blood-sucking lice of mammals, we sequenced the mt genomes of the domestic pig louse, Haematopinus suis, and the wild pig louse, H. apri, which diverged from human lice approximately 65 Ma. The 37 mt genes of the pig lice are on nine circular minichromosomes; each minichromosome is 3–4 kb in size. The pig lice have four genes per minichromosome on average, in contrast to two genes per minichromosome in the human lice. One minichromosome of the pig lice has eight genes and is the most gene-rich minichromosome found in the sucking lice. Our results indicate substantial variation in the rate and extent of mt genome fragmentation among different lineages of the sucking lice.
Keywords: mitochondrial genome, genome fragmentation, minichromosome, sucking lice
Introduction
The mitochondrial (mt) genomes of bilateral animals are typically 15–20 kb in size, consisting of a single circular chromosome with 37 genes: 13 protein-coding genes for oxidative phosphorylation, two ribosomal RNA genes, and 22 transfer RNA genes (Wolstenholme 1992; Boore 1999; Lavrov 2007). Extensively fragmented mt genomes, however, have been found recently in three species of sucking lice that infest humans. The human body louse, Pediculus humanus, and the human head louse, P. capitis, have their 37 mt genes on 20 circular minichromosomes; each minichromosome is 3–4 kb in size and has 1–3 genes. In the human pubic louse, Pthirus pubis, the 34 mt genes identified are on 14 minichromosomes; each minichromosome is 1.8–2.7 kb in size and has 1–5 genes (Shao et al. 2009, 2012).
Blood-sucking lice (class Insecta: suborder Anoplura) originated approximately 77 Ma (Light et al. 2010). There are approximately 540 described species of blood-sucking lice in 15 families, all parasitizing eutherian mammals (Kim and Ludwig 1978; Kim 1988; Durden and Musser 1994b). The human body louse and the human head louse are in the family Pediculidae; the human pubic louse is in the family Pthiridae. These two families of sucking lice separated approximately 7 Ma; all the three species of human lice in these two families have extensively fragmented mt genomes and share a common pattern of one protein-coding gene or rRNA gene per minichromosome except for two protein-coding genes, atp6 and atp8, which are on the same minichromosome (Shao et al. 2012). To understand the process of mt genome fragmentation in the blood-sucking lice, we sequenced the mt genomes of the domestic pig louse, Haematopinus suis, and the wild pig louse, H. apri. These two lice are obligate ectoparasites of pigs and are closely related to each other (Stimie and Van De Merwe 1968; Kadulski 1974; Girisgin et al. 2009). Haematopinus suis is only found on domestic pigs, Sus scrofa domesticus, whereas H. apri is only found on wild pigs, Sus scrofa scrofa (Durden and Musser 1994a). The two species of pig lice are in the family Haematopinidae, which diverged from the lineage leading to the human lice approximately 65 Ma (Light et al. 2010). We compared the mt genomes of the pig lice with those of the human lice; we found that the mt genomes of the pig lice are much less fragmented. Our results indicate substantial variation in the rate and extent of mt genome fragmentation among different lineages of the blood-sucking lice.
Materials and Methods
Sample Collection, DNA Extraction, and mt Genome Amplification
The domestic pig lice, H. suis, were collected at Murdoch University Veterinary Farm, Perth, Australia (sample B2311). The wild pig lice, H. apri, were collected in Nishinomiya City, Hyogo Prefecture, Japan (sample B2418). Total DNA was extracted from individual louse specimens with DNeasy Tissue Kit (QIAGEN). A 657-bp fragment of mt cox1 gene and a 452-bp fragment of mt rrnS gene of the pig lice were amplified initially by polymerase chain reaction (PCR) with primer pairs mtd6-mtd11 and 12SA-12SB (supplementary table S1, Supplementary Material online). These primers target conserved sequence motifs in the mt genomes of the pig lice. The cox1 and rrnS fragments were sequenced directly with AB3730xl 96-capillary sequencers at the Australian Genome Research Facilities (AGRF). Two pairs of pig-lice-specific primers, PGC1F-PGC1R and PG12SF-PG12SR, were designed from cox1 and rrnS genes (supplementary table S1, Supplementary Material online). PCRs with pig-lice-specific primers amplified the entire nad2-trnI-cox1-trnL2 minichromosome (∼4.2 kb) and the entire rrnS-trnC minichromosome (∼3.2 kb), respectively (figs. 1 and 2C). The PCR amplicons from these two minichromosomes were sequenced with AB3730xl 96-capillary sequencers at the AGRF; a primer-walking strategy was used with the primers PGseqF1-3 and PGseqR1-2 (supplementary table S1, Supplementary Material online). Sequences from the noncoding regions (NCRs) of nad2-trnI-cox1-trnL2 minichromosome and rrnS-trnC minichromosome were aligned with ClustalX (Larkin et al. 2007); a forward primer PLF1 and a reverse primer PLR were designed from perfectly conserved noncoding sequences adjacent to the 5′-end and the 3′-end, respectively, of the coding regions (fig. 3). The PCRs with PLF1 and PLR produced a mixture of amplicons ranging from 0.9 to 2.7 kb (fig. 2D), expected from the coding regions of the entire set of mt minichromosomes of the pig lice. The PCR amplicons generated with PLF1 and PLR were sequenced with next-generation platforms (see later).
