

Abstract
BACKGROUND:
The underlying mechanisms of hepatitis C virus (HCV) resistance to treatment are unknown. Signal transducers and activators of transcription (STAT) proteins play a critical role in antiviral defense.
OBJECTIVE:
To explore some of the mechanisms of HCV resistance to interferon, the expression of STAT1 and its negative regulators, protein inhibitor of activated STAT (PIAS1) and suppressor of cytokine signalling (SOCS3), in liver tissues of both inteferon responders and nonresponders in chronic HCV patients.
METHODS:
Sixty patients were divided into the following groups: group 1a comprised 38 treatment-responder chronic HCV patients; group 1b consisted of 22 treatment-nonresponder chronic HCV patients; and group 2 consisted of six control subjects. Liver biopsies were examined for histological scoring; STAT1, SOCS3 and PIAS1 expression was analyzed using Western blotting methods.
RESULTS:
STAT1 expression in the liver tissue of patients in group 1 was significantly increased compared with group 2 patients (P=0.001), while no significant difference in expression was observed between group 1a and group 1b patients (P=0.747). However, phosphorylated STAT1 protein was expressed at a significantly higher level in liver tissue of patients in group 1a compared with patients in group 1b (P=0.001). Western blot analysis of PIAS1 and SOCS3 protein expression in liver tissues from groups 1 and 2 revealed significantly increased expression in group 1 compared with group 2 (P=0.001). In addition, PIAS1 and SOCS3 protein expression was significantly higher in the liver tissues of patients in group 1b compared with patients in group 1a.
CONCLUSION:
Levels of STAT1 and/or the protein expression of its negative regulators, PIAS1 and SOCS3, may be a good predictor of response to therapy. These could be used as biomarkers that are easily detected by Western blotting or immunostaining during standard histopathological liver biopsy analysis.
Keywords: Chronic hepatitis C, HCV, HCV therapy response, IFN, IFN therapy, PIAS1, SOCS3, STAT1
Abstract
HISTORIQUE :
On ne connaît pas les mécanismes sous-jacents de la résistance au traitement du virus de l’hépatite C (VHC). Les protéines des transducteurs de signal et des activateurs de transcription (STAT) jouent un rôle essentiel dans la défense antivirale.
OBJECTIF :
Explorer certains mécanismes de la résistance du VHC à l’interféron, de l’expression de la STAT1 et de ses régulateurs négatifs, de l’inhibiteur protéique de la STAT activée (PIAS1) et du suppresseur de la signalisation des cytokines (SOCS3) dans les tissus hépatiques de patients atteints du VHC chronique répondant ou non à l’ interféron.
MÉTHODOLOGIE :
Soixante patients ont été répartis dans les groupes suivants : le groupe 1a se composait de 38 patients atteints du VHC chronique qui répondaient au traitement, le groupe 1b, de 22 patients atteints du VHC chronique qui ne répondaient pas au traitementet le groupe 2, de six sujets témoins. Les chercheurs ont examiné leur biopsie hépatique pour établir leur indice histologique et ont analysé l’expression des protéines STAT1, SOCS3 et PIAS1 au moyen du transfert Western.
RÉSULTATS :
L’expression de la STAT1 dans le tissu hépatique des patients du groupe 1 était significativement plus élevée que celle des patients du groupe 2 (P=0,001), mais il n’y avait pas de différence significative de l’expression entre les patients du groupe 1a et ceux du groupe 1b (P=0,747). Cependant, la protéine STAT1 phosphorylée était exprimée à un taux considérablement plus élevé dans les tissus hépatiques des patients du groupe 1a que dans ceux du groupe 1b (P=0,001). L’analyse de l’expression des protéines PIAS1 et SOCS3 des tissus hépatiques des groupes 1 et 2 par transfert Western a révélé une expression considérablement plus élevée dans le groupe 1 que dans le groupe 2 (P=0,001). En outre, l’expression des protéines PIAS1 et SOCS3 était considérablement plus élevée dans les tissus hépatiques des patients du groupe 1b que dans ceux des patients du groupe 1a.
CONCLUSION :
Le titrage de la STAT1 phosphorylée ou de l’expression protéique de ses régulateurs négatifs, les protéines PIAS1 et SOCS3, pour-rait être un bon prédicteur de la réponse au traitement. On pourrait l’utiliser comme biomarqueur facilement décelé par transfert Western ou par immunocoloration pendant l’analyse histopatologique standard de la biopsie hépatique.
