

Abstract
In our previous work, including analysis of more than 10,000 sera from control patients and patients with a variety of liver diseases, we have demonstrated that with the use of recombinant autoantigens, antimitochondrial autoantibodies (AMA) are only found in PBC and that a positive AMA is virtually pathognomonic of either PBC or future development of PBC. Although the mechanisms leading to the generation of AMA are enigmatic, we have postulated that xenobiotic-induced and/or oxidative modification of mitochondrial autoantigens is a critical step leading to loss of tolerance. This thesis suggests that a severe liver oxidant injury would lead to AMA production. We analyzed 217 serum samples from 69 patients with acute liver failure (ALF) collected up to 24 months post-ALF, compared with controls, for titer and reactivity with the E2 subunits of pyruvate dehydrogenase (PDC-E2), branched chain 2-oxo-acid dehydrogenase (BCOADC-E2) and 2-oxo-glutarate dehydrogenase (OGDC-E2). AMA were detected in 28/69 (40.6%) ALF patients with reactivity found against all of the major mitochondrial autoantigens. In addition, and as further controls, sera were also analyzed for autoantibodies to gp210, Sp100, centromere, chromatin, soluble liver antigen (SLA), tissue transglutaminase (tTG) and deaminated gliadin peptides (DGP) where the most frequently detected non-mitochondrial autoantibody was against tTG (57.1% of ALF patients). In conclusion, the strikingly high frequency of AMA in ALF supports the thesis that oxidative stress-induced liver damage may lead to AMA induction. The rapid disappearance of AMA in these patients provides further support for the contention that PBC pathogenesis requires additional factors including genetic susceptibility.
Keywords: Tolerance, Oxidative injury, Self-reactivity, Epitopes
Introduction
Environmental factors, in particular xenobiotics have been implicated in the etiology of PBC (1-3). Specifically, it has been demonstrated that in addition to the native mitochondrial autoantigens, antimitochondrial autoantibodies (AMA) also recognize xenobiotic modified E2 subunits of pyruvate dehydrogenase (PDC-E2) (4). Several such candidate xenobiotics include chemicals found in detergents, food additives and cosmetics (4-6). Interestingly, while immunization of experimental animals with xenobiotic modified E2 subunits induces true self-reacting AMA, it does not appear to be able to induce clinical liver disease (7). Fulminant hepatic failure, also known as acute liver failure (ALF) is a clinical condition that reflects both oxidant stresses and hepatic damage. ALF is characterized by severe and sudden liver cell dysfunction leading to coagulopathy and hepatic encephalopathy in previously healthy persons with no known underlying liver disease (8). While more than 43% of patients with ALF survive without a transplant, 28% die, and 29% undergo liver transplantation (9).
The hepatic insult secondary to acetaminophen (APAP, paracetamol, N-acetyl-p-aminophenol) consumption has a major oxidant component; toxic doses of APAP produce reactive oxygen and nitrogen species (ROS/RNS) and reactive metabolites in experimental animals or cultured cells (10, 11). Importantly, AMA induction and perpetuation appears sensitive to the oxidative state of the tissues involved (12-14).We hypothesized that in patients with acute liver failure, AMA may be induced by the combination of oxidative stress and liver cell death resulting in exposure of the immune system to both native and modified intracellular proteins. This transient induction of AMA could proceed to PBC if the immune response occurs in genetically susceptible subjects (15, 16).
The objective of this study was to make use of serum samples of patients with ALF obtained by the Acute Liver Failure Study Group to investigate if and to what extent these patients’ sera contain AMA (17). In addition, we have also analyzed sera for autoantibodies against a number of additional autoantigens including gp210, sp100, centromere, chromatin, soluble liver antigen (SLA), tissue transglutaminase (tT) and deaminated gliadin peptide (DGP). Here we report the results of our analysis of autoantibody reactivity of 217 serum samples collected post-ALF (days 1-7), at 12 months and at 24 months long term follow up (LTFU), from 69 subjects with ALF.
Materials and Methods
Sera
The US Acute Liver Failure Study Group comprises 23 sites around the United States that has developed a prospective registry of cases of ALF of all etiologies since 1998 (for more information and a list of the sites see www.acuteliverfailure.org). More than 1,100 patients have been enrolled with detailed clinical information; serum and DNA available on most patients. All patients enrolled meet criteria for encephalopathy (any grade) and coagulopathy (International Normalized Ratio [INR] ≥ 1.5). In this study, 217 serum samples from 69 subjects (46 females, 23 males) with a clinical diagnosis of ALF were selected to represent the standard etiologies associated with ALF and where for the most part follow-up samples had been obtained during initial hospitalization (days 1-7; all 69 patients), at 12-month post-ALF (n=27) and at 24 months post-ALF (n=14). Of the 217 serum samples, 176 samples were obtained from subjects between days 1-7 (86 samples obtained between days 1-4, 90 sera samples obtained between days 5- 7) for all 69 subjects, 27 samples at 12 months and 14 subjects at 24 months long term follow up (LTFU). The causes of ALF included APAP (n=26), drug induced liver injury (n=11), autoimmune hepatitis (n=9), hepatitis A virus (HAV) infection (n=3), hepatitis B virus (HBV) infection (n=4), indeterminate (n=6), miscellaneous (n=10). Sera from patients with PBC (n=15) and healthy volunteers (n=81) from our laboratory sera banks were used throughout as positive and negative controls as previously detailed (17).
