

Abstract
Clinically available red blood cells (RBCs) for transfusions are at high demand, but in vitro generation of RBCs from hematopoietic stem cells requires significant quantities of growth factors. Here, we describe the production of four human growth factors: erythropoietin (EPO), stem cell factor (SCF), interleukin 3 (IL-3), and insulin-like growth factor-1 (IGF-1), either as non-fused proteins or as fusions with a carrier molecule (lichenase), in plants, using a Tobacco mosaic virus vector-based transient expression system. All growth factors were purified and their identity was confirmed by western blotting and peptide mapping. The potency of these plant-produced cytokines was assessed using TF1 cell (responsive to EPO, IL-3 and SCF) or MCF-7 cell (responsive to IGF-1) proliferation assays. The biological activity estimated here for the cytokines produced in plants was slightly lower or within the range cited in commercial sources and published literature. By comparing EC50 values of plant-produced cytokines with standards, we have demonstrated that all four plant-produced growth factors stimulated the expansion of umbilical cord blood-derived CD34+ cells and their differentiation toward erythropoietic precursors with the same potency as commercially available growth factors. To the best of our knowledge, this is the first report on the generation of all key bioactive cytokines required for the erythroid development in a cost-effective manner using a plant-based expression system.
Introduction
The timely supply of blood, the only current source of red blood cells (RBCs, or erythrocytes), is critical in both military and civilian healthcare systems. RBC transfusion is a common intervention that is used for treating acute blood loss of more than 30% of blood volume as well as anemia caused by surgery, major burns, or cancer [1–5]. Transfusion of whole blood entirely depends on the availability of human donors with matching blood types. While the demand for whole blood is increasing, the supply continues to decline, worsening the shortage of blood available for transfusions. At present, there is no alternative source of RBCs to relieve this shortage. In vitro generation of clinically available RBCs from hematopoietic stem cells (HSCs) could be a promising approach for addressing this growing concern and for overcoming the limitations associated with the use of whole donor's blood. However, generation of RBCs from HSCs in culture requires significant quantities of growth and differentiation factors such as erythropoietin (EPO), stem cell factor (SCF), interleukin 3 (IL-3), and insulin-like growth factor-1 (IGF-1), making manufacturing at large scale cost prohibitive.
EPO, a hormone produced in kidneys, controls a steady-state erythropoiesis by stimulating differentiation and maturation of erythrocytes [6]. Mature human EPO consists of 166 amino acids (aa) and has a molecular weight (Mw) of 34 kDa, of which 40% is comprised by carbohydrates [7]. EPO contains three N-linked oligosaccharides at asparagine positions 24, 38, and 83 and one O-linked oligosaccharide at a serine position 126 [8–10]. N-linked oligosaccharides, especially those containing terminal sialic acids, are believed to play a role in secretion, folding, solubility, biological activity, and half life of recombinant EPO, but do not affect the interaction of EPO with its receptor in vitro [11–15].
SCF, a cytokine that binds to the c-Kit receptor (CD117) [16], plays an important role in hematopoiesis [17,18], spermatogenesis [19], and melanogenesis [20]. It is produced by stromal cells in fetal liver and adult bone marrow as well as by other cell types and organs [21]. SCF exists in soluble and transmembrane-anchored forms that are generated through an alternative splicing of RNA, leading to inclusion or exclusion of a proteolytic cleavage site [22]. Both soluble and transmembrane-anchored forms of SCF are biologically active [23]. Soluble human SCF is a 165-aa protein with a predicted Mw of 18.5 kDa [24,25]. Soluble SCF has an extensive secondary structure and circulates in the form of a non-covalently bound dimer [25,26]. In in vitro models, SCF directly accelerated the entry of CD34+ HSCs into the cell cycle and in combination with other cytokines, promoted HSC survival (but not self-renewal) [27,28] and colony formation [29]. Expression in mammalian cells results in both N- and O-linked glycosylation of SCF, which increases its Mw by about 30% [25,30].
IL-3 is a pleiotropic cytokine secreted by activated T cells, keratinocytes, natural killer cells, mast cells, endothelial cells, and monocytes, which stimulates the proliferation and differentiation of pluripotent HSCs and various lineage-committed progenitor cells [31,32]. Native human IL-3 represents an N-glycosylated protein of 15–17 kDa (133 aa) containing two putative sites at positions 15 and 70 for N-linked glycosylation and a single disulfide bond (Cys 16/84) [33,34]. While IL-3 is well known for inducing proliferation and differentiation and maintaining survival of myeloid cell lineages [31], it has also been shown to stimulate an early development of stem cells toward the erythropoietic maturation [35] as well as to augment the effect of EPO on late erythroid progenitors [36].
Human IGF-1, a polypeptide with a molecular mass of 7.6 kDa (70 aa) and a 50% structural homology with insulin [37], is secreted by the liver cells and is shown to be involved in the regulation of proliferation and differentiation of various cell types [38]. In a study in vivo, IGF-1 has been shown to stimulate erythropoiesis in hypophysectomized rats [39], and in a serum-free culture model, the cytokine was found to serve as a survival factor for human CD34+ HSCs during early erythropoiesis and to be involved in erythroid maturation at the final stages [40].
Up to date, the recombinant human hematopoietic growth factors have been produced in several heterologous expression systems, including bacteria, yeast, and insect and mammalian cell cultures. For example, EPO has been produced using engineered Escherichia coli [41], Pichia pastoris [42], Drosophila S2 cells [43], and several mammalian cell lines [7,44–46]. The soluble form of SCF has been expressed in E. coli as well as in Chinese hamster ovary (CHO) cells [25]. A dual form of SCF containing a full-length SCF polypeptide plus a truncated SCF polypeptide, linked by a 12-aa peptide, has been produced at large scale in the baculovirus-infected insect cell culture and demonstrated a significantly increased specific activity compared with SCF monomer expressed in E. coli [47]. IL-3 has been cloned and expressed in E. coli [48] and CHO cells [33]. Large-scale manufacturing utilizing the insect cell expression system demonstrated that full-length human IL-3 was produced and purified as a highly stable and soluble protein, with a specific biological activity several times higher than that of the commercially available IL-3 derived from E. coli [49]. IGF-1 has been manufactured at large scale using both E. coli [50] and Saccharomyces cerevisiae [51].
Although bacteria, yeast, and insect and mammalian cell cultures have been successfully utilized for the production of the hematopoietic growth and differentiation factors, each of these systems has limitations that are associated with target product safety, cost, scalability, and suitability for mammalian-type post-translational modifications of the target protein, thus indicating the need for alternative expression systems. The development of a robust, scalable, and cost-effective expression system is critical for the production of the quantities of human hematopoietic growth factors that are required for manufacturing RBCs to meet the healthcare demand. Recently, plants have emerged as a platform for the production of therapeutic proteins, vaccine antigens, and monoclonal antibodies. Advantages of plants as production systems include freedom from contamination with animal pathogens, high production capacity, and relatively low capital investment [52]. Plants also share many post-translational protein modifications with mammals, although there are some differences between these systems in the glycosylation patterns happening in the late Golgi apparatus [53]. The latter dictates the need to develop strategies for humanization of N-linked glycosylation pathway in plants [54].
Biological activity of plant-produced recombinant proteins has been demonstrated both in vitro and in vivo. Furthermore, several therapeutic products and vaccine candidates have successfully completed Phase 1, and some have progressed into a more advanced stage of clinical development [55]. To date, recombinant proteins in plants have been produced using stable nuclear and chloroplast transformation and transient expression, most commonly via infection with plant viral vectors [52]. Transient expression of target proteins in plants can be achieved by using a ‘launch’ vector, a hybrid vector combining elements of a Tobacco mosaic virus-based expression vector carrying target sequences and a binary plasmid of Agrobacterium tumefaciens [56]. Introduction of Agrobacteria harboring the launch vector into an entire plant by vacuum infiltration and amplification of the target gene sequence through the use of the viral replication machinery result in high levels of target protein accumulation in a short period of time [57]. The targets produced in plants using this transient expression system include subunit vaccine candidates against seasonal and pandemic influenza [58–62], plague [63,64], anthrax [65], and human papilloma virus-associated cancer [66,67]. These vaccine candidates have been shown to induce cellular and humoral immune responses conferring protection against pathogen challenge when tested in animal models [58,61,63,64,67].
Recombinant proteins produced in heterologous systems are often expressed at low levels, form insoluble aggregates, are unstable, and undergo rapid degradation [68–70]. Fusion or conjugation with a carrier protein may enhance stability and solubility; facilitate accumulation of a soluble target; and also aid in target protein purification. For example, a thermostable enzyme lichenase (β-1,3–1,4-glucanase) from Clostridium thermocellum can be tracked during purification by monitoring lichenase activity, and engineered versions of lichenase have been frequently used as carrier molecules to assist in the expression of target genes in prokaryotic and eukaryotic systems [71]. Musiychuk et al. [56] have engineered a modified version of lichenase, LicKM, and demonstrated accumulation of approximately 0.5 g of LicKM per kg of fresh leaf tissue within 6 days post infiltration. LicKM has been successfully fused to more than 30 target proteins from different pathogens, including antigens from respiratory syncytial virus, human immunodeficiency virus (HIV), plague, anthrax, influenza, and Trypanosoma brucei, ranging in size from small peptides to full-length proteins of approximately 83 kDa [56].
In this study, we have engineered, expressed in the plant system, purified, and characterized four human recombinant erythropoietic growth and differentiation factors. Targets were engineered either as individual proteins (EPO, SCF, IL-3, and IGF-1) or as fusions to LicKM (EPO, IL-3 and IGF-1), and expressed in Nicotiana benthamiana. We have demonstrated that all four of these plant-produced factors—LicKM-fused EPO, IL-3, and IGF-1 and non-fused SCF—are biologically active in vitro, stimulating proliferation of relevant cell lines. Furthermore, our plant-derived recombinant factors stimulated expansion of human umbilical cord-derived CD34+ HSCs and their differentiation toward erythropoietic precursors with the same potency as commercially available growth factors. Our results also support the feasibility of the launch vector-based expression system for large-scale production of various therapeutically significant recombinant proteins.