Fig. 1.—

Mitochondrial genomes of the domestic pig louse, Haematopinus suis, and the wild pig louse, H. apri. Genes are shown in blank arrows: cox1–3 for cytochrome c oxidase subunits 1–3; cob for cytochrome b; nad1–5 and nad4L for NADH dehydrogenase subunits 1–5 and 4L; rrnS and rrnL for small and large ribosome RNA subunits. tRNA genes are shown in triangles and are labeled with the single-letter abbreviations of their corresponding amino acids. The pseudo-trnV genes are 10 and 26 bp shorter than the full-length trnV genes. Sizes of genes of H. apri are in brackets if they are different from that of H. suis. The illustration of pig louse is courtesy of Hu Li and Wanzhi Cai.
Fig. 2.—
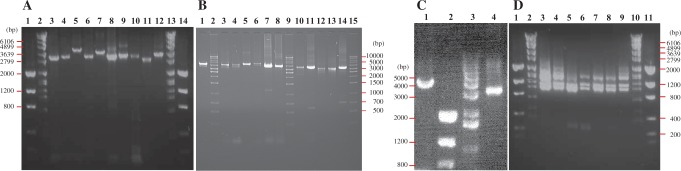
PCR verification of mt minichromosomes of Haematopinus suis and H. apri. (A) Lanes 1 and 14: low mass ladder (LML). Lanes 2 and 13: molecular weight marker VII (MWM). Lanes 3–10: amplicons from eight minichromosomes of H. suis (B2311), rrnS-trnC, trnL1-rrnL, nad2-trnI-cox1-trnL2, trnR-nad4L-nad6-trnM, trnK-nad4-atp8-atp6-trnN, trnE-cob-trnV, trnQ-nad1-trnT-trnG-nad3-trnW, and trnD-trnY-cox2-trnS1-trnS2-trnP-cox3-trnA. Lanes 11–12: PCR amplicons from two minichromosomes of H. apri (B2418), rrnS-trnC, and trnQ-nad1-trnT-trnG-nad3-trnW. Genes where PCR primers were designed are in bold. (B) Lane 1: amplicon from trnH-nad5-trnF minichromosome of H. suis (B2311). Lanes 2, 9, and 15: 1-kb ladder. Lanes 3–8 and 10–14: PCR amplicons from seven minichromosomes of H. apri (B2418), trnK-nad4-atp8-atp6-trnN, trnK-nad4-atp8-atp6-trnN, nad2-trnI-cox1-trnL2, nad2-trnI-cox1-trnL2, trnD-trnY-cox2-trnS1-trnS2-trnP-cox3-trnA, trnD-trnY-cox2-trnS1-trnS2-trnP-cox3-trnA, trnE-cob-trnV, trnH-nad5-trnF, trnR-nad4L-nad6-trnM, trnR-nad4L-nad6-trnM, and trnL1-rrnL. (C) Amplicons by pig-lice-specific primers, PGC1F-PGC1R and PG12SF-PG12SR, from the entire nad2-trnI-cox1-trnL2 minichromosome (4.5 kb, Lane 1) and the entire rrnS-trnC minichromosome (3.5 kb, Lane 4) of H. suis. Lane 2: LML. Lane 3: 1-kb Ladder. (D) Amplicons from the coding regions of mt minichromosomes of H. suis and H. apri. Lanes 1 and 11: LML. Lanes 2 and 10: MWM. Lanes 3–4: H. suis from Australia (B2311). Lanes 5–6: H. suis from Poland (B2419). Lanes 7–8: H. apri from Japan (B4218). Lane 9: H. suis from China (B2572).
Fig. 3.—

Alignment of the consensus sequences of the full-length NCRs of the mt minichromosomes of the pig lice, Haematopinus suis (B2311) and H. apri (B2418).