Clones
Recombinant glutathione fusion proteins containing the AMA epitopes of PDC-E2, BCOADCE2, OGDC-E2 and MIT3 (a triple hybrid containing the immunodominant epitopes of PDC-E2, BCOADC-E2 and OGDC-E2) were cloned and expressed in pGEX-4T-1. Briefly, a 414bp EcoRI fragment coding for the PDC-E2 amino acid residues 91-228 which encodes the inner lipoyl domain and part of the outer lipoyl domain was amplified by polymerase chain reaction, purified and cloned into the EcoRI site of pGEX-4T-1, transformed and expressed in E. coli DH5α. Likewise, the BCOADC-E2 epitope (amino acid residues 1-118) and OGDC-E2 epitope (amino acid residues 67-147) were amplified by polymerase chain reaction, cloned and expressed in the Bam HI site and Not-I site of pGEX-4T-1 respectively (17, 18). Successful cloning and expression were confirmed by DNA hybridization and immunoassay using monoclonal antibodies to PDC-E2, BCOADC-E2 and OGDC-E2 (19). An irrelevant control Met e I in plasmid pGEX-4T-1 was also purified as described (20)
Detection of AMA
All assays for AMA were performed blindly at two sites by P.L. at University of California, Davis and by H.M. at Teikyo University using similar methodology. Sera from subjects with ALF and controls were diluted 1:1,000, and analyzed for AMA reactivity against the triple hybrid MIT3 (17). ALF sera that were positive to MIT3 were absorbed against an irrelevant protein control, Met e I, (20) and then retested for reactivity against MIT3 and Met e I by immunoblotting. Briefly, recombinant MIT3 and Met e I were resolved individually by SDS-PAGE at 30 mA constant current. SDS-PAGE resolved proteins were transferred to nitrocellulose filters (Micron Separations Inc., Westboro, MA) and cut into 3mm strips, blocked with 3% milk in phosphate buffered saline (PBS) and probed with sera from patients with ALF for one hour at room temperature. After three 10 minute washes with PBS/0.05% tween 20 (PBS-tween), the filters were incubated with horse-radish peroxidase conjugated anti-human Ig, washed with PBS-tween and developed by chemiluminescence using the Supersignal West Pico Chemiluminescence Substrate (Pierce, Rockford, IL). Sera from AMA positive PBC patients and AMA negative controls were used as controls throughout. Sera that were positive to MIT3 after absorption but not the irrelevant protein Met e I, were scored as AMA positive. All ALF sera were also assayed for reactivity against recombinant proteins of PDC-E2, BCOADC-E2 and OGDC-E2 at 1:500 sera dilution by immunoblotting as described previously (17). AMA positive samples were assayed for titer to recombinant mitochondrial autoantigens by ELISA as described (21). Positive AMA reactivity is defined as more than 3X OD 405 ± SD than that of the healthy volunteers.
Detection of autoantibodies against gp210, Sp100, centromere, chromatin, soluble liver antigen (SLA), tissue transglutaminase (tTG) and deaminated gliadin peptides (DGP)
IgG to gp210, Sp100, centromere, chromatin, SLA, tTG and DGP were determined using ELISA kits (INOVA Diagnostics, San Diego, CA) to each of the autoantigens. Briefly, precoated ELISA plates were incubated with 100ul of diluted patient sera (1:100) for 30 minutes at room temperature and then washed three times with HRP wash buffer. The plates were then incubated with 100μl of HRP conjugated goat anti-human IgG for 30 minutes and washed three times with HRP wash buffer. Reactivity was detected by incubating with TMB in the dark for 30 minutes. The reaction was terminated by the addition of 100μl of stop solution and the absorbance read at 405nm. In all assays, low positive, high positive and negative controls were included.
Statistical Analysis
Autoantibody reactivity between ALF sera and normal sera against MIT3, gp210, Sp100, centromere, chromatin, SLA, tTG and DGP were analyzed by Student's t-test. The frequency of AMA positive sera between ALF sera and the estimated prevalence of PBC were compared statistically by Fisher's exact tests using SAS v. 9 software. The statistical differences between male and female ALF subjects were analyzed by Student's t-test. Values of p<0.05 were considered statistically significant.
Results
Serological autoantibody profile of ALF and healthy volunteers
In ALF subjects, the most frequent detected autoantibodies are directed against tTG and MIT3, with 36/69 (57.1%) and 23/69 (33.3%) of the ALF subjects reacted to tTG and MIT3 respectively. Only 1/69, 6/69, 3/69. 5/69, 1/69, 36/69 and 8/69 of the ALF subjects reacted with gp210, Sp100, centromere, chromatin, SLA, tTG and DGP respectively (Table 1). 0/81, 0/81, 2/81, 3/81, 0/81, 1/81, 1/81 and 3/81 of the normal sera reacted with MIT3, gp210, Sp100, centromere, chromatin, SLA, tTG and DGP respectively.
Table 1.
Autoantibody reactivity of ALF sera
| Antigen | Number of Reactive ALF Subjects (%) | Number of Reactive Normal Subjects (%) |
|---|---|---|
| MIT3 | 23/69 (33.3)* | 0/81 (0)* |
| Gp210 | 1/69 (1.4) | 0/81 (0) |
| Sp100 | 6/69 (8.7) | 2/81 (2.5) |
| Centromere | 3/69 (4.3) | 3/81 (3.7) |
| Chromatin | 5/69 (7.2) | 0/81 (0) |
| SLA | 1/69 (1.4) | 1/81 (1.2) |
| tTG | 36/69 (57.1)*# | 1/81 (1.2)* |
| DGP Screen | 8/69 (11.6) | 3/81 (3.7) |
p<0.0001 between ALF and control subjects, determined by Student's t test.