Materials and Methods
Cloning and expression in plants
The nucleotide sequences encoding EPO (GenBank accession number CAA26094), SCF (P21583.1), IL-3 (NM_000588), and IGF-1 (Q9NP10) were optimized for expression in N. benthamiana by GeneArt, Inc. The nucleotide sequence of LicKM (DQ776900) was optimized for expression in tobacco plants and synthesized by Entelechon GmbH. EPO, SCF, IL-3, and IGF-1 are naturally secreted proteins; thus, to facilitate the entry of targets into the plant secretory pathway, their native signal peptides were substituted by the signal peptide of the plant pathogenesis-related protein 1a (PR-1a) of Nicotiana tabacum [72]. To facilitate protein purification, all targets were engineered to carry a poly-histidine (6xHis) tag at the C-terminus (H variants). To test whether the inclusion of the 6xHis tag has a negative impact on the biological activity of the growth factors, variants without the 6xHis tag were also produced. In addition, variants with the endoplasmic reticulum (ER) retention sequence, KDEL, at the C-terminus (K variants), and without KDEL were designed to evaluate the potential influence of plant glycosylation on protein activity. Combined variants carrying both a 6xHis tag and KDEL (HK variants) were also produced for all targets.
To enhance protein expression, solubility, and stability, some targets were fused to LicKM. LicKM was engineered to contain PR-1a at the N-terminus and three potential fusion sites: N-terminal, C-terminal, and an internal fusion site in a surface loop [56]. In addition, LicKM contains a 6xHis tag and the KDEL sequence to facilitate target purification and retention in the ER, respectively [56]. EPO and IGF were engineered as fusions either into the surface loop (EPO-2E and IGF-1-2E variants) or to the C-terminus (EPO-3E and IGF-1-3E variants) of LicKM. IL-3 was engineered as an in-frame fusion into the surface loop of LicKM (IL-3-2E variant), while SCF fusions to LicKM were not generated. Each resulting target sequence was inserted into the launch vector pGR-D4 [61]. The engineered variants and schematic diagrams of the constructs are shown in Fig. 1A, B.
FIG. 1.

List of engineered target variants (A) and schematic diagrams of launch vector-based target expression constructs (B).
Each construct was introduced into A. tumefaciens strain GV3101 by electroporation, and the transformed bacteria were cultured in minimal medium for 24–48 h and used to infiltrate leaves of 4–6-week-old N. benthamiana as previously described [57].
Target protein expression was assessed by western blotting using an anti-tetra-His mouse monoclonal antibody (mAb; Qiagen) or target-specific mouse mAb as a primary antibody and a horseradish peroxidase (HRP)-labeled anti-mouse secondary antibody (Jackson ImmunoResearch). His-tagged influenza hemagglutinin (HAI) from A/Indonesia/05/05 (H5N1) strain of influenza virus purified in house was used as a standard for western blots developed with an anti-tetra-His mAb [62]. Human recombinant IL-3, IGF-1, SCF, and EPO proteins were purchased from R&D Systems to be used as standards on western blots developed with target-specific antibodies. Results were analyzed using the GeneGnome System (Syngene). The optimal accumulation for each target was analyzed to determine the day of harvest after agroinfiltration.
Protein purification
Infiltrated leaf tissues were harvested, homogenized (fresh or frozen), and extracted in a phosphate-based buffer. All constructs except EPO-2E contained 0.5% Triton X-100 in the extraction buffer. The extracts were clarified by centrifugation (15,900g for 15 min) and filtration through a polypropylene depth filter (3M-Cuno, PolyPro XL Capsule) followed by a 0.45/0.2 μm dead-end filtration (Sartorius Stedim Biotech) for EPO-2E and SCF-HK or just the 0.45/0.2 μm dead-end filtration for IGF-1-2E and IL-3-2E. Capture with immobilized metal affinity chromatography (IMAC) was similar for all targets and was followed by elution of the targets with 300 mM imidazole, except for EPO-2E, which was eluted with 250 mM imidazole. After IMAC capture, the polishing chromatography diverged between the targets. EPO-2E IMAC eluate was buffer exchanged into a 20 mM Bis-Tris, 75 mM NaCl, pH 7.0 buffer in preparation for passage over a HiTrap Capto Q column. At 75 mM NaCl, the target was in the flow-through fraction, which was concentrated and buffer exchanged into phosphate-buffered saline (PBS), pH 7.5. For SCF, the IMAC elution fraction was directly buffer exchanged into 50 mM Tris, pH 9.0 buffer, loaded onto a Hi Trap Capto diethylaminoethyl cellulose (DEAE) column, washed with the loading buffer, and eluted with 50 mM Tris, 150 mM NaCl, pH 9.0. The eluent was concentrated and buffer exchanged into PBS, pH 7.5.
IGF-1-2E and IL-3-2E were captured by IMAC, eluted with 300 mM imidazole-containing buffer, concentrated, and frozen for further processing over size exclusion chromatography (SEC). The frozen aliquots were thawed, centrifuged, and filtered prior to fractionation on a Superdex 75 SEC column equilibrated with PBS, pH 7.5. The process of fractionation on the SEC column was used to exchange buffer as well as to increase purity. Target-containing fractions from repeated injections were pooled and concentrated to yield the final samples. All samples were filtered through a 0.22 μm syringe filter (Nalgene) before aliquoting and storage at−70°C.
Target yields were determined by a densitometry analysis using a reducing 10% or 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) of targets versus bovine serum albumin (BSA) standards followed by Coomassie staining (Gel Code Blue; Thermo Fisher).
Protein characterization
Identity and solubility of target proteins were determined by the western blot analysis of total and soluble protein preparations, using an anti-tetra-His mouse mAb (Qiagen) or a target-specific mouse mAb as a primary antibody and an HRP-labeled anti-mouse secondary antibody. The results of these analyses were used to select the target variant with maximum expression and solubility levels.
Isolated proteins were analyzed for final concentration and yields by in-gel densitometry comparison of Coomassie-stained target bands with BSA standards. The identity of purified targets was determined by confirming the expected peptide sequence using tri-enzyme digest peptide mapping (MSBioworks). The solution state of the purified targets was evaluated using SEC.
Assessment of biological activity of growth factors in vitro
Specific biological activities of the plant-produced growth factors were investigated in proliferation assays using specific cell lines that are responsive to the individual cytokines. Commercially available recombinant EPO, SCF, IL-3, and IGF-1 (287-C, 255-SC, 203-IL, 291-G1; R&D Systems) equalized by protein content with our plant-produced growth factors served as standards.
TF1 cell proliferation assay
TF1, a human bone marrow-derived erythroleukemic cell line, was used to assess the activity of EPO, SCF, and IL-3. TF1 cell line was obtained from the American Type Culture Collection (ATCC) (CRL-2003, lot# 58266729) and maintained in RPMI 1640 culture media containing 10% fetal bovine serum (FBS) and 2 ng/mL granulocyte-macrophage colony stimulating factor (GM-CSF) according to ATCC instructions. Potency of the plant-produced growth factors (EPO, SCF, and IL-3) was determined by measuring their dose-dependent effects on TF1 cell proliferation. In 96-well plates, 4×103 TF1 cells per well in triplicate were cultured with increasing concentrations of EPO (0.04 to 400 ng/mL), SCF (0.2 to 200 ng/mL), or IL-3 (0.005 to 50 ng/mL) or similar concentrations of corresponding standards in the assay media (culture media without GM-CSF) for 24 h. The culture plates were centrifuged at termination; pellets were frozen at−80°C, thawed, and labeled with CyQuant® (C7026; Life Technologies Corp.); and fluorescent signals were measured according to the manufacturer's instructions. Changes in cell numbers following a 24 h culture with each cytokine were plotted against cytokine concentrations. The standard deviations for the measurements were within±10%. The potency was measured in terms of the effective concentration of an individual cytokine to cause a half-maximal increase in cell proliferation (EC50). Growth curves were fit with a 4-parameter logistic model (Y=((A-D)/1+(X/C)^b)+D), where A is a minimum asymptote, B is a slope factor, C is an inflection point, and D is a maximum asymptote, with limit on value of the minimum asymptote (A≤100) and limit on the slope value (B≤5). A normalized root mean squared error was determined for each fit. The EC50 values were then compared with those of the corresponding cytokine standards.
MCF7 cell proliferation assay
MCF7, a human breast carcinoma cell line, was used to evaluate potency of IGF-1. MCF7 was obtained from ATCC (HTB-22™, lot# 58412664) and maintained in Dulbecco's modified Eagle medium (DMEM) F12 media containing 10% FBS and 0.01 mg/mL bovine insulin in flat-well plates or culture flasks. Subcultures were performed according to the ATCC protocol after trypsinization. Cells were starved for 4–6 h before the potency assay in the media without FBS and insulin. To measure dose-dependent effects of IGF-1 on cell proliferation, 4×103 MCF7 cells per well were cultured in triplicate with increasing concentrations of either plant-produced IGF-1 or the corresponding standard (0.02–100 ng/mL) for 48 h in 96-well flat-bottomed plates. Cells were then frozen at−80°C before CyQuant labeling as described earlier. EC50 values for IGF-1 were measured similarly to the EC50 for EPO, SCF, and IL-3.
Erythroid progenitor cell expansion assay
Umbilical cord blood-derived CD34+ cells were obtained from AllCells and maintained in the StemSpan® Serum-Free Expansion Medium (StemCell Technologies) containing 1 μM glucocorticoids, 40 μg/mL lipids, 100 ng/mL SCF, 40 ng/mL IGF-1, and 25 ng/mL EPO. In culture variations, each of the standard cytokines obtained from R&D Systems was individually replaced with a plant-produced cytokine. An additional no cytokine control was maintained to monitor any spontaneous differentiation. The cultures were maintained over 4 weeks with intermittent subculture and passage into fresh media every 4 days. Cells were maintained at the following densities: 2×104–3×105/mL from day 0–7; 3×105–1×106 from day 7–14; 1–2×106/mL from day 14–21; and 1–3×106/mL from day 21–28. The experiment was repeated four times, and the representative data were presented. In each study, proliferation rates were measured in terms of fold expansion (final cell number/initial cell number), and population doublings estimated as ln (fold expansion)/ln (2) were plotted. The responsiveness of cells in culture to EPO was monitored by detection of EPO-R expression using antibodies to EPO-R (FAB307F, R&D Systems) by flow cytometry. The cell differentiation status was determined by surface expression of an erythroid marker Glycophorin A (also known as CD235a) (BD Biosciences) using flow cytometry. The enucleation status of CD235a+ cells was measured using TO-PRO®-3 (Life Technologies) nuclear staining at indicated intervals. CD235a+ TO-PRO-3− cells lacking nuclei are considered mature enucleated cells. Comparisons were made between cultures containing standard versus the plant-produced cytokines. Cells at various intervals were cytospotted onto slides and analyzed to evaluate the cell morphology microscopically after benzidine–Giemsa staining.
