

Abstract
Class IIa histone deacetylases (HDACs) move between skeletal muscle fibre cytoplasm and nuclei in response to various stimuli, suppressing activity of the exclusively nuclear transcription factor Mef2. Protein kinase A (PKA) phosphorylates class IIa HDACs in cardiac muscle, resulting in HDAC nuclear accumulation, but this has not been examined in skeletal muscle. Using HDAC4–green fluorescent protein (HDAC4-GFP) expressed in isolated skeletal muscle fibres, we now show that activation of PKA by the beta-receptor agonist isoproterenol or dibutyryl (Db) cAMP causes a steady HDAC4-GFP nuclear influx. The beta-receptor blocker propranolol or PKA inhibitor Rp-cAMPS blocks the effects of isoproterenol on the nuclear influx of HDAC4-GFP, and Rp-cAMPS blocks the effects of Db cAMP. The HDAC4-GFP construct having serines 265 and 266 replaced with alanines, HDAC4 (S265/266A)-GFP, did not respond to beta-receptor or PKA activation. Immunoprecipitation results show that HDAC4-GFP is a substrate of PKA, but HDAC4 (S265/266A)-GFP is not, implicating HDAC4 serines 265/266 as the site(s) phosphorylated by PKA. During 10 Hz trains of muscle fibre electrical stimulation, the nuclear efflux rate of HDAC4-GFP, but not of HDAC4 (S265/266)-GFP, was decreased by PKA activation, directly demonstrating antagonism between the effects of fibre stimulation and beta-adrenergic activation of PKA on HDAC4 nuclear fluxes. 8-CPT, a specific activator of Epac, caused nuclear efflux of HDAC4-GFP, opposite to the effect of PKA. Db cAMP increased both phosphorylated PKA and GTP-bound Rap1. Our results demonstrate that the PKA and CaMKII pathways play important opposing roles in skeletal muscle gene expression by oppositely affecting the subcellular localization of HDAC4.
Key points
Application of either the beta-adrenergic agonist isoproterenol, dibutyryl cAMP or specific PKA activator N6 benzoyl cAMP caused nuclear influx of wild-type (wt) HDAC4-GFP expressed in cultured adult skeletal muscle fibres, but caused no change in nuclear/cytoplasmic distribution of expressed ‘mut’ HDAC4-GFP mutated (S 265 and 266 to A) at the protein kinase A (PKA) phosphorylation site(s), demonstrating that PKA promotes HDAC4 nuclear influx by phosphorylation of HDAC4 at the PKA sites.
In non-transfected muscle fibres, myocyte enhancer factor 2 (MEF2)-driven luc reporter activity was decreased by application of isoproterenol, indicating that endogenous HDAC4 increased in fibre nuclei and suppressed MEF2 transcriptional activity. Levels of phosphorylated (i.e. active) PKA were elevated by exposure to dibutyryl (Db) cAMP.
Fibre repetitive electrical stimulation with 10 Hz trains caused a CaMKII dependent nuclear efflux of wt and mut HDAC4, which was partially decreased by Db cAMP for wt but not for mut HDAC4-GFP.
The specific activator 8-CPT of Epac caused efflux of both wt and mut HDAC4-GFP, which was eliminated by the CaMK inhibitor KN-93 or by buffering cytosolic Ca2+ using BAPTA-AM loading, both of which also eliminated a slow elevation of cytosolic Ca2+ during 8-CPT application.
Using a submaximally effective stimulus frequency of 4 Hz trains of electrical stimulation, Db cAMP increased the rate of nuclear influx of mut HDAC4-GFP, which cannot be phosphorylated by PKA but can be phosphorylated by CaMKII, which is here activated via the cAMP/Epac pathway.
Immunostain for active PKA or for GTP-bound RAP1, which is an indicator of Epac activation, showed responses consistent with the functional results above.
Introduction
Class IIa histone deacetylases (HDACs), including HDACs 4, 5, 7 and 9, bind to and suppress the transcriptional activity of myocyte enhancer factor 2 (MEF2), a major muscle gene transcription factor that is important for skeletal muscle fibre type determination (Bassel-Duby & Olson, 2006). The nuclear cytoplasmic distribution of class IIa HDACs, which in turn determines the degree of suppression of MEF2 by HDACs, is controlled by the interplay between kinases and phosphatases. Phosphorylation at serine 246, 467 and 632 of human HDAC4 (or serine 259 and 498 for human HDAC5) generates 14–3–3 binding sites. Binding of 14–3–3 results in HDAC4/5 nucleus to cytoplasm translocation and retention of HDAC4/5 in the cytoplasm (Grozinger & Schreiber, 2000; McKinsey et al. 2001). CaMKs were the first family of kinases shown to phosphorylate and thereby promote nuclear export of class IIa HDACs (McKinsey et al. 2000). Protein kinase D (PKD) was also found to directly phosphorylate class IIa HDACs and induce 14–3–3 binding and cytoplasmic accumulation (Vega et al. 2004), but PKD is not expressed in fast twitch skeletal muscle fibres (Kim et al. 2008). The protein phosphotases PP1 and PP2A, as counterparts of HDAC kinases, are both key phosphatases in the dephosphorylation of HDACs and their consequent nuclear translocation (Paroni et al. 2008).
Most kinases (CaMK, PKD, AMPK, SIK and DyrKB1) phosphorylate HDACs and enhance their nuclear efflux (Parra & Verdin, 2010; McGee & Hargreaves, 2011). In contrast, phosphorylation by protein kinase A (PKA) causes nuclear accumulation of HDACs in C2C12 myoblasts and vascular smooth muscle cells (Du et al. 2008; Gordon et al. 2009). Recent studies show that PKA phosphorylates HDAC5 at serine 280 (Ha et al. 2010; Chang et al. 2013), or HDAC4 at serine 265/266 (Helmstadter et al. 2011), which is localized between the phosphorylatable 14–3–3 binding sites critical for nuclear export of HDAC4 or 5. Phosphorylation of serine 280 of HDAC5 by PKA interrupts the binding of 14–3–3 without hindering phosphorylation at serine 259 and 498 by other kineses (Ha et al. 2010; Chang et al. 2013). Thus, it is anticipated that phosphorylation by PKA or CaMKII should produce antagonistic effects on nuclear/cytoplasmic distribution of class IIa HDACs, and such antagonistic effects have recently been observed in adult cardiomyocytes (Helmstadter et al. 2011; D. Bers, personal communication).
PKA is activated due to increased cAMP produced as a result of beta-adrenergic activation. In addition to activating PKA, cAMP can also activate Epac (Exchange protein directly activated by cAMP), which is modestly expressed in skeletal muscle (Kawasaki et al. 1998; de Rooij et al. 1998). Epacs are guanine nucleotide exchange factors (GEFs) for Rap1 and Rap2. Rap GTPases cycle between an inactive GDP-bound and an active GTP-bound state, with GEFs mediating the exchange of GDP for GTP (Metrich et al. 2010a, 2010b). In rat cardiac cells the specific pharmacological Epac activator 8-CPT can activate CaMKII and affect excitation–contraction (E-C) coupling without activating PKA (Pereira et al. 2007). Recent reports show that Epac activation induces nuclear efflux of HDAC4 and 5 in cardiomyocytes via CaMK (Metrich et al. 2010a, 2010b; Pereira et al. 2012).
In skeletal muscle cellular cAMP levels are modulated by beta-adrenergic input, which increases in vivo during the sympathetic ‘fight or flight’ response which may or may not accompany moderate exercise, but probably does accompany rigorous exercise. Relatively little information is available on the functional roles of PKA and Epac, both of which are activated by cAMP, in the regulation of the subcellular distribution of class II HDACs and consequent modulation of MEF2 transcriptional activity in skeletal muscle. This is especially important in situations in which an increase in cAMP could activate PKA, Epac or both.
Previous work from our laboratory has shown that the nuclear cytoplasmic localization of HDAC4 in skeletal muscle is regulated by moderate-intensity muscle activity, by alpha-adrenergic receptor activation and during high-intensity muscle activity by reactive oxygen species (Liu et al. 2005, 2009, 2012). Now we report that beta-adrenergic agonists or cAMP also modulate HDAC4–green fluorescent protein (HDAC4-GFP) localization in skeletal muscle, enhancing the nuclear influx of HDAC4 via activation of PKA and the resulting PKA-dependent phosphorylation of HDAC4. Mutation of serines at 265/266, the PKA phosphorylation sites of HDAC4, blocks the effects of PKA activation on HDAC4-GFP nuclear influx. These effects of adrenergic activation directly oppose the increased nuclear efflux of HDAC4 produced by moderate-intensity electrical stimulation. As a smaller secondary effect of beta-adrenergic activation on HDAC4 nuclear fluxes we also find that specific pharmacological activation of Epac alone leads to nuclear efflux of HDAC4 due to activation of CaMKII, indicating positive cross talk from the beta-adrenergic to the activity-dependent pathways modulating HDAC4 nuclear fluxes. However, the primary effect of adrenergic activation of PKA is to produce nuclear influx of HDACs, which partially antagonizes the nuclear efflux of HDACs due to CaMKII activation during moderately intensive repetitive muscle fibre activity.
Methods
Animals
CD1 mice were purchased from Charles River (Wilmington, MA, USA). All mice were housed in a pathogen-free area at the University of Maryland, Baltimore. Mice were killed according to authorized procedures of the Institutional Animal Care and Use Committee, University of Maryland, Baltimore, by regulated delivery of compressed CO2 overdose followed by cervical dislocation.
Infection of recombinant adenoviruses in muscle fibres
Single muscle fibres were enzymatically dissociated from flexor digitorum brevis (FDB) muscles of 4–5-week-old CD-1 mice and cultured as described previously (Liu et al. 2005). Isolated fibres were cultured on laminin-coated glass coverslips, each glued over a 10 mm-diameter hole through the centre of a plastic Petri dish. Fibres were cultured in minimal essential medium (MEM) containing 10% fetal bovine serum and 50 μg ml−1 gentamicin sulfate in 5% CO2 (37°C). Recombinant adenovirus (Ad5) containing HDAC4-GFP was produced as described previously (Liu et al. 2005) according to the methods of Hardy et al. (1997) with a cytomegalovirus (CMV) promoter. Recombinant adenovirus expressing HDAC4-GFP S265/266A was constructed with an AdEasy Adenoviral Vector System according to the manufacturer's instruction (Agilent Technologies, Santa Clara, CA, USA) with a CMV promoter and was a gift from Dr D. M. Bers (University of California Davis; Helmstadter et al. 2011). Virus infections were performed with about 108 particles per muscle fibre. The recombinant adenoviruses were added to the culture dishes with MEM without serum. After 1 h of infection, the medium was changed to virus-free MEM with serum for continuous culture.