Hepatitis C virus (HCV) infection has been recognized as a major public health problem worldwide. HCV infection is a significant cause of chronic liver disease, with frequent progression to cirrhosis and an elevated risk for the development of hepatocellular carcinoma (1). HCV infects more than 170 million people worldwide (3% of the global population) (2). Approximately 13% of Egyptians are positive for HCV antibodies and approximately 9.8% are HCV RNA positive. This means that almost 10% of the total population of Egypt is infected and are infectious to other people (3).
Among the six major HCV genotypes found worldwide, genotype 4 is the most prevalent in Egypt, with genotype 4a the dominant subtype (4). However, no effective vaccine is available, and current treatment consists of the combination of interferon-alpha (IFN-α) and a nucleotide analogue, ribavirin (RBV) (2). Unfortunately, patients infected with HCV genotype 4 show poor responses to IFN-α therapy compared with those with genotypes 2 and 3, but a similar response to those infected with HCV genotype 1b. Genotype 4 HCV RNA clearance has been recorded in only 22.2% of patients (5). The mechanism of viral resistance to IFN are not well defined. Thus, a major challenge to the HCV research community is to elucidate such mechanisms (2).
IFN is one of the most potent innate antiviral cytokines. Its activity against viruses is believed to depend on both activation of IFN stimulatory genes and modulation of the immune system. IFN intracellular signalling occurs mainly through the Janus kinase (JAK) family, and the JAK signal transducer and activator of transcription (STAT) signalling pathways (2). STATs are a family of cellular proteins that mediate transduction of extracellular signals from growth factors and cytokines, such as IFNs and interleukin-6, and directly regulate transcription (6). The interaction of these cytokines with specific cell surface receptors triggers activation of STATs through phosphorylation by the receptor-associated JAK kinases. Once phosphorylated, STATs homo- or heterodimerize and translocate into the nucleus where they regulate the transcription of STAT-responsible genes (7). It is well documented that the activation of STAT1, STAT2 and STAT3 is essential for the antiviral effect of IFNs (8,9).
Negative regulators of STATs include suppressor of cytokine signalling (SOCS) family members and protein inhibitor of activated STATs (PIAS). SOCS1 and SOCS3 prevent phosphorylation of STATs by binding to receptor-associated Jak kinases. PIAS1 and PIAS3 inhibit binding of STAT1 and STAT3, respectively, to the promoter of target genes thus preventing gene transcription (10,11).
STATs and their regulators are essential to antiviral mechanisms within HCV-infected cells and, to our knowledge, no similar studies have been conducted in Egyptian patients to date. The present study aimed to analyze the expression of STAT1 and its negative regulators, SOCS3 and PIAS1, in liver cells and to to detect any relationships between their expression.
METHODS
Although 112 patients were initially enrolled in the present study, only 60 HCV RNA-positive patients (36 men, 24 women) with ages ranging from 26 to 45 years fulfilled the conclusion criteria and continued to the end of the follow-up period.
Inclusion criteria
All patients demonstrated anti-HCV antibodies by ELISA and positive viral RNA using reverse transcriptase polymerase chain reaction. The diagnosis of chronic hepatitis C was further confirmed by positive liver biopsy. Only patients between 20 and 60 years of age, and with mild or moderate liver fibrosis according to the Ishak classification (3), were included.
Exclusion criteria
Patients who consumed alcohol, were coinfected with hepatitis B virus or HIV, or demonstrated the presence of other liver or systemic disease were excluded from the analysis. In addition, patients with a body mass index >30 kg/m2, hemoglobin <120 g/L, white blood cell count <3×109/L, platelet count <80×109/L, serum bilirubin >20.52 μmol/L, serum albumin <35 g/L, or refusal to participate in the study were also excluded. None of the patients had received antiviral therapy before sample collection.
Patients were followed up at outpatient clinics of the Tropical Medicine and Internal Medicine Departments, Tanta University Hospitals (Egypt) for 18 months during the period from February 2008 to August 2009, to assess treatment responders and nonresponders. Informed written consent was obtained from all patients. The present study was approved by the ethics committee of the institution and the recruited subjects gave their informed consent for genetic analysis. Normal liver samples were obtained from six patients (serving as control) at laparotomy performed for cholecystectomy in five cases and hiatal hernia in one case. The control subjects had not received anti-inflammatory therapy previous to the procedure and, in all cases, histological examination of the liver biopsies revealed normal liver architecture.
