

Abstract
The release of lentiviral vectors for clinical use requires the testing of vector material, production cells, and, if applicable, ex vivo-transduced cells for the presence of replication-competent lentivirus (RCL). Vectors derived from the nonprimate lentivirus equine infectious anemia virus (EIAV) have been directly administered to patients in several clinical trials, with no toxicity observed to date. Because EIAV does not replicate in human cells, and because putative RCLs derived from vector components within human vector production cells would most likely be human cell-tropic, we previously developed an RCL assay using amphotropic murine leukemia virus (MLV) as a surrogate positive control and human cells as RCL amplification/indicator cells. Here we report an additional RCL assay that tests for the presence of theoretical “equine-tropic” RCLs. This approach provides further assurance of safety by detecting putative RCLs with an equine cell-specific tropism that might not be efficiently amplified by the human cell-based RCL assay. We tested the ability of accessory gene-deficient EIAV mutant viruses to replicate in a highly permissive equine cell line to direct our choice of a suitable EIAV-derived positive control. In addition, we report for the first time the mathematical rationale for use of the Poisson distribution to calculate minimal infectious dose of positive control virus and for use in monitoring assay positive/spike control failures in accumulating data sets. No RCLs have been detected in Good Manufacturing Practice (GMP)-compliant RCL assays to date, further demonstrating that RCL formation is highly unlikely in contemporary minimal lentiviral vector systems.
Farley and colleagues report on a novel replication-competent lentivirus (RCL) assay that tests the theoretical possibility that non-primate lentivirus production might lead to the formation of RCLs with slow replication kinetics that may not be detected in human cell-based RCL assays. Additionally, the authors examine in detail the mathematical rationale for positive control virus dose setting and monitoring of assay control failures using Poisson distribution related mathematics.
Introduction
The development of lentiviral gene therapy vectors to treat genetic disorders in humans is becoming more widespread, with several such therapeutics currently proceeding through clinical trials in Europe and North America (DiGiusto et al., 2010; Galy and Thrasher, 2010; Sadelain et al., 2010; Porter et al., 2011; Campochiaro, 2012; Cartier et al., 2012). We have developed a highly engineered nonprimate lentiviral vector system based on equine infectious anemia virus (EIAV) that has an excellent safety profile in preclinical trials (Jarraya et al., 2009; and K.A. Mitrophanous, unpublished). Indeed, early in 2008 the first in vivo administration of a lentiviral vector in humans was performed with a vector based on EIAV (ProSavin®). This administration was performed as part of an ongoing phase 1 clinical trial for Parkinson's disease. The results to date in this trial, in which 15 people have been intrastriatally administered ProSavin, provide an encouraging indication that EIAV vectors are safe and well tolerated. More recently, phase 1 trials have been initiated using lentiviral vector-based treatments based on the EIAV platform for “wet” age-related macular degeneration (AMD) (RetinoStat; Campochiaro, 2012), Stargardt disease (StarGen™), and Usher syndrome type 1B (UshStat®), again showing the vector is safe and well tolerated.
The EIAV vector platform is designed to minimize the spontaneous formation of a replication-competent lentivirus (RCL) by splitting the vector system into three components. In addition, the system lacks all accessory genes (tat, rev, S2) and uses a self-inactivating long terminal repeat (LTR) configuration (Miyoshi et al., 1998). In addition, because EIAV does not infect or replicate within human cells, patients have no preexisting immunity to EIAV. Last, in contrast to gammaretroviral vectors, lentiviral vectors have no preference for integrating into proto-oncogene loci (Hacker et al., 2006; Biffi et al., 2011; Biasco et al., 2012).
Nevertheless, RCL testing of both clinical-grade lentiviral vector material and end-of-production cells (EOPCs) from the manufacturing process is currently a prerequisite for drug release. This testing approach takes into consideration the potential for unpredictable recombination of vector components with other nucleic acid sequences within the production cells, such as endogenous retroviruses and/or therapeutic gene sequences. The nonpermissive nature of human cell cultures to EIAV precludes the development of a human cell-based RCL assay coupled with an EIAV-derived positive control, and consequently we previously validated an assay using amphotropic murine leukemia virus (MLV) as a surrogate positive control and HEK293 cells as amplification cells (Miskin et al., 2006). However, after consultation with the Food and Drug Administration (FDA), and with a desire to comprehensively demonstrate the safety of the EIAV lentiviral vector system, we now report the development of an additional Good Manufacturing Practice (GMP)-compliant RCL assay based on equine cells and using an EIAV-derived positive control virus. The rationale for this additional assay was that it should be capable of detecting hypothetical “equine-tropic” RCLs that might not be amplified in the MLV/HEK293 assay. It is worth noting that the features conferring EIAV with an equine tropism have been removed from the minimal vector system, as it is currently configured; EIAV envelope, the EIAV accessory genes, and equine cyclin T1 (Bieniasz et al., 1999) are all absent from the production system.