Microscopy, image acquisition and analysis
To study the localization of HDAC4-GFP or HDAC4 (S265/266A)-GFP, FDB fibres were infected with adenovirus encoding HDAC4-GFP or HDAC4 (S265/266A)-GFP. Two days after infection, culture medium was changed to Ringer's solution (in mm: 135 NaCl, 4 KCl, 1 MgCl2, 10 Hepes, 10 glucose and 1.8 CaCl2, pH 7.4). The culture dish was mounted on an Olympus IX70 inverted microscope equipped with an Olympus FluoView 500 laser scanning confocal imaging system. Fibres were viewed with an Olympus 60× 1.2 NA water immersion objective and scanned at 2.0× zoom with constant laser power and gain.
All experiments (HDAC4 or 5-GFP fluorescence imaging and fibre stimulation and calcium measurements) were carried out at room temperature, 21–23°C.
The average fluorescence of pixels within a user specified area of interest (AOI) in each image was quantified using Image J. The nuclear fluorescence values at each time point were normalized by the nuclear fluorescence value of 0 min of that specific muscle fibre to obtain the N/N0 ratio. Results are expressed as the mean ± SEM. If an image of a fibre had more than one nucleus in focus, then all the nuclei in good focus were analysed and the multiple nuclei were treated equally.
Dibutyryl adenosine 3′,5′-cyclic monophosphate was purchased from Sigma (St Louis, MO, USA). Rp-BrcAMPS was purchased from Calbiochem (La Jolla, CA, USA). Anti-p-CaMKII (Thr 286) and anti-p-PKAα/β/γ catalytic subunit (Thr 198) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-active Rap1 was purchased from NewEast Biosciences (King of Prussia, PA, USA). 8CPT-2Me-cAMP (8-CPT) was purchased from Tocris (Bristol, UK).
Immunohistochemistry
FDB fibre cultures were fixed with 4% paraformeldehyde. The cultures were immunostained with primary antibodies against phosphorylated/activated PKA catalytic subunit (Thr 198, from Santa Cruz Biotechnology), active Rap1 (from NewEast Biosciences) which recognizes the GTP-bound Rap1, phosphorylated CaMKII (Thr 286, from Santa Cruz Biotechnology) or HDAC4 (from Santa Cruz Biotechnology).
Immunoprecipitation and Western blot analysis
After 2 days infection with HDAC4-GFP or HDAC4 (S265/266A)-GFP, FDB cultures were treated with dibutyryl cAMP for 1 h. After treatment, FDB muscle fibres were lysed on ice with M-PER Mammalian Protein Extraction Reagent (Thermo Scientific, Waltham, MA, USA). After centrifugation, the supernatant containing 300 μg of total protein was transferred to an eppendorf tube and around 3 μg immunoprecipitating antibody (anti-phospho-PKA substrate, from Cell Signaling Technology, Inc., Danvers, MA, USA) was added the supernatant. The mixture was on a rotator at 4°C overnight. The immunocomplex was captured by adding protein A/G agarose beads (Thermo Scientific) and was incubated for 3 h at 4°C. The beads were then rinsed with PBS.
The proteins from the precipitates were separated on 3–8% NuPAGE Tris-Acetate gel (Invitrogen, Carlsbad, CA, USA) and then transferred to PVDF membrane. The membrane was blotted with anti-GFP (Invitrogen) and fluorescent-conjugated secondary antibody. The immunoreactive bands were visualized on a GE Typhoon FLA-9500 imaging system.
Calcium recording
Culture medium was first changed to normal Ringer's solution. Fluo-4AM in DMSO was added to dishes to give a final concentration of 2 μm fluo-4 AM in Ringer's solution. After loading for 10 min, cultures were rinsed three times with Ringer's solution and equilibrated for 30 min before recording to allow dye conversion. Dye loading and calcium recording were carried out at room temperature (21–23°C). Muscle fibres were imaged by excitation at 488 nm. The emitted light was collected above 505 nm. For fluorescence data from fluo-4, the average fluorescence of pixels within user-specified AOIs in each image was quantified using Image J. At the end of some experiments, A23187 (1.0 μm in final concentration) was added the culture dishes to obtain Fmax values. A value of Fmax/F0 >4 was recorded. As Fmax/F0 is many times greater than the maximum change observed in the experiments, this indicates that we are well below saturation of fluo-4, and probably within the linear range of fluo-4.
MEF2 activity reporter assay
For MEF2 reporter assay, cultured muscle fibres were infected with adenovirus encoding MEF2-driven luciferase reporter (Wilkins et al. 2004) for 48 h. The cultures were then treated with isoproterenol, or with propranolol and isoproterenol, or not treated as control. The cultures were kept in the incubator for another 24 h. Cultures were then lysed in passive lysis buffer (Promega, Madison, WI, USA). Luciferase activity was determined with a luciferase assay kit (Promega).
Data analysis and statistics
All values are presented as means ± SEM. Statistical significance was tested with ANOVA or a t-test as appropriate. For all comparisons, the level of statistical significance was set at P < 0.05.
Results
Beta-adrenergic activation causes nuclear influx of HDAC4-GFP due to phosphorylation by PKA
We first examined the effects of the beta-adrenergic agonist isoproterenol on muscle fibres expressing HDAC4-GFP by monitoring the mean pixel fluorescence in one or more nuclei and in a relative large cytoplasmic area in a confocal fluorescence image (Fig. 1A). The nuclear fluorescence of HDAC4-GFP remained constant during a 30 min control period in muscle fibres under resting conditions (Fig. 1B). Subsequent application of isoproterenol (0.5 or 5 μm) to the culture dishes resulted in a steady and significant continuous increase in nuclear HDAC4-GFP during the 60 min observation period (Fig. 1B, triangle and circle). While 60 min exposure to 5.0 μm isoproterenol caused a 26 ± 3% increase in nuclear HDAC4-GFP, with a slope of 0.44 ± 0.03% min−1 (25 nuclei from 12 fibres of 2 mice), 0.5 μm isoproterenol caused a 17 ± 2% increase in nuclear HDAC4-GFP with a slope of 0.31 ± 0.03% min−1 (28 nuclei from 14 fibres of 2 mice). Thus, both 0.5 and 5 μm are well above the concentration for half-maximal effect of isoproteranol. Both the mean level after 60 min and the mean slope were significantly lower for 0.5 than for 5.0 μm isoproterenol.
Figure 1. Effects of isoproterenol or Db cAMP on the nuclear localization of HDAC4-GFP or HDAC4 (S265/266A)-GFP.
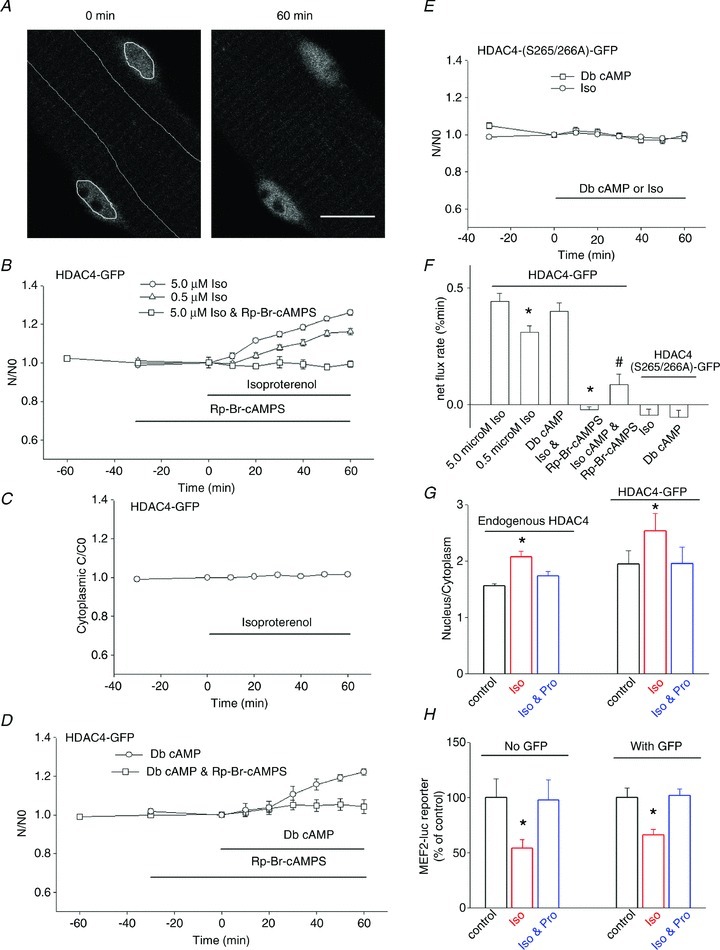
A, image of an FDB fibre expressing HDAC4-GFP, demonstrating how the nuclear and cytoplasmic AOI is defined and application of isoproterenol resulted in accumulation of HDAC4-GFP in nuclei. Scale bar, 10 μm. B, application of the beta-receptor agonist isoproterenol (0.5 or 5.0 μm) resulted in accumulation of HDAC4-GFP in the nuclei. Data were from 28 nuclei of 14 fibres of 2 mice for 0.5 μm, and 25 nuclei of 12 fibres of 2 mice for 5.0 μm isoproterenol. Data are presented as the ratio of nuclear HDAC4-GFP mean pixel fluorescence at different time point/nuclear mean pixel fluorescence at 0 min from the same individual fibre. If FDB fibres were incubated with Rp-Br-cAMPS, a competitive antagonist of PKA, before addition of isoproterenol the effects of isoproterenol were eliminated. Data were from 11 nuclei of 7 fibres of 2 mice. C, cytoplasmic HDAC4-GFP was constant in the presence of isoproterenol. Data were from 26 fibres of 4 mice. The error bars are smaller than the size of symbols in this figure. D, FDB fibres expressing HDAC4-GFP were treated with 500 mm dibutyryl cAMP (Db cAMP) for 60 min. Db cAMP resulted in a net increase of nuclear HDAC4-GFP. Nuclear fluorescence increased continuously during the 60 min period. Data were from 10 nuclei of 7 fibres of 2 mice. The effects of Db cAMP were blocked by pretreatment with Rp-Br-cAMPS. Data were from 9 nuclei of 6 fibres of 1 mouse. E, in FDB fibres expressing HDAC4 (S265/266A)-GFP, isoproterenol or Db cAMP did not affect the subcellular localization of HDAC4 (S265/266A)-GFP. Data were from 10 nuclei of 8 fibres of 2 mice for isoproterenol treatment and from 14 nuclei of 8 fibres of 2 mice for Db cAMP treatment. F, a comparison of the net flux rate of HDAC4-GFP or HDAC4 (S265/266A)-GFP. The net flux rates were obtained using linear fits to the data from each fibre in B, D, F and G. *P < 0.01 compared with 5.0 μm isoproterenol. #P < 0.01 compared with Db cAMP. G, (left 3 columns) nuclear/cytoplasmic (n/c) ratio of antibody stain of endogenous HDAC4, under resting conditions, treated with isoproterenol, and trteated with propranolol plus isoproterenol. Data were from 25 nuclei of 20 fibres, 35 nuclei of 20 fibres, and 26 nuclei of 20 fibres of 2 mice, respectively. The right three columns are n/c ratio of HDAC4-GFP expressing fibres and their response to isoproterneol or propranolol. Data were from 25 nuclei of 12 fibres of 2 mice, 25 nuclei of 12 fibres of 2 mice, and 10 nuclei of 7 fibres of 1 mouse, respectively. *P < 0.01, compared with control. H, MEF2 luciferase reporter activity was inhibited in fibres treated with isoproterenol. The inhibitory effects of isoproterenol were blocked by propranolol. *P < 0.01, compared with control. Infection of muscle fibres with adenovirus expressing GFP did not interfere with the effects of isoproterenol or propranolol (right three columns).