Reactivity to tTG was only transient. Only 1 subject remained tTG positive at 24 months post-ALF.
Detection of AMA by immunoblotting
AMA positive serum samples from subjects with ALF have the same AMA specificity as observed in sera from patients with PBC to recombinant PDC-E2, BCOADC-E2 and OGDC-E2 by immunoblot (Figure 1). AMA were detected in 28/69 (40.6%) ALF subjects. Serum samples from 22/69 and 20/69 subjects collected from days 1-4 and days 5-7 respectively were found to have AMA against one or more mitochondrial autoantigens. 8/27 subjects had AMA at 12 months post-ALF, which included two subjects who were not AMA positive until 12 months LTFU. One subject remained AMA positive even at 24 months LTFU (Table 2). At 1-4 days post ALF, 18/69, 6/69 and 7/69 reacted against PDC-E2, BCOADC-E2 and BCOADC-E2 respectively. At 5-7 days post-ALF, 16/69, 5/69 and 6/69 subjects reacted against PDC-E2, BCOADC-E2 and BCOADC-E2 respectively. At 12 months post-ALF, 4/27, 2/27 and 4/27 of ALF subjects reacted against PDC-E2, BCOADC-E2 and BCOADC-E2 respectively. 1/14 subjects remained AMA positive at 24 months post-ALF and reacted against OGDC only.
Figure 1.
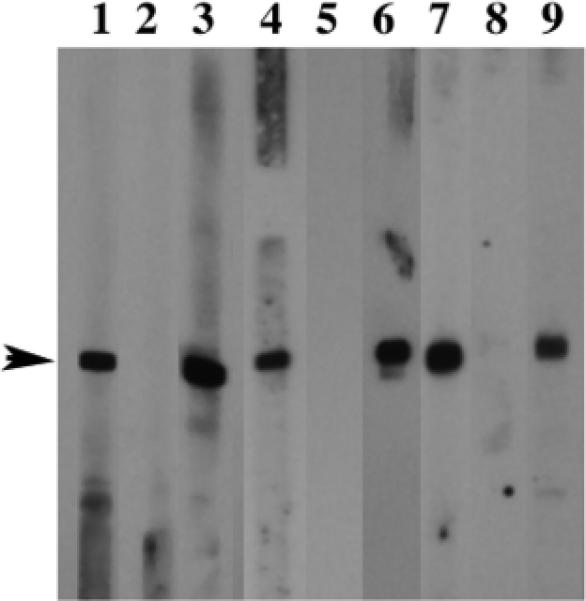
Ig reactivity to recombinant mitochondrial autoantigens. Purified recombinant PDC-E2 (lanes 1-3), BCOADC-E2 (lanes 4-6) and OGDC-E2 (lanes 7-9) were probed with sera from subjects with acute liver failure (lanes 1, 4 and 7); sera from healthy volunteers (lanes 2, 5 and 8) and sera from patients with PBC (lanes 3, 6 and 9) at 1: 250 dilution. Note that sera from ALF subjects and patients with PBC reacted to recombinant PDC-E2, BCOADC-E2 and OGDCE2. Sera from healthy volunteers did not react.
Table 2.
Time course of AMA reactivity and antigen specificity to PDC-E2, BCOADC-E2 and OGDC-E2
| Time of Sample Collection | No. of AMA + Subjects/ Total No. of subjects* | No. of subjects reacted with PDC-E2 | No. of subjects reacted with BCOADC-E2 | No. of subjects reacted with OGDC-E2 |
|---|---|---|---|---|
| Day 1-4 | 22/69 | 18/69 | 6/69 | 7/69 |
| Day 5-7 | 20/69 | 16/69 | 5/69 | 6/69 |
| 12 months | 8/27 | 4/27 | 2/27 | 4/27 |
| 24 months | 1/14 | 0/14 | 0/14 | 1/14 |
Two subjects who were previously AMA negative became AMA positive at 12 months
Antigen specificity and time course of AMA reactivity in AMA Positive ALF sera
AMA positive ALF subjects reacted to the major mitochondrial autoantigens including PDC-E2, BCOADC-E2 and OGDC-E2 in PBC (Tables 2 and 3). Among the 86 serum samples obtained from days 1-4 post-ALF, 28/86 reacted with PDC-E2, 10/86 reacted with BCOADC-E2 and 12/86 reacted with OGDC-E2. Among the 90 serum samples obtained from 5-7 days post-ALF, 25/90 recognized PDC-E2, 9/90 recognized BCOADC-E2 and 10/90 recognized OGDC-E2. Of the sera samples obtained from 27 subjects obtained at 12 months LTFU, 4/27, 2/27 and 4/27 remained reactive to PDC-E2, BCOADC-E2 and OGDC-E2 respectively. Only 1/14 serum samples obtained at 24 months post-ALF was AMA reactive and it recognized OGDC-E2 only (Table 2). PDC-E2 is the predominant antigen while a similar frequency of AMA reactivity against BCOADC-E2 and OGDC-E2 were detected from the sera samples. Sixteen subjects recognized PDC-E2 only, 3 recognized both PDC-E2 and BCOADC-E2 while 7 subjects recognized PDC-E2, BCOADC-E2 and OGDC-E2. 6 AMA positive ALF subjects were found to react with OGDC-E2 only. One subject was positive to both BCOADC-E2 and OGDC-E2. None reacted with BCOADC-E2 only (Table 3).