TaKaRa LA Taq kit was used in all PCR amplifications. Each PCR (25 µl) contained 0.25 µl of LA Taq, 2.5 µl of 10× Buffer, 2.5 µl of MgCl2 (25 mM), 4.0 µl of dNTP mixture (2.5 mM each), 1.0 µl of forward primer (10 µM), 1.0 µl of reverse primer (10 µM), 1.0 µl of DNA template, and 12.75 µl of Milli-Q water. PCR cycling conditions were 94 °C for 1 min, 30–40 cycles of 98 °C for 10 s, 45–65 °C (depending on primers) for 30 s, and 68 °C for 1–5 min (depending on target size, ∼1 min/kb), followed by 72 °C for 2–6 min. Negative controls were executed with each PCR experiment to detect DNA contamination and false-positive amplicons. PCR amplicons were checked by agarose-gel (1%) electrophoresis. The sizes of PCR amplicons were estimated by comparison with molecular markers. PCR amplicons used for sequencing were purified with Wizard SV Gel/PCR Clean-up System (Promega).
Next-Generation Sequencing of the Coding Regions of mt Minichromosomes
Purified PCR amplicons generated above with primers PLF1 and PLR from the coding regions of the mt minichromosomes of the domestic pig louse and the wild pig louse were sequenced initially with Roche GS FLX (454) platform at the AGRF and then with Illumina Hiseq 2000 platform at the Beijing Genomics Institute (BGI) for deeper coverages. Sequence reads were assembled de novo with Geneious 6.1.2 (Kearse et al. 2012). The assembly parameters were minimum overlap 100 bp and minimum match percentage 98% for Roche sequence reads; and minimum overlap 70 bp and minimum match percentage 99% for Illumina sequence reads. tRNA genes were identified with tRNA-Scan (Lowe and Eddy 1997) and ARWEN (Laslett and Canbäck 2008). Protein-coding genes and rRNA genes were identified with Basic Local Alignment Search Tool (Blast) searches of GenBank (Altschul et al. 1990; Cummings et al. 2002; McGinnis and Madden 2004). Blast searches did not identify nad6 gene of H. suis and H. apri; this gene was identified by 1) comparison of the hydrophilicity profile of its putative protein, NAD6, with those of Drosophila yakuba, Homo sapiens, Pth. pubis, and P. humanus (supplementary fig. S1, Supplementary Material online; Anderson et al. 1981; Clary and Wolstenholme 1985; Shao and Barker 2003; Shao et al. 2012) and 2) comparison of the conserved amino acid sequences of NAD6 of the pig lice with those of the human lice (supplementary fig. S2, Supplementary Material online). Identical sequences shared between mt genes were identified by Wordmatch (Rice et al. 2000).
Verification of mt Minichromosomes and Sequencing of the Full-Length NCRs of mt Minichromosomes
We verified by PCR the size and the circular organization of each mt minichromosome of the domestic pig louse and the wild pig louse obtained above by the assembly of the Roche and Illumina sequence reads. Outbound primers (forward and reverse; supplementary table S2, Supplementary Material online) were designed from the coding region of each minichromosome; the two primers in each pair were next to each other with a small gap or no gap in between. PCRs with these primers amplify each minichromosome in full or near-full length, that is, both coding region and NCR, if that minichromosome has a circular organization (figs. 1 and 2A–C). PCR set up, cycling conditions, agarose-gel electrophoresis, molecular size measurement, and amplicon purification were the same as described earlier.
To obtain full-length sequences of the NCRs of the mt minichromosomes, we sequenced individually the PCR amplicons from eight minichromosomes of the domestic pig louse (B2311) and two minichromosomes of the wild pig louse (B2418). Purified PCR amplicons were sequenced (pair end) with Illumina Hiseq 2000 platform at the BGI. Sequence reads were assembled de novo with Geneious (Kearse et al. 2012); the assembly parameters were minimum overlap 70 bp and minimum match percentage 99%. Alignment of NCR sequences was with ClustalX (Larkin et al. 2007).
Results
Mitochondrial Genomes of the Domestic Pig Louse, H. suis, and the Wild Pig Louse, H. apri
We obtained 1,386 and 775,493 sequence reads with Roche 454 and Illumina Hiseq platforms, respectively, from the mt genome of the domestic pig louse, H. suis. We also obtained 423 and 499,585 sequence reads with these two platforms, respectively, from the mt genome of the wild pig louse, H. apri (table 1). The sequence reads generated by Roche platform range from 100 to 500 bp in length, whereas those generated by Illumina platform are all 90-bp long. We assembled these sequence reads into contigs and identified all the 37 mt genes typical of bilateral animals and a pseudo-trnV gene in both H. suis and H. apri; these genes are on nine circular minichromosomes (figs. 1 and 2A and B; table 1). The distribution of these genes among the nine minichromosomes is identical between the domestic pig louse and the wild pig louse. Each minichromosome is 3–4 kb in size and consists of a coding region and an NCR. The coding region of each minichromosome is 0.9–2.7 kb in size and contains 2–8 genes. All the mt genes of both H. suis and H. apri have the same orientation of transcription relative to the NCR, except trnT, nad1, and trnQ, which have the opposite orientation of transcription relative to other genes (fig. 1). The nucleotide sequences of the mt minichromosomes of the pig lice have been deposited in GenBank under accession numbers KC814602–19.