Treatment and criteria for response to therapy
All patients fulfilled the criteria for IFN therapy, received pegylated IFN (peg-IFN alpha-2a [40 Kd, PEGASYS, F Hoffmann-La Roche, Switzerland]) at a dose of 180 mg once weekly plus RBV given at doses of 1000 mg/day if body weight was ≤75 kg and of 1200 mg/day if >75 kg. The duration of treatment was 48 weeks. Patients who did not achieve in serum HCV RNA level at week 12 of therapy a reduction of >2 log10 were considered to be treatment failures and discontinued HCV therapy prematurely (12,13). Similarly, patients with detectable serum HCV RNA at week 24 were also considered to represent treatment failures and stopped HCV treatment. Sustained virological response was defined as an undetectable HCV RNA level six months after the end of treatment. Viral load testing was repeated at 12 weeks to assess patients who achieved an early virological response, at the end of treatment to assess end-of-treatment virological response, and six months later to confirm a sustained virological response (14). Follow-up of patients was performed during treatment and for six months after treatment to detect responders and nonresponders. Follow-up of these patients during treatment and six months after treatment revealed 38 patients as responders and 22 as nonresponders.
The subjects were divided into the following groups:
-
Group 1: 60 chronic HCV patients who were subdivided into two groups according to treatment response.
○ Group 1a: 38 treatment-responder chronic HCV patients.
○ Group 1b: 22 treatment-nonresponder chronic HCV patients.
Group 2 (control group): Six subjects with normal liver biopsy taken during a laparotomy procedure.
All patients in the present study were subjected to the following:
Detailed history taking and full clinical examination.
Laboratory investigation including complete blood count, blood urea and serum creatinine, liver function tests, viral markers for HCV and hepatitis B using commercially available ELISA kits (Abbott GmbH, Germany), fasting and postprandial blood glucose level, antinuclear antibodies, thyroid stimulating hormone, alpha-fetoprotein, HIV and cytomegalovirus.
Quantitative estimation of HCV RNA level (15).
Body mass index
Fundus examination
Electrocardiography
Abdominal ultrasonography
Liver biopsy examination: Liver biopsies from all patients were reviewed for the purpose of the present study. The liver biopsies were divided into two samples: one was used for histological examination and the other was used for protein extraction.
Histological assessment
Paraffin-embedded sections of specimens were stained with hematoxy-lin and eosin, Masson’s trichrome and periodic acid-Schiff after diastase digestion. All specimens were evaluated using the Ishak modified histological activity index (3). Inflammation scoring ranged from 0 to 18 and included assessment of periportal hepatitis, confluent necrosis, focal necrosis and portal inflammation. Fibrosis scoring ranged from 0 to 6, distinguishing mild architectural changes (1 to 2) from fibrosis (3 to 4) and cirrhosis (5 to 6).
Protein extraction from liver tissue
Liver tissue was homogenized in 300 μL of Nonidet P-40 (Shell Chemical Co, USA) lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% Nonidet P-40, containing 1× protease inhibitor cocktail [Roche Diagnostic Corporation, USA]) for 1 h at 4°C. Cell debris was removed by centrifugation at 12,000 rpm for 15 min. The supernatant was collected and protein concentration was determined using the Bio-Rad reagent (Bio-Rad Laboratories, USA) (7).
Western blot analysis of STAT1, SOCS3 and PIAS1 expression
Equal amounts of liver protein extract (80 μg) were fractionated on 10% Bis-Tris gels (Novex NuPAGE precast mini gels, Invitrogen, USA) and transferred to polyvinylidene difluoride membranes (Bio-Rad, USA). Membranes were incubated with anti-STAT1 (Epitomics Inc, USA), anti-STAT1 phospho (ps727) (Epitomics Inc, USA), anti-SOCS3 (Cell Signaling Technologies, USA) and anti-PIAS1 primary antibodies (Abcam, USA). Binding of the primary antibodies was detected by incubating the membranes with the corresponding secondary antibody. The blots were then developed using an enhanced chemiluminescence system (Amersham, USA) according to the manufacturer’s instructions. Signals were visualized by exposure to x-ray film (X-Omat films, Kodak Company, USA). To ensure equal loading of the protein, the membranes were stripped in stripping buffer (1 M Tris, 20% SDS, and 0.8% beta-mercaptoethanol) at 55°C for 20 min, and incubated with antiactin (Sigma Chemical Co, USA) primary antibody. The actin signals were developed by sequential incubation of the membranes with the corresponding secondary antibody and enhanced chemiluminescence system followed by exposure to x-ray film. Signals were scanned using Alpha Imager 3.2 software (IS-1000 Digital Imaging System, USA) and normalized to that of actin (16) (Figures 1 and 2).
Figure 1.