As the cytoplasmic volume greatly exceeds the nuclear volume in these adult skeletal muscle fibres (Schachter et al. 2012), and thus constitutes an effectively infinite volume, the cytoplasmic fluorescence remained constant over the 1–2 h duration of our experiments (Fig. 1C) despite the clear changes in nuclear HDAC4-GFP (Fig. 1 and below). If the fibres were pretreated with the beta-receptor blocker propranolol (5 μm), application of isoproterenol did not change the nuclear cytoplasmic distribution of HDAC4-GFP (data not shown). To establish the role of PKA activation by cAMP in the isoproterenol-induced nuclear influx of HDAC4-GFP, we pretreated another group of muscle fibres with Rp-Br-cAMPS, which occupies the cAMP binding sites on PKA and thus prevents activation of the PKA holoenzyme by cAMP. Rp-Br-cAMPS completely eliminated the effects of isoproteranol on the nuclear accumulation of HDAC4-GFP (Fig. 1B, squares). These results indicate that cAMP, via activation of PKA, promotes a net nuclear influx of HDAC4-GFP in FDB muscle fibres.
To verify that the observed effects of isoproterenol were mediated via cAMP, we examined the effects of application of dibutyryl adenosine 3′,5′-cyclic monophosphate (Db cAMP), a cell permeant mimic of endogenous cAMP which can bind and activate both PKA and Epac (Holz et al. 2008; Poppe et al. 2008), by repeating the experiment of Fig. 1B, but now using Db cAMP instead of isoproteranol. Application of Db cAMP (500 μm) to muscle fibres expressing HDAC4-GFP resulted in continuous increase in nuclear HDAC4-GFP during the 60 min observation period (Fig. 1D, circles), similar to the response of HDAC4-GFP to isoproterenol (Fig. 1B). Pretreatment of another group of muscle fibres with the PKA-specific inhibitor Rp-Br-cAMPS fully blocked the effects of Db cAMP on the nuclear accumulation of HDAC4-GFP (Fig. 1D, squares). The mean rates of nuclear influx of HDAC4-GFP from the fibres in Fig. 1B and D are plotted in Fig. 1F (leftmost five bars). Application of Rp-Br-cAMPS essentially eliminated the HDAc4-GFP net nuclear influx due to isoproterenol or Db cAMP.
It has been previously reported that HDAC5 is a substrate of PKA (Ha et al. 2010; Chang et al. 2013). Phosphorylation of HDAC5 at serine 280 by PKA interrupts the association of HDAC5 with 14–3–3 and thereby inhibits nuclear export of HDAC5 and promotes nuclear retention (Ha et al. 2010; Chang et al. 2013). An alignment analysis shows that there is a potential PKA phosphorylation site at serine 265 and/or 266 in HDAC4 (D. Bers, personal communication; Helmstadter et al. 2011), equivalent to serine 280 in HDAC5. Therefore, to pursue the mechanism of nuclear accumulation of HDAC4 by cAMP we tested the effects of beta-adrenergic activation on HDAC4 (S265/266A)-GFP (Helmstadter et al. 2011), an HDAC4-GFP mutant construct having serines 265 and 266 replaced with alanines, which eliminates the possibility of phosphorylation of these residues. Under control resting conditions, the nuclear/cytoplasmic fluorescence ratio in fibres expressing HDAC4 (S265/266A)-GFP was not significantly different from fibres expressing wild-type (wt) HDAC4-GFP (1.86 ± 0.29 in 14 nuclei from 8 fibres expressing HDAC4-GFP vs. 2.19 ± 0.20 in 10 nuclei from 7 fibres expressing HDAC4 (S265/266A)-GFP, P > 0.05). Thus, possible phosphorylation at serine 265 or 266 does not seem to affect the subcellular localization of HDAC4-GFP under resting control conditions in skeletal muscle. However, in marked contrast to fibres expressing HDAC4-GFP, which exhibited brisk nuclear accumulation of fluorescence on exposure to isoproterenol, fibres expressing HDAC4 (S265/266A)-GFP did not exhibit any detectable changes in nuclear fluorescence (Fig. 1E) on exposure to isoproterenol, demonstrating that serines 265/266, and presumably phosphorylation at this site by PKA, are critical for the action of beta-receptor activation on the localization of HDAC4. In addition, treatment with Db cAMP did not affect the subcellular localization of HDAC4 (S265/266A)-GFP (Fig. 1E), confirming that serines 265/266 are critical for the action of the beta-adrenergic receptor → cAMP → PKA signalling pathway on the nuclear-cytoplasmic movement of HDAC4-GFP in skeletal muscle fibres.
We also used antibody staining to determine redistribution of endogenous HDAC4. Using this approach the nuclear/cytoplasmic (n/c) ratio of endogenous HDAC4 in the resting condition was 1.56 ± 0.04 (25 nuclei from 20 fibres of 2 mice, Fig. 1G). Treatment of fibres with isoproterenol caused a 33% increase in the n/c ratio. The beta-adrenergic blocker propranolol blocked the effects of isoproterenol on endogenous HDAC4 translocation (Fig. 1G). The n/c ratios attained using immunocytochemistry of endogenous HDAC4 agreed very closely with n/c ratios of HDAC4-GFP under control conditions.
We next measured MEF2-driven luciferase reporter activity in fibres with and without exposure to isoproterenol. We found that treatment of fibres with isoproterenol caused repression of MEF2-driven transcriptional activity (a 46% decrease in reporter activity, Fig. 1H), which is in agreement with our results that isoproterenol causes nuclear accumulation of HDAC4-GFP (resulting from activation of PKA), which suppresses MEF2 transcriptional activation.
We also performed the MEF2 reporter assay with FDB cultures infected either with MEF2 reporter alone or with MEF2 reporter simultaneously expressed with GFP adenovirus (both without HDAC4). Treatment with isoproterenol inhibited the MEF2 reporter activity by 34% in the presence of expressed GFP, which is similar to our result of a 46% decrease of MEF2 reporter activity without GFP expression (Fig. 1H).
To characterize the expression level of the HDAC4-GFP chimeras under our experimental conditions, we compared the cytoplasmic anti-HDAC4 fluorescence levels in fibres expressing HDAC4-GFP and in non-infected control fibres. We treated both sets of fibres with anti-HDAC4 primary antibody and conjugated Alexa-647 secondary antibody (which does not interfere with GFP emissions). Assuming that the primary antibody binds to endogenous and exogenous HDAC4 with the same affinity, we found that on average the fibres expressing HDAC4-GFP and endogenous HDAC4 are 4.8 times brighter than fibres expressing only endogenous HDAC4. Therefore, we conclude that HDAC4-GFP is expressed at a level 3.8-fold that of endogenous HDAC4.
We also examined the localization of HDAC4-GFP in cytoplasm. Muscle fibres expressing HDAC4-GFP were fixed with paraformaldehyde and immunostained with anti-alpha actinin antibody, which is located at the z-line in skeletal muscle. We found that the striated HDAC4-GFP pattern was co-localized at the z-line with alpha actinin (data not shown). Our result is in line with that of Gupta et al. (2008) that HDAC4 is located at the z-line in cardiac myocytes.
Activation status of PKA in muscle fibres under various conditions
To verify independently that Db cAMP did in fact activate PKA, we next examined the level of phosphorylated (and thus activated) PKA catalytic subunit in FDB cultures under various conditions using immunohistochemistry (Fig. 2A). FDB cultures were divided into three groups: control (maintained under resting conditions); exposure to 500 μm Db cAMP for 60 min; or preincubated with Rp-cAMPS for 30 min, followed by exposure to 500 μm Db cAMP for 60 min. The primary antibody used recognizes phosphorylated (i.e. activated) PKA catalytic subunit. Figure 2B shows that under resting conditions there is a basal level of phosphorylated PKA in both cytoplasm and nucleus. Incubation with Db cAMP resulted in a 1.39-fold increase in phospho PKA immunostain in the cytoplasm and a 1.48-fold increase in phospho PKA stain in the nuclei compared to fibres under resting conditions (Fig. 2B). Treatment of muscle cultures with Rp-cAMPS before addition of Db cAMP completely blocked the activation of PKA (Fig. 2B).
Figure 2. Monitoring PKA activation by immunostain.
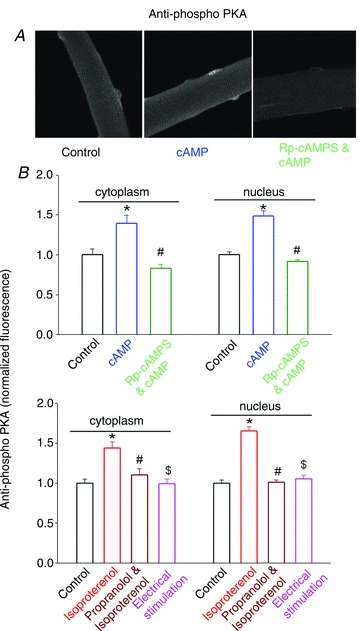
The activation status of PKA was monitored by quantifying the amount of phosphorylated PKA catalytic subunit (Thr 198). A, typical images of antibody stain of phospho PKA in different conditions. B, FDB fibres under resting conditions, treated with Db cAMP (500 mm for 60 min), or first treated with Rp-Br-cAMPS then followed by Db cAMP were fixed and stained with anti-phosphorylated PKA catalytic subunits (Thr 198), followed by fluorescent conjugated secondary antibody. Fluorescence was quantified with Image J. cAMP treatment significantly increased the amount of phosphorylated PKA catalytic subunits both in nucleus and in cytoplasm (*P < 0.01, compared with without cAMP treatment). Pre-incubation with Rp-Br-cAMPS blocked the effects of cAMP on PKA activation (#P < 0.01, compared with cAMP treated). The experiment was repeated three times with each experiment using one mouse. C, in another muscle culture, FDB fibres were under resting conditions, in isoproterenol (5 mm) for 60 min, first in 5 mm propranolol for 30 min followed by isoproterenol, or electrically stimulated with a 10 Hz train for 60 min, respectively. Isoproterenol significantly increased the immunostain of phosphorylated PKA (*P < 0.01, compared with control). Pretreatment with propranolol blocked the effects of isoproterenol (#P < 0.01, compared with isoproterenol treated). Electrical stimulation with 10 Hz trains did not change the immunostain of phospho PKA ($P > 0.05, compared with control). The experiment was repeated three times with each experiment using one mouse.