Table 1.
mt Minichromosomes of the Domestic Pig Louse, Haematopinus suis, and of the Wild Pig Louse, H. apri, Identified by Roche 454 Sequencing and Illumina Hiseq Sequencing
| Minichromosome | Size of Coding Region (bp) | Number of Roche 454 Sequence Reads | Number of Illumina Hiseq Sequence Reads |
|---|---|---|---|
| rrnS-C | 786 (788) | 316 (139) | 102,663 (85,912) |
| L1-rrnL-pesudo V | 1,283 (1,265) | 177 (NA) | 110,577 (52,308) |
| E-cob-V | 1,219 (1,220) | 257 (NA) | 47,509 (49,971) |
| Q-nad1-T-G-nad3-W | 1,516 (1,516) | 228 (191) | 81,895 (46,841) |
| nad2-I-cox1-L2 | 2,669 (2,669) | 198 (NA) | 55,828 (20,396) |
| D-Y-cox2-S1-S2-P-cox3-A | 1,851 (1,852) | 68 (NA) | 96,713 (34,512) |
| K-nad4-atp8-atp6 | 2,304 (2,305) | NA (NA) | 91,137 (31,359) |
| H-nad5-F | 1,779 (1,781) | NA (NA) | 81,301 (35,681) |
| R-nad4L-nad6-M | 931 (930) | 141 (93) | 107,870 (142,605) |
| Total | 14,338 (14,326) | 1,386 (423) | 775,493 (499,585) |
Note.—NA, not available. Numbers outside brackets are for H. suis and those in brackets are for H. apri.
We obtained the full-length sequences of the NCRs of eight mt minichromosomes of the domestic pig louse (sample B2311) and two minichromosomes of the wild pig louse (sample B2418). We also obtained partial sequences of the NCRs upstream and downstream of the coding regions of other minichromosomes of these two lice. The assembled NCRs of the mt minichromosomes of the pig lice range from 1.5 to 2.4 kb in size: rrnS-C minichromosome (B2311) and R-nad4L-nad6-M minichromosome (B2311) have the longest NCRs (2,370 and 2,363 bp, respectively), whereas nad2-I-cox1-L2 minichromosome (B2311) has the shortest one (1,495 bp) (note: minichromosomes are named after the genes they contain hereafter). The NCRs contain tandem repetitive sequences that vary in length (12–75 bp) and copy number (2–5 repeats). The NCRs of H. suis have 56.4–96.6% pairwise identity to each other and the two NCRs of H. apri have 79.8% pairwise identity to each other. There are nucleotide polymorphisms in the NCR within each type of minichromosome; the sequences shown in figure 3 are the consensus sequences. On the other hand, there are sequence motifs in the NCRs that are highly conserved between H. suis and H. apri (fig. 3). As in the human lice (Shao et al. 2012), a GC-rich motif (67 bp, 62.7% C and G) is downstream the 3′-end of the coding region and an AT-rich motif (86 bp, 77.9% A and T) upstream the 5′-end of the coding region, indicating functional significance of these motifs in the mt genomes of sucking lice.
Gene Arrangement in the mt Genomes of the Pig Lice and Other Parasitic Lice
Parasitic lice have the most rearranged mt genomes observed in insects and other animals (Shao et al. 2001, 2012; Covacin et al. 2006; Shao and Barker 2007; Cameron et al. 2011). Compared with the six species of parasitic lice whose mt genomes were sequenced in previous studies, the pig lice are among the least rearranged; nine mt genes of the pig lice retained the ancestral arrangement inferred for insects: atp8-atp6, H-nad5-F, cox1-L2, and G-nad3 (fig. 4). Only the screamer louse is comparable to the pig lice and has the same number of genes that retained the ancestral arrangement of insects: atp8-atp6, nad4-H-nad5, V-rrnL, and G-nad3 (Cameron et al. 2007). In the human head louse and the human body louse, atp8-atp6 is the only ancestral mt gene arrangement of insects retained (Shao et al. 2009, 2012). In addition to atp8-atp6, the ancestral arrangement of insects, G-nad3, is retained in the human pubic louse (Shao et al. 2012) and the pigeon louse (Covacin et al. 2006), whereas the ancestral arrangement of insects, nad4L-nad4, is retained in the wallaby louse (Shao et al. 2001).
Fig. 4.—

Comparison of mt gene arrangement among parasitic lice, the booklouse, Liposcelis bostrychophila, and the hypothetical ancestor of insects. “+” indicates presence, whereas “−” indicates absence of a gene arrangement. The suborder-level phylogeny of the parasitic lice (Phthiraptera) is after Lyal (1985) and Barker et al. (2003). The grouping of the Pediculus species with Pth. pubis is after Barker et al. (2003). The grouping of Bothriometopus macrocnemis with sucking lice is after Wei et al. (2012). The grouping of L. bostrychophila with parasitic lice is after Lyal (1985), Wei et al. (2012), and Li et al. (2013).