A number of nonhuman cell lines were evaluated for use as amplification cells, both for their ability to support EIAV replication and their practicability for long-term passaging and viable cell banking. In addition, we generated attenuated EIAV mutants, each functionally lacking one of the three accessory genes, and tested their replication in an attempt to model putative RCLs that might arise from the minimal vector system. The equine-tropic RCL assay (ET-RCL assay) described herein follows a format similar to that described for the MLV/HEK293 RCL assay: cell culture phase amplification of putative RCL within final drug product (FDP) or EOPCs, followed by an end-point analysis of reverse transcriptase activity in supernatants by quantitative RT-PCR (Miskin et al., 2006).
We also discuss in greater detail than has been published to date, our interpretation of FDA guidelines pertaining to positive control virus dose setting in replication-competent retrovirus (RCR)/RCL assays. As well as ensuring assay robustness (acceptable failure rate), the dose of virus must be set at an amount that demonstrates assay sensitivity. Because virus infection follows a Poisson distribution, the virus dose may be mathematically determined and empirically tested. Consequently, we suggest a mathematical rationale for monitoring and interpretation of positive control or spike/inhibition control failure rates.
Materials and Methods
ET-RCL assay principles
The current manufacturing approach for EIAV-based Investigational Medicinal Products (IMP) at Oxford BioMedica (OXB, Oxford, UK) is the production of crude vector material followed by downstream processing to purify and concentrate vector from several separate subruns; after analytical testing, appropriate batches are pooled and processed into a single FDP preparation. Each FDP is tested in one RCL assay and EOPCs from each of the subruns are tested in separate RCL assays. FDA guidelines state that RCL testing must be performed on 1% or 1×108 EOPCs (whichever is less) and 5% of the vector material or “test article” (Fig. 1) (Food and Drug Administration, 2006). Alternatively, a mathematically determined minimal volume of test article may be tested when generating greater than 6 liters of crude harvest material; this depends on an assumed RCL concentration and the degree of sensitivity of detection chosen for the assay (typically ≥95% detection rate). The sensitivity of RCL detection must be enhanced by an amplification phase, requiring a permissive cell type, followed by a sensitive indicator or end-point assay (Escarpe et al., 2003; Sastry et al., 2003, 2005; Segall et al., 2003); for a review see Sastry and Cornetta (2009).
FIG. 1.

Overview of equine infectious anemia virus (EIAV) vector manufacture and replication-competent lentivirus (RCL) testing. Typical manufacture of EIAV-based lentiviral vectors is initiated by triple transfection of vector components into adherent HEK293T cells in 24 CF10 Cell Factories per subrun. The end-of-production cells (EOPCs) from Cell Factories are pooled and stored at −80°C. Crude harvest vector material from three subruns is pooled, purified, and concentrated to yield the finished drug product. For drug release, 5% of final drug product (FDP) is tested in a single RCL assay. EOPCs (1×108) from each of the three subruns are tested for RCLs by coculture with amplification cells in three separate cocultivation RCL assays. Supernatant (Supnt) from the cocultures is used to inoculate a second round of RCL amplification. The end-point assay uses product-enhanced reverse transcriptase (PERT), a highly sensitive quantitative RT-PCR method, to detect RT associated with RCL. VSV-G, vesicular stomatitis virus glycoprotein G.
The highly sensitive product-enhanced reverse transcriptase (PERT) assay (a qRT-PCR assay) is used for the relative titration of vector lots and as an end-point assay for RCL detection (Martin-Rendon et al., 2002; Miskin et al., 2006). Testing for RT activity is considered to be the most sensitive and appropriate readout because, whatever its genomic configuration, by definition any RCL must possess RT activity. Because of the high sensitivity of the PERT assay and the inability to discriminate between RT from vector and putative RCL, the amplification phase (involving serial passage of inoculated permissive cells) is necessary to ensure removal of preexisting RT activity associated with the test article or EOPCs that would otherwise lead to false positive results.
Viruses and cell lines
The infectious strain of EIAV used in this study (and on which the vector system is based) was SPEIAV-19, obtained from S. Payne (Faculty of Genetics, Texas A&M University, College Station, TX). Strain SPEIAV-19 was isolated from cell culture adaptation of EIAVPV[A1/E] (Rwambo et al., 1990; Payne et al., 1994). Amphotropic MLV-4070A hybrid (VR1450) was purchased from the American Type Culture Collection (ATCC, Manassas, VA).