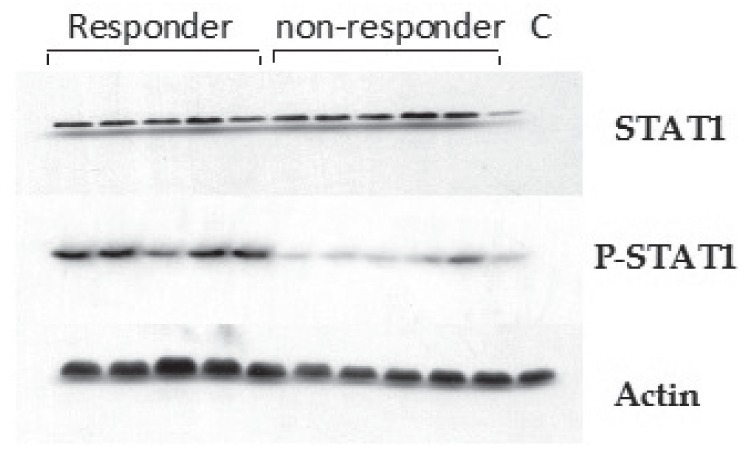
Western blot of signal transducer and activator of transcription (STAT)1 and phosphorylated STAT1 (P-STAT1). Total cell extracts were prepared from liver tissue and subjected to sodium dodecyl sulphate poly-acrylamide electrophoresis. Western blot analysis was performed using the corresponding antibody. To ensure equal loading, blots were stripped and incubated with antiactin antibody. C Control
Figure 2.
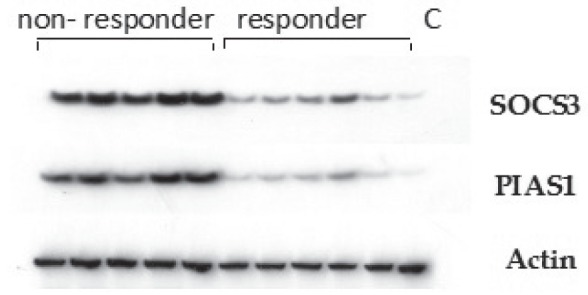
Western analysis of suppressor of cytokine signalling (SOCS)3 and protein inhibitor of activated signal transducer (PIAS)1 protein expression in liver tissue of interferon responders and nonresponders of chronic hepatitis C patients. C Control
Statistical analysis
Analysis of the data was performed using SPSS version 16 (IBM Corporation, USA). The unpaired Student’s t test was used to test statistical differences between two groups for quantitative data. Comparison of nonparametric quantitative data in two different groups using their mean rank was performed using the Mann-Whitney U test (Z) (17). Correlational analysis between independent variables was performed using the Spearman rank correlation test; P<0.05 was considered to be statistically significant.
RESULTS
A total of 112 patients were initially enrolled in the study; however, only 60 (34 men, 26 women) with ages ranging from 26 to 45 years were included in the analysis. These patients were subdivided into 38 HCV treatment responders (group 1a) and 22 HCV treatment nonresponders (group 1b), while six subjects with normal liver biopsies represented the control group (ie, group 2).
The clinical characteristics of the patients and controls are summarized in Table 1. Serum levels of aspartate aminotransferase and alanine aminotransferase were significantly higher in group 1 compared with group 2 (P<0.05), while no difference was observed between group 1 and group 2 with regard to age, body mass index, albumin and total serum bilirubin level (P>0.05).
TABLE 1.
Characteristics of hepatitis C virus (HCV) and control groups enrolled in the study
| Characteristics |
Group
|
||
|---|---|---|---|
| HCV (n=60) | Control (n=6) | P | |
| Age, years | 38.9±11.8 | 39. 4±13.5 | 0.922 |
| Sex, male/female, n/n | 36/24 | 4/2 | – |
| Body mass index, kg/m2 | 23.1±4.87 | 22.7±4.15 | 0.847 |
| AST, U/L | 52.55±11.41 | 24.17±5.84 | 0.001* |
| ALT, U/L | 61.22±14.74 | 26.17±4.16 | 0.001* |
| Albumin, g/L | 39.0±3.0 | 42±2.0 | 0.020 |
| Total bilirubin, mg/dL | 20.52±6.8 | 15.39±1.71 | 0.074 |
| Viral load, ×105 | 6.69±6.81 | – | – |
| STAT1 | 407.9±72.48 | 20.83±4.44 | 0.001* |
| STAT1-phosphorylated | 161.3±110.1 | 17.33±4.36 | 0.002* |
| PIAS1 | 503.4±306.1 | 26.67±7.99 | 0.001* |
| SOCS3 | 244.0±176.2 | 32.17±12.19 | 0.005* |
Data presented mean ± SD unless otherwise indicated.