Of the numerous derived mt gene arrangements observed in the pig lice and other parasitic lice, only Y-cox2 is present, and only present, in species from all the four suborders of the parasitic lice investigated, Anoplura, Ischnocera, Amblycera (fig. 4), and Rhynchophthirina (Shao R, Barker SC, Su Y, unpublished data). This derived gene arrangement supports the monophyly of the parasitic lice (order Phthiraptera) indicated by morphological and molecular studies (Lyal 1985; Li et al. 2013). A derived gene arrangement, nad1-Q, is present only in species from the suborders Anoplura and Ischnocera and supports, potentially, the closer relationship between these two suborders than either of them is to the suborder Amblycera (Yoshizawa and Johnson 2003; Murrell and Barker 2005; Wei et al. 2012; Li et al. 2013). Four derived gene arrangements, rrnS-C, L1-rrnL, nad2-I, and cox3-A, are present and only present in species of the suborder Anoplura (sucking lice) and provide strong support for the monophyly of this suborder (Lyal 1985; Barker et al. 2003; Murrell and Barker 2005; Light et al. 2010). The mt genome of the booklouse, Liposcelis bostrychophila, which is most closely related to parasitic lice, is also highly rearranged (Wei et al. 2012). The booklouse, however, does not share any derived mt gene arrangement with parasitic lice, indicating that mt gene rearrangement occurred independently in the lineage leading to the booklouse and the lineage leading to the parasitic lice.
Recombination of mt Genes in the Pig Lice
Fragmentation of mt genomes appears to facilitate recombination between mt genes (Shao et al. 2012). Evidence for recombination between mt genes has been reported in the human lice, P. humanus, P. capitis, and Pth. pubis (Shao et al. 2009, 2012; Shao and Barker 2011). Stretches of identical sequences (26–127-bp long) shared between genes on different minichromosomes provided unequivocal evidence for interminichromosome recombination in the human lice (Shao et al. 2009, 2012; Shao and Barker 2011). We found three pairs of mt genes in the pig lice, H. suis and H. apri, also share stretches of identical sequences much longer than expected by chance (table 2). trnL1 and trnL2 genes of the pig lice share three stretches of identical sequences, 9, 10, and 16-bp long, in the D-arm and AC-arm of their putative tRNAs (table 2 and fig. 5). Another pair of tRNA genes, trnT and trnP, share 26-bp identical sequence in the D-arm and AC-arm of their putative tRNAs, which is three times longer than that in other animal species. A pair of protein-coding genes, atp8 and nad2, share 25-bp identical sequence, which is 2–3 times longer than that in other animal species. With the exception of trnL1 and trnL2, the identities of the genes that share longer-than-expected identical sequences in the pig lice differ from those in the human lice (Shao et al. 2012). Together, there is evidence for recombination between 10 pairs of mt genes in the pig lice and the human lice (table 2). Recombination between mt genes in the sucking lice appears to occur regardless of gene identities, for example, it occurs between two protein-coding genes, between two tRNA genes, between a rRNA gene and a protein-coding gene, between a rRNA gene, and a tRNA gene, and between a protein-coding gene and a tRNA gene (table 2). Intriguingly, in all the 10 pairs of genes that share stretches of identical sequences longer than expected by chance, the two genes in the same pair are always on two different minichromosomes, indicating that recombination occurs only between genes on different types of minichromosomes.
Table 2.