Spectrophotometric assay for hemoglobin content analysis
Cell pellets containing 5×105 cells were collected in triplicates from cultures at desired time intervals, washed with ice-cold PBS, and frozen for further analysis using Drabkin's reagent (D5941; Sigma Aldrich). Frozen cells were thawed, resuspended in 200 μL of the reagent (prepared as per manufacturer's instructions) in 96-well assay plates, and incubated at room temperature for 15–20 min. The absorbance values at the wavelength of 540 nm were measured using a spectrophotometric plate reader Spectramax M3 (Molecular Devices). Standards were maintained with known concentrations of hemoglobin and/or defined numbers of mature RBCs for reference, and the content in cell samples was estimated and plotted as an average (Avg [pg/cell] Hb).
Benzidine–Giemsa staining and imaging
Approximately 5×104 cells per culture were centrifuged onto Poly-l-lysine coated slides (95042-270; VWR Scientific) at 500 RPM for 2 min (Cytospin 4; Thermo Shandon) and air dried. For benzidine–Giemsa staining, cells were fixed in ice-cold methanol for 2 min, stained with 3,3-diaminobenzidine (D4418-50; Sigma Aldrich) and Giemsa stain (26149-01; Sigma Aldrich) according to the manufacturer's recommendations, air dried, and stored at 4°C until microscopic examination and imaging using inverted microscope Leica model DM IRB (Micro-tech Optical NE, Inc.).
Colony-forming cell assays
CD34+ cells were plated in methylcellulose-based medium, MethoCult™ H4230 (StemCell Technologies), containing 1% methylcellulose, 30% FBS, and 1% BSA. Human recombinant cytokines from R&D Systems were added at the following concentrations: 50 ng/mL SCF, 20 ng/mL GM-CSF, 20 ng/mL IL-3, 20 ng/mL IL-6, 20 ng/mL granulocyte CSF, and 3 units/mL EPO. In variations, the standard cytokines were replaced with the respective plant-produced cytokines for IL-3, SCF, and EPO. Cells were plated in triplicates at 500 cells per 35-mm plate and incubated in a humidified incubator at 37°C with 5% CO2 for 12 days. Hematopoietic colony-forming units were identified and scored by morphological criteria, using an inverted microscope, as colony-forming unit granulocyte, macrophage (CFU-GM), CFU erythrocyte (CFU-E), burst-forming unit erythrocyte (BFU-E), and multipotential CFU granulocyte, erythrocyte, monocyte, and megakaryocyte (CFU-GEMM).
Results
Expression of recombinant human EPO, SCF, IL-3, and IGF-1 in N. benthamiana
Expression of EPO, SCF, IL-3, and IGF-1 in N. benthamiana was assessed using the western blot analysis as described in the Materials and Methods section.
The expression levels of EPO, EPO-H, EPO-K, and EPO-HK were low as determined by western blotting using either an anti-tetra-His or an anti-EPO mAb (∼10–15 mg/kg of fresh leaf tissue with an estimated solubility of 50%, data not shown). In contrast, EPO-LicKM fusions demonstrated increased levels of expression and solubility: for EPO-2E, 52 mg/kg of leaf tissue with an estimated solubility of 60% and for EPO-3E, 44 mg/kg of leaf tissue with 90% solubility (Fig. 2A, B).
FIG. 2.

Western blot analysis of erythropoietin (EPO)-LicKM fusion products expression. (A) EPO-3E. M–Magic marker; 1–EPO-3E (clone 1)–soluble protein; 2–EPO-3E (clone 1)–total protein; 3–EPO-3E (clone 2)–soluble protein; 4–EPO-3E (clone 2) total protein. (B) EPO-2E. M–Magic marker; 1–EPO-2E soluble protein; 2–EPO-2E soluble protein; 3–EPO-2E total protein. An anti-tetra-His mAb was used for detection.
Similar to EPO, expression levels of IGF-1 and IGF-1-H (data not shown), IGF-1-K, and IGF-1-HK in plants were low (25 mg/kg of leaf tissue with an estimated solubility of ∼5%, Fig. 3A, B). In addition to low expression levels, IGF-1-K (Fig. 3B) and IGF-1-HK (data not shown) had, in SDS-PAGE under non-reducing conditions, mobility different than predicted (Mw>100 kDa). IGF-1 fused into the surface loop of LicKM (IGF-1-2E variant), on the other hand, had significantly increased expression and solubility levels (250 mg/kg leaf tissue with an estimated solubility of ∼30%, Fig. 3C). Similarly, fusion of IGF-1 to the C-terminus of LicKM (IGF-1-3E variant) showed enhanced expression and solubility levels compared with those of both non-fusion variants (IGF-1-HK and IGF-1-K). However, the accumulation levels and solubility of IGF-1-3E were lower (50 mg/kg leaf tissue with an estimated solubility of ∼15%, Fig. 3C) compared with IGF-1-2E.
FIG. 3.

(A) Western blot analysis of non-fused insulin-like growth factor (IGF) expression using an anti-tetra-His mAb for detection. M–Magic marker; 1–IGF-1-HK (clone 4); 2–IGF-1-HK (clone 6) (B) Western blot analysis of non-fused IGF-1 expression using an anti-IGF-1 mAb for detection and run under non-reducing conditions. M–Magic marker; 1–IGF-1-K–soluble protein. (C) Western blot analysis of IGF-1-LicKM fusion products expression using an anti-tetra-His antibody for detection. M–Magic marker; 1–10 ng HAI standard [hemagglutinin from A/Indonesia/05/05 (H5N1) strain of influenza virus]; 2–25 ng HAI standard; 3–50 ng HAI standard; 4–IGF-1-2E–soluble protein; 5–IGF-1-2E–total protein; 6–IGF-1-3E–soluble protein; 7–IGF-1-3E–total protein.
The expression levels of IL-3 and IL-3-H (data not shown), IL-3-HK, and IL-3-K were 20 mg/kg of leaf tissue, with an estimated solubility of 70% (Fig. 4A, B). Fusion of IL-3 into the surface loop of LicKM (IL-3-2E variant) resulted in a noticeable increase in target expression and solubility (144 mg/kg of leaf tissue with an estimated solubility of ∼90%, Fig. 4C).
FIG. 4.

(A) Western blot analysis of non-fused interleukin 3 (IL-3) expression using an anti-tetra-His mAb for detection. M–Magic marker; 1–IL-3-HK (clone 1); 2–IL-3-HK (clone 2). (B) Western blot analysis of non-fused IL-3 expression using an anti-IL-3 mAb for detection. M–Magic marker; 1–IL-3-K–soluble protein; 2–IL-3-K–total protein. (C) Western blot analysis of IL-3-LicKM fusion product expression using an anti-tetra-His mAb for detection. M–Magic marker; 1–HAI [hemagglutinin from A/Indonesia/05/05 (H5N1) strain of influenza virus] standard 15 ng; 2–HAI standard 30 ng; 3–HAI standard 60 ng; 4–IL-3-2E (clone 1); 5–IL-3-2E (clone 2).
SCF and SCF-H (data not shown), SCF-HK, and SCF-K were expressed at high levels (200 mg/kg of leaf tissue) with an estimated solubility of 100% (Fig. 5A, B). Detection with either an anti-tetra-His or an anti-SCF mAb resulted in multiple bands (at ∼22, 24 and 27 kDa). To determine whether these bands represent different glycosylation forms, SCF-HK was treated with PNGaseF, an enzyme that removes N-linked glycans. After the treatment with PNGaseF, only a 22 kDa band could be observed, suggesting that 24 and 27 kDa bands represented different glycosylation forms of the protein (Fig. 5C) which collapsed into a 22 kDa band after the removal of glycans.
FIG. 5.
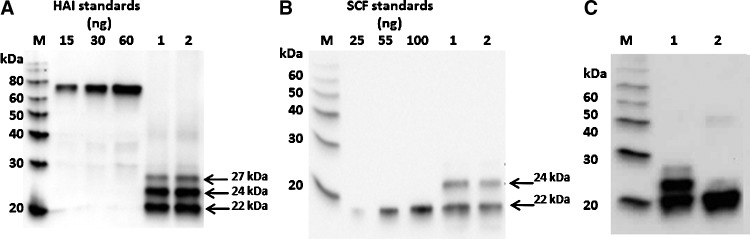
Western blot analysis of non-fused stem cell factor (SCF) product expression. (A) Detection with an anti-tetra-His mAb. M–Magic marker; 1–SCF-HK (clone 1); 2–SCF-HK (clone 2) (B) Detection with an anti-SCF mAb. M–Magic marker; 1–SCF-K–soluble protein; 2–SCF-K–total protein. (C) PNGase F treatment of purified SCF-HK, detected with an anti-SCF mAb. M–Magic marker; 1–non-treated SCF-HK; 2–PNGase F-treated SCF-HK.
Purification and characterization of plant-produced EPO, SCF, IL-3, and IGF-1
EPO-2E, IL-3-2E, IGF-1-2E, and SCF-HK were chosen, based on the results of the expression analysis and preliminary activity testing (data not shown), for further purification and characterization. All targets were captured from clarified homogenate using IMAC and further purified by ion exchange chromatography and SEC.
EPO-2E resulting from the IMAC/Capto-Q process is shown in Fig. 6A. A slightly diffuse main band is observed for the glycosylated EPO-2E when separated on a 10% SDS-PAGE and stained with Coomassie. The purified target migrated at ∼55 kDa, which is in agreement with the theoretical Mw of 45.7 kDa and allowing for added glycosylation. The elution profile for EPO-2E fractionated over a Superdex 200 SEC column is shown in Fig. 7A. The target, identified by the western blot analysis of the fractions (data not shown), was shown to be in the main peak eluting at 14.8 mL. Estimation of the size for this elution volume is consistent with an EPO-2E monomer. Peptide mapping resulted in combined 100% sequence coverage by liquid chromatography-tandem mass spectrometry (LC-MS-MS) analysis of target peptides after digestion with trypsin, elastase, or chymotrypsin (data not shown). The results of peptide mapping confirmed the target identity for the protein detected by the western blot analysis.