The beta-adrenergic receptor agonist isoproterenol also increased the immuno stain of phospho PKA, by 1.7-fold in nuclei and by 1.4-fold in cytoplasm, presumably through activation of adenosine cyclase and the resulting increased cAMP. The beta-adrenergic receptor antagonist propranolol blocked the effects of isoproterenol (Fig. 2C). We also tested whether muscle fibre activity altered the activation of PKA using fibre direct electrical stimulation. Electrical stimulation at 10 Hz for 5 s every 50 s (‘10 Hz trains’) over a period of 60 min, which we have previously found to activate CaMKII and to cause nuclear efflux of HDAC4-GFP (Liu et al. 2005), did not result in any changes in phospho PKA in either nucleus or cytoplasm (Fig. 2C). The absence of changes in PKA phosphorylation in electrically stimulated muscle fibres indicates that PKA is not activated by muscle fibre activity, at least not with the 10 Hz train muscle activity pattern used here.
HDAC4 is a substrate for phosphorylation by PKA at serine 265 and/or 266
To analyse the mechanisms by which PKA regulates the subcellular localization of HDAC4-GFP, we infected FDB muscle fibres with HDAC4-GFP and then treated the cultures with N6-benzoyl cAMP, a specific PKA activator. The muscle cultures were lysed, and the lysate was immunoprecipitated with an antibody which reacts with phospho PKA substrate. Western blot analysis with anti-GFP antibody shows that HDAC4-GFP is in the precipitate, demonstrating that HDAC4-GFP is a substrate of PKA in FDB muscles (Fig. 3A, left lane). When FBD muscle cultures were infected with HDAC4 (S265/266A)-GFP, there was no band from the lysate in Western blot analysis, demonstrating that HDAC4 (S265/266A)-GFP is not a substrate of PKA (Fig. 3A, right lane). Alignment of HDAC4 and 5 of human and mouse shows that the consensus phosphorylation motif for PKA substrates (R/K)XX(S*/T*) is conserved in human and mouse HDAC4 and 5 (Fig. 3B). Figure 3C presents a cartoon representation of the location of the residues phosphorylated by PKA or by CaMKII, a kinase which when activated phosphorylates HDAC4 and promotes its nuclear efflux (see below).
Figure 3. HDAC4 is a substrate of PKA.
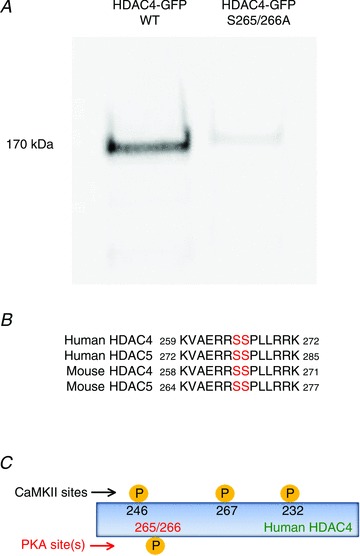
A, muscle fibres were infected with wt HDAC4-GFP or HDAC4 (S265/266A)-GFP. After 2 days in culture, muscle cultures were treated with Db cAMP. Total cell lysates were first incubated with anti-phospho-PKA substrate antibodies for immunoprecipitation then immunobloted with anti-GFP antibodies. The band is consistent with the molecular weight of HDAC4-GFP. The experiment was repeated three times, each time using two mice. B, alignment of amino acid sequences surrounding HDAC4 serines 265 and 266, showing the PKA phosphorylation motif is conserved in human and mouse HDAC4 and 5. C, cartoon showing the phosphorylation sites of CaMKII and PKA in HDAC4.
Specific activation of PKA promotes nuclear influx of wt but not S265/266A HDAC4-GFP
As cAMP and Db cAMP can activate both PKA and Epac (Christensen et al. 2003; Poppe et al. 2008), we next tested the effects of the selective PKA activator N6-benzoyl cAMP on the localization of HDAC4-GFP and HDAC4 (S265/266A)-GFP. N6-Benzoyl cAMP caused a brisk net increase of nuclear HDAC4-GFP (Fig. 4), which is similar to the effect of Db cAMP, but did not affect the localization of HDAC4 (S265/266A)-GFP (Fig. 4), further demonstrating that specific activation of PKA causes nuclear influx of HDAC4-GFP, and that serines 265/266 are critical sites for this effect of PKA. Application of N6-benzoyl cAMP to fibres expressing HDAC5-GFP, the fluorescent fusion protein of GFP with HDAC isoform 5, which also has the residues for phosphorylation by PKA, caused a nuclear influx of HDAC5-GFP (Fig. 4) which was similar to that of HDAC4-GFP (Fig. 4).
Figure 4. Effects of the specific PKA activator N.
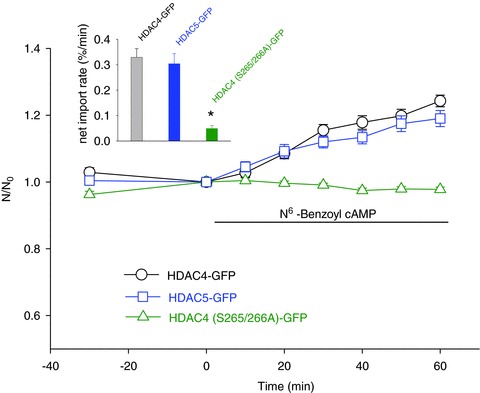
6-benzoyl cAMP on the localization of HDAC4-GFP or HDAC5-GFP FDB fibres expressing wt HDAC4-GFP were treated with N6-benzoyl cAMP, which caused a net nuclear influx of HDAC4-GFP in the 60 min observation period (circle). In FDB fibres expressing HDAC4 (S265/266A)-GFP, PKA activation by the specific PKA activator N6-benzoyl cAMP did not affect the nuclear localization of HDAC4 (S265/266A)-GFP (triangle). In fibres expressing HDAC5-GFP, N6-benzoyl cAMP resulted in an accumulation of nuclear HDAC5-GFP (square). Data are from 16 nuclei of 10 fibres of 2 mice for HDAC4-GFP, 15 nuclei of 11 fibres of 2 mice for HDAC4 (S265/266A)-GFP, and 17 nuclei of 13 fibres of 2 mice for HDAC5-GFP, respectively. The inset shows the net influx rates of HDAC4-GFP, HDAC4 (S265/266A)-GFP or HDAC5-GFP (from left to right), obtained by linear fits of the time course data from the same figure. *P < 0.01, compared with HDAC4-GFP.
Activation of PKA decreases the nuclear efflux of HDAC4 produced by repetitive muscle fibre activity
The above results suggest that the beta-adrenergic receptor-cAMP-PKA signalling pathway enhances HDAC4-GFP nuclear localization by increasing HDAC4-GFP nuclear influx and/or decreasing nuclear efflux. We have previously shown that repetitive electric field stimulation can activate CaMKII in muscle fibres, resulting in net nuclear efflux of HDAC4-GFP (Liu et al. 2005), an effect opposite to that observed here during PKA activation. This is of important physiological interest as in vivo muscle fibre activity, which is initiated by motor neuron action potentials, may be accompanied by adrenergic activity. We therefore next investigated whether PKA activation has any effects on the nuclear efflux of HDAC4-GFP triggered by 10 Hz trains of electric field stimulation. The results clearly demonstrate that application of Db cAMP (500 μm) during stimulation decreased the rate of loss of nuclear HDAC4-GFP fluorescence during repetitive 10 Hz trains of fibre field stimulation (Fig. 5A). Using linear fits to the data from each fibre, the mean net export rate during the stimulation was 0.57 ± 0.04% min−1 (15 nuclei from 7 fibres) and 0.23 ± 0.04% min−1 (11 nuclei from 8 fibres) for fibres in the absence or presence of cAMP, respectively (Fig. 5A, inset). Thus, the presence of Db cAMP significantly slowed the net efflux of nuclear HDAC4-GFP triggered by muscle activity and the resulting activation of CaMKII.
Figure 5. Activation of PKA antagonizes the HDAC4-GFP nuclear efflux triggered by electrical stimulation.
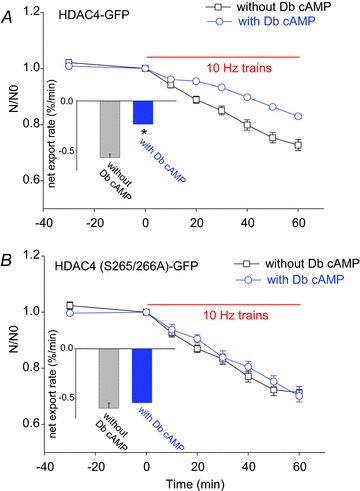
A, FDB muscle fibres expressing wt HDAC4-GFP were electrically stimulated with 10 Hz trains in the absence (square, 15 nuclei of 7 fibres of 3 mice) or presence (circle, 11 nuclei of 8 fibres of 3 mice) of Db cAMP. The decay of nuclear wt HDAC4-GFP was fitted with a linear fit. Db cAMP significantly slowed the slope of nuclear wt HDAC4-GFP decay. B, in fibres expressing HDAC4 (S265/266A)-GFP, Db cAMP did not affect the decay of nuclear HDAC4 (S265/266A)-GFP triggered by 10 Hz train stimulation. Data are from 10 nuclei of 5 fibres of 2 mice in the absence of Db cAMP and 12 nuclei of 6 fibres of 2 mice in the presence of Db cAMP. The insets show the net export rate in fibres expressing HDAC4-GFP or HDAC4 (S265/266A)-GFP in the absence or presence of Db cAMP. *P < 0.05, compared with the group of fibres without Db cAMP.