Statistically significant (ie, P<0.05). ALT Alanine aminotransferase; AST Aspartate aminotransferase; PIAS Protein inhibitor of activated suppressor of cytokine signalling (STAT); SOCS Suppressor of cytokine signalling
Comparison between group 1a and group 1b showed a significant difference with respect to serum alanine aminotransferase level (P<0.05), while no difference was observed between group 1a and group 1b with respect to age, body mass index, aspartate aminotransferase, serum albumin, viral load or histological findings (P>0.05) (Table 2).
TABLE 2.
Characteristics of hepatitis C virus (HCV) patients
|
HCV group (n=60)
|
|||
|---|---|---|---|
| Characteristics | Group 1a (responders [n=38]) | Group 1b (nonresponders [n=22])) | P |
| Age, years | 38.6 ±0.8 | 39.3 ±10.9 | 0.573 |
| Sex, male/female, n/n | 23/15 | 13/9 | – |
| Body mass index, kg/m2 | 22.8±11.35 | 23.3±5.11 | 0.846 |
| AST, U/L | 50.91±11.15 | 53.5±11.6 | 0.401 |
| ALT, U/L | 55.05±13.07 | 64.79±14.62 | 0.012* |
| Albumin, g/L | 39.0±13.0 | 38.0±6.0 | 0.735 |
| Total bilirubin, mg/dL | 18.81±1.7 | 20.52±6.84 | 0.146 |
| Viral load, ×105 | 7.37±8.06 | 6.3±6.06 | 0.562 |
| Histological finding | |||
| Activity score | 4.0±1.81 | 4.1± 2.86 | 0.869 |
| Fibrosis score | 1.8±0.77 | 2.2±0.8 | 0.061 |
| STAT1 | 411.9±77.5 | 405.5±40.37 | 0.747 |
| STAT1-phosphorylated | 281±80.8 | 91.95±46.46 | 0.001* |
| PIAS1 | 142.0±52.41 | 712.7±158.8 | 0.001* |
| SOCS3 | 45.82±16.43 | 358.7±111.8 | 0.001* |
Data presented as mean ± SD unless otherwise indicated.
Statistically significant (ie, P<0.05). ALT Alanine aminotransferase; AST Aspartate aminotransferase; BMI Body mass index; PIAS Protein inhibitor of activated suppressor of cytokine signalling (STAT); SOCS Suppressor of cytokine signalling
To examine the activation state of the JAK-STAT signalling pathway in chronic HCV patients and analyze its effect on their response to antiviral therapy, STAT1 protein expression was examined in pre-treatment liver tissue by Western blotting. STAT1 expression was significantly increased in the liver tissue of group 1 compared with group 2 patients (P=0.001). However, no difference was observed between group 1a and group 1b patients (P=0.747). The present study demonstrated a positive correlation between STAT1 protein expression and viral load in the IFN responder (ie, group 1a) (r=0.651; P=0.001) (Figures 3 and 4) and nonresponder patients (group 1b) (r=0.735; P=0.001). No correlation was found between STAT1 expression and serum levels of the measured liver enzymes.
Figure 3.
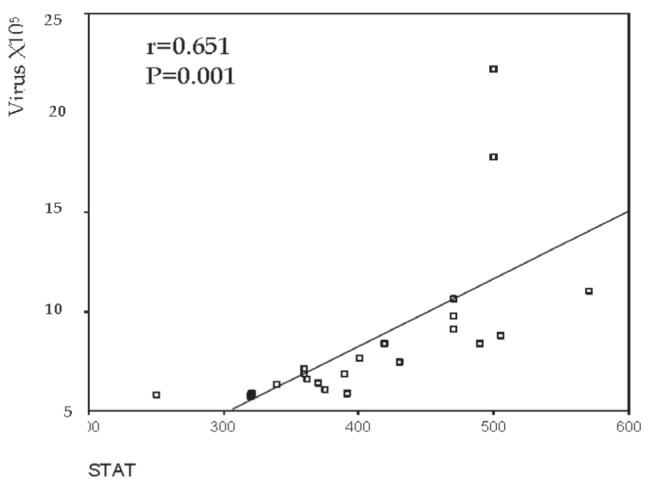
Correlation between signal transducer and activator of transcription1 (STAT1) and viral load in treatment-responder patients (ie, group 1a)
Figure 4.