The Longest Stretches of Identical Sequence Shared by mt Genes in the Pig Lice and the Human Lice, Which Have Fragmented mt Genomes, and Four Other Species of Bilateral Animals That Have the Typical Unfragmented mt Genomes
| Pairs of Genes | The Longest Stretches of Identical Sequence Shared (bp) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Hs | Ha | Pc | Pp | Ph | Bm | Cb | Hm | Dy | ||
| trnL1 | trnL2 | 16, 10, 9 | 16, 10, 9 | 33, 32 | 35, 32 | 33, 32 | 7 | 6 | 7 | 10 |
| trnT | trnP | 26 | 26 | 7 | NA | 7 | 6 | 8 | 8 | 9 |
| atp8 | nad2 | 25 | 25 | 10 | 8 | 10 | 10 | 14 | 12 | 14 |
| nad4 | nad5 | 12 | 12 | 127, 30 | NA | 127, 30 | 13 | 15 | 15 | 16 |
| rrnL | nad5 | 11 | 10 | 99 | 10 | 99 | 12 | 14 | 13 | 15 |
| cox1 | nad4L | 11 | 11 | 10 | 29 | 10 | 13 | 11 | 14 | 13 |
| rrnL | nad2 | 10 | 11 | 26 | 10 | 26 | 13 | 11 | 14 | 13 |
| atp8 | trnG | 6 | 6 | 26 | 9 | 26 | 10 | 11 | 11 | 12 |
| trnR | trnG | 5 | 5 | 28, 14 | 32, 26 | 28, 14 | 5 | 6 | 7 | 6 |
| trnI | trnT | 6 | 6 | 6 | 16 | 6 | 6 | 5 | 7 | 7 |
Note.—Hs, Haematopinus suis (domestic pig louse); Ha, H. apri (wild pig louse); Pc, Pediculus capitis (human head louse); Pp, Pthirus pubis (human pubic louse); Ph, P. humanus (human body louse); Bm, Bothriometopus macrocnemis (screamer louse); Cb, Campanulotes bidentatus (pigeon louse); Hm, Heterodoxus macropus (wallaby louse); Dy, Drosophila yakuba (fruit fly). Stretches of shared identical sequences much longer than expected are in bold.
Fig. 5.—


The putative secondary structures of mt tRNAs of Haematopinus suis (Hs) and H. apri (Ha). Identical or near-identical sequences shared between trnL1 and trnL2 genes, and between trnT and trnP genes, are indicated in red boxes.
Secondary Structures of the mt tRNAs of the Pig Lice
All the putative tRNAs of the pig lice, H. suis and H. apri, have the cloverleaf-shaped secondary structure except trnS1(tct), which lacks the D-arm (fig. 5), as in other parasitic lice (Shao et al. 2012) and bilateral animals (Sprinzl et al. 1998; Ohtsuki et al. 2002). trnN of the pig lice, however, has an unusual large D-arm loop with 15 nt in H. suis and 16 nt in H. apri (fig. 5), rendering this tRNA gene undetectable by tRNA-search programs (see details in Materials and Methods). In the three human lice, trnQ has an atypical structure and lacks the D-arm (Shao et al. 2012). In the pig lice, however, trnQ has the conventional cloverleaf-shaped structure. Apparently, the loss of the D-arm occurred in the lineage leading to the human lice but not in the lineage leading to the pig lice. In the human lice, trnL1 and trnL2 have identical secondary structures except for the nucleotides at the third anti-codon positions (Shao et al. 2012). In the pig lice, trnL1 and trnL2 have near-identical sequences (and secondary structures) at the D-arm and the AC-arm but differ at the T-arm and the AA-arm (fig. 5). trnR and trnG also have highly similar sequences in the human lice but differ substantially in the pig lice, as in other parasitic lice and animals (Shao et al. 2001; Covacin et al. 2006; Cameron et al. 2007). trnT and trnP, however, went the opposite way: They differ substantially in the human lice but have near-identical sequences at the D-arm and the AC-arm in the pig lice (fig. 5).
Discussion
There are approximately 540 described species of blood-sucking lice in 15 families (suborder Anoplura) that parasitize eutherian mammals (Kim and Ludwig 1978; Kim 1988; Durden and Musser 1994b). Before this study, complete or near-complete mt genomes have been sequenced for three species of sucking lice from two families: 1) the human body louse, P. humanus, and the human head louse, P. capitis, from the family Pediculidae; and 2) the human pubic louse, Pth. pubis, from the family Pthiridae (Shao et al. 2009, 2012). Parts of mt genomes have been sequenced for the chimpanzee louse, P. schaeffi, from the family Pediculidae, and a monkey louse, Pedicinus ancoratus, from the family Pedicinidae (Shao et al. 2009). The fact that all these sucking lice and the pig lice have mt genes on minichromosomes indicates that fragmentation of mt genomes already started in the most recent common ancestor of blood-sucking lice, which lived approximately 77 Ma (Light et al. 2010).
The human body louse, P. humanus, has an extensively fragmented mt genome with the 37 mt genes on 20 types of minichromosomes, so did the human head louse, P. capitis (Shao et al. 2009, 2012). The human pubic louse also has an extensively fragmented mt genome and shares the same pattern of one protein-coding or rRNA gene per minichromosome (except one minichromosome with two protein-coding genes, atp6 and atp8) with the human body louse and the head louse (Shao et al. 2012). Not all sucking lice, however, have extensively fragmented mt genomes. The mt genomes of the pig lice, H. suis and H. apri, are much less fragmented than those of the human lice. In the human lice, each minichromosome has a maximum of five genes with no more than one protein-coding or rRNA gene except one minichromosome with two protein-coding genes, atp8-atp6. In the pig lice, however, four of the nine minichromosomes have two protein-coding genes and one minichromosome has three protein-coding genes, atp8-atp6-nad4. Further, one of the minichromosomes of the pig lice has eight genes, D-Y-cox2-S1-S2-P-cox3-A, and is the most gene-rich minichromosome found in sucking lice to date. The 37 mt genes we identified in the pig lice are on nine minichromosomes, that is, 4.0 genes per minichromosome on average, in comparison to 2.1 genes per mt minichromosome in the human body louse and the head louse, and 2.4 genes per minichromosome in the human pubic louse (Shao et al. 2012). Clearly, fragmentation of mt genome occurred at a much slower pace in the lineage leading to the pig lice than in the lineage leading to the human lice. We conclude that the rate of mt genome fragmentation varies substantially among different lineages of sucking lice. It remains to be determined in future studies the extent of mt genome fragmentation in other lineages of blood-sucking lice and the biological factors that may affect the rate of genome fragmentation.