In other fibres expressing HDAC4 (S265/266A)-GFP, which cannot be phosphorylated by PKA, electric field stimulation with 10 Hz trains caused the same rate of decline of nuclear fluorescence either in the presence or in the absence of 500 μm Db cAMP (Fig. 5B). As determined by linear fits, in contrast to muscle fibres expressing HDAC4-GFP, in muscle fibres expressing HDAC4 (S265/266A)-GFP Db cAMP did not significantly affect the mean net export rate during 30 min of stimulation (Fig. 5B). The mean net export rate during the stimulation was 0.59 ± 0.05% min−1 (10 nuclei from 5 fibres) and 0.54 ± 0.07% min−1 (12 nuclei from 6 fibres) for fibres in the absence or presence of Db cAMP, respectively (Fig. 5B, inset). With or without Db cAMP, HDAC4 (S265/266A)-GFP responded to muscle activity with a nuclear efflux rate that was similar to that observed using wt HDAC4-GFP in the absence of added Db cAMP, suggesting that serines 265/266 in HDAC4 are critical for cAMP modulation of the nuclear cytoplasm shuttling of HDAC4 and that the cAMP-PKA pathway can functionally antagonize the calcium-CaMKII pathway in the HDAC4 nuclear cytoplasmic shuttling.
Specific activator of Epac causes nuclear efflux of both wt HDAC4-GFP and HDAC4 (S265/266)-GFP SA mutant
Thus far we have examined the two signalling pathways beta-adrenergic receptor → cAMP → PKA → HDAC4 nuclear influx, and muscle fibre activity → myoplasmic Ca2+→ CaMKII → HDAC4 nuclear efflux. In our next experiments we looked for evidence of cross talk from the beta-adrenergic receptor initiated pathway to the activity initiated pathway mediated by cAMP → Epac → CaMKII. To determine whether such cross talk was feasible, we examined the effects of 8-CPT, a specific Epac activator, on the subcellular distribution of HDAC4-GFP at a concentration of 8-CPT that is reported to activate only Epac while having no effect on PKA. As shown in Fig. 6A, after 30 min under resting conditions with stable nuclear HDAC4-GFP, addition of 5 μm 8-CPT caused a constant decline of nuclear HDAC4-GFP during a 60 min observation period. In fibres preincubated with KN-93, a CaMKII blocker, the effects of 8-CPT on nuclear HDAC4-GFP were eliminated (Fig. 6B), suggesting that CaMKII is involved in the nuclear efflux of HDAC4-GFP caused by 8-CPT. To further examine the roles of calcium and CaMKII in the nuclear efflux of HDAC4-GFP by 8-CPT, fibres expressing HDAC4-GFP were first loaded with 15 μm of the calcium chelator BAPTA-AM for 20 min, then rinsed and incubated at room temperature for 30 min. Figure 6C shows that BAPTA antagonized the effects of 8-CPT on HDAC4-GFP, presumably through buffering the calcium increase that would otherwise be caused by 8-CPT. Results from both Fig. 6B and C and suggest that calcium-CaMKII plays an important role in the pathway from Epac activation to HDAC4-GFP nuclear efflux.
Figure 6. 8-CPT causes a net nuclear efflux of wt HDAC4-GFP or HDAC4 (S265/266A)-GFP.
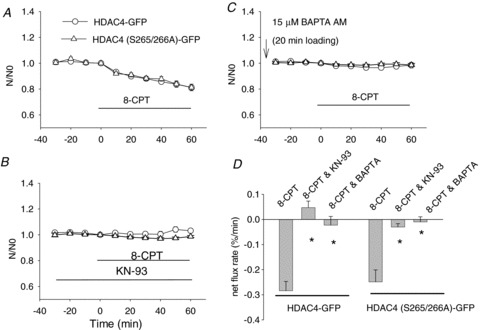
A, in muscle fibres expressing wt HDAC4-GFP, Epac activator 8-CPT caused a net nuclear efflux of HDAC4-GFP or HDAC4 (S265/266A)-GFP. B, in the presence of the CaMKII inhibitor KN-93, 8-CPT did not affect the nuclear localization of wt HDAC4-GFP or HDAC4 (S265/266A)-GFP. C, if muscle fibres were incubated with the calcium chelator BAPTA-AM, addition of 8-CPT did not affect the nuclear localization of wt HDAC4-GFP or HDAC4 (S265/266A)-GFP. Data are from 16 nuclei of 10 fibres of 2 mice for HDAC4-GFP, and 14 nuclei of 11 fibres of 2 mice for HDAC4 (S265/266A)-GFP in A; 9 nuclei of 5 fibres of 1 mouse for HDAC4-GFP, and 12 nuclei of 12 fibres for HDAC4 (S265/266A)-GFP of 2 mice in B; and 13 nuclei of 9 fibres of 2 mice for HDAC4-GFP, and 17 nuclei of 10 fibres of 2 mice for HDAC4 (S265/266A)-GFP in C. D, summary of net flux rate using data of linear fit from A, B and C. *P < 0.01, compared with the group of fibres with 8-CPT alone.
We further tested if the HDAC4 mutation S265/266A interrupts the effects of 8-CPT on the localization of HDAC4-GFP. As anticipated and as shown in Fig. 6A, compared to muscle fibres expressing HDAC4-GFP, fibres expressing HDAC4 (S265/266A)-GFP exhibited almost identical responses to 8-CPT. Both the CaMKII inhibitor KN-93 and the calcium buffer BAPTA blocked the nuclear efflux of HDAC4 (S265/266A)-GFP triggered by 8-CPT (Fig. 6B and C). These results establish that phosphorylation of HDAC4 at serines 265 and 266 is not involved in the response of HDAC4 to Epac, which is believed to act by activating CaMKII.
Epac activation causes elevated cellular calcium concentration and CaMKII activation
As the effects of 8-CPT on HDAC5-GFP are believed to be calcium related (Pereira et al. 2012) we next monitored the cellular calcium in FBD fibres treated with 8-CPT. FDB fibres loaded with the calcium indicator fluo-4 were imaged every 10 min. The resting calcium level was stable for 30 min before the addition of 8-CPT. Adding 5 μm 8-CPT triggered a gradual increase of cellular calcium, which lasted for the entire 60 min observation period (Fig. 7A).
Figure 7. Epac activator 8-CPT increases cellular calcium and activates CaMKII.
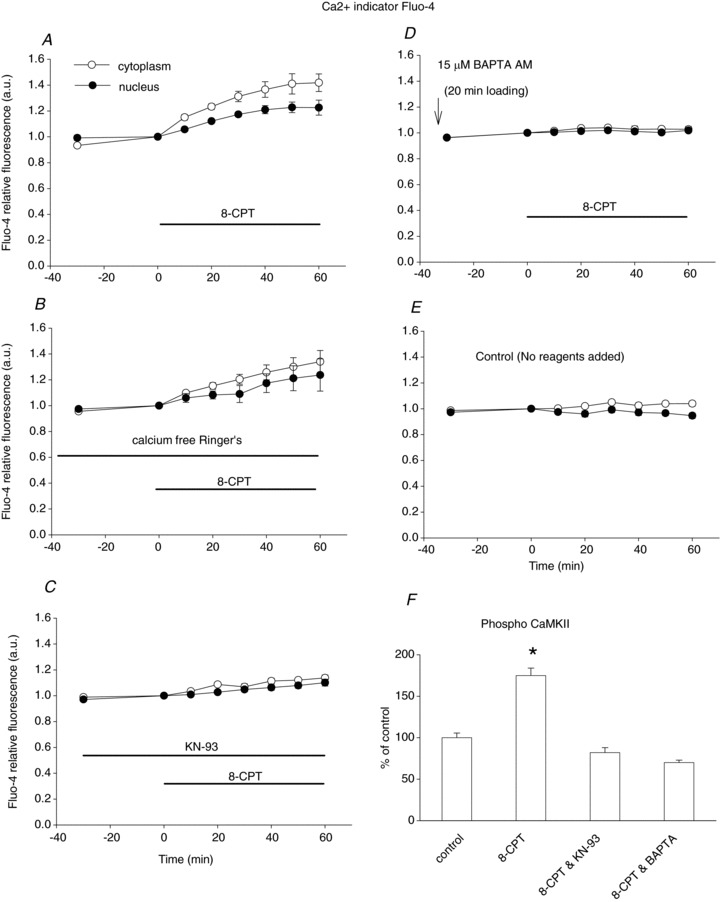
A, muscle fibres were loaded with the calcium-sensitive dye Fluo-4AM. Images were quantified and background subtracted. First, FDB fibres were imaged for 30 min to obtain the baseline resting calcium level. Addition of 8-CPT resulted in steady elevation of calcium both in nucleus and in cytoplasm. Data are from 10 nuclei of 9 muscle fibres of 2 mice. B, in fibres in calcium-free Ringer's solution, 8-CPT caused a similar increase in resting calcium. Data are from 11 nuclei of 11 muscle fibres of 2 mice. C, CaMKII inhibitor KN-93 largely antagonized the calcium elevation caused by 8-CPT. Data are from 20 nuclei of 12 muscle fibres of 2 mice. D, if muscle fibres are first loaded with calcium chelator BAPTA-AM, addition of 8-CPT did not cause any changes in cellular or nuclear calcium. Data are from 12 nuclei of 9 muscle fibres of 2 mice. E, cellular calcium was stable for the 90 min observation period. Data are from 17 nuclei of 12 muscle fibres of 2 mice. F, the activation status of CaMKII was monitored by immunostain with antibody to activated (autophosphorylated) CaMKII. The fluorescence of immunostain was quantified and normalized to control. 8-CPT significantly increased the amount of activated CaMKII (P < 0.01, compared with control). Pretreatment with KN-93 blocked the activation of CaMKII by 8-CPT (P < 0.05, compared with control). Pre-loading muscle fibres with BAPTA-AM also antagonized CaMKII activation by 8-CPT (P < 0.01, compared with control). Data are from 28, 33, 31 and 34 muscle fibres of 2 mice, from left to right, respectively.
We also performed experiments with calcium-free Ringer's solution containing 100 μm EGTA, as shown in Fig. 7B. 8-CPT caused a similar increase in fluorescence (i.e. increased cellular calcium) as in calcium-containing solution. This suggests that the increased calcium came from internal stores, probably the sarcoplasmic reticulum, as there should be minimal calcium influx in the calcium-free solution. Our results suggest that the increased intracellular free Ca2+ results from an increased ryanodine recepor (RyR) leak, as reported by Pereira et al. (2013) or from slowed sarcoplasmic reticulum Ca2+ uptake. In CaMKII inhibitor KN-93-treated fibres (Fig. 7C), the increase in cellular calcium caused by 8-CPT was largely inhibited (from a 40% increase in cellular calcium in fibres without KN-93 to a 10% increase in fibres with KN-93).