92BR cells (lot CB1621; European Collection of Cell Cultures [ECACC], Porton Down, UK) and DH82 cells (lot 08E028; ECACC) were maintained in Eagle's minimal essential medium (EMEM)–15% (v/v) fetal bovine serum (FBS)–2 mM l-glutamine–1% (v/v) nonessential amino acids (NEAA). ED (NBL-6) cells (CCL-57, lot 58255369; ATCC) were maintained in EMEM–10% (v/v) FBS–2 mM l-glutamine–1% (v/v) NEAA. EML-3C cells and NIH3T3(ELR1/cyc) cells were kind gifts under material transfer agreements (MTAs) from R. Montelaro (Center of Vaccine Research, University of Pittsburgh, Pittsburgh, PA) and were maintained in Dulbecco's modified Eagle's medium (DMEM)–20% (v/v) FBS–4 mM l-glutamine–1% (v/v) NEAA and DMEM–10% (v/v) FBS–4 mM l-glutamine–1% (v/v) NEAA, respectively. Cf2Th cells (CRL-1430; ATCC) were maintained as per EML-3C cells. D17 and HEK293T cells were maintained in DMEM–10% (v/v) FBS–2 mM l-glutamine–1% (v/v) NEAA.
Virus infections and vector transductions
All work involving EIAV and attenuated EIAV was carried out at Biosafety Level 3 (BSL3) containment according to U.K. Department for Environment, Food, and Rural Affairs (DEFRA) regulations. All infections and transductions occurred in the presence of Polybrene at a final concentration of 8 μg/ml. For comparison of EIAV replication in permissive cell lines, 1 day before infection with EIAV, NIH3T3(ELR1/cyc), 92BR, ED, and Cf2Th cells were seeded at 2.7×105 cells per replicate well in a 6-well plate format. On the day of viral inoculation, medium was removed and cultures were incubated for 24 hr with 0.5 ml of medium containing either EIAV (multiplicity of infection [MOI] of 0.01) or MLV (MOI of 0.003) or were mock-infected (negative control). NIH3T3(ELR1/cyc), 92BR, ED, and Cf2Th cultures were subsequently up-scaled to 10-cm2 dish format and passaged at 1:5–8, 1:4–6, 1:3–4 and 1:8–10 split ratios, respectively. Culture supernatants were sampled before passage, filtered (pore size, 0.45 μm), and stored at −80°C.
For determination of the minimal infectious dose of the positive control stock of EIAVΔS2, 92BR cells were seeded into 24-well plates at 2.9×104 cells per well 20 hr before infection. On the day of inoculation, virus was diluted to 100 PERT-predicted transduction units (TU)/ml (prediction based on RT activity associated with a standard EIAV-green fluorescent protein [GFP] vector of known biological titer) in medium and then a serial 4-fold dilution series from 12.5 to 0.05 PERT-predicted TU/ml generated. Media in the 24-well cultures were replaced with 0.4-ml volumes of diluted virus to generate eight replicates at each concentration of the dilution series (i.e., 5, 1.25, 0.31, 0.08, or 0.02 PERT-predicted TU/well). Infected cultures were incubated for 20 hr before 0.4-ml volumes of fresh medium replaced the inoculums and cultures were incubated for a further 9 days, whereupon samples were taken for PERT analysis.
A vesicular stomatitis virus glycoprotein G (VSV-G)-pseudotyped EIAV-GFP vector preparation was used to transduce ∼2×105 92BR or HEK293T cells per well in 48-well plate format at an equivalent MOI of 1 in the presence of Polybrene (8 μg/ml). Approximately 48 hr posttransduction, digital images were taken under ultraviolet light.
Construction of mutant EIAV proviral DNA and accessory gene plasmids
EIAV mutant proviral plasmid DNA was derived from pSPEIAV-19 (Payne et al., 1994), which contained an SC101 low-copy origin of replication to avoid unwanted rearrangement of DNA in bacteria (Cunningham et al., 1993). Subsequent reference to nucleotide positions are relative to the R-to-R sequence of EIAV in the 7988-nucleotide SPEIAV-19 genome.
EIAVΔtat: A cytosine base was inserted between positions 168 and 169 in the tat1 exon to disrupt the atypical CTG translation start codon. To generate a TGA stop codon in the tat2 exon, a T-to-A mutation at position 4953 was generated. Nucleotide changes were introduced into pSPEIAV-19 by gene synthesis (GeneArt/Life Technologies, Carlsbad, CA) of a 1052-bp DNA fragment (encompassing the 5′ LTR, mutated tat1 exon, and a portion of GAG) and replacement of the sequence between the two BbvCI sites. Similarly, synthesis of a 671-bp DNA fragment (containing the mutated tat2 exon) was carried out and cloned into the NcoI–BlpI sites.