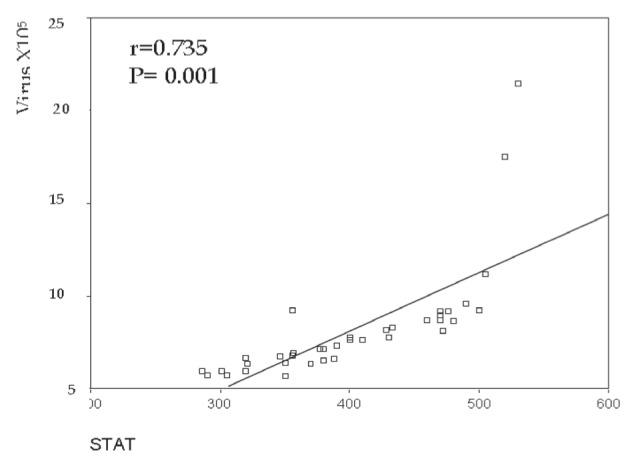
Correlation between signal transducer and activator of transcription (STAT)1 and viral load in treatment nonresponder patients (ie, group 1b)
Because the active forms of STAT proteins involved in targeted gene transcription are phosphorylated, the expression of phosphorylated STAT1 using specific anti-STAT1 phosphoantibody, which detects STAT1 phosphorylation at serine residue 727, was tested. Phosphorylated STAT1 protein was expressed at a significantly higher level in the liver tissue of group 1a compared with group 1b patients (P=0.001) (Table 2), with no correlation between phosphorylated STAT1 expression and viral load in group 1a (r=0.249; P=0.263) and group 1b (r= −0.155; P=0.353).
Western blotting of PIAS1 and SOCS3 protein expression in liver tissues from group 1 and group 2 revealed significantly increased expression in group 1 compared with group 2 (P=0.001). PIAS1 and SOCS3 protein expression were significantly higher in the livers of group 1b patients (P=0.001) compared with group 1a patients (Table 2).
PIAS1 showed no correlation with measured liver enzyme levels or viral load in group 1a (r=0.375) or group 1b (r= −0.225; P=0.175). Moreover, SOCS3 showed no correlation with measured liver enzymes or viral load in group 1a (r= −0.138; P=0.539) or group 1b (r= −0.222; P=0.181).
DISCUSSION
Approximately one-half of chronic HCV patients do not respond to current IFN/RBV combination therapy. The response rates of therapy vary depending on viral and host factors. However, factors determining responsiveness are not well understood. With the exception of some clinical and biochemical factors (the degree of inflammation and degree of fibrosis), there are no other markers that serve as valid predictors of response to therapy (18).Therefore, assessment of biomarkers that predict the response to IFN in HCV patients is necessary.
In the present study, we analyzed the status of STAT1, the key regulator of IFN signalling and protein expression of its negative regulators PIAS1 and SOCS3 in the liver tissue of pretreatment chronic HCV patients using Western blotting techniques (18).
STAT proteins are part of the signalling pathway for IFN. Dysregulation of STAT expression is involved in cellular resistance to viral infection (19). Several studies have shown that activation of STATs pathways are predictive of treatment outcome in chronic HCV infection (20,21). The present work showed increased STAT1 protein expression in HCV-infected liver tissue relative to control tissue, with a positive correlation with viral load but no differences between IFN responder and nonresponder patients. In accordance with our findings, Bautista et al (19) and Kim et al (20) demonstrated overexpression of the STAT proteins STAT1, STAT2, and STAT5 in HCV-infected liver tissue relative to healthy donors, with no difference between IFN responders and nonresponders; however, the increased level of STAT proteins paralleled the degree of liver injury. In another study, STAT1 messenger RNA expression correlated positively with a higher inflammatory score in chronic HCV-infected liver tissue (7). In contrast, a previous study reported decreased STAT1 expression in cells transfected with HCV genomic constructs. This discrepancy was largely due to the forced cellular expression of HCV proteins in the absence of viral replication in this in vitro study (22).
Interestingly, the activated form of STAT1 (ie, phosphorylated STAT1) showed significant overexpression in IFN responder patients, with nearly blocked expression in nonresponders. These observations are consistent with the antiviral activity of STAT proteins. Supporting this finding, Larrea et al (7) used an HCV replicon system to show that HCV replication blocked activation of STAT1, STAT2 and STAT3 after activation with IFN-α. This indicates interference of HCV with the IFN-α-induced JAK-STAT signalling cascade. Helbig et al (23) confirmed this finding and reported that IFN-α-mediated STAT activation was blocked in hepatic cell lines containing an HCV genomic replicon. In a study to identify the HCV proteins that block STAT signalling (24), the NS3/4a protein has been shown to inhibit phosphorylation of STAT1 at serine residue 727. These studies suggest that HCV proteins play an important role in the escape of HCV from the IFN system.
The observation that STAT1 phosphorylation is blocked in treatment nonresponders prompted us to investigate STAT1 activation inhibitors. Among the suppressors of the JAK-STAT signalling pathway are the SOCS3 and PIAS1 proteins. SOCS3, a member of SOCS family, has been shown to inhibit IFN-mediated antiviral activities and IFN signalling (1). The members of PIAS are special inhibitors of STATs that inhibit STAT-mediated gene activation by blocking the DNA binding activity of STATs. PIAS1 is an exclusive inhibitor of STAT1 (1).