If fibres were pre-loaded with BAPTA-AM, the effects of 8-CPT on cellular calcium were minimal, suggesting that cytosolic and nuclear calcium was buffered by BAPTA (Fig. 7D). The results of calcium recording with 8-CPT and BAPTA are in line with the results on the effects of 8-CPT and BAPTA on the localization of HDAC4-GFP, demonstrating that increasing cellular calcium underlies the effects of 8-CPT.
To test for changes in CaMKII phosphorylation, muscle fibres were incubated in Ringer's alone, 8-CPT, KN-93 and 8-CPT, or BAPTA plus 8-CPT before fixation and staining for activated (autophosphorylated) CaMKII. Staining for activated CaMKII was 1.75-fold stronger in muscle fibres incubated with 8-CPT (P < 0.01). Pre-treatment of muscle fibres with KN-93 or BAPTA-AM blocked the activation of CaMKII by 8-CPT (Fig. 7F), further confirming that calcium and CaMKII play crucial roles in the action of 8-CPT.
Cross talk from the beta-adrenergic pathway to the activity-dependent pathway for HDAC nuclear efflux
As 8-CPT activation of Epac produces very similar nuclear efflux of both HDAC4-GFP and HDAC4 (S265/266A)-GFP, indicating a lack of involvement of PKA-dependent phosphorylation, we next looked carefully for evidence that the beta-adrenergic signalling pathway might exhibit cross talk with the activity-dependent pathway activating CaMKII, possibly via cAMP activation of Epac. Here we used only HDAC4 (S265/266A)-GFP to completely avoid any effects of PKA phosphorylation of wt HDAC4. Figure 5B has shown that exposure to Db cAMP had no effect on the nuclear efflux rate of HDAC4 (S265/266A)-GFP during 10 Hz trains of electrical stimulation, indicating a lack of such cross talk. However, if 10 Hz trains of stimulation was already sufficient to produce near maximal activation of CaMKII, then adding Db cAMP could not further increase the rate of HDAC (S265/266A)-GFP nuclear efflux. In fact, when we decreased the stimulation frequency during the trains to 4 Hz, but kept the train duration at 5 s and the train application rate at once every 50 s (called ‘4 Hz trains’), we found that exposure to Db cAMP increased the rate of nuclear efflux during 4 Hz trains of electrical stimulation, and that the augmented rate of nuclear efflux was very similar to the rate of nuclear efflux produced by 10 Hz trains (Fig. 8). This provides an indication that there is some component of positive cross talk from the beta-adrenergic pathway to the activity-dependent pathway via cAMP. The mechanism(s) underlying this effect will be considered in the Discussion. However, it should be noted that the major interaction between the beta-adrenergic pathway and the activity-dependent pathway is that the nuclear influx of HDAC4 generated by beta-adrenergic activation of PKA opposes the muscle activity-dependent nuclear efflux of HDAC generated by the activity-dependent activation of CaMKII.
Figure 8. Comparison of net export rates of HDAC4 (S265/266A)-GFP in fibres stimulated with 10 Hz trains, 4 Hz trains or 4 Hz trains plus Db cAMP.
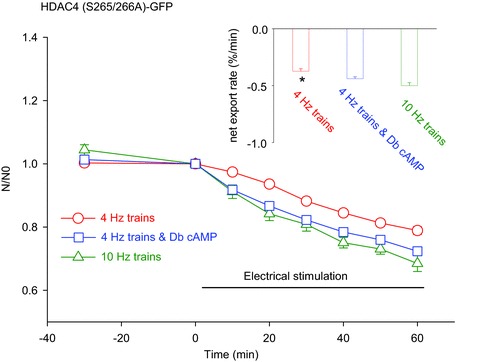
Four hertz train stimulation in the presence of Db cAMP caused faster net export of HDAC4 (S265/266A)-GFP than that of 4 Hz in the absence of Db cAMP. Data are from 17 nuclei of 13 fibres of 2 mice (10 Hz trains), 23 nuclei of 14 fibres of 2 mice (4 Hz trains), and 17 nuclei of 12 fibres of 2 mice (4 Hz trains plus Db cAMP). The inset shows the net export rate of HDAC4 (S265/266A)-GFP obtained by linear fit of the time course data. *P < 0.05, compared with 4 Hz in the presence of Db cAMP.
Monitoring both PKA activation and Epac activation in the same muscle fibres
Because in the above experiments we employed either a PKA activator or Epac activator, or both, we examined the activation status of PKA and Epac with immunohistochemistry to selectively monitor the activated forms of PKA and RAP1 using antibodies specific to phosphorylated (activated) PKA catalytic subunits (p-PKA) or specific to GTP-bound RAP1 (RAP1-GTP) in the same muscle fibres. Rap1 is a member of the small GTPases Rap family, which is downstream of Epac in the signal transduction pathway (Metrich et al. 2010a, 2010b). The active form of Rap1 (GTP-bound Rap1) was used as an indicator of Epac activation (Enserink et al. 2002). In Db cAMP-treated fibres the activated forms of PKA or RAP1 are both significantly higher than in control fibres (Fig. 9), showing that Db cAMP activated both PKA and Epac and is thus a non-selective activator. In the N6-benzoyl cAMP-treated fibres, only the amount of p-PKA was significantly elevated, with no changes in RAP1-GTP. In contrast, in the 8-CPT-treated group, RAP1-GTP was increased with no significant change in p-PKA. These results suggest that in our experimental conditions, Db-cAMP activates both Epac and PKA, whereas N6-benzoyl cAMP or 8-CPT activates either PKA or Epac, respectively.
Figure 9. Monitoring Epac and PKA activation.
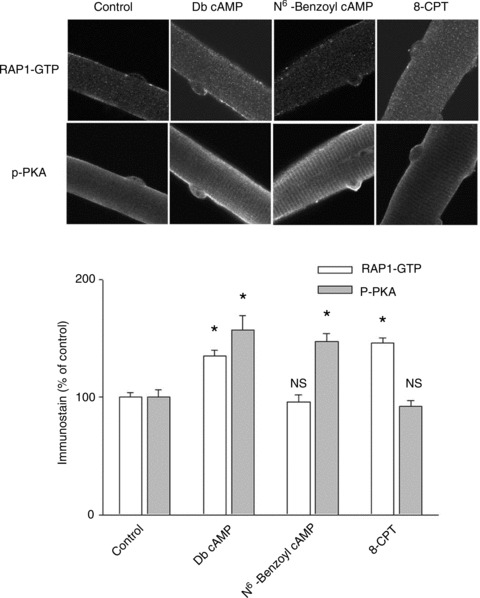
Muscle fibres were treated as indicated and immunostained for both phosphorylated PKA catalytic subunit and GTP-bound RAP1, which reflect activated PKA or activated Epac, respectively. In Db cAMP-treated fibres, both activated PKA and activated Epac are enhanced significantly. In N6-benzoyl cAMP-treated fibres, only activated PKA is increased, while in the same fibres there is no change in the activation of PKA. In 8-CPT-treated fibres, only activated Epac is increased, with no changes in the activation of PKA (NS: P > 0.05; *P < 0.01 compared with control). Data are from 21 to 27 fibres in each group of 3 mice.
Discussion
In this study we present the first report that phosphorylation of HDAC4 by PKA after beta-adrenergic activation causes net HDAC4 nuclear influx, which is opposite to the HDAC4 net nuclear efflux that occurs as a result of repetitive muscle fibre activity and which was previously shown to be due to phosphorylation of HDAC4 by CaMKII (McKinsey et al. 2000). Our group has previously characterized the activity dependence of nuclear efflux of HDAC4 in response to moderate-intensity repetitive fibre field stimulation which is mediated by CaMKII-dependent phosphorylation of HDAC4 (Liu et al. 2005). Now we show that PKA activation and the resulting phosphorylation of HDAC4 at other sites causes nuclear influx of HDAC4. Furthermore, application of beta-adrenergic agonist or Db cAMP during repetitive electrical stimulation retards the rate of nuclear efflux of HDAC4-GFP compared with the efflux in the absence of PKA activity. These observations directly demonstrate the opposing effects of the beta-adrenergic and the activity-dependent signalling pathways on HDAC4 nuclear movement. The beta-adrenergic signalling pathway to HDAC4 (beta-adrenergic receptor → cAMP → PKA → HDAC4 phosphorylation at the PKA sites) causes HDAC4 nuclear influx. In contrast, the muscle fibre activity signalling pathway (muscle activity → ryanodine recepor/Ca2+ release channel (RyR) → Ca2+→ CaMKII → HDAC4 phosphorylation at the CaMKII sites) causes HDAC4 nuclear efflux.
Figure 10 presents a cartoon representation of the signalling pathways underlying our observations. The heavy arrows represent HDAC4 nuclear efflux mediated by 14–3–3 (McKinsey et al. 2001) and Crm1 via the nuclear export system (right curved arrow) and via the importin-mediated nuclear import system (left curved arrow). At the left is shown the beta-adrenergic pathway leading through cAMP, and PKA to PKA-dependent phosphorylation of HDAC4 at its PKA phosphorylation sites (Fig. 10, lower left), with the resulting potentiation of HDAC4 nuclear influx. At the right is shown the muscle activity-dependent signalling pathway leading through RyR, Ca2+ and CaMKII to CaMKII-dependent phosphorylation of HDAC4 at its CaMKII phosphorylation sites (Fig. 10, lower right), and the resulting potentiation of HDAC4 nuclear efflux. The relative balance of activation in the beta-adrenergic and the activity-dependent pathways will determine the net direction and rate of movement of HDAC4 into or out of the muscle fibre nuclei. Thus, adrenergic signalling will counteract the effects of moderate-intensity exercise in promoting muscle fibre endurance and the slow fibre type.
Figure 10.
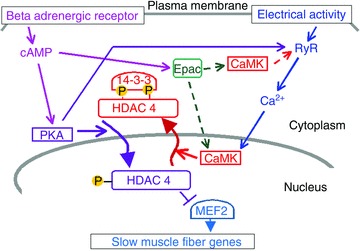
Regulation of the localization of class II HDACs by the PKA and CaMKII pathways
Even though circulating epinephrine is increased early in moderate exercise (Galbo et al. 1977), the resulting activation of muscle beta-adrenergic receptors could be relatively modest, with important levels of activation requiring greater adrenergic output. In this case, moderate exercise without strong adrenergic activity could promote fibre remodelling toward the slow phenotype due to HDAC4 nuclear efflux. In contrast, during a fight or flight response, the much more intense adrenergic response accompanying the muscle activity could counteract the effects of activity on HDAC4 nuclear efflux, and thus negate the remodelling response that would otherwise occur due to the activity-dependent HDAC4 nuclear efflux.