EIAVΔrev: Two stop codons were introduced into the 5′ region of the rev2 exon, which encodes all domains essential for function (Fridell et al., 1993; Harris et al., 1998), without disrupting the envelope primary amino acid sequence in the overlapping open reading frame. This was achieved by two C-to-T mutations at positions 7035 and 7056; a 1433-bp DNA fragment containing the mutations was synthesized and cloned into pSPEIAV-19 via BsmBI–BstXI sites. Changes in the tat and rev exons were confirmed by sequencing of purified fragments of plasmid DNA.
EIAVΔS2: The pSPEIAVΔS2 plasmid had been constructed previously (Yoon, 1999); the translation initiation codon was replaced with a TAG stop codon (AT to TA at position 5080-1) and a T>C mutation at position 5127 resulted in an M16T in S2 in order to stop potential downstream translation initiation (the envelope primary amino acid sequence was unaffected).
The rev open reading frame was codon-optimized (CO) for human expression, resulting in 73% sequence identity with wild-type rev. The gene sequence was assembled using short oligonucleotides and cloned into pCI-Neo. The tat open reading frame was codon-optimized for human expression, resulting in 66% sequence identity with wild-type tat. The sequence was synthesized by GeneArt and cloned into pCI-Neo.
Pseudo-infection of 92BR cells with EIAV and mutant EIAV
Approximately 1.5×106 92BR cells were seeded into replicate 10-cm2 dishes the day before transfection. Cultures were transfected with 10 μg of total DNA in 0.5-ml volumes of 2.4 mM polyethylenimine (PEI, 25 kDa)–0.15 M NaCl, 7 μg of genome plasmid, 1.5 μg of each appropriate accessory gene plasmid, and 0, 1.5, or 3 μg of pBlueScript were used to normalize DNA input. Twenty hours posttransfection the culture supernatant fluid was harvested, filtered (pore size, 0.45 μm), and stored for PERT analysis. Six milliliters of fresh medium was added to each dish and cultures were incubated until 40 hr posttransfection, when samples were taken as described previously. Cultures where then passaged for 41 days, splitting 1:6 to a final volume of 10 ml in 10-cm2 dishes, and taking samples for PERT analysis before passage. To assess transfection efficiency, a control transfection with 10 μg of pONY8.9NCZ (a cytomegalovirus promoter [CMVp]-driven, LacZ-expressing EIAV vector genome) was included in parallel; 20 hr posttransfection this culture was fixed in 38% (v/v) formalin and stained with 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) solution.
Predicting virus infection probability using Poisson distribution
The probability of virus infection (p[positive]) follows a Poisson distribution as stated within FDA guidelines (Food and Drug Administration, 2006) and denoted by Eq. (1) (see Terms and Equations). Equation (1) may be rearranged to Eq. (3), solving for test volume (Vt), or to Eq. (4), solving for virus concentration (c). Because concentration is also a function of volume [Eq. (5)], Eqs. (4) and (5) are equivalent only when the entire inoculated volume is tested. Therefore, Vt becomes an irrelevant factor and Eq. (6) may be solved to find Eq. (7). Thus, the required number of infectious units (IU) of any virus to achieve a particular probability of infection rate (p) is a function of the natural log of 1 – p, which effectively considers the probability of noninfection (p[negative]) [Eq. (8)] (see Grigorov et al., 2011). Under the same conditions, the probability of infection can be predicted when a known number of IU has been inoculated [Eq. (2)]. Note that these simplified equations should be applied only under preset experimental conditions: assuming the number of permissive cells is not limiting, the maximal tissue culture volume per area in which a single infectious unit can be amplified must be determined empirically, and this volume-to-area ratio must not be increased to accommodate larger test volumes—instead, the number of test vessel replicates (i.e., area) must be increased.
Terms and equations
Infection rate or probability of infection (p)
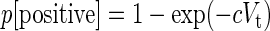 |
(1) |
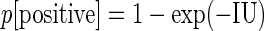 |
(2)* |
Test volume (Vt)
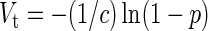 |
(3) |
Concentration (c)
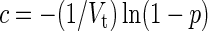 |
(4) |
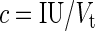 |
(5) |
Infectious units required for a given detection rate (IU[p])
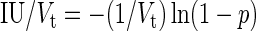 |
(6) |
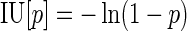 |
(7)* |
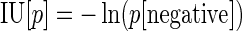 |
(8)* |
*When entire inoculated test volume is assayed under empirically determined conditions.