Table 3.
Antigen Specificity of ALF sera to Mitochondrial Autoantigens*
| Antigen | No. Reactive/Total subjects* |
|---|---|
| PDC-E2 only | 16/69 |
| BCOADC-E2 only | 0/69 |
| OGDC-E2 only | 6/69 |
| PDC-E2 and BCOADC-E2 | 3/69 |
| PDC and OGDC-E2 | 0 |
| BCOADC-E2 and OGDC-E2 | 1/69 |
| PDC-E2, BCOADC-E2 and OGDC-E2 | 7/69 |
determined by immunoblot against recombinant mitochondrial autoantigens.
The titers to PDC-E2 ranged from 1:80 to 1:2560 in samples obtained from days 1-4, and 1:80 to 1:1280 in samples obtained from days 5-7 and at 1:160 to 1:640 at 12 months post-ALF. The titers to BCOADC-E2 ranged from 1:640-1:2560 in samples obtained from days 1-4 and at 1:160 to 1:1280 in samples obtained from days 5-7. Two serum samples remained reactive to BCOADC-E2 at 12 months post-ALF with titers of 1:640 and 1:1280. The titers to OGDC-E2 ranged from 1:80-2560 in samples obtained from days 1-4 and at 1:80 to 1:1280 at days 5-7. Four serum samples remained reactive to OGDC-E2 at 12 months and 24 months with a titer of 1:80 to 1:1280. One serum sample was reactive to OGDC-E2 with a titer of 1:640 even at 12 months post-ALF (Figure 2).
Figure 2.
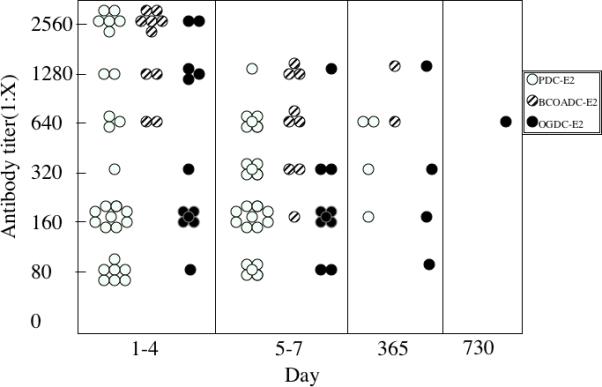
Antibody titer of ALF sera against PDC-E2, BCOADC-E2 and OGDC-E2 obtained at days 1-4, 5-7 days, 12 month and 24 months post ALF. AMA positive ALF sera were serial diluted from 1:40 to 1:2560 and analyzed for their reactivity against recombinant proteins of PDC-E2, BCOADC-E2 and OGDC-E2 by ELISA. Positive sera and negative sera were used throughout as controls. Titer was defined as the highest sera dilution with positive AMA reactivity.
AMA is not limited to any specific ALF diagnosis and is detected in both male and female ALF subjects
Though limited by the small sample size in each group of ALF subjects, our data do not relate the detection of AMA to any particular causal agent as AMA were detected in 9/26 of APAP, 5/11 of drug induced liver injury, 4/9 of autoimmune hepatitis, 2/3 of Hepatitis A, 1/4 of Hepatitis B patients and 4/10 subjects with ALF due to other liver diseases. In addition, 3/6 subjects with unknown causes of liver failure were also found to be AMA positive (Table 4). AMA reactivity was not different between females and males with 20/46 (43.3%) of females and 8/23 (34.7%) of male ALF subjects. The absence of a sex difference in AMA is striking as the female to male ratio in PBC approaches 9 to 1.
Table 4.
Diagnosis and gender analyses of acute liver failure subjects with positive AMA reactivity.
| Diagnoses | Number of Subjects | Number AMA + Female* | Number of AMA + Male* | Total number of AMA positive subjects/number of subjects (%) |
|---|---|---|---|---|
| Acetaminophen Poisoning | 26 | 8/21 | 1/5 | 9/26 (34.6) |
| Drug Induced Liver Injury | 11 | 4/6 | 1/5 | 5/11 (45.5) |
| Autoimmune Hepatitis | 9 | 2/6 | 2/3 | 4/9 (44.4) |
| Hepatitis A | 3 | 1/2 | 1/1 | 2/3 (66.7) |
| Hepatitis B | 4 | 1/2 | 0/2 | 1/4 (25) |
| Miscellaneous** | 10 | 2/6 | 2/4 | 4/10 (40) |
| Indeterminate# | 6 | 2/3 | 1/3 | 3/6 (50) |
| Total | 69 | 20/46 | 8/23 | 28/69 (40.6) |
Expressed as number of reactive sample /number of samples.
Includes known causes such as pregnancy-related, cancer, heat stroke, shock, non Hepatitis A or Hepatitis B viruses.
Clinical cause unknown.
Prevalence of AMA in ALF subjects compared to prevalence of PBC
The frequency of AMA positive sera in our sample of ALF subjects is much higher than what is expected from the normal population. For example, the 95% confidence interval for the frequency of positive AMA in the group of ALF patients studied is 0.2895-0.5213,(p<0.0001) which is significantly higher than the prevalence of PBC AMA, which varies from 19.1 per million to 402 per million (22, 23). Similarly, the 95% confidence limits for the frequency of AMA positive subjects/total number of subjects at 1-7 days, 12 months, 24 months are 0.2621-0.4908, 0.1237-0.4681, and -0.0639-0.2059 respectively, with p<0.0001.