In addition to the parallel but opposing beta-adrenergic-dependent and activity-dependent pathways to HDAC4 phosphorylation, but at different sites and resulting in opposite nuclear movements of HDAC4 (above), there is also positive cross talk from the adrenergic pathway to the activity-dependent pathway. cAMP, an intermediate in the adrenergic pathway, can activate Epac, which in turn may activate CaMKII by a multi-step process represented by the dashed arrows, possibly via phospholipase C (PLC) activation and inositol trisphosphate (IP3) production and local Ca2+ release (Pereria et al. 2012), possibly via diacyl glycerol production and PLC activation of CaMKII (Oestreich et al. 2007, 2009), which have both been proposed to occur in cardiac myocytes, or possibly via alternative pathways. Our results with the selective Epac activator 8-CPT indicate that Epac may be present and functional in these skeletal muscle fibres, so a cross talk pathway from the beta-adrenergic signalling pathway to the activity-dependent pathway via Epac is present in skeletal muscle fibres. Our present results also establish that CaMKII is involved downstream of cAMP/Epac activation of Ca2+ release in resting skeletal muscle fibres, and that extracellular Ca2+ is not involved in the Epac signalling in skeletal muscle fibres. A second possible positive cross talk pathway from the beta-adrenergic to the activity-activated pathway could be PKA-dependent phosphorylation of the ryanodine receptor/Ca2+ release channel (Fig. 10, long kinked arrow from PKA), which would increase Ca2+ release in response to the muscle action potential (Liu et al. 1997; Andersson et al. 2012). The increase in nuclear efflux rate of HDAC4 (S265/266A)-GFP, which cannot be phosphorylated by PKA, that we observed when fibres stimulated by 4 Hz trains were exposed to Db cAMP (Fig. 8) might be accounted for by either or both of these cross talk pathways.
Although our results were obtained in isolated cultured adult skeletal muscle fibres, which lack both motor neuron and autonomic innervation, they have important implications for muscle gene regulation in intact animals. Extrapolated to the in vivo situation in a functioning animal, our findings imply that a given pattern of repetitive voluntary muscle activity will be less effective in de-repressing slow fibre gene expression when adrenergic output is high as during a ‘fight or flight response’, and HDAC4 nuclear efflux is consequently suppressed by PKA, than when adrenergic outflow is low. To our knowledge, this constitutes the first report of interplay between adrenergic activity and motor neuron activity on skeletal muscle fibre gene regulation. Studying the effects of cAMP on HDAC4 shuttling in skeletal muscle is physiologically relevant, as strong activation of beta-adrenergic activation occurs during exercise, which results in elevation of cAMP level in skeletal muscle cells. For example, it is reported that epinephrine is released into the circulation during exercise (Galbo et al. 1977) and intramuscular cAMP levels increase within minutes of treadmill running (Goldfarb et al. 1989).
Catecholamines may also be potentially elevated in long-term heart failure patients experiencing increased adrenergic activation, where the percentage of slow twitch type I fibers was reduced and the percentage of type IIb fast twitch fibers was increased (Sullivan et al. 1990). However, whether this fiber type change is caused by activation of PKA and nuclear accumulation of HDAC4/5 calls for further study.
Previous studies from our group showed that HDAC4, but not HDAC5, translocates from nucleus to cytoplasm in response to moderately intensive repetitive muscle activity (one 5 s train of 10 Hz stimuli every 50 s; Liu et al. 2005, 2012) due to CaMKII activation. We also previously found that both HDAC4 and 5 move out of fibre nuclei in response to Nox2-dependent reactive oxygen species production during more intensive fibre stimulation (Liu et al. 2012) and in response to alpha-adrenergic activation of PKD in slow but not fast fibres (Liu et al. 2009). In this study we now find that activation of beta-adrenergic signalling cascades results in nuclear influx of HDAC4 through activation of PKA, and that activation of PKA by N6-benzoyl cAMP causes net nuclear influx of both HDAC4-GFP and HDAC5-GFP. The net nuclear influx of HDACs observed here with beta-adrenergic activation of PKA is opposite to the phosphorylation-dependent nuclear efflux of HDACs observed previously with other stimuli. Results with mutant HDAC4 show that PKA-dependent phosphorylation occurs at HDAC4 residues 265 and/or 266, which are phosphorylated by PKA but not by the kinases activated by the other stimuli studied previously.
Our results with HDAC4 immunocytochemistry in fibres expressing HDAC4-GFP and in non-transfected muscle fibres demonstrate that the observed movements of HDAC4-GFP in response to activation of PKA reflect similar movements of endogenous HDAC4, and that HDAC4-GFP expression causes a 3.8-fold increase in the total level of endogenous HDAC4 plus expressed HDAC4-GFP in our fibres.
It was reported previously that activation of the PKA pathway in vascular smooth muscle cells resulted in an enhanced nuclear accumulation of HDAC4 by inhibiting salt-inducible kinase 1, without direct interaction between HDAC4 and PKA (Gordon et al. 2009). In the present report we demonstrate direct phosphorylation of HDAC4 by PKA. Our immunoprecipitation data demonstrate that wt HDAC4 is a substrate of PKA. The consensus phosphorylation motif for PKA substrates (R/K)XX(S*/T*) is well conserved in both human and mouse HDAC4 and 5 (Montminy, 1997; see Fig. 3). In HDAC5, phosphorylation by PKA at serine 280 can interrupt the binding between HDAC5 and 14–3–3 and thus block the nuclear efflux of HDAC5 (Ha et al. 2010; Chang et al. 2013). Whether phosphorylation of HDAC4 causes the interruption in binding between HDAC4 and 14–3–3 requires further investigation.
As cAMP or Db cAMP can activate Epac as well as PKA, we also employed specific activators of PKA or Epac to dissect the different roles of PKA or Epac in the localization of HDAC4. While an increase of cAMP due to application of beta-adrenergic agonist or the application of Db cAMP results in nuclear accumulation of HDAC4-GFP due to activation of PKA, artificial activation of Epac with 8-CPT causes nuclear efflux of HDAC4-GFP, so cAMP itself can potentially induce opposite changes in HDAC4 nuclear cytoplasmic distribution. Using antibodies recognizing activated PKA or Rap1, we found that the activation of both PKA and Epac are enhanced if Db cAMP is applied to muscle cultures. In addition, by monitoring HDAC4 (S265/266A)-GFP, which cannot be phosphorylated by PKA, we observed increased nuclear efflux of HDAC4 (S265/266A)-GFP when Db cAMP was applied to fibres stimulated using 4 Hz trains, a stimulation pattern which is submaximal for full activation of HDAC4 (S265/266A) nuclear efflux.
Epac1 is modestly expressed in skeletal muscle (Kawasaki et al. 1998; de Rooij et al. 1998). In skeletal muscle, the cAMP/Epac pathway is involved in the inhibitory effects of epinephrine on proteolysis (Baviera et al. 2010). In this respect, PKA and Epac work in a collaborative way to inhibit proteolysis in skeletal muscle. In cardiac myocytes, 8-CPT activates Epac and downstream CaMKII to modulate excitation–contraction coupling in a PKA-independent manner (Pereira et al. 2007). It was also reported that in rat cardiomyocytes application of 8-CPT triggered a nuclear efflux of HDAC5 through the PLC/IP3/Ca2+/CaMKII pathway (Pereira et al. 2012). In this report we found that application of cAMP can activate both Epac and PKA as monitored by active Rap1 (GTP-bound Rap1) and phospho PKA. However, there is no indication that the localization of HDAC4 is affected by the Epac pathway if Db cAMP is applied to muscle fibres. We observed only Epac- and CaMK-mediated nuclear efflux of HDAC4 when a specific Epac activator is used. The reason for this difference needs further investigation. There were reports that cAMP binds to the regulatory subunits of PKA with higher affinity than Epac (Dao et al. 2006). However, if using holoenzyme of PKA, it was found that PKA holoenzyme and Epac1 have similar cAMP affinity (Dao et al. 2006). Since in living cells the holoenzyme is the form of PKA, in physiological conditions cAMP should activate PKA and Epac equally. Thus, we do not believe that the lack of effects on Epac by Db cAMP seen here is due to different affinities of PKA and Epac to cAMP.
In conclusion, the interaction between the PKA and CaMKII pathways, and their respective phosphorylation of HDAC4 at different sites occurs when beta-adrenergic activation and muscle activity occur in the same time frame. Our results show that PKA can partially antagonize the effects of muscle activity on HDAC4 nuclear efflux, and that beta-adrenergic activation can thereby minimize the derepression of MEF2-driven gene expression during a given bout of muscle activity or exercise.
Acknowledgments
We are grateful to Dr Donald M. Bers (University of California Davis) for supplying the adeno virus for HDAC4 (S265/266A)-GFP and for sharing unpublished results.
Glossary
- AOI
area of interest
- CMV
cytomegalovirus
- Db cAMP
dibutyryl adenosine 3′,5′-cyclic monophosphate
- FDB
flexor digitorum brevis
- GEFs
guanine nucleotide exchange factors
- HDACs
histone deacetylases
- MEF2
myocyte enhancer factor 2
- MEM
minimal essential medium
- n/c
nuclear/cytoplasmic ratio
- PKA
protein kinase A
- PKD
protein kinase D
- PLC
phospholipase C
- RyR
ryanodine recepor
Additional information
Competing interests
No conflict of interest, financial or otherwise, are declared by the authors.
Author contribution
Y.L. and M.F.S. conceived and designed the research; Y.L. performed the experiments; Y.L. and M.F.S. analysed the data; Y.L. and M.F.S. wrote the manuscript; Y.L. and M.F.S. approved the final version of the manuscript.
Funding
This work was supported by research grant AR056477 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases, NIH.