FIG. 6.

Coomassie-stained sodium dodecyl sulfate polyacrylamide gel electrophoresis analysis of purified targets. (A) EPO-2E, (B) SCF-HK, (C) IGF-1-2E, and (D) IL-3-2E. Molecular weight markers (M) are on the left with the 50 and 20 kDa markers indicated. The target band is indicated with the arrowhead and where multiple bands exist due to glycosylation, the multiple bands are bracketed. The asterisks indicate the band(s) used for concentration determination.
FIG. 7.

Analytical size exclusion chromatography of isolated proteins. Absorbance is shown as normalized A280 values. Where a prominent single peak is not obvious, the target elution peak was emphasized with a bar above the elution profile. (A) EPO-2E was tested using a Superdex 200 column. Superdex 75 columns were used for (B) SCF-HK, (C) IGF-1-2E, and (D) IL-3-2E.
SCF-HK was isolated by IMAC capture followed by polishing using a DEAE column. Purified protein was analyzed using 15% SDS-PAGE stained with Coomassie, as shown by the bracketed portion of the lane in Fig. 6B. SCF-HK is a glycoprotein containing multiple potential N-linked glycosylation sites [25]. Subsequently, the protein purified from plants represents the mixture of several glycoforms, as suggested by the PNGaseF treatment (Fig. 5C). The Mw (19.8 kDa), deduced from the aa sequence, is in agreement with the migration of the target bands, as observed on SDS-PAGE (Fig. 6B). SCF-HK was detected by the western blot analysis (data not shown) of the main peak eluting at 9.8 mL from a Superdex 75 column (Fig. 7B). Estimation of the size for this elution volume indicates an oligomer of ∼4–5 molecules and was consistent for multiple batches of SCF-HK. Peptide mapping of two bands indicated with an asterisk in Fig. 6B resulted in combined sequence coverage for both bands of 100% by the LC-MS-MS analysis of target peptides after digestion with trypsin, elastase, or chymotrypsin (data not shown).
Both IGF-1-2E and IL-3-2E were purified using initial IMAC capture followed by SEC. This process resulted in the samples shown in Fig. 6C, D for IGF-1-2E and IL-3-2E, respectively. The target band observed in Fig. 6C is in agreement with the predicted size (34.9 kDa) of IGF-1-2E. Bands indicated by brackets in Fig. 6C were identified as IGF-1-2E in the western blot analysis (data not shown), and the band marked with an asterisk was sequenced by tri-enzyme digest peptide mapping. The peptide map resulted in 100% combined sequence coverage (data not shown). Analytical SEC over a Superdex 75 column showed a single main peak with a retention volume, 12.4 mL, which is equivalent to a half of the nominal Mw compared with SEC Mw standards (Fig. 7C). The delayed retention relative to standards and broad peak base may indicate some interaction with the SEC column matrix, causing the delayed elution.
IL-3-2E had a predicted Mw of 42.3 kDa, which is in agreement with what was observed by the Coomassie-stained SDS-PAGE analysis (Fig. 6D, marked by the bracket). The protein band marked with asterisk was subjected to tri-enzyme digest peptide mapping. As with the previous samples, the LC-MS-MS peptide mapping resulted in 100% combined sequence identification (data not shown). Examination using an analytical Superdex 75 column fractionation demonstrated a single prominent peak eluting at 11.0 mL, which is consistent with a monomer when compared with SEC Mw standards (Fig. 7D).
Biological activity of plant-produced EPO, SCF, IGF-1, and IL-3 in cell proliferation assays
Biological activity of plant-produced EPO-2E, SCF-HK, and IL-3-2E, and IGF-1-2E has been demonstrated in vitro using the TF1 and MCF-7 cell proliferation assays, respectively, as described in the Materials and Methods section (Table 1). EPO-2E and SCF-HK had comparable activities with EC50s within a close range of the corresponding fresh commercially available standards from R&D Systems (EPO-2E: 0.4 vs. 0.3 ng/mL; SCF-HK: 6.4 vs. 6.1 ng/mL). Activity of IL-3-2E was lower compared with the standard (1.2 vs. 0.1 ng/mL). IGF-1-2E also had slightly lower activity compared with the standard, with an estimated EC50 of 1.7 ng/mL versus 0.2 ng/mL.
Table 1.
Assessment of Biological Activity of Plant-Produced Cytokines In Vitro in Cell Proliferation Assays
| R&D Systems | EC50 (ng/mL) | NRMSE | FhCMB | EC50 (ng/mL) | NRMSE |
|---|---|---|---|---|---|
| SCF | 6.1 | 5% | SCF-HK | 6.4 | 22% |
| EPO | 0.3 | 11% | EPO-2E | 0.4 | 10% |
| IL-3 | 0.1 | 6% | IL-3-2E | 1.2 | 5% |
| IGF-1 | 0.2 | 14% | IGF-1-2E | 1.7 | 12% |
Dose-dependent cytokine effect on cell proliferation was assessed by increasing concentrations of EPO-2E, SCF-HK, or IL-3-2E added to TF1 cells, or increasing concentrations of IGF-1-2E added to MCF-7 cells. EC50 values for the plant-produced cytokines calculated from the data obtained in the linear dose-response range were compared with the EC50 of corresponding commercial standards.
NRMSE, normalized root mean squared error; EPO, erythropoietin; SCF, stem cell factor; IL-3, interleukin 3; IGF-1, insulin-like growth factor-1.
All four plant-produced growth factors have been shown to stimulate the expansion from CD34+ cells and their cytokine-responsive differentiation toward erythropoietic precursors with similar potency as commercially available growth factors. The cells cultured in media without added cytokines did not proliferate and lost 90% viability, indicating the lack of spontaneous expansion and differentiation in the absence of essential mitogens. Therefore, this particular culture was terminated within 7 days of initiation. When CD34+ cells were cultured over 4 weeks in the erythroid differentiation media containing either plant-produced EPO-2E or SCF-HK, or their commercial standards, expansion rates were comparable (Fig. 8A). Replacement of commercial standards in the media formulation with plant-produced cytokines IL-3-2E or IGF-1-2E produced similar growth rates in erythroid cultures (Fig. 8B).
FIG. 8.

Effect of EPO-2E, SCF-HK, IL-3-2E, and IGF-1-2E on CD34+ cell proliferation and differentiation. (A, B) Cumulative population doublings of CD34+ cell cultures in the presence of plant-produced cytokines or the commercial standards are plotted against the culture period in days. (C) EPO-R expression in cells cultured in media with EPO-2E versus commercial standard EPO. (D, E) Cell differentiation at the end of cell culture evaluated by CD235a expression measured by flow cytometry. (F, G) Enucleated cell numbers (expressed as CD235a+ TO-PRO-3−) plotted against time in culture.
Cells showed similar responses in terms of EPO-R up-regulation to plant-derived EPO and commercial EPO (all other growth factors were from the commercial source in both cultures) as shown in Fig. 8C. The EPO-R levels increased progressively with days in culture, peaking at day 14 and decreasing thereafter. When the differentiation of cultured CD34+ cells was examined, the percentage of CD235a+ cells was shown to increase progressively during the course of 2–3 weeks, reaching a maximum of about 80%–90%. Notably, CD235a expression levels in the cultures in which the commercial EPO, SCF, IL-3, or IGF-1 was replaced with a corresponding plant-produced cytokine preparation were comparable to those with the standard cytokines (Fig. 8D, E). Enucleation was measured and represented in terms of the percent cells that expressed CD235a and were negative for nuclear dye TO-PRO-3. The results (Fig. 8F, G) demonstrated comparable enucleation (ie, CD235a+ TO-PRO-3− cells). The percent increased gradually with time in culture as cells differentiated, and by the end of a 4-week culture period, there were 20%–40% enucleated cells in culture variations, where either commercial or plant-produced cytokines were used. The cells that expressed CD235a but were still nucleated (CD235a+ TO-PRO-3+) also increased in a similar fashion with comparable percentages among the variations tested (∼60%–70% at peak, data not shown) with no remarkable differences. Some lots of the plant-produced IL-3-2E and IGF-1-2E produced slightly higher levels of enucleated cells (CD235a+ TO-PRO-3−) (∼10%, data not shown) compared with the standards (Fig. 8G), with correspondingly higher numbers of CD235a+ TO-PRO-3+ cells (∼10%). These experiments were repeated four times, and the data presented are from a representative study. The culture replicates showed approximately 5%–10% variance in expansion rates. CD235a expression and enucleation rates showed a 10%–15% variance within an experiment, which are represented as deviations in plots (Fig. 8A–G). Furthermore, an approximately 20% variation was noted from one experiment to another in CD235a expression levels and enucleation, which may be attributed to the batch and donor variability of the cell stocks used in cultures.
The hemoglobin content of cells at various culture time intervals was also determined. The data obtained indicate a similar trend with an increase in hemoglobin content with progression of culture, in all variations tested, including the commercial cytokines or replacement of individual mitogens with the plant-derived forms. A steep increase in hemoglobin was noted between days 20 and 28, with erythrogenic differentiation, and reached a peak value with an average of 30 pg per cell at day 28 (Fig. 9A). The visual examination of pellets at later time points (days 23 and 28) showed red coloration, indicating hemoglobin accumulation in pellets from all cultures (Fig. 9B), with no significant differences in the variations (Fig. 10).
FIG. 9.

(A) Hemoglobin content of cells cultured in media with the commercial cytokine mix compared with cocktails where individual cytokines were replaced with respective plant-derived cytokines as indicated. (B) Images of cell pellets from combinations of media as in (A), from days 23 and 28 of differentiation.
FIG. 10.

Microscopic images of benzidine–Giemsa staining for samples from cord blood-derived CD34+ cells cultured in media containing R&D commercial cytokines (A) or in media with replacement of each individual cytokine with plant-produced EPO-2E (B), SCF-HK (C), IL-3-2E (Lots 1KgFP or P3) (D, E), or IGF-1-2E (Lots P1, P2 or RA) (F, G, H) collected on days 7 and 23. Hemoglobinized erythroblasts and reticulocytes show benzidine-positive staining (brown), Giemsa stains nuclei blue/purple. Magnification: 40×.