Supplementary Material
Supplementary tables S1 and S2 and figures S1 and S2 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank Qianqian Yang, Cath Covacin, Lisa Le Strange, and Matsuo Kobayashi for assistance. They thank the anonymous reviewers for comments that greatly improved this manuscript. This work was supported by the Australian Research Council (DP0662755 to R.S. and DP120100240 to R.S. and S.C.B). R.S. also acknowledges the funding support from the Australian Government for an Australia-China Science & Research Fund Group Mission visit to China (ACSRF00980).
Literature Cited
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Anderson S, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- Barker SC, Whiting M, Johnson KP, Murrell A. Phylogeny of the lice (Insecta, Phthiraptera) inferred from small subunit rRNA. Zool Scr. 2003;32:407–414. [Google Scholar]
- Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron S, Johnson K, Whiting M. The mitochondrial genome of the screamer louse Bothriometopus (Phthiraptera: Ischnocera): effects of extensive gene rearrangements on the evolution of the genome. J Mol Evol. 2007;65:589–604. doi: 10.1007/s00239-007-9042-8. [DOI] [PubMed] [Google Scholar]
- Cameron SL, Yoshizawa K, Mizukoshi A, Whiting MF, Johnson KP. Mitochondrial genome deletions and minicircles are common in lice (Insecta: Phthiraptera) BMC Genomics. 2011;12:394. doi: 10.1186/1471-2164-12-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clary DO, Wolstenholme DR. The mitochondrial DNA molecular of Drosophila yakuba: nucleotide sequence, gene organization, and genetic code. J Mol Evol. 1985;22:252–271. doi: 10.1007/BF02099755. [DOI] [PubMed] [Google Scholar]
- Covacin C, Shao R, Cameron S, Barker SC. Extraordinary number of gene rearrangements in the mitochondrial genomes of lice (Phthiraptera: Insecta) Insect Mol Biol. 2006;15:63–68. doi: 10.1111/j.1365-2583.2005.00608.x. [DOI] [PubMed] [Google Scholar]
- Cummings L, et al. Genomic BLAST: custom-defined virtual databases for complete and unfinished genomes. FEMS Microb Lett. 2002;216:133–138. doi: 10.1111/j.1574-6968.2002.tb11426.x. [DOI] [PubMed] [Google Scholar]
- Durden LA, Musser GG. The mammalian hosts of the sucking lice (Anoplura) of the world: a host-parasite list. Bull Soc Vector Ecol. 1994a;19:130–168. [Google Scholar]
- Durden LA, Musser GG. The sucking lice (Insecta, Anoplura) of the world: a taxonomic checklist with records of mammalian hosts and geographical distributions. Bull Amer Mus Nat Hist. 1994b:1–90. [Google Scholar]
- Girisgin O, Girisgin AO, Sonmez F, Akyol CV. Occurrence of Haematopinus suis Linnaeus, 1758 (Insecta, Anopluridae) on a wild boar (Sus scrofa) Turk J Vet Anim Sci. 2009;33:529–530. [Google Scholar]
- Johnson KP, Whiting MF. Multiple genes and the monophyly of Ischnocera (Insecta: Phthiraptera) Mol Phylo Evol. 2002;22:101–110. doi: 10.1006/mpev.2001.1028. [DOI] [PubMed] [Google Scholar]
- Kadulski S. Occurrence of Haematopinus apri Gour. (Anoplura) on wild boar Sus scrofa L. in Poland. Acta Parasitol Pol. 1974;22:219–228. [Google Scholar]
- Kearse M, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KC. Evolutionary parallelism in Anoplura and eutherian mammals. In: Service MW, editor. Biosystematics of haematophagous insects. Oxford: Oxford University Press; 1988. pp. 91–114. [Google Scholar]
- Kim KC, Ludwig HW. The family classification of the Anoplura. Syst Entomol. 1978;3:249–284. [Google Scholar]
- Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Laslett D, Canbäck B. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics. 2008;24:172–175. doi: 10.1093/bioinformatics/btm573. [DOI] [PubMed] [Google Scholar]
- Lavrov DV. Key transitions in animal evolution: a mitochondrial DNA perspective. Integr Comp Biol. 2007;47:734–743. doi: 10.1093/icb/icm045. [DOI] [PubMed] [Google Scholar]