Test article ET-RCL assay protocol
Tissue culture flasks (T150) were seeded with 4.3×106 92BR cells per flask and the next day the supernatant fluid from each flask was replaced with 8 ml of complete medium containing Polybrene at 8 μg/ml. The 10 test flasks were inoculated with a 5% volume of Final Drug Product (FDP); typically, EIAV vector manufacture from 72 Nunc CF10 Cell Factories (Thermo Fisher Scientific, Waltham, MA) resulted in approximately 30 ml of FDP, of which 1.5 ml was tested, that is, 150 μl per flask (or 0.5% of the FDP). The positive and 1× spike flasks each received 3.9 IU of EIAVΔS2, and the 10× spike flask received 39 IU of EIAVΔS2; both spike flasks were transduced with 150 μl (0.5%) of FDP. The next day cultures were observed for evidence of cytopathic effect before all cultures were transferred to T225 flasks. Once 80–100% confluent, cultures were passaged by 1:2 split ratio (passage 1) followed by 1:4 split ratios in T225 flasks (passages 2–7) until passage 8, whereupon the amplification phase was terminated. Filtered cell-free supernatant samples were taken immediately prepassage at all passage points. Viable cell banks were taken at passages 1 and 8, and end-point PERT analysis was performed on passage 8 supernatant samples. The assay was performed to GMP in a BSL3 containment laboratory according to U.K. DEFRA regulations.
EOPC ET-RCL assay protocol
Cocultures were initiated by seeding tissue culture flasks (T225) with 1×107 92BR cells and 1×107 EOPCs (for 10× test flasks [1×108 EOPCs in total] and 1× spike flasks) or 1×107 HEK293T (for negative and positive flasks) in 35 ml of complete medium containing Polybrene at 8 μg/ml. The next day the supernatant fluid from each flask was replaced with 12 ml of complete medium containing Polybrene at 8 μg/ml. Positive and 1× spike flasks each received 3.9 IU of EIAVΔS2, and the 10× spike flask received 39 IU of EIAVΔS2. Cultures were incubated for another day before an additional 12 ml of complete medium was added to each flask and cultures were incubated for a further 6 days. After this cocultivation stage, approximately 24 ml of culture medium from each flask was filtered (pore size, 0.20 μm), and Polybrene was added to a final concentration of 8 μg/ml and inoculated into T225 flasks seeded the day before at 1×107 92BR cells per flask. The next day cultures were supplemented with an additional 17 ml of complete medium. Once 80–100% confluent, cultures were passaged by 1:4 split ratio until passage 7, following the same format as described for the test article assay. End-point PERT analysis was performed on passage 7 supernatant samples. The assay was performed to GMP in a BSL3 containment laboratory according to U.K. DEFRA regulations.
Reverse transcriptase and viral RNA detection assays
The product-enhanced reverse transcriptase (PERT) assay was used to quantify RT activity in filtered (pore size, 0.45 μm) culture supernatant that had been mixed with equal quantities of particle disruption buffer, and 25-μl PERT reactions were tested as described in greater detail elsewhere (Martin-Rendon et al., 2002). Viral RNA extracted from 140 μl of filtered (pore size, 0.45 μm) culture supernatants was analyzed in 25-μl reactions by qPCR, as described by Martin-Rendon and colleagues (2002). The lower limits of detection for PERT and pRT-PCR were 0.739 PERT-predicted TU/ml and 100 copies/ml, respectively.
Results
Selection of 92BR cells as the ET-RCL assay amplification cell line
A good candidate for an RCL amplification cell line is one that is (1) able to support efficient replication of the parental virus on which the vector system is based, (2) of the same mammalian species that is used for vector production (because a putative RCL will most likely replicate in these cells), and (3) permissive for a human-tropic RCL. In the case of EIAV vector manufacturing, the human cell line HEK293T is used to produce vector, usually by transient triple transfection of vector component-encoding plasmid DNA (see Fig. 1). No human cell line is able to support replication of EIAV because of the lack of receptor(s) (e.g., equine lentivirus receptor-1 [ELR1]; Zhang et al., 2005) and the low activity of the LTR enhancer/promoter in human cells because of the inability of equine Tat to recruit human cyclin T1 to the transcription complex (Bieniasz et al., 1999). Our efforts in developing an EIAV-permissive HEK293-based cell line coexpressing equine cyclin-T1 (eCT1) (Miskin et al., 2006) and equine lentivirus receptor-1 (ELR1) (our unpublished data) have been met with major challenges that, although interesting, are beyond the scope of this paper to describe.