Benzidine–Giemsa staining of the cells revealed comparable morphology at all stages among the culture variations tested, with no remarkable differences (Fig. 10). Cells at earlier stages of expansion (day 0 to 10) appeared larger with blue staining of nuclei by Giemsa, as shown for day 7 cells, which did not contain much hemoglobin and, hence, did not stain brown. Past day 10, a gradual increase in the hemoglobin content was noted. At later stages of differentiation (day 23), several brown (benzidine-stained) hemoglobin-positive cells were observed with a mix of large immature cells, orthochromatic erythroblasts with condensed nuclei, and a few enucleated red cells (Fig. 10), albeit no remarkable differences between the variations were observed, suggesting comparable differentiation states when replacing the commercial cytokines with the plant-derived cytokines.
In order to further evaluate the potency of the plant-derived EPO, SCF, and IL-3, we compared these cytokines with the respective commercial forms in inducing colony formation on the semi-solid methylcellulose-based medium in vitro. After plating cells on methylcellulose in the presence of cytokine variations, BFU-E and CFU-E colonies were evaluated microscopically, and the average number of colonies for each type was determined (Fig. 11). The cells responded similarly to the plant-derived cytokines and the commercial cytokine cocktails. No colonies were noted in plates without cytokine cocktail, demonstrating the specific effect of cytokine stimulation. Clonogenicity of the particular media as described earlier was compared by an assessment of CFU-E/BFU-E, CFU-GM, and CFU-GEMM (Table 2). Similar potencies were observed for the plant-derived cytokines and the commercial cytokines.
FIG. 11.

Hematopoetic colony-forming assay. Burst-forming unit erythrocyte (BFU-E) and colony-forming unit erythrocyte (CFU-E) were evaluated at 12 days after plating cord blood-derived CD34+ cells in methylcellulose, using R&D commercial or plant-derived cytokine mix or individually replacing commercial cytokines with corresponding plant-derived cytokines as indicated.
Table 2.
CFUs and Clonogenicity Measured 12 Days After Plating Cells on Methylcellulose
| |
CFU composition |
Clonogenicity |
||
|---|---|---|---|---|
| Sample | %BFU-E/CFU-E | %CFU-GM | %CFU-GEMM | %Total colonies/cells plated |
| Plant-derived cytokine mix | 34.8±7.1 | 62.7±7.1 | 2.5±0.5 | 16.5±3.4 |
| R&D standard cytokine mix | 28.8±9.4 | 68.1±10.4 | 3.1±0.3 | 19.1±1.6 |
| SCF-HK | 33.2±2.4 | 63.7±2.4 | 3.1±0.4 | 18.4±2.0 |
| IL-3-2E–Lot P3 | 37.1±6.7 | 59.4±6.7 | 3.5±0.3 | 11.5±1.2 |
| EPO-2E | 27.1±3.5 | 67.8±3.5 | 5.1±1.3 | 12.1±2.8 |
The average of four determinations is shown.
BFU-E, burst-forming unit erythrocyte; CFU-E, colony-forming unit erythrocyte; CFU-GM, colony-forming unit granulocyte, macrophage; CFU-GEMM, colony-forming unit granulocyte, erythrocyte, monocyte, megakaryocyte.
Discussion
In previous studies conducted at FhCMB, the launch vector-based plant transient expression system has been used for producing a wide range of target proteins at levels ranging from 50 mg to more than 1 g per kg of fresh leaf biomass [58,61,62,64,66,73]. Here, we extended the utility of this system to the field of hematopoietic growth factors, and have successfully expressed four human growth factors—EPO, SCF, IGF-1, and IL-3—either as non-fused proteins or as fusions with LicKM in N. benthamiana. All target sequences were optimized for the expression in tobacco plants, contained a plant signal peptide (PR-1a) in place of native signal peptides, and at the C-terminus carried a 6xHis affinity purification tag and the KDEL ER retention sequence that was added to evaluate the potential influence of plant glycosylation on protein activity. Plants differ significantly from mammals in the modifications that protein N-glycans undergo in the Golgi, specifically trimming of mannose residues from high-mannose oligosaccharides and transfer of additional sugars resulting in complex-type N-glycans. In contrast, the oligosaccharide chains produced in the rough ER [eg, Man8(GlcNAc)2] are similar between plants and mammals [73]. Therefore, addition of the KDEL sequence retains proteins in the ER, resulting in the mammalian-type glycosylation of the target.
Of the four targets, EPO, IGF-1, and IL-3 were also fused into the surface loop of LicKM (in these cases, C-terminal 6xHis and KDEL were parts of LicKM) to enhance their expression, solubility, and stability. The expression levels of LicKM-fused EPO, IGF-1, and IL-3 were 44, 250 and 144 mg/kg of leaf tissue, respectively, with solubility of 90%, ∼30%, and ∼90%, respectively. Non-fused SCF was expressed at a high level of 250 mg/kg of leaf tissue with 100% solubility. Identity of the target proteins was confirmed by the western blot analysis and tri-enzyme digest peptide mapping. All plant-expressed growth factors were glycosylated, and the LicKM fusions (EPO-2E, IGF-1-2E, and IL-3-2E) were present as monomers, whereas the non-fused target (SCF-HK) eluted from a SEC column at a volume that was consistent with a tetramer or pentamer. In contrast to non-fused EPO and non-fused IGF-1, their LicKM fusion variants were not toxic to plants (EPO-2E showed light symptoms by 6–7 days post infiltration) (data not shown), expressed at high levels, and exhibited biological activity in vitro comparable or superior to commercially available standards in cytokine-sensitive cell proliferation assays. Plants expressing non-fused SCF, non-fused IL-3, or LicKM-fused IL-3 (IL-3-2E) showed no signs of morphological or physiological retardation over the period of expression. Similar to LicKM-fused EPO-2E and IGF-1-2E, non-fused SCF and LicKM-fused IL-3-2E expressed at high levels and exhibited biological activity in vitro comparable or superior to commercially available standards.
To our knowledge, this is the first report on the production of recombinant human SCF and IL-3 in plants. However, there have been several reports on the production of recombinant human EPO and IGF in different plant systems. Matsumoto et al. [74] expressed non-fused, full-length human EPO, including its native signal peptide in transgenic BY2 tobacco cell culture. EPO was detected in both cell extracts and protoplast culture medium at the level of ∼1 ng/mL and exhibited biological activity in vitro, but was not able to penetrate cell wall and was not secreted into the culture medium of BY2 cell line. In another expression system, moss Physcomitrella patens mutant lacking the plant-specific core-bound α1,3-fucose and β1,2-xylose in N-glycans, EPO containing a plant-derived signal peptide was found to be expressed and secreted by the transiently transformed protoplasts into the culture medium at the level of up to 0.5 μg/mL. When expressed by transgenic Physcomitrella patens lines, EPO was secreted through cell wall into culture medium and over 6 days, accumulated approximately 250 μg/g dry weight of moss [75]. In transgenic tobacco plants, high-level expression of non-fused human EPO containing its native signal peptide has been shown to affect plant morphology, causing retarded vegetative growth and male sterility [76]. However, by modifying the tobacco transformation system (eg, reducing cytokinin 6-BA concentration and changing a strain of Agrobacterium) and the expression cassette (flanking the EPO gene with the His tag and KDEL sequences), Musa et al. [77] have developed transgenic tobacco overexpressing EPO with no plant health abnormality. Another study of a healthy EPO-expressing transgenic tobacco (using another type of tobacco and different regulatory elements in the expression cassette) has been recently reported by Sperb et al. [78]. This study, however, did not report on the EPO yield and its biological activity. Conley et al. [79] have expressed EPO in transiently and stably transformed tobacco plants and demonstrated its cytoprotective activity in an in vitro model of kidney epithelial cell death. In an effort to enhance the yield of recombinant EPO in tobacco plant tissue, various approaches were explored, and targeting to the ER by means of attaching KDEL demonstrated the greatest impact on EPO accumulation, resulting in a yield of approximately 0.05% of total soluble protein (TSP) in tobacco leaves. An additional advantage of the ER-targeted EPO was the lack of complex glycans possessing high immunogenicity. Among other tested approaches, the codon optimization provided no benefit, EPO native signal peptide was processed more efficiently compared with tobacco signal peptide, and glycosylation was very important for EPO stability in plant tissue. Contrary to our findings with LicKM fusion, others reported that fusion to the elastin-like polypeptide (ELP) carrier protein had a negligible effect on EPO accumulation, which the authors explain by a potential toxicity of the EPO-ELP fusion to the plant [76,79]. This finding is also in contradiction with the potent enhancing effect of fusion to ELP on the expression of human IL-10 and murine IL-4 [80], spider silk protein [80,81], full-size anti-HIV-1 antibody 2F5 [82] in tobacco leaves, and single-chain variable antibody fragments in tobacco seeds [83]. Fusion to other carrier molecules, including ubiquitin [84], β-glucuronidase [85,86], and human immunoglobulin α-chains [87], have also been shown to enhance target protein accumulation in plants.
Transgenic plants have also been used to produce human IGF-1. After codon optimization for expression in plants and introducing plant regulatory elements and plant signal sequence into the expression constructs, Panahi et al. [88] have achieved an accumulation of IGF-1 at the levels of 26 ng/mg protein in transgenic tobacco leaf extracts and 113 ng/mg protein in transgenic rice leaf extracts, corresponding to 0.02% and 0.03% of TSP, respectively. The expressed protein stimulated in vitro proliferation of SH-SY5Y, an IGF-sensitive human neuroblastoma cell line, indicating biological activity and stability of IGF-1 [88]. In another study, higher levels of IGF-1 expression in tobacco were achieved by targeting the construct to chloroplasts and using optimal regulatory elements, and were further enhanced by illumination of plants with continuous light and during leaf maturation (up to 32% of TSP). In contrast, codon optimization did not enhance the expression [89]. Chloroplast-derived IGF-1 carrying Staphylococcus aureus Z tag exhibited strong in vitro biological activity and high stability, as shown by stimulation of human megakaryoblastic HU-3 cell line [89].
