

Abstract
Damage to endothelial glycocalyx impairs vascular barrier function and may contribute to progression of chronic vascular disease. An early indicator is microalbuminuria resulting from glomerular filtration barrier damage. We investigated the contributions of hyaluronic acid (HA) and chondroitin sulfate (CS) to glomerular microvascular endothelial cell (GEnC) glycocalyx and examined whether these are modified by vascular endothelial growth factors A and C (VEGFA and VEGFC). HA and CS were imaged on GEnCs and their resynthesis was examined. The effect of HA and CS on transendothelial electrical resistance (TEER) and labeled albumin flux across monolayers was assessed. Effects of VEGFA and VEGFC on production and charge characteristics of glycosaminoglycan (GAG) were examined via metabolic labeling and liquid chromatography. GAG shedding was quantified using Alcian Blue. NDST2 expression was examined using real-time PCR. GEnCs expressed HA and CS in the glycocalyx. CS contributed to the barrier to both ion (TEER) and protein flux across the monolayer; HA had only a limited effect. VEGFC promoted HA synthesis and increased the charge density of synthesized GAGs. In contrast, VEGFA induced shedding of charged GAGs. CS plays a role in restriction of macromolecular flux across GEnC monolayers, and VEGFA and VEGFC differentially regulate synthesis, charge, and shedding of GAGs in GEnCs. These observations have important implications for endothelial barrier regulation in glomerular and other microvascular beds.
The apical side of endothelial cells is coated with an endothelial surface layer (ESL) composed of a surface-anchored glycocalyx which is itself composed of negatively charged proteoglycans [proteins with glycosaminoglycan (GAG) side chains] and glycoproteins (glycosylated proteins) and a more loosely associated layer of adsorbed plasma proteins.1 The endothelial glycocalyx is 200 nm to 2 μm thick (depending on the vascular bed and also on the visualization technique).2
The glycocalyx mediates shear, attenuates leukocyte and platelet adhesion,2 regulates systemic vascular permeability,3–6 and allows free passage of solutes but limits passage of charged macromolecules.7 The ESL is damaged in reperfusion injury, inflammation and trauma, hypervolemia, atherosclerosis, and diabetes (summarized by Becker et al2). ESL thickness is reduced by hyperglycemic infusions in healthy subjects, correlating with endothelial dysfunction and an increase in vascular permeability,8 and in type 1 diabetes correlating with microalbuminuria,9 suggesting a direct link between endothelial ESL dysfunction and this dysfunction of the glomerular filtration barrier. In mice, infusions of the GAG-specific enzymes hyaluronidase, chondroitinase, and heparinase reduced the glomerular endothelial cell (GEnC) ESL thickness, resulting in reduced charge selectivity and increased macromolecular passage (proteinuria).10 In addition, proteinuria is accompanied with a loss of charge selectivity and a reduction in the core proteins decorin, fibromodulin, and versican in nonobese diabetic mice.11 Salmon et al12 demonstrated an age-related reduction in ESL in glomerular (and other) microvessels of Munich Wistar Frömter rats; this was accompanied by an increase in glomerular albumin permeability, which could be rescued by intravenous injections of lectin. Finally, using doxorubicin (Adriamycin) to induce proteinuria in mice, Jeansson et al13 demonstrated increased fractional clearance of larger molecules in cooled, isolated, perfused kidneys, as well as reduced charge in the glomerular filtration barrier; this was accompanied by reduced synthesis of some core proteins and GAGs in the isolated glomeruli and reduced thickness of the ESL. Taken together, these studies strongly suggest that the ESL also regulates permeability in the glomerular filtration barrier. The glomerular filtration barrier is a tightly regulated filter that restricts macromolecular protein passage while allowing filtration of water and small solutes. Dysfunction of this barrier results in proteinuria, a hallmark of kidney disease. The glomerular filtration barrier consists of a trilayer of GEnCs, a glomerular basement membrane and glomerular epithelial cells (podocytes) whose foot processes interdigitate around the glomerular microvessels. Each of these layers contributes to the regulation of glomerular macromolecular permeability.1,14 The GEnC contribution is increasingly thought to be largely dependent on the ESL.10,15,16
We have previously studied the GEnC ESL in vitro in a human conditionally immortalized (ci) cell line and, using electron microscopy, revealed the presence of an ESL layer measuring 200 nm, which is consistent with recent sophisticated measurements of endothelial glycocalyx in vivo.17 We confirmed the presence of proteoglycan core proteins, as previously demonstrated on human GEnCs,18 and of heparan sulfate (HS), a sulfated GAG.19 Enzymatic removal of HS increased macromolecular protein passage across a monolayer by 40%, whereas electrical resistance (a measure of pathways open to water and small molecules) was not affected. Furthermore, we have shown that high-glucose conditions reduce GEnC ESL and lead to a corresponding increase in macromolecular protein passage across GEnC monolayers,20 demonstrating that the GEnC glycocalyx is present in vitro and plays a functional role.
Glomerular enzyme infusion studies by Jeansson et al10 implicated, in addition to removal of HS, removal of hyaluronic acid (HA) and chondroitin sulfate (CS) in increased fractional albumin clearance. HA and CS are also thought to play a role in systemic macromolecular permeability.21 In contrast to other GAGs, HA is not synthesized in the Golgi apparatus, but rather at the plasma membrane (by HA synthases 1 to 3), and it is not attached to core proteins (reviewed by Genasetti et al22). Furthermore, HA is unbranched and unsulfated, and therefore it does not have a strong negative charge. CS is sulfated on assembly within the Golgi apparatus, where it becomes attached to one of its core proteins (eg, aggrecan or versican), forming a proteoglycan. In the present study, we aimed to determine the contribution of HA and CS to the GEnC glycocalyx through in vitro studies using unique human ciGEnCs and selective enzymes.
Vascular endothelial growth factor A (VEGFA), originally called vascular permeability factor, is a powerful angiogenic growth factor that has profound effects on vascular endothelial behavior (as summarized by Tammela et al23), including GEnC maintenance,24 repair,25 and permeability.26 VEGFA is highly expressed by podocytes.27 Another member of the VEGF family of proteins, VEGFC, is a lymphangiogenic growth factor that can stimulate similar pathways to those of VEGFA in both lymphatic and vascular endothelial cells.28 VEGFC also is expressed by podocytes.29 Podocyte–endothelial signaling through VEGF is vital for GEnC maintenance and permeability regulation. It has been postulated that the effects of VEGFA on microvessel permeability are due in part to partial degradation of the glycocalyx.30 In the present study, we aimed to determine whether VEGFs can modify the GEnC glycocalyx. Our central hypothesis was that CS and HA contribute to GEnC barrier maintenance and that they are modified by VEGFs.
Materials and Methods
Binding Proteins, Primary and Secondary Antibodies, Enzymes and Recombinant Proteins, and GAG
Biotinylated hyaluronic acid binding protein (HABP) [AMS Biotechnology (Europe), Abingdon, UK] was used in conjunction with fluorescein isothiocyanate (FITC)–conjugated avidin (Vector Laboratories, Peterborough, UK; Burlingame, CA). When 10 μg/mL HABP was used, cells were fixed in 100% methanol and blocked in 0.1% bovine serum albumin (BSA; Sigma-Aldrich, Gillingham, UK) for 1 hour at room temperature; otherwise, 4% paraformaldehyde (Sigma-Aldrich) and 5% BSA block was used. Mouse IgM anti-CS (4 μg/mL) was used in conjunction with 1:200 Alexa Fluor 488–conjugated anti-mouse IgM (Santa Cruz Biotechnology, Santa Cruz, CA) and mouse IgG anti–PECAM-1 (R&D Systems, Abingdon, UK; Minneapolis, MN) was used in conjunction with 1:200 Alexa Fluor 568–conjugated anti-mouse IgG (Life Technologies–Invitrogen, Paisley, UK; Carlsbad, CA). Cells were incubated in primary antibodies/binding proteins for 1 hour at room temperature or overnight at 4°C. Cell nuclei were counterstained with 1 μg/mL DAPI (Life Technologies–Invitrogen) for 5 minutes at room temperature. Bovine testicular hyaluronidase (Sigma-Aldrich) was reconstituted in 20 mmol/L sodium phosphate, pH 7, at a concentration of 100 mg/mL and was stored in aliquots at −20°C. Cells and tissue were incubated in hyaluronidase for 1 hour at 37°C. Chondroitin ABC lyase (chondroitinase; AMS Biotechnology) was reconstituted in 0.1% BSA at a concentration of 10 U/mL and was stored in aliquots at −20°C. Cells and tissue were incubated in chondroitinase for 2 hours at 37°C. Human recombinant VEGFA and VEGFC (R&D Systems) were reconstituted in 0.1% BSA at a concentration of 100 μg/mL. VEGFA was used at a concentration of 1 nmol/L and VEGFC at 10 nmol/L, as described previously.31 HA potassium salt from human umbilical cord (∼750 kDa) and CS sodium salt from shark cartilage (both from Sigma-Aldrich) were resuspended in PBS.