Discussion
Our laboratory has had extensive experience with the use of recombinant autoantigens and in studies of AMA reactivity in large numbers of sera, we have demonstrated that when recombinant antigens are used for immunodiagnosis, they are strikingly specific for PBC and that such specificity and sensitivity is dramatically increased with the use of ELISA and/or immunoblot assays compared to immunofluorescence (21, 24, 25). In our initial analysis and our observations that AMAs were indeed found in acute liver failure, we decided to replicate the data in a second site. Hence, the duplicate analysis both in Davis and Japan and we note that both sites had identical results. Recent work has suggested that post-translational modification of the mitochondrial 2-OADC lipoyl moiety is a possible mechanism in the initiation of AMA in PBC (6, 26). The 2-OADC lipoyl domains are structurally exposed on the periphery of the molecule and mobile, resembling a swinging arm that facilitates electron transfer. In light of the structural configuration of the lipoyl domains in the 2-oxo-acid dehydrogenase complex and the biochemical characteristics of lipoic acid, i.e., its ability to alternate between oxidized and reduced state in its electron transport function, 2-OADC E2s are highly susceptible to chemical modification through endogenous or exogenous influence that may affect the redox state of the cells (13).
In this study, we analyzed 217 individual serum samples obtained from 69 patients with ALF, where catastrophic damage to hepatocytes and presumably bile duct epithelial cells has occurred. In this group of ALF patients, 28/69 subjects developed AMA to one or more of the mitochondrial autoantigens in PBC; only 1 subject remained AMA positive at 24 months post-ALF. It is not known whether this subject developed PBC, as no further samples were available for testing. Although an extensive long term follow up program is in place, to date we have had difficulty getting patients that do not have a long term illness back for care, except those who have received transplants. Limitations include, of course, that there is no follow up on those who die. In addition, most acetaminophen cases, in our experience, cannot be located because they are seldom found at the same address after one year.
AMA reactivity to each of the mitochondrial autoantigens remained rather constant from day 1-7, but decreased sharply by 12 months post-ALF (Table 3) and only 1 subject remained positive to a single mitochondrial autoantigen (OGDC-E2) by 24 months post-ALF. The rapid induction of AMA in ALF subjects suggests that liver injury can trigger the transient production of AMA. The AMA titer in AMA positive ALF subjects ranged from 1:80 to 1:2560 to PDC-E2, 1:160 to 1:2560 to BCOADC-E2 and 1:80 to 1:2560 to OGDC-E2 by ELISA, which is significantly lower than the AMA titer usually observed in patients with PBC (27). Clearly, the frequency of AMA positive sera among ALF subjects in this study is higher than many logs higher those observed in the general population.
Of interest to the data herein is our inclusion of autoimmune hepatitis. Such cases were included primarily as an additional group of for study. However, the percentage of AMA positive subjects from the AIH group is similar to the other included ALF patients. Autoantibodies from AIH subjects do not appear to be directly involved in liver injury prior to acute liver failure in the AMA positive AIH subjects. Although there is no clear evidence of a higher incidence of AMA among various ALF diagnoses, it is interesting to note that AMA was found in almost 35% of APAP poisoning subjects. Acetaminophen overdose is the most frequent cause of ALF (28). APAP toxicity involves a major oxidant component generating ROS (10, 11). Furthermore, the generation of ROS is associated with mitochondrial damage in murine hepatocytes incubated with APAP (29). Our data that AMA are found in almost 35% % of the sera from APAP poisoning subjects suggests that the 2-OADC E2 lipoyl domain is a likely target of APAP induced ROS/RNS. It is known that a decrease in glutathione and CYP2E1 are risk factors in toxic liver injury and both CYP2E1 and glutathione play important roles in APAP metabolism (30, 31). Human HepG2 cells that over-express CYP2E1 have been shown to have elevated glutathione levels for protection against oxidative stress (32) and a recent study in patients with alcoholic hepatitis has shown that both GSH and CYP2E1 mRNA were reduced by 70-80% (33). The effect of APAP on the level of GSH is of importance in the context of this study as the lipoyl domain of PDC-E2 is modified by glutathionylation during apoptosis with this modification blocking PDC-E2 recognition by AMA (12). The lipoyl domain of PDC-E2 is modified by glutathionylation during apoptosis with this modification blocking PDC-E2 recognition by AMA. This phenomenon is of significant importance in PBC because biliary epithelial cells do not glutathionylate PDC-E2 during apoptosis while other liver cells do (12). Although it appears to be a logical assumption that APAP severity may be correlated with GSH depletion and hence a higher titer of AMA by presentation of available non-glutathionylated PDC-E2 to the immune system, the titers of AMA observed did not correlate with disease severity in subjects with APAP. However, it is possible that there is a threshold level of antigen involved in the breaking of tolerance and subsequent disease development. Similarly, while the data herein point to a role for oxidative stress in inducing AMAs, we do not wish to imply that oxidative stress is an absolute necessity for the pathophysiology of PBC. The patients studied reflect an extreme example of hepatic inflammation and oxidative stress and therefore there are likely multiple pathways that may be involved in the generation of autoantibodies. Similarly, it should be noted that in addition to acetaminophen-induced liver injury, oxidative stress induced injury is implicated in other acute liver failure diseases (34-37). In this study, in addition to AMA, a number of other autoantibodies are also detected in ALF patients. Thus, it remains a possibility that the induction of autoantibodies is an epiphenomenon in patients with ALF.