Non-radioactive methods based on the use of cell lines that were responsive to cytokines have been instrumental in measuring biological activity of cytokines [90]. In this study, we used TF1 (responsive to EPO, SCF, and IL-3) and MCF7 (responsive to IGF-1) cell lines to assess cell proliferation rates and successfully evaluated the potency of the plant-produced cytokines. The calculated EC50 values of standards were then correlated with their potency to support erythroid cell cultures derived from umbilical cord blood CD34+ cells. To the best of our knowledge, this is the first report on the generation of all the key cytokines required for the erythroid development in a cost-effective manner using plants as a production platform. Biological activity of our growth factors estimated here in terms of EC50s was lower or within the range cited from commercial sources and literature (EPO: 1.4–2 ng/mL, SCF: 2–10 ng/mL, IL-3: 0.1–0.25 ng/mL, IGF-1: 1.5–1.7 ng/mL).
Biological activity of EPO has been shown to be dependent on its glycosylation status. Glycosylation was demonstrated to increase the persistence of EPO, resulting in a prolonged and increased biological response in vivo, and overcoming reduced receptor-binding activity [91]. The authors overexpressed recombinant human EPO and its glycosylation analog with an altered N-linked carbohydrate content, darbapoetin, in CHO cells and compared them to elucidate the relationship between the carbohydrate content and biological activity. The in vitro measurement of biological activity performed using the UT7-EPO cell line resulted in the EC50 value in the range of 12–60 pM depending on the carbohydrate content of the protein. EPO-2E produced in our study showed an EC50 value of 0.3 ng/mL, or 6.7 pM, which is within the range of activity mentioned earlier for CHO cell-produced EPO.
The cost of media used for RBC production these days represents a significant challenge to making ex vivo manufacturing cost effective. The culture media used in this process requires substantial fortification with high-cost growth factors and has been referred to as liquid gold [92]. Plant-produced cytokines might have a significant impact on cost reduction of the media and, thus, facilitate lower-cost production of RBCs or other HSC-derived cell types.
Acknowledgments
The authors thank Natasha Kushnir of Fraunhofer USA Center for Molecular Biotechnology for editorial assistance and James Edinger and Ellen Baum of Celgene Cellular Therapeutics for critically reviewing the article and their comments. This work was supported by the Defense Advanced Research Projects Agency grant # FA 9550-08-1-0392.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Lelubre C. Vincent JL. Red blood cell transfusion in the critically ill patient. Ann Intensive Care. 2011;1:43. doi: 10.1186/2110-5820-1-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sakr Y. Lobo S. Knuepfer S. Esser E. Bauer M. Settmacher U. Barz D. Reinhart K. Anemia and blood transfusion in a surgical intensive care unit. Crit Care. 2010;14:R92. doi: 10.1186/cc9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calabrich A. Katz A. Management of anemia in cancer patients. Future Oncol. 2011;7:507–517. doi: 10.2217/fon.11.24. [DOI] [PubMed] [Google Scholar]
- 4.Sharma S. Sharma P. Tyler LN. Transfusion of blood and blood products: indications and complications. Am Fam Physician. 2011;83:719–724. [PubMed] [Google Scholar]
- 5.Curinga G. Jain A. Feldman M. Prosciak M. Phillips B. Milner S. Red blood cell transfusion following burn. Burns. 2011;37:742–752. doi: 10.1016/j.burns.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 6.Jelkmann W. Erythropoietin: structure, control of production, and function. Physiol Rev. 1992;72:449–489. doi: 10.1152/physrev.1992.72.2.449. [DOI] [PubMed] [Google Scholar]
- 7.Lin FK. Suggs S. Lin CH. Browne JK. Smalling R. Egrie JC. Chen KK. Fox GM. Martin F, et al. Cloning and expression of the human erythropoietin gene. Proc Natl Acad Sci U S A. 1985;82:7580–7584. doi: 10.1073/pnas.82.22.7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sasaki H. Bothner B. Dell A. Fukuda M. Carbohydrate structure of erythropoietin expressed in Chinese hamster ovary cells by a human erythropoietin cDNA. J Biol Chem. 1987;262:12059–12076. [PubMed] [Google Scholar]
- 9.Sasaki H. Ochi N. Dell A. Fukuda M. Site-specific glycosylation of human recombinant erythropoietin: analysis of glycopeptides or peptides at each glycosylation site by fast atom bombardment mass spectrometry. Biochemistry. 1988;27:8618–8626. doi: 10.1021/bi00423a017. [DOI] [PubMed] [Google Scholar]
- 10.Takeuchi M. Takasaki S. Miyazaki H. Kato T. Hoshi S. Kochibe N. Kobata A. Comparative study of the asparagine-linked sugar chains of human erythropoietins purified from urine and the culture medium of recombinant Chinese hamster ovary cells. J Biol Chem. 1988;263:3657–3663. [PubMed] [Google Scholar]
- 11.Delorme E. Lorenzini T. Giffin J. Martin F. Jacobsen F. Boone T. Elliott S. Role of glycosylation on the secretion and biological activity of erythropoietin. Biochemistry. 1992;31:9871–9876. doi: 10.1021/bi00156a003. [DOI] [PubMed] [Google Scholar]
- 12.Fukuda MN. Sasaki H. Lopez L. Fukuda M. Survival of recombinant erythropoietin in the circulation: the role of carbohydrates. Blood. 1989;73:84–89. [PubMed] [Google Scholar]
- 13.Yamaguchi K. Akai K. Kawanishi G. Ueda M. Masuda S. Sasaki R. Effects of site-directed removal of N-glycosylation sites in human erythropoietin on its production and biological properties. J Biol Chem. 1991;266:20434–20439. [PubMed] [Google Scholar]
- 14.Spivak JL. Hogans BB. The in vivo metabolism of recombinant human erythropoietin in the rat. Blood. 1989;73:90–99. [PubMed] [Google Scholar]
- 15.Takeuchi M. Inoue N. Strickland TW. Kubota M. Wada M. Shimizu R. Hoshi S. Kozutsumi H. Takasaki S. Kobata A. Relationship between sugar chain structure and biological activity of recombinant human erythropoietin produced in Chinese hamster ovary cells. Proc Natl Acad Sci U S A. 1989;86:7819–7822. doi: 10.1073/pnas.86.20.7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geissler EN. Liao M. Brook JD. Martin FH. Zsebo KM. Housman DE. Galli SJ. Stem cell factor (SCF), a novel hematopoietic growth factor and ligand for c-kit tyrosine kinase receptor, maps on human chromosome 12 between 12q14.3 and 12qter. Somat Cell Mol Genet. 1991;17:207–214. doi: 10.1007/BF01232978. [DOI] [PubMed] [Google Scholar]
- 17.Palacios R. Nishikawa S. Developmentally regulated cell surface expression and function of c-kit receptor during lymphocyte ontogeny in the embryo and adult mice. Development. 1992;115:1133–1147. doi: 10.1242/dev.115.4.1133. [DOI] [PubMed] [Google Scholar]
- 18.Ogawa M. Matsuzaki Y. Nishikawa S. Hayashi S. Kunisada T. Sudo T. Kina T. Nakauchi H. Nishikawa S. Expression and function of c-kit in hemopoietic progenitor cells. J Exp Med. 1991;174:63–71. doi: 10.1084/jem.174.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshinaga K. Nishikawa S. Ogawa M. Hayashi S. Kunisada T. Fujimoto T. Nishikawa S. Role of c-kit in mouse spermatogenesis: identification of spermatogonia as a specific site of c-kit expression and function. Development. 1991;113:689–699. doi: 10.1242/dev.113.2.689. [DOI] [PubMed] [Google Scholar]
- 20.Nishikawa S. Kusakabe M. Yoshinaga K. Ogawa M. Hayashi S. Kunisada T. Era T. Sakakura T. Nishikawa S. In utero manipulation of coat color formation by a monoclonal anti-c-kit antibody: two distinct waves of c-kit-dependency during melanocyte development. EMBO J. 1991;10:2111–2118. doi: 10.1002/j.1460-2075.1991.tb07744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Broudy VC. Stem cell factor and hematopoiesis. Blood. 1997;90:1345–1364. [PubMed] [Google Scholar]
- 22.Huang EJ. Nocka KH. Buck J. Besmer P. Differential expression and processing of two cell associated forms of the kit-ligand: KL-1 and KL-2. Mol Biol Cell. 1992;3:349–362. doi: 10.1091/mbc.3.3.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toksoz D. Zsebo KM. Smith KA. Hu S. Brankow D. Suggs SV. Martin FH. Williams DA. Support of human hematopoiesis in long-term bone marrow cultures by murine stromal cells selectively expressing the membrane-bound and secreted forms of the human homolog of the steel gene product, stem cell factor. Proc Natl Acad Sci U S A. 1992;89:7350–7354. doi: 10.1073/pnas.89.16.7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin FH. Suggs SV. Langley KE. Lu HS. Ting J. Okino KH. Morris CF. McNiece IK. Jacobsen FW, et al. Primary structure and functional expression of rat and human stem cell factor DNAs. Cell. 1990;63:203–211. doi: 10.1016/0092-8674(90)90301-t. [DOI] [PubMed] [Google Scholar]
- 25.Arakawa T. Yphantis DA. Lary JW. Narhi LO. Lu HS. Prestrelski SJ. Clogston CL. Zsebo KM. Mendiaz EA. Wypych J. Langley KE. Glycosylated and unglycosylated recombinant-derived human stem cell factors are dimeric and have extensive regular secondary structure. J Biol Chem. 1991;266:18942–18948. [PubMed] [Google Scholar]