Cell Culture
Human ciGEnCs were developed and fully characterized in detail, as described previously.32
Immunofluorescence
The ciGEnCs were treated with 250 μg/mL hyaluronidase or 0.1 mU/mL chondroitinase, or were left untreated, and then were fixed and blocked. Cells were incubated with or without HABP, with anti-CS, or with matched concentration of normal mouse IgM (Santa Cruz Biotechnology). After a wash, cells were incubated in avidin-fluorescein or AF488 conjugated secondary antibodies, and the nuclei were counterstained with DAPI and mounted using Vectashield mounting medium (Vector Laboratories). Cells were imaged using either a Leica SP2 confocal microscope or a Leica AF600 LX wide-field fluorescence microscope (Leica Microsystems, Wetzlar, Germany). Confocal Z-stacks were reconstructed into a three-dimensional image using Volocity software version 5 (PerkinElmer, Waltham, MA).
Specificity of Hyaluronidase and Chondroitinase
The cross-reactivity of hyaluronidase for CS and of chondroitinase for HA was tested by treating differentiated ciGEnCs with each enzyme. Cells were incubated in a 10-fold serial dilution of hyaluronidase or chondroitinase. Cells were then fixed and stained for CS or HA, and the nuclei were counterstained. The fluorescence intensity was quantified on a fluorescence plate reader (Wallac 1420 Victor2; PerkinElmer) using 485-nm excitation and 530-nm emission filters (fluorescein) or 360-nm excitation and 460-nm emission filters (umbelliferone). Readings were normalized to DAPI and expressed as fold change relative to the untreated condition.
Quantitative HA and CS Recovery
ciGEnCs in a black 96-well plate (Appleton Woods, Birmingham, UK) were washed then incubated in serum-free endothelial basal medium (EBM-2; Lonza, Walkersville, MD) containing vehicle, 1 μg/mL hyaluronidase, and 1 mU/mL heat-inactivated chondroitinase or 1 mU/mL chondroitinase. A portion of the cells was fixed immediately (background); the remainder was left for a further 24 hours at 37°C in serum-free medium containing vehicle. These cells were washed, fixed, blocked, and stained for HA or CS, and the nuclei were counterstained. The fluorescence signal was quantified as described above, with fluorescence data expressed as fold change relative to background. Representative images were captured as described above.
Endohm Chamber TEER
Transendothelial electrical resistance (TEER), a measure of ion flux, is inversely related to the fractional area of pathways open to water and small molecules across a cell monolayer. TEER was measured as described previously.31 GEnCs were seeded at 100,000/cm2 and thermoswitched from 33° to 37° C at 70% confluency. This ensured that when proliferation ceased the cells formed a tight, but not overcrowded, monolayer. Experiments were performed in serum-free EBM-2 medium (for hyaluronidase treatment) or 1% EBM-2 (for chondroitinase treatment). Hyaluronidase, chondroitinase, heat-inactivated chondroitinase, or vehicle was used at a final concentration of hyaluronidase ranging from 0 to 1 mg/mL or of chondroitinase ranging from 0 to 10 mU/mL; TEER was measured again at 6 hours.
Electrical Cell-Substrate Impedance Sensing
An electrical cell-substrate impedance sensing (ECIS) system (Applied BioPhysics, Troy, NY) was used for real-time measurements of TEER, as described previously.19,20,31 GEnCs were seeded at 100,000/cm2 and thermoswitched from 33° to 37° C at 70% confluency. Cells were treated with vehicle or with 10 or 100 μg/mL CS or HA in serum-free medium. Resistance was expressed as fold change relative to vehicle.
FITC-BSA Passage
Transmonolayer permeability to macromolecules was assessed by measuring passage of FITC-labeled BSA (FITC-BSA; Sigma-Aldrich) across the monolayer, essentially as described previously.19,20,31 Treatments were performed as for the Endohm chamber measurements described above in the presence of 100 μg/mL FITC-BSA.
6-[3H]Glucosamine Labeling of Cells
GEnCs were incubated in 20 μCi/mL of 6-[3H]glucosamine (PerkinElmer), as described previously,20,33 in the presence of vehicle, VEGFA, or VEGFC in complete medium for 48 hours. Secreted GAG [conditioned medium (CM)] was removed, and cells were treated with 20 mmol/L ammonium hydroxide (Sigma-Aldrich) to remove cell-associated GAG (lysate) without removing underlying matrix. Samples were incubated in an equal volume of 200 μg/mL pronase in pronase buffer (100 mmol/L Tris-HCl pH 8 and 0.05% sodium azide, Sigma-Aldrich) for 24 hours at room temperature, to digest GAG from core proteins. Lysates were centrifuged at 580 × g to remove debris. All samples were then concentrated using spin columns with a 3-kDa cutoff (Merck Millipore, Darmstadt, Germany; EMD Millipore, Billerica, MA).
Ion Exchange and Size-Exclusion Chromatography Protocol
The concentrated samples were diluted in fresh urea buffer [7 mol/L urea (Fisher Scientific UK Ltd, Loughborough, UK), 0.05 mol/L sodium acetate, pH 6 (Sigma-Aldrich)] and injected onto a HiTrap diethylaminoethanol (DEAE)–Sepharose anion exchange column (GE Healthcare, Chalfont St Giles, UK). Samples were subjected to a salt gradient up to 0.15 mol/L NaCl for 10 minutes, then up to 0.3 mol/L, 0.4 mol/L, 0.5 mol/L, 0.6 mol/L, and 0.7 mol/L NaCl (Sigma-Aldrich) over 10 minutes each, and then a final gradient up to 2 mol/L NaCl for 5 minutes at a flow rate of 0.6 mL/minute. Debris was eluted in 0.15 mol/L NaCl, unsulfated GAG was eluted in 0.3 mol/L NaCl, and sulfated GAG was eluted in 0.4 to 0.5 mol/L NaCl (low), 0.5 to 0.6 mol/L NaCl (medium), and 0.6 to 0.7 NaCl (highly sulfated). Samples of fractions and of stock sample (experimental input) were mixed with 70% ethanol and then with scintillation fluid, and were read on a scintillation counter (Packard 2200CA; PerkinElmer). The quantity of counts injected onto the column were calculated.
Size-Exclusion Chromatography
Ion exchange fraction pairs were treated with 50 μg/mL HA (Sigma-Aldrich), heparin sodium, and CS (from shark cartilage; Sigma-Aldrich), mixed with three volumes of ice-cold 95% ethanol/1% potassium acetate (Sigma-Aldrich) on ice, then centrifuged at 2400 × g at 4°C. Supernatant was discarded; the pellet was dried, resuspended, and split into hyaluronidase digested and undigested portions in hyaluronidase buffer (sodium acetate, 0.05% sodium azide and 0.15 mol/L NaCl, pH 6), then diluted in fresh urea buffer and injected onto a Sephacryl S-500 size-exclusion column (GE Healthcare). The flow rate of the size-exclusion column was kept at 0.5 mL/minute, and a total of 55 fractions were collected at 63 to 228 minutes after injection.