Several EIAV-permissive primary cell types have been reported in the literature: fetal equine kidney (FEK) (Montelaro et al., 1982; Payne et al., 1987), equine monocyte-derived macrophage (eMDM) (Payne et al., 1994), and equine dermis (ED or NBL-6) (Malmquist et al., 1973); the latter primary cell type is capable of 20–30 passages in vitro, albeit with a relatively slow doubling time. Immortalized cell lines that are claimed to support EIAV replication include EML-3C (equine monocyte) (Fidalgo-Carvalho et al., 2009), Cf2Th (canine thymus) (Bouillant et al., 1986), 92BR (donkey testis), DH82 (canine monocyte) (Hines and Maury, 2001), D17 (canine osteosarcoma) (Beisel et al., 1993), NIH3T3(ELR1/cyc) (mouse fibroblast cells engineered to express ELR1 and eCT1; Zhang and Montelaro, 2009), and FEA (feline embryonic fibroblast) (Derse et al., 1987).
Of the readily available cell types, we chose to evaluate EML-3C, 92BR, DH82, Cf2Th, ED, and NIH3T3(ELR1/cyc) cells for their respective growth characteristics and also their capacity for long-term culture in vitro with multiple passages. We discounted D17 cells because these cells were poorly transduced by an EIAV envelope-“pseudotyped” GFP vector (data not shown). EML-3C and DH82 cells were also discounted because of slow growth rate and semiadherence, respectively (data not shown). We therefore evaluated EIAV replication in ED, 92BR, NIH3T3(ELR1/cyc), and Cf2Th cells, using MLV as a comparison (Fig. 2A). Infection with EIAV (SPEIAV-19) or MLV (MLV-4070A hybrid) was initiated at a low multiplicity of infection (MOIs of 0.01 and 0.003, respectively) in six-well cultures and passaged for 16–20 days, taking supernatants for PERT analysis before further passage in tissue culture at each time point. The highest titers of EIAV were observed in 92BR cultures at 16–20 days postinfection. The titers observed for EIAV in 92BR cells were approximately 10-fold higher than were observed for ED cells, although ED cells might have achieved equivalent titers had the time course continued. The differences between these two cell lines could potentially be attributed to the slower growth rate of ED cells, although MLV infection had reached maximal titers in all cultures by day 13 postinfection. Surprisingly, no detectable EIAV infection of Cf2Th cells was observed at the end of the time course, and only minimal RT activity was detected in EIAV-inoculated NIH3T3(ELR1/cyc) cultures, in contrast to published work. Subsequent to this result, we were advised that the ELR1 receptor component expressed in NIH3T3(ELR1/cyc) cells may be unstable (R. Montelaro, personal communication) and so a cell-spreading infection of this cell type from low MOI may not be robust.
FIG. 2.

Evaluation of EIAV-permissive cells for use in amplification of equine-tropic replication-competent lentivirus (ET-RCL). (A) Four cell lines were preselected for evaluation on the basis of reported literature, growth characteristics, and availability. Wild-type EIAV (EIAV) and MLV-4070A (MLV) were inoculated into small-scale cultures at multiplicities of infection (MOIs) of 0.01 and 0.003, respectively, and infection was monitored over 16–20 days by PERT assay of supernatants sampled before each passage. Uninfected cultures were included in parallel (NEG). Each data marker represents accumulation of RT in cultures from the previous passage point, thus representing steady state RT levels (n=2); lines between data points are therefore displayed for clarity purposes. (B) HEK293T cells and (C) 92BR cells were transduced with a VSV-G-pseudotyped EIAV-GFP vector.
Next we decided to evaluate 92BR cell transduction by a VSV-G-pseudotyped EIAV vector to ensure this cell line was capable of being infected with a putative RCL enveloped with VSV-G (the most likely envelope to be encoded by an RCL derived from the vector system). A GFP-expressing VSV-G-pseudotyped vector transduced both HEK293T and 92BR cells efficiently (Fig. 2B and C, respectively). Because the 92BR cell line was capable of supporting growth of EIAV from low MOI to yield high titers within a short time frame, had flexible cell-culturing properties (split ratios of between 1:4 to 1:8, twice per week), and was highly permissive for VSV-G-mediated entry, it was selected for further evaluation as the amplification cell line of choice for the new assay.