- 26.Zsebo KM. Wypych J. McNiece IK. Lu HS. Smith KA. Karkare SB. Sachdev RK. Yuschenkoff VN. Birkett NC, et al. Identification, purification, and biological characterization of hematopoietic stem cell factor from buffalo rat liver—conditioned medium. Cell. 1990;63:195–201. doi: 10.1016/0092-8674(90)90300-4. [DOI] [PubMed] [Google Scholar]
- 27.Leary AG. Zeng HQ. Clark SC. Ogawa M. Growth factor requirements for survival in G0 and entry into the cell cycle of primitive human hemopoietic progenitors. Proc Natl Acad Sci U S A. 1992;89:4013–4017. doi: 10.1073/pnas.89.9.4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li CL. Johnson GR. Stem cell factor enhances the survival but not the self-renewal of murine hematopoietic long-term repopulating cells. Blood. 1994;84:408–414. [PubMed] [Google Scholar]
- 29.Bernstein ID. Andrews RG. Zsebo KM. Recombinant human stem cell factor enhances the formation of colonies by CD34+and CD34+lin- cells, and the generation of colony-forming cell progeny from CD34+lin- cells cultured with interleukin-3, granulocyte colony-stimulating factor, or granulocyte-macrophage colony-stimulating factor. Blood. 1991;77:2316–2321. [PubMed] [Google Scholar]
- 30.Lu HS. Clogston CL. Wypych J. Parker VP. Lee TD. Swiderek K. Baltera RF., Jr Patel AC. Chang DC. Brankow DW, et al. Post-translational processing of membrane-associated recombinant human stem cell factor expressed in Chinese hamster ovary cells. Arch Biochem Biophys. 1992;298:150–158. doi: 10.1016/0003-9861(92)90106-7. [DOI] [PubMed] [Google Scholar]
- 31.Whetton AD. Dexter TM. The mode of action of interleukin 3 in promoting survival, proliferation, and differentiation of hemopoietic progenitor cells. Lymphokines. 1988;15:355–374. [Google Scholar]
- 32.Mangi MH. Newland AC. Interleukin-3 in hematology and oncology: current state of knowledge and future directions. Cytokines Cell Mol Ther. 1999;5:87–95. [PubMed] [Google Scholar]
- 33.Yang YC. Ciarletta AB. Temple PA. Chung MP. Kovacic S. Witek-Giannotti JS. Leary AC. Kriz R. Donahue RE. Wong GG. Clark SC. Human IL-3 (multi-CSF): identification by expression cloning of a novel hematopoietic growth factor related to murine IL-3. Cell. 1986;47:3–10. doi: 10.1016/0092-8674(86)90360-0. [DOI] [PubMed] [Google Scholar]
- 34.Yang YC. Clark SC. Structure of the gene for human interleukin-3 or multi-CSF. Prog Clin Biol Res. 1987;251:3–11. [PubMed] [Google Scholar]
- 35.Böhmer RM. Stem Cells IL-3-dependent early erythropoiesis is stimulated by autocrine transforming growth factor beta. Stem Cells. 2004;22:216–224. doi: 10.1634/stemcells.22-2-216. [DOI] [PubMed] [Google Scholar]
- 36.Umemura T. al-Khatti A. Donahue RE. Papayannopoulou T. Stamatoyannopoulos G. Effects of interleukin-3 and erythropoietin on in vivo erythropoiesis and F-cell formation in primates. Blood. 1989;74:1571–1576. [PubMed] [Google Scholar]
- 37.Daughaday WH. Rotwein P. Insulin-like growth factors I and II. Peptide, messenger ribonucleic acid and gene structures, serum, and tissue concentrations. Endocr Rev. 1989;10:68–91. doi: 10.1210/edrv-10-1-68. [DOI] [PubMed] [Google Scholar]
- 38.Jones JI. Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995;16:3–34. doi: 10.1210/edrv-16-1-3. [DOI] [PubMed] [Google Scholar]
- 39.Kurtz A. Zapf J. Eckardt KU. Clemons G. Froesch ER. Bauer C. Insulin-like growth factor I stimulates erythropoiesis in hypophysectomized rats. Proc Natl Acad Sci U S A. 1988;85:7825–7829. doi: 10.1073/pnas.85.20.7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ratajczak J. Zhang Q. Pertusini E. Wojczyk BS. Wasik MA. Ratajczak MZ. The role of insulin (INS) and insulin-like growth factor-I (IGF-I) in regulating human erythropoiesis. Studies in vitro under serum-free conditions—comparison to other cytokines and growth factors. Leukemia. 1998;12:371–381. doi: 10.1038/sj.leu.2400927. [DOI] [PubMed] [Google Scholar]
- 41.Lee-Huang S. Cloning and expression of human erythropoietin cDNA in Escherichia coli. Proc Natl Acad Sci U S A. 1984;81:2708–2712. doi: 10.1073/pnas.81.9.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hamilton SR. Davidson RC. Sethuraman N. Nett JH. Jiang Y. Rios S. Bobrowicz P. Stadheim TA. Li H, et al. Humanization of yeast to produce complex terminally sialylated glycoproteins. Science. 2006;313:1441–1443. doi: 10.1126/science.1130256. [DOI] [PubMed] [Google Scholar]
- 43.Kim YK. Shin HS. Tomiya N. Lee YC. Betenbaugh MJ. Cha HJ. Production and N-glycan analysis of secreted human erythropoietin glycoprotein in stably transfected Drosophila S2 cells. Biotechnol Bioeng. 2005;92:452–461. doi: 10.1002/bit.20605. [DOI] [PubMed] [Google Scholar]
- 44.Jacobs K. Shoemaker C. Rudersdorf R. Neill SD. Kaufman RJ. Mufson A. Seehra J. Jones SS. Hewick R. Fritsch EF, et al. Isolation and characterization of genomic and cDNA clones of human erythropoietin. Nature. 1985;313:806–810. doi: 10.1038/313806a0. [DOI] [PubMed] [Google Scholar]
- 45.Powell JS. Berkner KL. Lebo RV. Adamson JW. Human erythropoietin gene: high level expression in stably transfected mammalian cells and chromosome localization. Proc Natl Acad Sci U S A. 1986;83:6465–6469. doi: 10.1073/pnas.83.17.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goto M. Akai K. Murakami A. Hashimoto C. Tsuda E. Ueda M. Kawanishi C. Takahashi N. Ishimoto A. Chiba H. Sasaki R. Production of recombinant human erythropoietin in mammalian cells: host-cell dependency of the biological activity of the cloned glycoprotein. Bio/Technology. 1988;6:67–71. [Google Scholar]
- 47.Han J. Yan X. Zhu J. Zhi X. Zang Y. Shen B. Qin J. Expression of a novel recombinant dual human stem cell factor in insect cells. Protein Expr Purif. 2003;31:311–317. doi: 10.1016/s1046-5928(03)00214-6. [DOI] [PubMed] [Google Scholar]
- 48.Yang YC. Clark SC. Cloning of the human interleukin-3 gene. Lymphokines. 1988;15:375–391. [Google Scholar]
- 49.Ding H. Griesel C. Nimtz M. Conradt HS. Weich HA. Jäger V. Molecular cloning, expression, purification, and characterization of soluble full-length, human interleukin-3 with a baculovirus-insect cell expression system. Protein Expr Purif. 2003;31:34–41. doi: 10.1016/s1046-5928(03)00138-4. [DOI] [PubMed] [Google Scholar]
- 50.Joly JC. Leung WS. Swartz JR. Overexpression of Escherichia coli oxidoreductases increases recombinant insulin-like growth factor-I accumulation. Proc Natl Acad Sci U S A. 1998;95:2773–2777. doi: 10.1073/pnas.95.6.2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Elliott S. Fagin KD. Narhi LO. Miller JA. Jones M. Koski R. Peters M. Hsieh P. Sachdev R. Rosenfeld RD. Yeast-derived recombinant human insulin-like growth factor I: production, purification, and structural characterization. J Protein Chem. 1990;9:95–104. doi: 10.1007/BF01024990. [DOI] [PubMed] [Google Scholar]
- 52.Yusibov V. Rabindran S. Recent progress in the development of plant derived vaccines. Expert Rev Vaccines. 2008;7:1173–1183. doi: 10.1586/14760584.7.8.1173. [DOI] [PubMed] [Google Scholar]
- 53.Gomord V. Fitchette AC. Menu-Bouaouiche L. Saint-Jore-Dupas C. Plasson C. Michaud D. Faye L. Plant-specific glycosylation patterns in the context of therapeutic protein production. Plant Biotechnol J. 2010;8:564–587. doi: 10.1111/j.1467-7652.2009.00497.x. [DOI] [PubMed] [Google Scholar]
- 54.Castilho A. Gattinger P. Grass J. Jez J. Pabst M. Altmann F. Gorfer M. Strasser R. Steinkellner H. N-glycosylation engineering of plants for the biosynthesis of glycoproteins with bisected and branched complex N-glycans. Glycobiology. 2011;21:813–823. doi: 10.1093/glycob/cwr009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yusibov V. Streatfield SJ. Kushnir N. Clinical development of plant-produced recombinant pharmaceuticals: vaccines, antibodies and beyond. Hum Vaccin. 2011;7:313–321. doi: 10.4161/hv.7.3.14207. [DOI] [PubMed] [Google Scholar]
- 56.Musiychuk K. Stephenson N. Bi H. Farrance CE. Orozovic G. Brodelius M. Brodelius P. Horsey A. Ugulava N, et al. A launch vector for the production of vaccine antigens in plants. Influenza Other Respi Viruses. 2007;1:19–25. doi: 10.1111/j.1750-2659.2006.00005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Green BJ. Fujiki M. Mett V. Kaczmarczyk J. Shamloul M. Musiychuk K. Underkoffler S. Yusibov V. Mett V. Transient protein expression in three Pisum sativum (green pea) varieties. Biotechnol J. 2009;4:230–237. doi: 10.1002/biot.200800256. [DOI] [PubMed] [Google Scholar]
- 58.Mett V. Musiychuk K. Bi H. Farrance CE. Horsey A. Ugulava N. Shoji Y. de la Rosa P. Palmer GA, et al. A plant-produced influenza subunit vaccine protects ferrets against virus challenge. Influenza Other Respi Viruses. 2008;2:33–40. doi: 10.1111/j.1750-2659.2008.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shoji Y. Chichester JA. Bi H. Musiychuk K. de la Rosa P. Goldschmidt L. Horsey A. Ugulava N. Palmer GA. Mett V. Yusibov V. Plant-expressed HA as a seasonal influenza vaccine candidate. Vaccine. 2008;26:2930–2934. doi: 10.1016/j.vaccine.2008.03.045. [DOI] [PubMed] [Google Scholar]