Fractions and stock sample (experimental input) were mixed with scintillation fluid and were read on a scintillation counter. Both the ion exchange column and the size-exclusion column were pre-equilibrated in 7 mol/L urea before sample was injected.
Quantitative Real-Time PCR
For quantitative real-time PCR, GEnCs were treated with vehicle, VEGFA or VEGFC for 1 hour in complete medium. mRNA was extracted using TRIzol reagent (Life Technologies–Invitrogen) according to the manufacturer’s instructions. The quality and quantity were checked using a nanospectrophotometer (Geneflow, Lichfield, UK), and 2 μg was reverse-transcribed using a high-capacity RNA-to-cDNA kit (Life Technologies–Applied Biosciences) according to the manufacturer’s instructions. Primers were used to amplify bifunctional heparan sulfate N-deacetylase/N-sulfotransferase 2 (NDST2), an enzyme that adds sulfate groups to HS: NDST2 forward 5′-AGCACCGCAAAGAGTTCTGG-3′ and reverse 5′-TGTTGAGCCTCATCTGGTCAG-3′; housekeeping gene GAPDH forward 5′-AAGGTGAAGGTCGGAGTCAAC-3′ and reverse 5′-GGGGTCATTGATGGCAACAATA-3′. The efficiency of the primers was tested using a 10-fold serial dilution from 10 to 1 × 107 of GEnC cDNA (data not shown). Real-time PCR was performed using a StepOne 96-well plate real-time PCR system (Life Technologies–Applied Biosystems, Foster City, CA) with an initial hold for 10 minutes at 95°C, followed by 40 cycles of 15 seconds at 95°C and 1 minute at 60°C, and then a melt curve for 15 seconds at 95°C, 1 minute at 60°C, and 15 seconds at 95°C. The 2−ΔΔCT value (relative fold change) was calculated.
Alcian Blue Assay
GEnCs were treated with vehicle, VEGFA, or VEGFC in serum-free medium for 48 hours. The medium was removed from cells and was concentrated using 95% ethanol, as described above. The pellet was resuspended in PBS and incubated with Alcian Blue (a cationic dye that binds to charged GAG; Sigma-Aldrich), as described previously.34 Absorbance was read at 490 nm on a fluorescence plate reader. GAG release was quantified using a standard curve of CS (from shark cartilage; Sigma-Aldrich) ranging from 0 to 160 μg/mL.
TACE Activation Assay
GEnCs were treated with vehicle, VEGFA, or VEGFC in complete medium for 24 hours. The medium was removed and activation of ADAM17 [alias TNF-alpha-converting enzyme (TACE)] was measured in cell lysate using a fluorimetric SensoLyte 520 TACE (α-secretase) activity assay kit (AnaSpec, Fremont, CA) according to the manufacturer’s instructions. In brief, cells were lysed and incubated at 4°C for 10 minutes. The cell suspension was centrifuged for 10 minutes at 2500 × g at 4°C; the supernatant was then removed, warmed to room temperature, and incubated with the TACE substrate solution for 60 minutes. The assay buffer was used instead of lysate as a background control. The fluorescence intensity was measured by excitation at 490 nm and emission at 535 nm. Background readings were subtracted from experimental readings, and then were expressed as fold change relative to vehicle.
Statistical Analysis
All statistical analyses were performed on a minimum of three separate experiments. In experiments with multiple treatments, a one-way analysis of variance was used with Dunnett’s multiple comparison post hoc test, unless indicated otherwise. If two variables were being tested, then a two-way analysis of variance was used with Bonferroni post hoc testing. A P value of <0.05 was considered statistically significant. Data are expressed as means ± SEM.
Results
HA and CS Are Expressed by GEnCs in Vitro
Confocal imaging and three-dimensional reconstruction of GEnCs with HABP and anti-CS demonstrated presence of both HA and CS on the cell surface of GEnCs, as expected for the glycocalyx (Figure 1, A and E). Omission of HABP (Figure 1B) or use of normal mouse IgM (Figure 1F) yielded minimal staining, compared with normal HA staining (Figure 1C) and normal CS staining (Figure 1G), as did pretreatment with hyaluronidase (Figure 1D) and chondroitinase (Figure 1H), thus demonstrating specificity of staining.
Figure 1.

HA and CS form part of the cell surface layer of ciGEnCs. ciGEnCs grown on coverslips were left untreated (A–C, E–G) or were treated with 250 μg/mL hyaluronidase (D) or 0.1 mU/mL chondroitinase (H). The cells were then fixed and immunostained with biotinylated HABP (A, C, and D) (green) or no HABP (B) in conjunction with streptavidin–Alexa Fluor 488 and anti-CS (green) (B, G, and F) or matched IgM control (F) in conjunction with anti-mouse IgM Alexa Fluor 488 and counterstained with DAPI (blue). Three-dimensional images were reconstructed from confocal Z-stacks (A, B, E, and F) or images were acquired with an upright fluorescence microscope (C, D, G, and H).
Specificity of Hyaluronidase and Chondroitinase
Hyaluronidase and chondroitinase were used as tools to remove HA and CS, respectively. Hyaluronidase catabolizes HA,35 but it also catabolizes CS at a slower rate36; chondroitinase catabolizes CS, but also HA at a lower pH. Chondroitinase significantly removed CS at concentrations down to 0.01 mU/mL, but it significantly removed HA only at the maximum concentration used (Figure 2, A and B). Hyaluronidase significantly removed HA at concentrations down to 0.01 μg/mL, but it significantly removed CS only down to the 100 μg/mL concentration (Figure 2, C and D). In these experiments, 1 mg hyaluronidase equates to approximately 700 U activity. In subsequent experiments, 1 μg/mL hyaluronidase and 1 mU/mL chondroitinase were used.
Figure 2.

Specificity of hyaluronidase and chondroitinase. ciGEnCs, grown on 96-well plates, were treated with chondroitin ABC lyase for 2 hours at 37°C (A and B) or with hyaluronidase for 1 hour at 37°C (C and D) and then were fixed and immunostained with either biotinylated HABP (B and C) or anti-CS (A and D). The last concentration at which enzyme significantly affected fluorescence intensity is indicated by an arrow. Data were analysed using one way ANOVAs [overall P < 0.0001 (A), P < 0.001 (B), P < 0.0001 (C), P < 0.01 (D)]. Data are expressed as means ± SEM. n = 6. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001, Dunnett’s multiple comparison post hoc test.
GEnCs Synthesize HA and CS
Fetal calf serum in GEnC medium contains GAGs, which can be adsorbed to the glycocalyx. To confirm synthesis of both HA and CS by GEnCs, recovery assays were developed. Hyaluronidase minimized HABP signal (Figure 3A). After 24 hours, there was a 1.7 ± 0.14-fold recovery in HABP signal, compared with a 2.1 ± 0.12-fold change in untreated cells (Figure 3, B, C, and G). Fetal calf serum (5%) was used as a positive control, demonstrating a 2.18 ± 0.21-fold change relative to background (data not shown). Chondroitinase minimized CS staining (Figure 3D). After 24 hours, there was a 1.9 ± 0.19-fold recovery in signal, compared with a 2.44 ± 0.19-fold change with vehicle (Figure 3, E–H). The recovery of HABP and anti-CS over time demonstrates that ciGEnCs have the cellular machinery to resynthesize HA and CS in vitro.
Figure 3.

ciGEnCs synthesize CS and HA in vitro. ciGEnCs, grown on 96-well plates were treated with either vehicle or 1 mg/mL hyaluronidase for 1 hour at 37°C (A–C) or with 1 mU/mL chondroitinase or 1 mU/mL heat-inactivated chondroitinase for 2 hours at 37°C (D–F). Cells were then washed extensively and a portion was fixed (A and D), with the remainder left for a further 24 hours in serum-free medium (B, C, E, and F). These cells were then fixed, stained for HA (A–C) or CS (D–F), and counterstained with DAPI. The fluorescence intensity of HA (G) and CS (H) was quantified using a plate reader and was normalized to that of DAPI. Data were analyzed using one way ANOVAs (overall P < 0.0001) and expressed as means ± SEM. n = 8. Bonfferoni post hoc tests (versus vehicle) indicated on graphs. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001.