In summary, the demonstration of the high frequency of AMA induction by ALF, albeit transient, provides putative associations of oxidative damage in the pathophysiology of PBC. This result along with the absence of any gender related differences demonstrated by this study suggests that the perpetuation of the AMA response and development of PBC will be a multi-factorial process which would include an oxidative insult in the context of a specific accompanying etiological scenario comprised of genetic susceptibility and appropriate innate and adaptive immune responses (38). Future studies should address this issue and could be studied by administration of drugs that induce ALF in the recently described murine models of human PBC (39-41).
Acknowledgments
Financial Support: This work is supported in part by National Institutes of Health grants DK39588 and DK067003. The Acute Liver Failure Study Group is funded by the National Institutes of Health/NIDDK grant DK058369, William M. Lee, M.D., Principal Investigator.
List of Abbreviations
- AIH
autoimmune hepatitis
- ALF
acute liver failure
- AMA
antimitochondrial autoantibodies
- APAP
Acetaminophen
- BCOADC-E2
E2 subunit of branched chain 2-oxo-acid dehydrogenase
- DGP
deaminated gliadin peptides
- LTFU
long term follow up
- OGDC-E2
E2 subunit of 2-oxoglutarate dehydrogenase
- PBC
primary biliary cirrhosis
- PBS
phosphate buffered saline
- PDCE2
E2 subunit of pyruvate dehydrogenase
- SLA
soluble liver antigen
- tTG
tissue transglutaminase
Footnotes
*Members and institutions participating in the Acute Liver Failure Study Group: W.M. Lee, MD, Julie Polson, MD, Carla Pezzia, University of Texas Southwestern, Dallas, TX; Anne Larson, MD, University of Washington, Seattle, WA; Timothy Davern, MD, University of California, San Francisco, CA; Paul Martin, MD, Mount Sinai School of Medicine, NY, NY; Timothy McCashland, MD, University of Nebraska, Omaha, NE; J. Eileen Hay, MD, Mayo Clinic, Rochester, MN; Natalie Murray, MD, Baylor University Medical Center, Fort Worth, TX; A. Obaid Shakil, MD, University of Pittsburgh, Pittsburgh, PA; Andres Blei, MD, Northwestern University, Chicago, IL; Atif Zaman, MD, University of Oregon, Portland, OR; Steven Han, MD, University of California, Los Angeles, CA; Robert Fontana, MD, University of Michigan, Ann Arbor, MI; Brendan McGuire, MD, University of Alabama, Birmingham, AL; Ray Chung, MD, Massachusetts General Hospital, Boston, MA; Alastair Smith, MB, ChB, Duke University Medical Center, Durham, NC; Michael Schilsky, MD, Cornell/Columbia University, NY, NY; Adrian Reuben, MBBS, Medical University of South Carolina, Charleston, SC; Santiago Munoz, MD, Albert Einstein Medical Center, Philadelphia, PA; Rajender Reddy, MD, University of Pennsylvania, Philadelphia, PA; R. Todd Stravitz, MD, Virginia Commonwealth University, Richmond, VA; Lorenzo Rossaro, MD, University of California at Davis, Sacramento, CA; Raj Satyanarayana, MD, Mayo Clinic, Jacksonville, FL; and Tarek Hassanein, MD, University of California, San Diego, CA.
References
- 1.Powell JJ, Van de Water J, Gershwin ME. Evidence for the role of environmental agents in the initiation or progression of autoimmune conditions. Environ Health Perspect. 1999;107(Suppl 5):667–672. doi: 10.1289/ehp.99107s5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leung PS, Coppel RL, Gershwin ME. Etiology of primary biliary cirrhosis: the search for the culprit. Semin Liver Dis. 2005;25:327–336. doi: 10.1055/s-2005-916324. [DOI] [PubMed] [Google Scholar]
- 3.Rieger R, Gershwin ME. The X and why of xenobiotics in primary biliary cirrhosis. J Autoimmun. 2007;28:76–84. doi: 10.1016/j.jaut.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amano K, Leung PS, Rieger R, Quan C, Wang X, Marik J, Suen YF, et al. Chemical xenobiotics and mitochondrial autoantigens in primary biliary cirrhosis: identification of antibodies against a common environmental, cosmetic, and food additive, 2-octynoic acid. J Immunol. 2005;174:5874–5883. doi: 10.4049/jimmunol.174.9.5874. [DOI] [PubMed] [Google Scholar]
- 5.Amano K, Leung PS, Xu Q, Marik J, Quan C, Kurth MJ, Nantz MH, et al. Xenobiotic-induced loss of tolerance in rabbits to the mitochondrial autoantigen of primary biliary cirrhosis is reversible. J Immunol. 2004;172:6444–6452. doi: 10.4049/jimmunol.172.10.6444. [DOI] [PubMed] [Google Scholar]