- Li HL, et al. Mitochondrial genomes of two barklice, Psococerastis albimaculata and Longivalvus hyalospilus (Psocoptera: Psocomorpha): contrasting rates in mitochondrial gene rearrangement between major lineages of Psocodea. PLoS One. 2013;8:e61685. doi: 10.1371/journal.pone.0061685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light JE, Smith VS, Allen JM, Durden LA, Reed DL. Evolutionary history of mammalian sucking lice (Phthiraptera: Anoplura) BMC Evol Biol. 2010;10:292. doi: 10.1186/1471-2148-10-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe T, Eddy S. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyal CHC. Phylogeny and classification of the Psocodea, with particular reference to the lice (Psocodea: Phthiraptera) Syst Entomol. 1985;10:145–165. [Google Scholar]
- McGinnis S, Madden TL. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004;32:W20–W25. doi: 10.1093/nar/gkh435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell A, Barker SC. Multiple origins of parasitism in lice: phylogenetic analysis of SSU rDNA indicates that the Phthiraptera and Psocoptera are not monophyletic. Parasitol Res. 2005;97:274–280. doi: 10.1007/s00436-005-1413-8. [DOI] [PubMed] [Google Scholar]
- Ohtsuki T, Kawai G, Watanabe K. The minimal tRNA: unique structure of Ascaris suum mitochondrial tRNASerUCU having a short T arm and lacking the entire D arm. FEBS Lett. 2002;514:37–43. doi: 10.1016/s0014-5793(02)02328-1. [DOI] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Shao R, Barker SC. The highly rearranged mitochondrial genome of the plague thrips, Thrips imaginis (Insecta: Thysanoptera): convergence of two novel gene boundaries and an extraordinary arrangement of rRNA genes. Mol Biol Evol. 2003;20:362–370. doi: 10.1093/molbev/msg045. [DOI] [PubMed] [Google Scholar]
- Shao R, Barker SC. Mitochondrial genomes of parasitic arthropods: implications for studies of population genetics and evolution. Parasitology. 2007;134:153–167. doi: 10.1017/S0031182006001429. [DOI] [PubMed] [Google Scholar]
- Shao R, Barker SC. Chimeric mitochondrial minichromosomes of the human body louse, Pediculus humanus: evidence for homologous and non-homologous recombination. Gene. 2011;473:36–43. doi: 10.1016/j.gene.2010.11.002. [DOI] [PubMed] [Google Scholar]
- Shao R, Campbell NJH, Barker SC. Numerous gene rearrangements in the mitochondrial genome of the wallaby louse, Heterodoxus macropus (Phthiraptera) Mol Biol Evol. 2001;18:858–865. doi: 10.1093/oxfordjournals.molbev.a003867. [DOI] [PubMed] [Google Scholar]
- Shao R, Kirkness EF, Barker SC. The single mitochondrial chromosome typical of animals has evolved into 18 minichromosomes in the human body louse, Pediculus humanus. Genome Res. 2009;19:904–912. doi: 10.1101/gr.083188.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao R, Zhu X-Q, Barker SC, Herd K. Evolution of extensively fragmented mitochondrial genomes in the lice of humans. Genome Biol Evol. 2012;4:1088–1101. doi: 10.1093/gbe/evs088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprinzl M, Horn C, Brown M, Ioudovitch A, Steinberg S. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 1998;26:148–153. doi: 10.1093/nar/26.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stimie M, Van De Merwe S. A revision of the genus Haematopinus Leach (Phthiraptera: Anoplura) Zool Anz. 1968;180:182–220. [Google Scholar]
- Wei DD, Shao R, Yuan ML, Dou W, Barker SC, Wang JJ. The multipartite mitochondrial genome of Liposcelis bostrychophila: insights into the evolution of mitochondrial genomes in bilateral animals. PLoS One. 2012;7:e33973. doi: 10.1371/journal.pone.0033973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolstenholme DR. Genetic novelties in mitochondrial genomes of multicellular animals. Curr Opin Genet Dev. 1992;2:918–925. doi: 10.1016/s0959-437x(05)80116-9. [DOI] [PubMed] [Google Scholar]
- Yoshizawa K, Johnson KP. Phylogenetic position of Phthiraptera (Insecta: Paraneoptera) and elevated rate of evolution in mitochondrial 12S and 16S rDNA. Mol Phylogenet Evol. 2003;29:102–114. doi: 10.1016/s1055-7903(03)00073-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.