Contribution of HA and CS to the Barrier to Protein Passage and to TEER across GEnC Monolayers
We next investigated the functional contribution of HA and CS to the GEnC barrier by measuring TEER (using both ECIS and Endohm chamber approaches) and macromolecular protein passage (FITC-labeled BSA) after removal. Hyaluronidase had a significant effect on both TEER and FITC-BSA passage only at 1 g/mL hyaluronidase, a dose that does not target HA specifically (Figure 4, A and B). ciGEnCs treated with hyaluronidase as above were stained with VE-cadherin. Hyaluronidase did not affect cell–cell junctions (Figure 4C).
Figure 4.

The functional contribution of HA to ciGEnC barrier properties. A: ciGEnCs were grown on tissue culture inserts or on 96-well ECIS arrays, and the electrical resistance was measured. Cells were then treated with hyaluronidase for 1 hour at 37°C or with HA (ECIS only); the electrical resistance was measured again and normalized to serum-starved conditions. B: The passage of FITC-labeled BSA across the monolayer was then monitored for two consecutive hours. The fluorescence intensity of the aliquots was quantified, and data were expressed as cumulative FITC-BSA over time. C: Monolayer integrity of ciGEnCs, at the doses of hyaluronidase shown in A and B, was assessed by VE-cadherin immunostaining. D: On the ECIS arrays, HA or vehicle was added directly to the monolayer, and resistance was measured in real time over 24 hours. Data are expressed as means ± SEM. A and D analyzed by one way ANOVA (overall P < 0.001 and P < 0.0001 respectively) and B analysed by repeated measures ANOVA (overall P < 0.0001). Bonfferoni post hoc tests indicated on graphs. ∗P < 0.05, ∗∗∗P < 0.001.
If HA contributes directly to GEnC barrier properties, then absorption of HA should increase GEnC resistance immediately. Surprisingly, the addition of 100 μg/mL HA had no effect on GEnC resistance; however, addition of 10 μg/mL HA induced a significant delayed increase after approximately 6 hours, suggesting that the effects of HA on GEnC resistance were not from the action of absorbing HA (Figure 4D). Taken together, these results suggest that HA does not make a direct contribution to either TEER or to the barrier to macromolecular passage.
In contrast to hyaluronidase, chondroitinase reduced ciGEnC TEER at 0.1 mU/mL (a fold decrease of 0.74 ± 0.052; P < 0.01) (Figure 5A), a concentration that does not remove HA. Curiously, the concentration at which macromolecular passage was affected was higher, at 1 mU/mL (2 ± 0.48-fold increase at 2 hours) (Figure 5B), whereas VE-cadherin junctional staining was not affected by chondroitinase (Figure 5C). The addition of 100 μg/mL CS induced an immediate increase in monolayer resistance, as assessed by ECIS (Figure 5D), suggesting that the absorption of CS contributed physically to the barrier, whereas the response to 10 μg/mL CS was not significant, suggesting a dose response to CS. Taken together, these results suggest that CS does contribute to GEnC barrier properties.
Figure 5.

The functional contribution of CS to ciGEnC barrier properties. A: ciGEnCs were grown on tissue culture inserts or on ECIS arrays. Baseline electrical resistance was measured before treatment with chondroitinase at 37°C or with CS. On inserts, the electrical resistance was normalized to baseline conditions. n = 5. B: The passage of FITC-labeled BSA across the monolayer was then monitored for three consecutive hours from 2 hours of treatment. The fluorescence intensity of the aliquots was quantified, and data were expressed as cumulative FITC-BSA over time. n = 4. C: Monolayer integrity of ciGEnCs at the various doses of chondroitinase was assessed by VE-cadherin immunostaining. n = 4. D: On the ECIS arrays, CS or vehicle was added directly to the monolayer, and resistance was measured in real time over 24 hours. n = 4. A analyzed by one way ANOVA (overall P < 0.001), B analysed by repeated measures ANOVA (overall P < 0.05) and C by unpaired t-test (overall P < 0.001). Data are expressed as means ± SEM. Bonfferoni post hoc tests indicated on graphs. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001.
VEGFC Affects Charge of GAGs in GEnCs
Given the importance of VEGF in determining GEnC behavior, including barrier properties,31 we used [3H]glucosamine and chromatography to examine whether VEGFA and VEGFC modify GAG metabolism. This approach ensures that only GAGs synthesized in the presence of treatment will be labeled over a 48-hour period. There was no difference in the disintegration counts per minute (dpm) of CM or lysate between experiments loaded onto the ion exchange column (input) (Figure 6, A and B). In minimally charged fractions (eluted in 0.3 mol/L NaCl), VEGFC induced a significant increase in the amount of radiolabeling in GEnCs (CM plus lysate), but VEGFA did not (P < 0.01) (Figure 6C). Further analysis demonstrated that the increased level of radiolabel occurred in the CM, but not in the cell lysate (P < 0.01) (Figure 6D). When hyaluronidase-digested samples were injected onto a size-exclusion column, the high molecular weight peak (<2000 kDa) shifted to a low molecular weight peak, confirming that the noncharged fractions related to the linear, unsulfated GAG HA (Figure 6E).
Figure 6.

The effect of VEGFA and VEGFC on uncharged GAGs. A and B: GEnCs incubated with vehicle, VEGFA, or VEGFC [3H]glucosamine radiolabel were separated into CM and lysate. Samples were loaded onto a DEAE ion exchange column; fractions were eluted in a NaCl gradient, collected, and read on a scintillation counter. Total disintegration counts are shown for CM (A) and lysate (B), demonstrating equal experimental input. C and D: Total counts are shown for the fraction eluted in 0.3 mol/L NaCl (minimal charge) (C) and separated into CM and lysate (D). E: Uncharged ion exchange fractions were either digested in hyaluronidase (low molecular weight HA) or left undigested (high molecular weight HA) and injected onto a size-exclusion column. Dashed line divides LMW from HMW. Data are expressed as means ± SEM. n = 4. Data were analyzed by one way ANOVA (C, overall P < 0.05) or two way ANOVA (D, overall P < 0.05). Bonfferoni post hoc tests are indicated on graphs. ∗∗P < 0.01 versus vehicle (C), versus CM vehicle (D). MW, molecular weight.
There was no significant change in the amount of charged GAGs in CM or lysate of cells between experiments (Figure 7A), although there were significantly more charged GAGs in CM than in lysate in all experiments (P < 0.01). These results show that VEGFs have no effect on overall synthesis of charged GAGs. Data from CM and lysate were separated into low, medium, and high sulfated fractions, depending on the NaCl concentration at elution. In the CM, VEGFC induced a greater proportion of highly charged GAGs, compared with vehicle (Figure 7B), even though the overall amount was the same (Figure 7A), but there was no difference in the degree of charge between conditions in the cell lysate (Figure 7C). A summary of the combined CM and lysate data (Figure 7D) shows that a preponderance (76 ± 8%) of total charged GAGs were highly charged in the presence of VEGFC (P < 0.01); in contrast, with vehicle the spread was even (32% low, 30% medium, and 38% high), and with VEGFA there was no change from vehicle in total charged GAGs (11% low, 42% medium, and 37% high).
Figure 7.