Evaluation of attenuated EIAV replication in 92BR cells
Figure 3 displays schematics of the proviral DNA and accessory gene-complementation plasmids that were used in this work, as well as the vector system components. The minimal EIAV vector system lacks all accessory genes. Tat function, and the requirement for eCT1, are negated by the use of a strong constitutive promoter to drive vector genome expression, and the configuration of an open reading frame (ORF) immediately downstream of the packaging signal confers rev independence to the vector RNA during production, and therefore the Rev-responsive element (RRE) is not required within EIAV vector genomes (FJ Wilkes, Oxford BioMedica Ltd). There is strong evidence that S2 functions as an immune system modulator in vivo (Li et al., 2000; Covaleda et al., 2010), and S2 is required neither for virus replication in vitro (Li et al., 1998) nor for vector activity (Mitrophanous et al., 1999). Because it is not envisaged that a putative RCL could “regain” the tat and rev ORFs during vector production but that a recombinant may partially lose the sequences that functionally replace tat and rev in the vector system, it might be argued that a “slow-growing” RCL could be produced. To evaluate attenuated EIAVs and to choose a suitable positive control for the ET-RCL assay, proviral DNAs encoding EIAVΔtat and EIAVΔrev were constructed. Mutations within appropriate accessory gene-encoding exons were engineered to ensure that no functional accessory protein would be produced. Because an EIAV derivative in which S2 had been knocked out was already available (Yoon, 1999) and had been demonstrated to replicate with similar kinetics to wild-type EIAV in vitro (Li et al., 1998), there was no requirement to generate an S2 complementation plasmid.
FIG. 3.

DNA constructs generated in this study and EIAV vector system components. The EIAV genome structure is simple, encoding only three accessory genes: tat, rev, and S2. To reduce the chance of mutation reversion occurring during infection studies, mutant EIAV proviral DNAs each contained two mutations to abrogate expression of the respective accessory protein. Color coding of these genomes is maintained in subsequent figures. F-shift, frameshift mutation; Stop, translation stop codon; ATG-KO, translation start knockout; ψ, packaging signal; RRE, Rev-responsive element; ORF, open reading frame; Pro, promoter; IN, synthetic intron; wPRE, woodchuck hepatitis virus posttranscriptional regulatory element; SIN, self-inactivating LTR; CO, codon-optimized; LTR, long terminal repeat.
To evaluate the capacity of the three mutant EIAV variants to produce viral particles in a single round of replication, we mimicked infection of 92BR cell cultures at an MOI of 0.1 by achieving a transfection efficiency of ∼10% with proviral DNA (parallel transfection of a lacZ plasmid control followed by X-Gal staining allowed retrospective assessment of transfection efficiency; data not shown). Cotransfection of single accessory gene expression plasmids with appropriate proviral DNA was carried out to test whether deficiencies in the mutant EIAV variants could be rescued. Culture supernatants at 20 and 40 hr posttransfection were sampled and subjected to PERT analysis (Fig. 4). Transfection of 92BR cells with wild-type EIAV proviral DNA resulted in ∼1×102 PERT-predicted TU/ml in supernatant at 20 hr posttransfection, indicating the level of virion production from “first round”-infected cells. This level of virion production increased 100-fold by 40 hr posttransfection, perhaps reflecting maximal production from first round–infected cells and possibly some second round–infected cells. pEIAVΔS2-transfected 92BR cultures produced viral particle titers similar to that of EIAV, as measured by PERT, and in agreement with published data (Li et al., 1998). Interestingly, no RT activity was observed in pEIAVΔtat-transfected cultures until 40 hr posttransfection, at which time virion titers were similar to wild-type EIAV. Cultures cotransfected with pEIAVΔtat/pTat-CO produced wild-type virus levels of virions at 20 hr posttransfection, demonstrating that the deficiency in EIAVΔtat could be rescued by tat provided in trans. EIAVΔrev produced 10-fold fewer particles than EIAV at 20 hr posttransfection, and cotransfection with pRev-CO only partially rescued titers at this time point. Only the additional cotransfection of pTat-CO enabled equivalent particle production from pEIAVΔrev-transfected cultures at 20 hr posttransfection. We demonstrated that pEIAVΔrev was capable of expressing tat by checking the integrity of both tat exons by sequencing (data not shown), therefore indicating that pTat-CO was not complementing a tat deficiency in EIAVΔrev proviral DNA.
FIG. 4.

Evaluation of virion titers in pseudo-infected 92BR cultures 20–40 hr posttransfection (p.t.) with proviral DNA. Cultures of 92BR cells were transfected with EIAV/EIAV-mutated proviral plasmid DNAs alone or with the stated combinations of accessory gene plasmids under conditions resulting in 10% transfection efficiency, and RT was measured in supernatants by PERT assay 20 and 40 hr posttransfection (n=2).