To determine the role of PIAS1 and SOCS3 in blocking the signalling pathway of IFN and therapy resistance in chronic HCV infection, we analyzed the expression of the STAT1 inhibitors, PIAS1 and SOCS3 in the liver tissue of pretreatment HCV patients. Results showed overexpression of both PIAS1 and SOCS3 in HCV patients resistant to IFN therapy, with no correlation with viral load or serum levels of liver enzymes. Consistent with this observation, Zhu et al (24), Kim et al (20) and Miyaaki et al (25) reported that IFN-resistant HCV-infected hepatic cells exhibited enhanced SOCS3 expression and that blocking SOCS3 partially restores IFN sensitivity. Moreover, post-treatment relapse was associated with increased expression of SOCS3 and PIAS3 (20). The authors concluded that SOCS3 expression in the liver before initiation of IFN therapy may be a useful predictor of HCV clearance by IFN. In another study (19), intense PIAS1 immunostaining was observed in IFN-resistant HCV-infected hepatic cells compared with sensitive cells.
CONCLUSION
The present study demonstrated the blocked activation of the STAT1 protein and a significant abundance of the STAT1 inhibitors SOCS3 and PIAS1 in the liver tissue of chronic HCV patients resistant to IFN/RBV therapy. These observations suggest that alteration of JAK/STAT signalling, either by blocking the activation of STAT proteins or the upregulation of the STAT-negative regulators SOCS3 and PIAS1, represent a potential mechanism by which HCV resists IFN therapy and, therefore, levels of STAT1, SOCS3 and PIAS1 expression could be used as pretreatment predictors of response to therapy in HCV patients.
Footnotes
DISCLOSURES: The authors have no financial disclosures or conflicts of interest to declare. The protocol for the research project was approved by Tanta Faculty of Medicine Ethics Committee.
REFERENCES
- 1.Ye L, Wang X, Wang S, et al. CD56+ T cells inhibit hepatitis C virus replication in human hepatocytes. Hepatology. 2009;49:753–62. doi: 10.1002/hep.22715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu H, Nelson DR, Crawford JM, Liu C. Defective JaK-Stat activation in hepatoma cells is associated with hepatitis C viral INF- α resistance. J Interferon and Cytokine Res. 2005;25:528–39. doi: 10.1089/jir.2005.25.528. [DOI] [PubMed] [Google Scholar]
- 3.Mohamed MK. Epidemiology of HCV in Egypt. Afro-Arab Liver J. 2004;3:41–52. [Google Scholar]
- 4.Abdel-Hamid M, El-Daly M, Molnegren V, et al. Genetic diversity in hepatitis C virus in Egypt and possible association with hepatocellular carcinoma. J Gen Virol. 2007;88:1526–31. doi: 10.1099/vir.0.82626-0. [DOI] [PubMed] [Google Scholar]
- 5.El-Zayadi A, Simmonds P, Dabbous H, Prescott L, Selim O, Andy A. Response to interferon α of Egyptian patients infected with hepatitis C virus genotype 4. J Viral Hepat. 2007;1:261–4. doi: 10.1111/j.1365-2893.1996.tb00052.x. [DOI] [PubMed] [Google Scholar]
- 6.Ihle JN. The Stat family in cytokine signaling. Curr Opin Cell Biol. 2001;13:211–17. doi: 10.1016/s0955-0674(00)00199-x. [DOI] [PubMed] [Google Scholar]
- 7.Larrea E, Aldabe R, Fernandez-Rodriguez CM, Ametzarra A, Civeira MP, Prieto J. Altered expression and activation of signal transducers and activators of transcription [STATs] in hepatitis C virus infection: In vivo and in vitro studies. Gut. 2006;55:1188–96. doi: 10.1136/gut.2005.070060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang CH, Murti A, Peffefer LM. STAT3 complement defects in an interferon resistant cell line: Evidence for an essential role for STAT3 in interferon signaling and biological activities. Proc Natl Acad Sci USA. 1998;95:5568–72. doi: 10.1073/pnas.95.10.5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heim MH. Intracellular signaling and antiviral effects of interferons. Dig Liver Dis. 2000;32:257–63. doi: 10.1016/s1590-8658(00)80831-2. [DOI] [PubMed] [Google Scholar]