The effect of VEGFA and VEGFC on GAG charge. A: GEnCs incubated with vehicle, VEGFA, or VEGFC with [3H]glucosamine radiolabel to measure GAG metabolism and were separated into cell lysate and CM. Samples were loaded onto a DEAE ion exchange column; fractions were eluted in a NaCl gradient, collected, and read on a scintillation counter. Total disintegration counts are shown for CM and lysate. B and C: Counts are shown for low (0.4 to 0.5 mol/L NaCl), medium (0.5 to 0.6 mol/L NaCl), and high (0.6 to 0.7 mol/L NaCl) charged fractions in CM (B) and lysate (C). D: Percentage of low, medium, or high sulfated fractions in CM and lysate combined. mRNA was extracted from GEnCs treated with vehicle, VEGFA, or VEGFC for 1 hour. Real-time PCR analysis of NDST2 expression was quantified and normalized to GAPDH. Data are expressed as means ± SEM (A, B, C, and E). One-way analysis of variance (A and E, overall P < 0.01 and P < 0.05 respectively) or two-way analysis of variance (B and D, overall P < 0.01 and P < 0.05 respectively). n = 4. Bonfferoni post hoc tests indicated on graphs. ∗P < 0.05 versus vehicle (E); ∗∗P < 0.01 CM versus lysate (A), high versus low (B), or VEGFC versus vehicle (D).
Because it appeared that VEGFC was modifying the degree of charge of GAG and not the overall amount, we examined whether VEGFC increases the expression of enzymes responsible for adding sulfate groups to GAG (NDST1 and NDST2). cDNA was reverse-transcribed from mRNA extracted from GEnCs stimulated with vehicle, VEGFA, or VEGFC for 1 hour. cDNA was amplified using primers specific for human NDST2 or GAPDH and NDST2 normalised first to GAPDH, then VEGFC treatment to vehicle (2−ΔΔCT). There was a significant increase in NDST2 cDNA in GEnCs stimulated with VEGFC, compared with vehicle (1.6 ± 0.07-fold; P < 0.05) (Figure 7E). There was no significant effect on NDST1 (data not shown).
VEGFA Induces Shedding of Charged GAGs by GEnCs, Potentially by Activation of ADAM17
We have here demonstrated that VEGFC increases the metabolism of highly charged GAGs in GEnC medium, but that it does not affect the amount of charged GAGs overall. To understand whether VEGFC can target the shedding of highly charged GAGs, we used an Alcian Blue colorimetric assay to measure shedding. Standard-curve calculation demonstrated linear sensitivity of the Alcian Blue assay down to 20 μg/mL CS (Figure 8A). Surprisingly, VEGFC had no effect on GAG secretion, but VEGFA increased it, compared with vehicle and VEGFC (P < 0.05) (Figure 8B). To identify a potential mechanism of VEGFA-induced GAG shedding, we measured activation of ADAM17 using a fluorescence activation kit. VEGFA induced a significant increase in ADAM17 activation in cell lysate after 24 hours, compared with vehicle (Figure 8C). Taken together, these results suggest that, although neither VEGFA nor VEGFC increases the synthesis of charged GAGs, VEGFA actively induces GAG shedding, potentially through ADAM17 activation, whereas VEGFC increases the population of highly charged GAGs by GEnCs.
Figure 8.
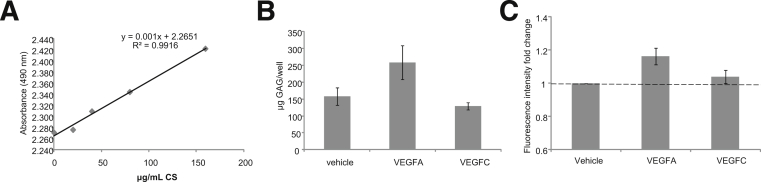
VEGFA induces the shedding of charged GAGs by GEnCs. GEnCs were stimulated with vehicle, VEGFA, or VEGFC for 48 hours. GAGs released from the cell were collected in the cell medium and were ethanol-precipitated. Samples were incubated in an Alcian Blue solution for 15 minutes and absorbance was measured at 490 nm. A: Representative standard curve for CS. B: The quantity of GAG equivalent to μg/mL CS produced per well in each cell fraction for each condition. C: GEnCs treated for 24 hours were lysed and assayed for TACE activation (fluorescence intensity fold change). Data are expressed as means ± SEM (B and C). n = 5. Data were analyzed using one way ANOVAs (overall P < 0.05 for B and C). Bonfferoni post hoc tests not significant and not indicated.
Discussion
We have demonstrated that both HA and CS are expressed on the cell surface of ciGEnCs and that ciGEnCs are capable of synthesizing these GAGs. Targeted HA removal did not affect solute flux or macromolecular passage, although the addition of 10 μg/mL HA did increase TEER, albeit after a delay, in a manner similar to that seen in human lung endothelial cells.37 The higher dose of HA (100 μg/mL) did not increase GEnC integrity, probably because of disorganization of the endothelial cells.38 Targeted removal of CS increased TEER and increased macromolecular passage in GEnCs. This contrasts with what we had previously found with HS removal, which had no effect on TEER in GEnCs but which increased albumin flux.19 In the present study, we saw no evidence of chondroitinase affecting cellular junctions (VE-cadherin), and the enzyme concentrations used did not affect the number of cell nuclei (data not shown), confirming that the enzymes had no effect on cell detachment. Taken together, these data indicate preservation of the monolayer, although it may be that there were subtle junctional effects not detectable with this approach. Addition of CS to the monolayer had an immediate effect on GEnC resistance, suggesting a physical contribution to the barrier. Similarly, when exogenous CS was added to bladder urothelial cells after acid damage, it induced an immediate restoration of barrier function.39 Taken together, these data suggest that CS contributes directly to GEnC barrier properties, but HA does not.
In our in vitro studies, we used TEER as a measure of pathways open to water and to small molecules and used FITC-BSA assays as a measure of macromolecular passage. It is important to note that, although these assays provide an index of hydraulic conductivity and protein permeability in vitro, the extent to which they are representative of the behavior of the glomerular endothelium in vivo is uncertain. Nonetheless, we have previously found good concordance between in vitro and ex vivo findings (eg, for angiopoietin-126,34), at least in terms of the direction and relative size of the effect, if not in the absolute values. The analysis of passage of both size and charge of molecules though the GEnC monolayer under flow (ie, grown within an ECIS capillary) would allow a direct comparison with the in vivo situation. For example, do smaller proteins sieve freely though the GEnC monolayer, and would neutral albumin sieve more readily than charged albumin, as observed in vivo13?
In this investigation of the effects of VEGFA and VEGFC on GAG metabolism, we have demonstrated that VEGFC significantly increased production of HA. Further analysis clarified that the majority of VEGFC-induced HA was secreted. Under control conditions, the majority of charged GAGs (eg, HS, CS, and dermatin sulfate) synthesized within 48 hours were secreted into the medium, whereas the majority of HA remained cell-associated, suggesting that sulfated GAGs may be turned over and released at a higher rate than HA in GEnCs. Of note, there was no significant difference in the amount of charged cell-associated or secreted GAGs induced by VEGFA or VEGFC. Interestingly, however, the greatest proportion of the GAGs released by VEGFC was highly charged, suggesting an increased number of sulfate groups. We went on to confirm that VEGFC, in contrast to VEGFA, increased the expression of NDST2 mRNA, an enzyme that induces N-sulfation of GAG side chains during elongation.40 These results suggest that VEGFC directly affects the degree of sulfation (charge) of GAGs. The degree of sulfation contributes to the overall negative charge of the ESL, which restricts macromolecular protein passage. We have previously shown that VEGFC, in contrast to VEGFA, reduces macromolecular passage across GEnC monolayers,31 and we suggest that the reduced protein permeability is due to the increased negative charge of the GEnC ESL.