- 6.Rieger R, Leung PS, Jeddeloh MR, Kurth MJ, Nantz MH, Lam KS, Barsky D, et al. Identification of 2-nonynoic acid, a cosmetic component, as a potential trigger of primary biliary cirrhosis. J Autoimmun. 2006;27:7–16. doi: 10.1016/j.jaut.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 7.Leung PS, Quan C, Park O, Van de Water J, Kurth MJ, Nantz MH, Ansari AA, et al. Immunization with a xenobiotic 6-bromohexanoate bovine serum albumin conjugate induces antimitochondrial antibodies. J Immunol. 2003;170:5326–5332. doi: 10.4049/jimmunol.170.10.5326. [DOI] [PubMed] [Google Scholar]
- 8.Lee WM. Acute liver failure. N Engl J Med. 1993;329:1862–1872. doi: 10.1056/NEJM199312163292508. [DOI] [PubMed] [Google Scholar]
- 9.Lee WM. Acute liver failure in the United States. Semin Liver Dis. 2003;23:217–226. doi: 10.1055/s-2003-42641. [DOI] [PubMed] [Google Scholar]
- 10.Adamson GM, Harman AW. Oxidative stress in cultured hepatocytes exposed to acetaminophen. Biochem Pharmacol. 1993;45:2289–2294. doi: 10.1016/0006-2952(93)90201-7. [DOI] [PubMed] [Google Scholar]
- 11.Lores Arnaiz S, Llesuy S, Cutrin JC, Boveris A. Oxidative stress by acute acetaminophen administration in mouse liver. Free Radic Biol Med. 1995;19:303–310. doi: 10.1016/0891-5849(95)00023-q. [DOI] [PubMed] [Google Scholar]
- 12.Odin JA, Huebert RC, Casciola-Rosen L, LaRusso NF, Rosen A. Bcl-2-dependent oxidation of pyruvate dehydrogenase-E2, a primary biliary cirrhosis autoantigen, during apoptosis. J Clin Invest. 2001;108:223–232. doi: 10.1172/JCI10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mao TK, Davis PA, Odin JA, Coppel RL, Gershwin ME. Sidechain biology and the immunogenicity of PDC-E2, the major autoantigen of primary biliary cirrhosis. Hepatology. 2004;40:1241–1248. doi: 10.1002/hep.20491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu CT, Eiserich JP, Ansari AA, Coppel RL, Balasubramanian S, Bowlus CL, Gershwin ME, et al. Myeloperoxidase-positive inflammatory cells participate in bile duct damage in primary biliary cirrhosis through nitric oxide-mediated reactions. Hepatology. 2003;38:1018–1025. doi: 10.1053/jhep.2003.50407. [DOI] [PubMed] [Google Scholar]
- 15.Aoki CA, Roifman CM, Lian ZX, Bowlus CL, Norman GL, Shoenfeld Y, Mackay IR, et al. IL-2 receptor alpha deficiency and features of primary biliary cirrhosis. J Autoimmun. 2006;27:50–53. doi: 10.1016/j.jaut.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 16.Gershwin ME, Selmi C, Worman HJ, Gold EB, Watnik M, Utts J, Lindor KD, et al. Risk factors and comorbidities in primary biliary cirrhosis: a controlled interview-based study of 1032 patients. Hepatology. 2005;42:1194–1202. doi: 10.1002/hep.20907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moteki S, Leung PS, Coppel RL, Dickson ER, Kaplan MM, Munoz S, Gershwin ME. Use of a designer triple expression hybrid clone for three different lipoyl domain for the detection of antimitochondrial autoantibodies. Hepatology. 1996;24:97–103. doi: 10.1002/hep.510240117. [DOI] [PubMed] [Google Scholar]
- 18.Leung PS, Iwayama T, Prindiville T, Chuang DT, Ansari AA, Wynn RM, Dickson R, et al. Use of designer recombinant mitochondrial antigens in the diagnosis of primary biliary cirrhosis. Hepatology. 1992;15:367–372. doi: 10.1002/hep.1840150302. [DOI] [PubMed] [Google Scholar]
- 19.Migliaccio C, Nishio A, Van de Water J, Ansari AA, Leung PS, Nakanuma Y, Coppel RL, et al. Monoclonal antibodies to mitochondrial E2 components define autoepitopes in primary biliary cirrhosis. Journal of Immunology. 1998;161:5157–5163. [PubMed] [Google Scholar]
- 20.Leung PS, Chu KH, Chow WK, Ansari A, Bandea CI, Kwan HS, Nagy SM, et al. Cloning, expression, and primary structure of Metapenaeus ensis tropomyosin, the major heat-stable shrimp allergen. J Allergy Clin Immunol. 1994;94:882–890. doi: 10.1016/0091-6749(94)90156-2. [DOI] [PubMed] [Google Scholar]
- 21.Mattalia A, Quaranta S, Leung PS, Bauducci M, Van de Water J, Calvo PL, Danielle F, et al. Characterization of antimitochondrial antibodies in health adults. Hepatology. 1998;27:656–661. doi: 10.1002/hep.510270303. [DOI] [PubMed] [Google Scholar]
- 22.Kim WR, Lindor KD, Locke GR, 3rd, Therneau TM, Homburger HA, Batts KP, Yawn BP, et al. Epidemiology and natural history of primary biliary cirrhosis in a US community. Gastroenterology. 2000;119:1631–1636. doi: 10.1053/gast.2000.20197. [DOI] [PubMed] [Google Scholar]
- 23.Watson RG, Angus PW, Dewar M, Goss B, Sewell RB, Smallwood RA. Low prevalence of primary biliary cirrhosis in Victoria, Australia. Melbourne Liver Group. Gut. 1995;36:927–930. doi: 10.1136/gut.36.6.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miyakawa H, Tanaka A, Kikuchi K, Matsushita M, Kitazawa E, Kawaguchi N, Fujikawa H, et al. Detection of antimitochondrial autoantibodies in immunofluorescent AMA-negative patients with primary biliary cirrhosis using recombinant autoantigens. Hepatology. 2001;34:243–248. doi: 10.1053/jhep.2001.26514. [DOI] [PubMed] [Google Scholar]