References
- Andersson DC, Betzenhauser MJ, Reiken S, Umanskaya A, Shiomi T, Marks AR. Stress-induced increase in skeletal muscle force requires protein kinase A phosphorylation of the ryanodine receptor. J Physiol. 2012;590:6381–6387. doi: 10.1113/jphysiol.2012.237925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassel-Duby R, Olson EN. Signaling pathways in skeletal muscle remodeling. Annu Rev Biochem. 2006;75:19–37. doi: 10.1146/annurev.biochem.75.103004.142622. [DOI] [PubMed] [Google Scholar]
- Baviera AM, Zanon NM, Navegantes LC, Kettelhut IC. Involvement of cAMP/Epac/PI3K-dependent pathway in the antiproteolytic effect of epinephrine on rat skeletal muscle. Mol Cell Endocrinol. 2010;315:104–112. doi: 10.1016/j.mce.2009.09.028. [DOI] [PubMed] [Google Scholar]
- Chang CW, Lee L, Yu D, Dao K, Bossuyt J, Bers DM. Acute β-adrenergic activation triggers nuclear import of histone deacetylase 5 and delays Gq-induced transcriptional activation. J Biol Chem. 2013;288:192–204. doi: 10.1074/jbc.M112.382358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen AE, Selheim F, de Rooij J, Dremier S, Schwede F, Dao KK, Martinez A, Maenhaut C, Bos JL, Genieser HG, Døskeland SO. cAMP analog mapping of Epac1 and cAMP kinase. Discriminating analogs demonstrate that Epac and cAMP kinase act synergistically to promote PC-12 cell neurite extension. J Biol Chem. 2003;278:35394–35402. doi: 10.1074/jbc.M302179200. [DOI] [PubMed] [Google Scholar]
- Dao KK, Teigen K, Kopperud R, Hodneland E, Schwede F, Christensen AE, Martinez A, Doskeland SO. Epac1 and cAMP-dependent protein kinase holoenzyme have similar cAMP affinity, but their cAMP domains have distinct structural features and cyclic nucleotide recognition. J Biol Chem. 2006;281:21500–21511. doi: 10.1074/jbc.M603116200. [DOI] [PubMed] [Google Scholar]
- de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- Du M, Perry RL, Nowacki NB, Gordon JW, Salma J, Zhao J, Aziz A, Chan J, Siu KW, McDermott JC. Protein kinase A represses skeletal myogenesis by targeting myocyte enhancer factor 2D. Mol Cell Biol. 2008;28:2952–2970. doi: 10.1128/MCB.00248-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, Genieser HG, Døskeland SO, Blank JL, Bos JL. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol. 2002;4:901–906. doi: 10.1038/ncb874. [DOI] [PubMed] [Google Scholar]
- Galbo H, Richter EA, Hilsted J, Holst JJ, Christensen NJ, Henriksson J. Hormonal regulation during prolonged exercise. Ann N Y Acad Sci. 1977;301:72–80. doi: 10.1111/j.1749-6632.1977.tb38187.x. [DOI] [PubMed] [Google Scholar]
- Gordon JW, Pagiatakis C, Salma J, Du M, Andreucci JJ, Zhao J, Hou G, Perry RL, Dan Q, Courtman D, Bendeck MP, McDermott JC. Protein kinase A-regulated assembly of a MEF2·HDAC4 repressor complex controls c-Jun expression in vascular smooth muscle cells. J Biol Chem. 2009;284:19027–19042. doi: 10.1074/jbc.M109.000539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb AH, Bruno JF, Buckenmeyer PJ. Intensity and duration of exercise effects on skeletal muscle cAMP, phosphorylase, and glycogen. J Appl Physiol. 1989;66:190–194. doi: 10.1152/jappl.1989.66.1.190. [DOI] [PubMed] [Google Scholar]
- Grozinger CM, Schreiber SL. Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14–3–3-dependent cellular localization. Proc Natl Acad Sci USA. 2000;97:7835–7840. doi: 10.1073/pnas.140199597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta MP, Samant SA, Smith SH, Shroff SG. HDAC4 and PCAF bind to cardiac sarcomeres and play a role in regulating myofilament contractile activity. J Biol Chem. 2008;283:10135–10146. doi: 10.1074/jbc.M710277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha CH, Kim JY, Zhao J, Wang W, Jhun BS, Wong C, Jin ZG. PKA phosphorylates histone deacetylase 5 and prevents its nuclear export, leading to the inhibition of gene transcription and cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2010;107:15467–15472. doi: 10.1073/pnas.1000462107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy S, Kitamura M, Harris-Stansil T, Dai Y, Phipps ML. Construction of Adenovirus vectors through Cre-lox recombination. J Virol. 1997;71:1842–1849. doi: 10.1128/jvi.71.3.1842-1849.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmstadter KG, Erickson JR, Dao K, Bossuyt J, Bers DM. β-Adrenergic activation enhances histone deacetylase 4 nuclear localization via PKA and counters CAMKII-dependent effects in adult rabbit cardiac myocytes. Biophys J. 2011;100:80a. [Google Scholar]
- Holz GG, Chepurny OG, Schwede F. Epac-selective cAMP analogs: new tools with which to evaluate the signal transduction properties of cAMP-regulated guanine nucleotide exchange factors. Cell Signal. 2008;20:10–20. doi: 10.1016/j.cellsig.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- Kim MS, Fielitz J, McAnally J, Shelton JM, Lemon DD, McKinsey TA, Richardson JA, Bassel-Duby R, Olson EN. Protein kinase D1 stimulates MEF2 activity in skeletal muscle and enhances muscle performance. Mol Cell Biol. 2008;28:3600–3609. doi: 10.1128/MCB.00189-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Kranias EG, Schneider MF. Regulation of Ca2+ handling by phosphorylation status in mouse fast- and slowtwitch skeletal muscle fibres. Am J Physiol. 1997;273:C1915–1924. doi: 10.1152/ajpcell.1997.273.6.C1915. [DOI] [PubMed] [Google Scholar]
- Liu Y, Randall WR, Schneider MF. Activity-dependent and -independent nuclear fluxes of HDAC4 mediated by different kinases in adult skeletal muscle. J Cell Biol. 2005;168:887–897. doi: 10.1083/jcb.200408128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Contreras M, Shen T, Randall WR, Schneider MF. α-Adrenergic signalling activates protein kinase D and causes nuclear efflux of the transcriptional repressor HDAC5 in cultured adult mouse soleus skeletal muscle fibres. J Physiol. 2009;587:1101–1115. doi: 10.1113/jphysiol.2008.164566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Hernández-Ochoa EO, Randall WR, Schneider MF. NOX2-dependent ROS is required for HDAC5 nuclear efflux and contributes to HDAC4 nuclear efflux during intense repetitive activity of fast skeletal muscle fibres. Am J Physiol Cell Physiol. 2012;303:C334–347. doi: 10.1152/ajpcell.00152.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee SL, Hargreaves M. Histone modifications and exercise adaptations. J Appl Physiol. 2011;110:258–263. doi: 10.1152/japplphysiol.00979.2010. [DOI] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN. Identification of a signal-responsive nuclear export sequence in class II histone deacetylases. Mol Cell. Biol. 2001;21:6312–6321. doi: 10.1128/MCB.21.18.6312-6321.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Lu J, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408:106–111. doi: 10.1038/35040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Métrich M, Berthouze M, Morel E, Crozatier B, Gomez AM, Lezoualc’h F. Role of the cAMP-binding protein Epac in cardiovascular physiology and pathophysiology. Pflugers Arch. 2010a;459:535–546. doi: 10.1007/s00424-009-0747-y. [DOI] [PubMed] [Google Scholar]
- Métrich M, Laurent AC, Breckler M, Duquesnes N, Hmitou I, Courillau D, Blondeau JP, Crozatier B, Lezoualc’h F, Morel E. Epac activation induces histone deacetylase nuclear export via a Ras-dependent signalling pathway. Cell Signal. 2010b;22:1459–1468. doi: 10.1016/j.cellsig.2010.05.014. [DOI] [PubMed] [Google Scholar]
- Montminy M. Transcriptional regulation by cyclic AMP. Annu Rev Biochem. 1997;66:807–822. doi: 10.1146/annurev.biochem.66.1.807. [DOI] [PubMed] [Google Scholar]
- Oestreich EA, Wang H, Malik S, Kaproth-Joslin KA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV. Epac-mediated activation of phospholipase Cɛ plays a critical role in β-adrenergic receptor-dependent enhancement of Ca2+ mobilization in cardiac myocytes. J Biol Chem. 2007;282:5488–5495. doi: 10.1074/jbc.M608495200. [DOI] [PubMed] [Google Scholar]
- Oestreich EA, Malik S, Goonasekera SA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV. Epac and phospholipase Cɛ regulate Ca2+ release in the heart by activation of protein kinase Cɛ and calcium-calmodulin kinase II. J Biol Chem. 2009;284:1514–1522. doi: 10.1074/jbc.M806994200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paroni G, Cernotta N, Dello Russo C, Gallinari P, Pallaoro M, Foti C, Talamo F, Orsatti L, Steinkühler C, Brancolini C. PP2A regulates HDAC4 nuclear import. Mol Biol Cell. 2008;19:655–667. doi: 10.1091/mbc.E07-06-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra M, Verdin E. Regulatory signal transduction pathways for class IIa histone deacetylases. Curr Opin Pharmacol. 2010;10:454–460. doi: 10.1016/j.coph.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Pereira L, Métrich M, Fernández-Velasco M, Lucas A, Leroy J, Perrier R, Morel E, Fischmeister R, Richard S, Bénitah JP, Lezoualc’h F, Gómez AM. The cAMP binding protein Epac modulates Ca2+ sparks by a Ca2+/calmodulin kinase signalling pathway in rat cardiac myocytes. J Physiol. 2007;583:685–694. doi: 10.1113/jphysiol.2007.133066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira L, Ruiz-Hurtado G, Morel E, Laurent AC, Métrich M, Domínguez-Rodríguez A, Lauton-Santos S, Lucas A, Benitah JP, Bers DM, Lezoualc’h F, Gómez AM. Epac enhances excitation-transcription coupling in cardiac myocytes. J Mol Cell Cardiol. 2012;52:283–291. doi: 10.1016/j.yjmcc.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira L, Cheng H, Lao DH, Na L, van Oort RJ, Brown JH, Wehrens XH, Chen J, Bers DM. Epac2 mediates cardiac β1-adrenergic-dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia. Circulation. 2013;127:913–922. doi: 10.1161/CIRCULATIONAHA.12.148619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppe H, Rybalkin SD, Rehmann H, Hinds TR, Tang XB, Christensen AE, Schwede F, Genieser HG, Bos JL, Doskeland SO, Beavo JA, Butt E. Cyclic nucleotide analogs as probes of signalling pathways. Nat Methods. 2008;5:277–278. doi: 10.1038/nmeth0408-277. [DOI] [PubMed] [Google Scholar]
- Schachter TN, Shen T, Liu Y, Schneider MF. Kinetics of nuclear-cytoplasmic translocation of Foxo1 and Foxo3A in adult skeletal muscle fibres. Am J Physiol Cell Physiol. 2012;303:C977–C990. doi: 10.1152/ajpcell.00027.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MJ, Green HJ, Cobb FR. Skeletal muscle biochemistry and histology in ambulatory patients with long-term heart failure. Circulation. 1990;81:518–527. doi: 10.1161/01.cir.81.2.518. [DOI] [PubMed] [Google Scholar]
- Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, McKinsey TA. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol. 2004;24:8374–8385. doi: 10.1128/MCB.24.19.8374-8385.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR, Molkentin JD. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]