VEGFC significantly increased the metabolism of highly sulfated GAGs, the majority of which were released into the medium by 48 hours. The question remained whether VEGFC targeted the shedding of highly sulfated proteoglycans (ie, HS proteoglycans) or whether more of the highly sulfated GAGs were present in the medium simply because more were available. To dissect this possibility, we quantified the secretion of charged GAGs in medium over time in response to VEGFA and VEGFC. Surprisingly, VEGFC had no additional effect to vehicle on GAG secretion, but VEGFA significantly increased the secretion of charged GAGs. Thus, VEGFC metabolizes more highly sulfated GAGs, compared with control, whereas VEGFA induces GAG shedding. At first glance, this appears to contradict the data presented in Figure 7A, which do not show increased GAG release into the medium of VEGFA-treated GEnCs. However, what these data actually show is that VEGFA does not increase the metabolic production of charged GAGs in the medium, compared with control, but does induce shedding of established GAGs. Proteoglycans are cleaved from the plasma membrane by matrix metalloproteinases (sheddases). VEGFA is known to increase shedding by ADAM17,41 which is a sheddase.42 We therefore investigated and confirmed that VEGFA, but not VEGFC, increases ADAM17 activation in GEnCs. This is consistent with the VEGFA-induced shedding of GAGs and suggests that ADAM17 may be involved. Thus, GAG shedding may account for the increase in molecular passage promoted by VEGFA in GEnCs.31 In support of this notion, Fu and Shen30 calculated that the effects of VEGFA on microvessel permeability are likely due in part to partial degradation of the ESL, and Suarez et al43 demonstrated that VEGFA165 increased the secretion, but not synthesis, of CS and HS into cell medium.
VEGFA is vital for normal glomerular function, and dysregulation of VEGFA (ie, increased or decreased expression) is associated with the development of many glomerulopathies.44 It may be that disordered glycocalyx regulation is part of the mechanism of the glomerulopathic effect. In diabetic nephropathy, for example, VEGFA levels are transiently increased, leading to proteinuria44; VEGFA blockade can ameliorate the progression of diabetic nephropathy, but such blockade is not good for long-term kidney physiology.45 GAGs are shed systemically in early diabetes,8,9 and there is evidence that targeted GAG therapy can reduce proteinuria in diabetic patients (and thus protect the glomerular ESL).46 VEGFC signals through the same receptor (VEGFR2) as does VEGFA in GEnCs,31 and it can induce similar maintenance signals as VEGFA.28 Understanding how barrier properties of GEnCs differ between VEGFC and VEGFA (ie, through glycocalyx modification) should allow identification of a potential therapeutic strategy to pharmacologically manipulate VEGFC expression to maintain critical glomerular function without the renotoxic effects associated with total VEGFA signaling blockade. Thus, the present study needs to be taken forward into animal models to confirm the physiological relevance of these findings.
Furthermore, understanding which GAG components contribute functionally to the glycocalyx barrier in the glomerular microvasculature (which is a microvascular bed with an easy clinical readout, microalbuminuria, that may indicate ESL damage), and understanding what may regulate the glycocalyx barrier should help in understanding the role of GAG components in the general vasculature system. Thus, it is important to relate this work to whole glomeruli. The gross morphology of the ESL is thought to differ between vascular beds; however, its regularity over fenestrated (including glomerular) and continuous capillaries is surprisingly similar (as discussed by Salmon and Satchell47).
We suggest that HA does not contribute directly and physically to barrier function in GEnCs in vitro. In contrast to our present findings, Jeansson et al10 demonstrated that hyaluronidase infusion in mice glomeruli led to a decrease in ESL thickness and a subsequent increase in fractional clearance of albumin. In their experiments, however, the hyaluronidase concentration was high enough to potentially act on both HA and CS. Zeng et al,48 however, in an in vitro study in rat fat pad endothelial cells, found that removal of HS, HA, and CS individually had no effect on ESL thickness. This suggests that the density of ESL may be affected, although the authors did not determine whether pore size or protein permeability was affected; interestingly, only removal of both CS and HA together resulted in increased absorption of albumin into the ESL.48 In a study in high-salt-perfused rat kidneys, albumin clearance increased 12-fold but ESL thickness was unchanged, and fractional clearance for Ficoll radii of 55 Å (a measure of the large pore fraction) increased fourfold with no change for Ficoll radii of Å.49 These findings suggest that the density of the ESL was affected, not its thickness.
Henry and Duling21 found that HA contributed to luminal permeation in microvessels of hamster cremaster muscle. Using Streptomyces hyaluronidase, they showed that plasma HA was increased by hyaluronidase treatment, as was the penetration of FITC-dextran 70 into apical ESL of different microvessels; however, they did not demonstrate whether plasma CS levels were also affected, although they did demonstrate that a combination of both HA and CS given after enzyme treatment rescued the effect.21 Thus, the role of HA on capillary permeability was not isolated from that of CS in their study either, which highlights the importance of determining the specificity of GAG enzymes for their substrates in individual systems. In contrast, Landsverk et al50 found that Streptomyces hyaluronidase administered to awake Golden Syrian hamsters increased plasma HA concentrations up to 100-fold, but did not reduce plasma volume. In this model, therefore, there was no vascular leakage caused by HA, which is consistent with our present results. It is important to note, however, that these other studies are all from in vivo situations, which does not allow a direct comparison with our in vitro studies.
HA does not have the same negative charge properties associated with it as do other GAGs,22 which perhaps explains why we saw no immediate effect of HA on barrier properties. HA does play an important role other than forming a physical barrier to macromolecules. Singleton et al37,51 examined the contribution of HA to endothelial barrier function in vitro and described the cell signaling effects of HA as resulting in increased barrier function over time (ie, indirectly). Given that in our study VEGFC modulated HA, it would be interesting to explore further the role of HA in GEnCs.
The regulation of GAGs by VEGFs may be important not only in glomerular physiology but also in aspects of vascular physiology in which GAGs are known to be important (eg, in angiogenesis52) and in the pathology of various vascular diseases in which there is GAG remodeling (eg, in atherosclerosis,53 inflammation and ischemia reperfusion,54 and airway cystic fibrosis,55 to name but a few). Nonetheless, the effects of VEGFs on the GEnC ESL may be unique, so the effects of VEGFs on other systems would have to be addressed individually.
In conclusion, GEnCs synthesize both CS and HA, although only CS contributes directly to barrier function. To our knowledge, ours is the first study of the regulation of the endothelial glycocalyx by VEGFs. Both VEGFA and VEGFC increase HA synthesis: VEGFC metabolizes more highly charged GAGs, and VEGFA induces the shedding of charged GAGs. Taken together, our results suggest that GAG synthesis, sulfation, and shedding may be a mechanism by which VEGFs control protein passage. This understanding has important implications for glomerular filtration control in health and disease and for permeability alterations in other vascular beds.
Acknowledgments
We thank Prof. Jerry Turnbull (University of Liverpool) and Dr. Sally Stringer (University of Manchester) for their advice with the chromatography work.
Footnotes
Supported by a Medical Research Council grant (G0500053 to R.R.F., G.I.W., P.W.M., and S.C.S.), a British Heart Foundation Basic Science Intermediate Fellowship (FS/10/017/28249 to R.R.F. and S.B.), and a Diabetes UK grant (10/0004003 to R.R. and S.C.S.).