- 10.Liao J, Fu Y, Shuai K. Distinct roles of the NH2- and COOH-terminal domains of the protein inhibitor of the activated signal transducer and activator of transcription [STAT]1 [PIAS1] in cytokine induced PIAS1-STAT1 interaction. Proc NatI Acad Sci USA. 2000;97:5267–72. doi: 10.1073/pnas.97.10.5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blindanbacher A, Duong FH, Hunziker L, et al. Expression of hepatitis C virus proteins inhibits interferon alpha signaling in the liver of transgenic mice. Gastroenterology. 2003;124:1465–75. doi: 10.1016/s0016-5085(03)00290-7. [DOI] [PubMed] [Google Scholar]
- 12.Alberti A, Clumeck N, Collins S, et al. Short statement of the first European Consensus Conference on the treatment of chronic hepatitis B and C in HIV co-infected patients. J Hepatol. 2005;42:615–24. doi: 10.1016/j.jhep.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 13.Strader DB, Wright T, Thomas DL, et al. Diagnosis, management, and treatment of hepatitis C. Hepatology. 2004;39:1147–71. doi: 10.1002/hep.20119. [DOI] [PubMed] [Google Scholar]
- 14.Veldt BJ, Heathcote EJ, Wedemeyer H, et al. Sustained virologic response and clinical outcomes in patients with chronic hepatitis C and advanced fibrosis. Ann Intern Med. 2007;147:677–84. doi: 10.7326/0003-4819-147-10-200711200-00003. [DOI] [PubMed] [Google Scholar]
- 15.Berg T, Meller AR, Platz KP, et al. Dynamics of virus C viremia early after orthotopic liver transplantation indicates extrahepatic tissues as the predominant site of virus C replication. Hepatology. 1999;29:245–9. doi: 10.1002/hep.510290121. [DOI] [PubMed] [Google Scholar]
- 16.Eid MA, Lewis RW, Kumar MV. Mifepristone pretreatment overcomes resistance of prostate cancer cells to tumor necrosis factor a-related apoptosis-inducing ligand [TRAIL] Mol Cancer Ther. 2002;1:831–40. [PubMed] [Google Scholar]
- 17.Aviva P, Caroline S. Glossary of terms (Appendix D) In: Medical Statistics at a Glance. 2nd edn. Oxford: Blackwell Publishing; 2005. pp. 144–152. [Google Scholar]
- 18.Ehrmann J, Aiglova K, Palas J, Kumpel P. The roles of STAT proteins in the regulation of the response to the interferon alpha therapy in chronic hepatitis C. Vnitr Lek. 2006;52:167–72. [PubMed] [Google Scholar]
- 19.Bautista D, Bermudez-Silva FJ, Lasarte JJ, Rodriguez-Fonseca E. Liver expression of proteins controlling interferon-mediated signaling as predictive factors in the response to therapy in patients with hepatitis C virus infection. J Pathol. 2007;213:347–55. doi: 10.1002/path.2214. [DOI] [PubMed] [Google Scholar]
- 20.Kim KA, Lin W, Shao RX, et al. Hepatic SOCS3 expression is strongly associated with non-response to therapy and race in HCV and HCV/HIV infection. J Hepatol. 2009;50:705–11. doi: 10.1016/j.jhep.2008.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyaaki H, Ichikawa T, Nakao K, et al. Predictive value of the phosphorylation of signal transducers and activators of transcription in the outcome of interferon therapy for chronic hepatitis C. Intervirology. 2008;51:384–9. doi: 10.1159/000205264. [DOI] [PubMed] [Google Scholar]
- 22.Lin W, Choe WH, Hiasa Y, et al. Hepatitis C virus expression suppresses interferon signaling by degrading STAT1. Gastroenterology. 2005;128:1034–41. doi: 10.1053/j.gastro.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 23.Helbig KJ, Yip E, McKartney EM, Erye NS, Beard MR. A screening method for identifying disruptions in interferon signaling reveals HCV NS3/4a disrupts Stat1 phosphorylation. Antiviral Res. 2008;77:169–76. doi: 10.1016/j.antiviral.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 24.Zhu H, Nelson DR, Crawford JM, Liu C. Defective Jak-Stat activation in hepatoma cells in associated with hepatitis C viral IFN-α resistance. J Interferon Cytokin Res. 2005;25:528–39. doi: 10.1089/jir.2005.25.528. [DOI] [PubMed] [Google Scholar]
- 25.Miyaaki H, Ichikawa T, Nakao K, et al. Predictive value of suppressor of cytokine signal 3 [SOCS3] in the outcome of interaction therapy in chronic hepatitis C. Hepatol Res. 2009;39:850–5. doi: 10.1111/j.1872-034X.2009.00529.x. [DOI] [PubMed] [Google Scholar]