- 60.Shoji Y. Farrance CE. Bi H. Shamloul M. Green B. Manceva S. Rhee A. Ugulava N. Roy G, et al. Immunogenicity of hemagglutinin from A/Bar-headed Goose/Qinghai/1A/05 and A/Anhui/1/05 strains of H5N1 influenza viruses produced in Nicotiana benthamiana plants. Vaccine. 2009;27:3467–3470. doi: 10.1016/j.vaccine.2009.01.051. [DOI] [PubMed] [Google Scholar]
- 61.Shoji Y. Bi H. Musiychuk K. Rhee A. Horsey A. Roy G. Green B. Shamloul M. Farrance CE, et al. Plant-derived hemagglutinin protects ferrets against challenge infection with the A/Indonesia/05/05 strain of avian influenza. Vaccine. 2009;27:1087–1092. doi: 10.1016/j.vaccine.2008.11.108. [DOI] [PubMed] [Google Scholar]
- 62.Shoji Y. Chichester JA. Jones M. Manceva SD. Damon E. Mett V. Musiychuk K. Bi H. Farrance C, et al. Plant-based rapid production of recombinant subunit hemagglutinin vaccines targeting H1N1 and H5N1 influenza. Hum Vaccin. 2011;7(Suppl):41–50. doi: 10.4161/hv.7.0.14561. [DOI] [PubMed] [Google Scholar]
- 63.Mett V. Lyons J. Musiychuk K. Chichester JA. Brasil T. Couch R. Sherwood R. Palmer GA. Streatfield SJ. Yusibov V. A plant-produced plague vaccine candidate confers protection to monkeys. Vaccine. 2007;25:3014–3017. doi: 10.1016/j.vaccine.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 64.Chichester JA. Musiychuk K. Farrance CE. Mett V. Lyons J. Mett V. Yusibov V. A single component two-valent LcrV-F1 vaccine protects non-human primates against pneumonic plague. Vaccine. 2009;27:3471–3474. doi: 10.1016/j.vaccine.2009.01.050. [DOI] [PubMed] [Google Scholar]
- 65.Chichester JA. Musiychuk K. de la Rosa P. Horsey A. Stevenson N. Ugulava N. Rabindran S. Palmer GA. Mett V. Yusibov V. Immunogenicity of a subunit vaccine against Bacillus anthracis. Vaccine. 2007;25:3111–3114. doi: 10.1016/j.vaccine.2007.01.068. [DOI] [PubMed] [Google Scholar]
- 66.Massa S. Franconi R. Brandi R. Muller A. Mett V. Yusibov V. Venuti A. Anti-cancer activity of plant-produced HPV16 E7 vaccine. Vaccine. 2007;25:3018–3021. doi: 10.1016/j.vaccine.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 67.Venuti A. Massa S. Mett V. Vedova LD. Paolini F. Franconi R. Yusibov V. An E7-based therapeutic vaccine protects mice against HPV16 associated cancer. Vaccine. 2009;27:3395–3397. doi: 10.1016/j.vaccine.2009.01.068. [DOI] [PubMed] [Google Scholar]
- 68.Park JS. Han KY. Lee JH. Song JA. Ahn KY. Seo HS. Sim SJ. Kim SW. Lee J. Solubility enhancement of aggregation-prone heterologous proteins by fusion expression using stress-responsive Escherichia coli protein, RpoS. BMC Biotechnol. 2008;8:15. doi: 10.1186/1472-6750-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fox JD. Waugh DS. Maltose-binding protein as a solubility enhancer. Methods Mol Biol. 2003;205:99–117. doi: 10.1385/1-59259-301-1:99. [DOI] [PubMed] [Google Scholar]
- 70.Carter J. Zhang J. Dang TL. Hasegawa H. Cheng JD. Gianan I. O'Neill JW. Wolfson M. Siu S, et al. Fusion partners can increase the expression of recombinant interleukins via transient transfection in 2936E cells. Protein Sci. 2010;19:357–362. doi: 10.1002/pro.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goldenkova IV. Musiĭchuk KA. Piruzian ES. [A thermostable Clostridium thermocellum lichenase-based reporter system for studying the gene expression regulation in prokaryotic and eukaryotic cells] Mol Biol (Mosk) 2002;36:868–876. . Russian. [PubMed] [Google Scholar]
- 72.Matsuoka M. Yamamoto N. Kano-Murakami Y. Tanaka Y. Ozeki Y. Hirano H. Kagawa H. Oshima M. Ohashi Y. Classification and structural comparison of full-length cDNAs for pathogenesis-related proteins. Plant Physiol. 1987;85:942–946. doi: 10.1104/pp.85.4.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rayon C. Lerouge P. Faye L. The protein N-glycosylation in plants. J Exp Bot. 1998;49:1463–1472. [Google Scholar]
- 74.Matsumoto S. Ikura K. Ueda M. Sasaki R. Characterization of a human glycoprotein (erythropoietin) produced in cultured tobacco cells. Plant Mol Biol. 1995;27:1163–1172. doi: 10.1007/BF00020889. [DOI] [PubMed] [Google Scholar]
- 75.Weise A. Altmann F. Rodriguez-Franco M. Sjoberg ER. Bäumer W. Launhard H. Kietzmann M. Gorr G. High-level expression of secreted complex glycosylated recombinant human erythropoietin in the Physcomitrella Delta-fuc-t Delta-xyl-t mutant. Plant Biotechnol J. 2007;5:389–401. doi: 10.1111/j.1467-7652.2007.00248.x. [DOI] [PubMed] [Google Scholar]
- 76.Cheon BY. Kim HJ. Oh KH. Bahn SC. Ahn JH. Choi JW. Ok SH. Bae JM. Shin JS. Overexpression of human erythropoietin (EPO) affects plant morphologies: retarded vegetative growth in tobacco and male sterility in tobacco and Arabidopsis. Transgenic Res. 2004;13:541–549. doi: 10.1007/s11248-004-2737-3. [DOI] [PubMed] [Google Scholar]
- 77.Musa TA. Hung C-Y. Darlington DE. Sane DC. Xie J. Overexpression of human erythropoietin in tobacco does not affect plant fertility or morphology. Plant Biotechnol Rep. 2009;3:157–165. [Google Scholar]
- 78.Sperb F. Werlang IC. Margis-Pinheiro M. Basso LA. Santos DS. Pasquali G. Molecular cloning and transgenic expression of a synthetic human erythropoietin gene in tobacco. Appl Biochem Biotechnol. 2011;165:652–665. doi: 10.1007/s12010-011-9283-2. [DOI] [PubMed] [Google Scholar]
- 79.Conley AJ. Mohib K. Jevnikar AM. Brandle JE. Plant recombinant erythropoietin attenuates inflammatory kidney cell injury. Plant Biotechnol J. 2009;7:183–199. doi: 10.1111/j.1467-7652.2008.00389.x. [DOI] [PubMed] [Google Scholar]
- 80.Patel J. Zhu H. Menassa R. Gyenis L. Richman A. Brandle J. Elastin-like polypeptide fusions enhance the accumulation of recombinant proteins in tobacco leaves. Transgenic Res. 2007;16:239–249. doi: 10.1007/s11248-006-9026-2. [DOI] [PubMed] [Google Scholar]
- 81.Scheller J. Henggeler D. Viviani A. Conrad U. Purification of spider silk-elastin from transgenic plants and application for human chondrocyte proliferation. Transgenic Res. 2004;13:51–57. doi: 10.1023/b:trag.0000017175.78809.7a. [DOI] [PubMed] [Google Scholar]
- 82.Floss DM. Sack M. Stadlmann J. Rademacher T. Scheller J. Stöger E. Fischer R. Conrad U. Biochemical and functional characterization of anti-HIV antibody-ELP fusion proteins from transgenic plants. Plant Biotechnol J. 2008;6:379–391. doi: 10.1111/j.1467-7652.2008.00326.x. [DOI] [PubMed] [Google Scholar]
- 83.Scheller J. Leps M. Conrad U. Forcing single-chain variable fragment production in tobacco seeds by fusion to elastin-like polypeptides. Plant Biotechnol J. 2006;4:243–249. doi: 10.1111/j.1467-7652.2005.00176.x. [DOI] [PubMed] [Google Scholar]
- 84.Hondred D. Walker JM. Mathews DE. Vierstra RD. Use of ubiquitin fusions to augment protein expression in transgenic plants. Plant Physiol. 1999;119:713–724. doi: 10.1104/pp.119.2.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gil F. Brun A. Wigdorovitz A. Catalá R. Martínez-Torrecuadrada JL. Casal I. Salinas J. Borca MV. Escribano JM. High-yield expression of a viral peptide vaccine in transgenic plants. FEBS Lett. 2001;488:13–17. doi: 10.1016/s0014-5793(00)02405-4. [DOI] [PubMed] [Google Scholar]
- 86.Dus Santos MJ. Wigdorovitz A. Trono K. Ríos RD. Franzone PM. Gil F. Moreno J. Carrillo C. Escribano JM. Borca MV. A novel methodology to develop a foot and mouth disease virus (FMDV) peptide-based vaccine in transgenic plants. Vaccine. 2002;20:1141–1147. doi: 10.1016/s0264-410x(01)00434-0. [DOI] [PubMed] [Google Scholar]
- 87.Obregon P. Chargelegue D. Drake PM. Prada A. Nuttall J. Frigerio L. Ma JK. HIV-1 p24-immunoglobulin fusion molecule: a new strategy for plant-based protein production. Plant Biotechnol J. 2006;4:195–207. doi: 10.1111/j.1467-7652.2005.00171.x. [DOI] [PubMed] [Google Scholar]
- 88.Panahi M. Alli Z. Cheng X. Belbaraka L. Belgoudi J. Sardana R. Phipps J. Altosaar I. Recombinant protein expression plasmids optimized for industrial E. coli fermentation and plant systems produce biologically active human insulin-like growth factor-1 in transgenic rice and tobacco plants. Transgenic Res. 2004;13:245–259. doi: 10.1023/b:trag.0000034619.21613.d0. [DOI] [PubMed] [Google Scholar]
- 89.Daniell H. Ruiz G. Denes B. Sandberg L. Langridge W. Optimization of codon composition and regulatory elements for expression of human insulin like growth factor-1 in transgenic chloroplasts and evaluation of structural identity and function. BMC Biotechnol. 2009;9:33. doi: 10.1186/1472-6750-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hintz-Obertreis P. Krumwieh D. Seiler FR. Development of a rapid, highly sensitive, non-radioactive assay system for hematopoietic growth factors. Behring Inst Mitt. 1991;90:99–103. [PubMed] [Google Scholar]
- 91.Elliott S. Egrie J. Browne J. Lorenzini T. Busse L. Rogers N. Ponting I. Control of rHuEPO biological activity: the role of carbohydrate. Exp Hematol. 2004;32:1146–1155. doi: 10.1016/j.exphem.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 92.Timmins NE. Nielsen LK. Blood cell manufacture: current methods and future challenges. Trends Biotechnol. 2009;27:415–422. doi: 10.1016/j.tibtech.2009.03.008. [DOI] [PubMed] [Google Scholar]