References
- 1.Haraldsson B., Nyström J., Deen W.M. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev. 2008;88:451–487. doi: 10.1152/physrev.00055.2006. [DOI] [PubMed] [Google Scholar]
- 2.Becker B.F., Chappell D., Bruegger D., Annecke T., Jacob M. Therapeutic strategies targeting the endothelial glycocalyx: acute deficits, but great potential. Cardiovasc Res. 2010;87:300–310. doi: 10.1093/cvr/cvq137. [DOI] [PubMed] [Google Scholar]
- 3.Malik A.B., Lynch J.J., Cooper J.A. Endothelial barrier function. J Invest Dermatol. 1989;93(2 Suppl):62S–67S. doi: 10.1111/1523-1747.ep12581072. [DOI] [PubMed] [Google Scholar]
- 4.Clough G. Relationship between microvascular permeability and ultrastructure. Prog Biophys Mol Biol. 1991;55:47–69. doi: 10.1016/0079-6107(91)90011-g. [DOI] [PubMed] [Google Scholar]
- 5.Curry F.R. Microvascular solute and water transport. Microcirculation. 2005;12:17–31. doi: 10.1080/10739680590894993. [DOI] [PubMed] [Google Scholar]
- 6.Regele H.M., Fillipovic E., Langer B., Poczewki H., Kraxberger I., Bittner R.E., Kerjaschki D. Glomerular expression of dystroglycans is reduced in minimal change nephrosis but not in focal segmental glomerulosclerosis. J Am Soc Nephrol. 2000;11:403–412. doi: 10.1681/ASN.V113403. [DOI] [PubMed] [Google Scholar]
- 7.van den Berg B.M., Nieuwdorp M., Stroes E.S., Vink H. Glycocalyx and endothelial (dys) function: from mice to men. Pharmacol Rep. 2006;(58 Suppl):75–80. [PubMed] [Google Scholar]
- 8.Nieuwdorp M., van Haeften T.W., Gouverneur M.C., Mooij H.L., van Lieshout M.H., Levi M., Meijers J.C., Holleman F., Hoekstra J.B., Vink H., Kastelein J.J., Stroes E.S. Loss of endothelial glycocalyx during acute hyperglycemia coincides with endothelial dysfunction and coagulation activation in vivo. Diabetes. 2006;55:480–486. doi: 10.2337/diabetes.55.02.06.db05-1103. [DOI] [PubMed] [Google Scholar]
- 9.Nieuwdorp M., Mooij H.L., Kroon J., Atasever B., Spaan J.A., Ince C., Holleman F., Diamant M., Heine R.J., Hoekstra J.B., Kastelein J.J., Stroes E.S., Vink H. Endothelial glycocalyx damage coincides with microalbuminuria in type 1 diabetes. Diabetes. 2006;55:1127–1132. doi: 10.2337/diabetes.55.04.06.db05-1619. [DOI] [PubMed] [Google Scholar]
- 10.Jeansson M., Haraldsson B. Morphological and functional evidence for an important role of the endothelial cell glycocalyx in the glomerular barrier. Am J Physiol Renal Physiol. 2006;290:F111–F116. doi: 10.1152/ajprenal.00173.2005. [DOI] [PubMed] [Google Scholar]
- 11.Jeansson M., Granqvist A.B., Nyström J.S., Haraldsson B. Functional and molecular alterations of the glomerular barrier in long-term diabetes in mice. Diabetologia. 2006;49:2200–2209. doi: 10.1007/s00125-006-0319-z. [DOI] [PubMed] [Google Scholar]
- 12.Salmon A.H., Ferguson J.K., Burford J.L., Gevorgyan H., Nakano D., Harper S.J., Bates D.O., Peti-Peterdi J. Loss of the endothelial glycocalyx links albuminuria and vascular dysfunction. J Am Soc Nephrol. 2012;23:1339–1350. doi: 10.1681/ASN.2012010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jeansson M., Björck K., Tenstad O., Haraldsson B. Adriamycin alters glomerular endothelium to induce proteinuria. J Am Soc Nephrol. 2009;20:114–122. doi: 10.1681/ASN.2007111205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jarad G., Miner J.H. Update on the glomerular filtration barrier. Curr Opin Nephrol Hypertens. 2009;18:226–232. doi: 10.1097/mnh.0b013e3283296044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ballermann B.J. Contribution of the endothelium to the glomerular permselectivity barrier in health and disease. Nephron Physiol. 2007;106:p19–p25. doi: 10.1159/000101796. [DOI] [PubMed] [Google Scholar]
- 16.Satchell S.C., Tooke J.E. What is the mechanism of microalbuminuria in diabetes: a role for the glomerular endothelium? Diabetologia. 2008;51:714–725. doi: 10.1007/s00125-008-0961-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arkill K.P., Neal C.R., Mantell J.M., Michel C.C., Qvortrup K., Rostgaard J., Bates D.O., Knupp C., Squire J.M. 3D reconstruction of the glycocalyx structure in mammalian capillaries using electron tomography. Microcirculation. 2012;19:343–351. doi: 10.1111/j.1549-8719.2012.00168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Björnson A., Moses J., Ingemansson A., Haraldsson B., Sörensson J. Primary human glomerular endothelial cells produce proteoglycans, and puromycin affects their posttranslational modification. Am J Physiol Renal Physiol. 2005;288:F748–F756. doi: 10.1152/ajprenal.00202.2004. [DOI] [PubMed] [Google Scholar]
- 19.Singh A., Satchell S.C., Neal C.R., McKenzie E.A., Tooke J.E., Mathieson P.W. Glomerular endothelial glycocalyx constitutes a barrier to protein permeability. J Am Soc Nephrol. 2007;18:2885–2893. doi: 10.1681/ASN.2007010119. [DOI] [PubMed] [Google Scholar]
- 20.Singh A., Fridén V., Dasgupta I., Foster R.R., Welsh G.I., Tooke J.E., Haraldsson B., Mathieson P.W., Satchell S.C. High glucose causes dysfunction of the human glomerular endothelial glycocalyx. Am J Physiol Renal Physiol. 2011;300:F40–F48. doi: 10.1152/ajprenal.00103.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henry C.B., Duling B.R. Permeation of the luminal capillary glycocalyx is determined by hyaluronan. Am J Physiol. 1999;277:H508–H514. doi: 10.1152/ajpheart.1999.277.2.H508. [DOI] [PubMed] [Google Scholar]
- 22.Genasetti A., Vigetti D., Viola M., Karousou E., Moretto P., Rizzi M., Bartolini B., Clerici M., Pallotti F., De Luca G., Passi A. Hyaluronan and human endothelial cell behavior. Connect Tissue Res. 2008;49:120–123. doi: 10.1080/03008200802148462. [DOI] [PubMed] [Google Scholar]
- 23.Tammela T., Enholm B., Alitalo K., Paavonen K. The biology of vascular endothelial growth factors. Cardiovasc Res. 2005;65:550–563. doi: 10.1016/j.cardiores.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 24.Kitamoto Y., Takeya M., Tokunaga H., Tomita K. Glomerular endothelial cells are maintained by vascular endothelial growth factor in the adult kidney. Tohoku J Exp Med. 2001;195:43–54. doi: 10.1620/tjem.195.43. [DOI] [PubMed] [Google Scholar]
- 25.Ostendorf T., Kunter U., Eitner F., Loos A., Regele H., Kerjaschki D., Henninger D.D., Janjic N., Floege J. VEGF(165) mediates glomerular endothelial repair. J Clin Invest. 1999;104:913–923. doi: 10.1172/JCI6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Satchell S.C., Anderson K.L., Mathieson P.W. Angiopoietin 1 and vascular endothelial growth factor modulate human glomerular endothelial cell barrier properties. J Am Soc Nephrol. 2004;15:566–574. doi: 10.1097/01.asn.0000115397.22519.03. [DOI] [PubMed] [Google Scholar]
- 27.Kretzler M., Schröppel B., Merkle M., Huber S., Mundel P., Horster M., Schlöndorff D. Detection of multiple vascular endothelial growth factor splice isoforms in single glomerular podocytes. Kidney Int Suppl. 1998;67:S159–S161. doi: 10.1046/j.1523-1755.1998.06733.x. [DOI] [PubMed] [Google Scholar]
- 28.Bahram F., Claesson-Welsh L. VEGF-mediated signal transduction in lymphatic endothelial cells. Pathophysiology. 2010;17:253–261. doi: 10.1016/j.pathophys.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 29.Foster R.R., Satchell S.C., Seckley J., Emmett M.S., Joory K., Xing C.Y., Saleem M.A., Mathieson P.W., Bates D.O., Harper S.J. VEGF-C promotes survival in podocytes. Am J Physiol Renal Physiol. 2006;291:F196–F207. doi: 10.1152/ajprenal.00431.2005. [DOI] [PubMed] [Google Scholar]