- 25.Oertelt S, Rieger R, Selmi C, Invernizzi P, Ansari AA, Coppel RL, Podda M, et al. A sensitive bead assay for antimitochondrial antibodies: Chipping away at AMA-negative primary biliary cirrhosis. Hepatology. 2007;45:659–665. doi: 10.1002/hep.21583. [DOI] [PubMed] [Google Scholar]
- 26.Long SA, Van de Water J, Gershwin ME. Antimitochondrial antibodies in primary biliary cirrhosis: the role of xenobiotics. Autoimmun Rev. 2002;1:37–42. doi: 10.1016/s1568-9972(01)00020-9. [DOI] [PubMed] [Google Scholar]
- 27.Surh CD, Coppel R, Gershwin ME. Structural requirement for autoreactivity on human pyruvate dehydrogenase-E2, the major autoantigen of primary biliary cirrhosis. Implication for a conformational autoepitope. J Immunol. 1990;144:3367–3374. [PubMed] [Google Scholar]
- 28.Schiodt FV, Atillasoy E, Shakil AO, Schiff ER, Caldwell C, Kowdley KV, Stribling R, et al. Etiology and outcome for 295 patients with acute liver failure in the United States. Liver Transpl Surg. 1999;5:29–34. doi: 10.1002/lt.500050102. [DOI] [PubMed] [Google Scholar]
- 29.Ferret PJ, Hammoud R, Tulliez M, Tran A, Trebeden H, Jaffray P, Malassagne B, et al. Detoxification of reactive oxygen species by a nonpeptidyl mimic of superoxide dismutase cures acetaminophen-induced acute liver failure in the mouse. Hepatology. 2001;33:1173–1180. doi: 10.1053/jhep.2001.24267. [DOI] [PubMed] [Google Scholar]
- 30.Mendel-Hartvig I, Nelson BD, Loof L, Totterman TH. Primary biliary cirrhosis: further biochemical and immunological characterization of mitochondrial antigens. Clin Exp Immunol. 1985;62:371–379. [PMC free article] [PubMed] [Google Scholar]
- 31.Rumack BH. Acetaminophen misconceptions. Hepatology. 2004;40:10–15. doi: 10.1002/hep.20300. [DOI] [PubMed] [Google Scholar]
- 32.Cederbaum AI, Wu D, Mari M, Bai J. CYP2E1-dependent toxicity and oxidative stress in HepG2 cells. Free Radic Biol Med. 2001;31:1539–1543. doi: 10.1016/s0891-5849(01)00743-2. [DOI] [PubMed] [Google Scholar]
- 33.Lee TD, Sadda MR, Mendler MH, Bottiglieri T, Kanel G, Mato JM, Lu SC. Abnormal hepatic methionine and glutathione metabolism in patients with alcoholic hepatitis. Alcohol Clin Exp Res. 2004;28:173–181. doi: 10.1097/01.ALC.0000108654.77178.03. [DOI] [PubMed] [Google Scholar]
- 34.Cederbaum AI. CYP2E1--biochemical and toxicological aspects and role in alcohol-induced liver injury. Mt Sinai J Med. 2006;73:657–672. [PubMed] [Google Scholar]
- 35.Colmenero J, Bataller R, Sancho-Bru P, Bellot P, Miquel R, Moreno M, Jares P, et al. Hepatic expression of candidate genes in patients with alcoholic hepatitis: correlation with disease severity. Gastroenterology. 2007;132:687–697. doi: 10.1053/j.gastro.2006.12.036. [DOI] [PubMed] [Google Scholar]
- 36.Dey A, Cederbaum AI. Alcohol and oxidative liver injury. Hepatology. 2006;43:S63–74. doi: 10.1002/hep.20957. [DOI] [PubMed] [Google Scholar]
- 37.Fujii H, Takahashi T, Matsumi M, Kaku R, Shimizu H, Yokoyama M, Ohmori E, et al. Increased heme oxygenase-1 and decreased delta-aminolevulinate synthase expression in the liver of patients with acute liver failure. Int J Mol Med. 2004;14:1001–1005. [PubMed] [Google Scholar]
- 38.Gershwin ME, Ansari AA, Mackay IR, Nakanuma Y, Nishio A, Rowley MJ, Coppel RL. Primary biliary cirrhosis: an orchestrated immune response against epithelial cells. Immunol Rev. 2000;174:210–225. doi: 10.1034/j.1600-0528.2002.017402.x. [DOI] [PubMed] [Google Scholar]
- 39.Irie J, Wu Y, Wicker LS, Rainbow D, Nalesnik MA, Hirsch R, Peterson LB, et al. NOD.c3c4 congenic mice develop autoimmune biliary disease that serologically and pathogenetically models human primary biliary cirrhosis. J Exp Med. 2006;203:1209–1219. doi: 10.1084/jem.20051911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oertelt S, Lian ZX, Cheng CM, Chuang YH, Padgett KA, He XS, Ridgway WM, et al. Anti-mitochondrial antibodies and primary biliary cirrhosis in TGF-beta receptor II dominant-negative mice. J Immunol. 2006;177:1655–1660. doi: 10.4049/jimmunol.177.3.1655. [DOI] [PubMed] [Google Scholar]
- 41.Wakabayashi K, Lian ZX, Moritoki Y, Lan RY, Tsuneyama K, Chuang YH, Yang GX, et al. IL-2 receptor alpha(−/−) mice and the development of primary biliary cirrhosis. Hepatology. 2006;44:1240–1249. doi: 10.1002/hep.21385. [DOI] [PubMed] [Google Scholar]