- 30.Fu B.M., Shen S. Structural mechanisms of acute VEGF effect on microvessel permeability. Am J Physiol Heart Circ Physiol. 2003;284:H2124–H2135. doi: 10.1152/ajpheart.00894.2002. [DOI] [PubMed] [Google Scholar]
- 31.Foster R.R., Slater S.C., Seckley J., Kerjaschki D., Bates D.O., Mathieson P.W., Satchell S.C. Vascular endothelial growth factor-C, a potential paracrine regulator of glomerular permeability, increases glomerular endothelial cell monolayer integrity and intracellular calcium. Am J Pathol. 2008;173:938–948. doi: 10.2353/ajpath.2008.070416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Satchell S.C., Tasman C.H., Singh A., Ni L., Geelen J., von Ruhland C.J., O’Hare M.J., Saleem M.A., van den Heuvel L.P., Mathieson P.W. Conditionally immortalized human glomerular endothelial cells expressing fenestrations in response to VEGF. Kidney Int. 2006;69:1633–1640. doi: 10.1038/sj.ki.5000277. [DOI] [PubMed] [Google Scholar]
- 33.Sörensson J., Björnson A., Ohlson M., Ballermann B.J., Haraldsson B. Synthesis of sulfated proteoglycans by bovine glomerular endothelial cells in culture. Am J Physiol Renal Physiol. 2003;284:F373–F380. doi: 10.1152/ajprenal.00257.2002. [DOI] [PubMed] [Google Scholar]
- 34.Salmon A.H., Neal C.R., Sage L.M., Glass C.A., Harper S.J., Bates D.O. Angiopoietin-1 alters microvascular permeability coefficients in vivo via modification of endothelial glycocalyx. Cardiovasc Res. 2009;83:24–33. doi: 10.1093/cvr/cvp093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Itano N. Simple primary structure, complex turnover regulation and multiple roles of hyaluronan. Cardiovasc Res. 2008;144:131–137. doi: 10.1093/jb/mvn046. [DOI] [PubMed] [Google Scholar]
- 36.Stern R., Jedrzejas M.J. Hyaluronidases: their genomics, structures, and mechanisms of action. Chem Rev. 2006;106:818–839. doi: 10.1021/cr050247k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singleton P.A., Dudek S.M., Ma S.F., Garcia J.G. Transactivation of sphingosine 1-phosphate receptors is essential for vascular barrier regulation. Novel role for hyaluronan and CD44 receptor family. J Biol Chem. 2006;281:34381–34393. doi: 10.1074/jbc.M603680200. [DOI] [PubMed] [Google Scholar]
- 38.West D.C., Kumar S. The effect of hyaluronate and its oligosaccharides on endothelial cell proliferation and monolayer integrity. Exp Cell Res. 1989;183:179–196. doi: 10.1016/0014-4827(89)90428-x. [DOI] [PubMed] [Google Scholar]
- 39.Hauser P.J., Buethe D.A., Califano J., Sofinowski T.M., Culkin D.J., Hurst R.E. Restoring barrier function to acid damaged bladder by intravesical chondroitin sulfate. J Urol. 2009;182:2477–2482. doi: 10.1016/j.juro.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pikas D.S., Eriksson I., Kjellén L. Overexpression of different isoforms of glucosaminyl N-deacetylase/N-sulfotransferase results in distinct heparan sulfate N-sulfation patterns. Biochemistry. 2000;39:4552–4558. doi: 10.1021/bi992524l. [DOI] [PubMed] [Google Scholar]
- 41.Swendeman S., Mendelson K., Weskamp G., Horiuchi K., Deutsch U., Scherle P., Hooper A., Rafii S., Blobel C.P. VEGF-A stimulates ADAM17-dependent shedding of VEGFR2 and crosstalk between VEGFR2 and ERK signaling. Circ Res. 2008;103:916–918. doi: 10.1161/CIRCRESAHA.108.184416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manon-Jensen T., Itoh Y., Couchman J.R. Proteoglycans in health and disease: the multiple roles of syndecan shedding. FEBS J. 2010;277:3876–3889. doi: 10.1111/j.1742-4658.2010.07798.x. [DOI] [PubMed] [Google Scholar]
- 43.Suarez E.R., Nohara A.S., Mataveli F.D., de Matos L.L., Nader H.B., Pinhal M.A. Glycosaminoglycan synthesis and shedding induced by growth factors are cell and compound specific. Growth Factors. 2007;25:50–59. doi: 10.1080/08977190701272701. [DOI] [PubMed] [Google Scholar]
- 44.Foster R.R. The importance of cellular VEGF bioactivity in the development of glomerular disease. Nephron Exp Nephrol. 2009;113:e8–e15. doi: 10.1159/000228078. [DOI] [PubMed] [Google Scholar]
- 45.Zachary I., Gliki G. Signaling transduction mechanisms mediating biological actions of the vascular endothelial growth factor family. Cardiovasc Res. 2001;49:568–581. doi: 10.1016/s0008-6363(00)00268-6. [DOI] [PubMed] [Google Scholar]
- 46.Blouza S., Dakhli S., Abid H., Aissaoui M., Ardhaoui I., Ben Abdallah N., Ben Brahim S., Ben Ghorbel I., Ben Salem N., Beji S., Chamakhi S., Derbel A., Derouiche F., Djait F., Doghri T., Fourti Y., Gharbi F., Jellouli K., Jellazi N., Kamoun K., Khedher A., Letaief A., Limam R., Mekaouer A., Miledi R., Nagati K., Naouar M., Sellem S., Tarzi H., Turki S., Zidi B., Achour A., DAVET (Diabetic Albuminuria Vessel Tunisia Study Investigators) Efficacy of low-dose oral sulodexide in the management of diabetic nephropathy. J Nephrol. 2010;23:415–424. [PubMed] [Google Scholar]
- 47.Salmon A.H., Satchell S.C. Endothelial glycocalyx dysfunction in disease: albuminuria and altered microvascular permeability. J Pathol. 2012;226:562–574. doi: 10.1002/path.3964. [DOI] [PubMed] [Google Scholar]
- 48.Zeng Y., Ebong E.E., Fu B.M., Tarbell J.M. The structural stability of the endothelial glycocalyx after enzymatic removal of glycosaminoglycans. PLoS One. 2012;7:e43168. doi: 10.1371/journal.pone.0043168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fridén V., Oveland E., Tenstad O., Ebefors K., Nyström J., Nilsson U.A., Haraldsson B. The glomerular endothelial cell coat is essential for glomerular filtration. Kidney Int. 2011;79:1322–1330. doi: 10.1038/ki.2011.58. [DOI] [PubMed] [Google Scholar]
- 50.Landsverk S.A., Tsai A.G., Cabrales P., Intaglietta M. Impact of enzymatic degradation of the endothelial glycocalyx on vascular permeability in an awake hamster model. Crit Care Res Pract. 2012;2012:842545. doi: 10.1155/2012/842545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Singleton P.A., Mirzapoiazova T., Guo Y., Sammani S., Mambetsariev N., Lennon F.E., Moreno-Vinasco L., Garcia J.G. High-molecular-weight hyaluronan is a novel inhibitor of pulmonary vascular leakiness. Am J Physiol Lung Cell Mol Physiol. 2010;299:L639–L651. doi: 10.1152/ajplung.00405.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fuster M.M., Wang L. Endothelial heparan sulfate in angiogenesis. Prog Mol Biol Transl Sci. 2010;93:179–212. doi: 10.1016/S1877-1173(10)93009-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karangelis D.E., Kanakis I., Asimakopoulou A.P., Karousou E., Passi A., Theocharis A.D., Triposkiadis F., Tsilimingas N.B., Karamanos N.K. Glycosaminoglycans as key molecules in atherosclerosis: the role of versican and hyaluronan. Curr Med Chem. 2010;17:4018–4026. doi: 10.2174/092986710793205354. [DOI] [PubMed] [Google Scholar]
- 54.Annecke T., Fischer J., Hartmann H., Tschoep J., Rehm M., Conzen P., Sommerhoff C.P., Becker B.F. Shedding of the coronary endothelial glycocalyx: effects of hypoxia/reoxygenation vs ischaemia/reperfusion. Br J Anaesth. 2011;107:679–686. doi: 10.1093/bja/aer269. [DOI] [PubMed] [Google Scholar]
- 55.Reeves E.P., Bergin D.A., Murray M.A., McElvaney N.G. The involvement of glycosaminoglycans in airway disease associated with cystic fibrosis. ScientificWorldJournal. 2011;11:959–971. doi: 10.1100/tsw.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]