To further evaluate the impact of the accessory gene knockout mutations on virus function, these transfected 92BR cultures were passaged for 41 days and samples were taken for PERT analysis at regular intervals (Fig. 5). The pGagPol-CO-transfected culture was included to model RT activity expressed from episomal or stably integrated (nonviral) DNA as the time course proceeded. All pEIAV-transfected cultures became maximally infected by day 9 posttransfection as judged by the plateau of RT activity in supernatants (maximal titer, 5×105 PERT-predicted TU/ml). pEIAVΔS2-transfected cultures followed a similar pattern. However, the RT activity in pEIAVΔtat- or pEIAVΔrev-transfected cultures decayed at a similar rate as that of pGagPol-CO over this time until steady state RT activity was observed from day 9 to day 41 posttransfection, on the order of 103 and 102 PERT-predicted TU/ml, respectively. No RT activity could be detected within pGagPol-transfected cultures beyond 22 days posttransfection, implying that episomal DNA had been lost from cells and that nonvirus-mediated stable integration of plasmid DNA had not occurred at a frequency sufficient to express detectable RT. There was evidence that pTat-CO and pRev-CO provided low levels of virion rescue for their respective mutants during the first 21 days, which is best explained by gradual loss of episomal expression and/or Tat/Rev protein decay (modeled by pGagPol-CO). The difference in steady state RT activity of EIAVΔtat and EIAVΔrev compared to EIAV at day 41 posttransfection was on the order of 2.5 and 4 logs, respectively. In contrast to the rescue of virion production by pTat-CO and pRev-CO observed in the first 20 hr posttransfection, the long-term benefit of trans-complementation was minimal by day 41 posttransfection.
FIG. 5.

Evaluation of EIAVΔtat, EIAVΔrev, and EIAVΔS2 replication in 92BR cultures subsequent to transfection of proviral DNA. Cultures of 92BR cells were transfected with EIAV/EIAV-mutated proviral plasmid DNAs alone or with the stated combinations of accessory gene plasmids under conditions resulting in 10% transfection efficiency, and RT was measured in supernatants over a 41-day period by PERT (n=2). The inclusion of the pGagPol-CO transfection allowed the monitoring of RT expressed from nonintegrated DNA and loss of plasmid DNA from the cultures. Each data marker represents accumulation of RT in cultures from the previous passage point, thus representing steady state RT levels; lines between data points are therefore displayed for clarity purposes.
Selection of EIAVΔS2 as the positive control virus
Although these data provide strong evidence that the tat-deleted and rev-deleted EIAV mutants are highly attenuated, the measure of RT by PERT analysis gives only a measure of physical particle titers. Given that stable, albeit low, RT activity was detected in pEIAVΔtat- and pEIAVΔrev-pseudo-infected cultures, these data imply that some integration-proficient virus was produced from these mutants by 40 hr posttransfection. Although the attempt to model infection at an MOI of 0.1 was achieved in terms of numbers of cells transfected, the copy number of proviral DNA per transfected cell would likely have been substantially greater than 1. High copies of proviral DNA per cell may have allowed virion rescue of the attenuated viruses, and a small fraction of these virions must have contained viral RNA that led to a single round of authentic infection. Because GagPol expression is derived from full-length, packageable genomic viral RNA, we hypothesized that the RNA titers (RNA copy number, measured by qRT-PCR) of tat and rev EIAV mutants might be additionally attenuated. To test this, we quantified both viral RNA and RT activity in supernatants from day 34 posttransfection from the time course experiment (Supplementary Fig. S1; supplementary data are available online at http://www.liebertpub.com/hgtb). The RT activity associated with the supernatants was in agreement with Fig. 5. However, viral RNA could not be detected, using highly sensitive real-time qRT-PCR, in supernatants from EIAVΔtat- or EIAVΔrev-infected 92BR cultures, which was in contrast to that of EIAVΔS2 or EIAV, which produced titers of >5×108 copies/ml. The observed differences between these two mutants suggest that the lack of functional Rev is even more attenuating than the lack of Tat; basal EIAV LTR activity in infected 92BR cells enables low-level empty virus-like particle (VLP) production, but the lack of Rev ensures that the majority of unspliced GagPol viral RNA is not transported from the nucleus, leading to extremely low VLP production, with no evidence of the presence of packaged genomic RNA in either case.
Taken together, these data indicated that EIAVΔtat and EIAVΔrev are essentially nonfunctional mutants, and therefore unsuitable and impractical for use as positive controls in the ET-RCL assay. Therefore EIAVΔS2 was selected and a frozen stock of multiple single-use aliquots was prepared for further characterization and use in future ET-RCL assays.
Mathematical and empirical determination of the minimal infectious dose of EIAVΔS2
We next sought to determine the minimal infectious dose (MID) of EIAVΔS2 required to achieve infection of 92BR cultures at a rate of 98% (hereafter referred to as the MIDp=0.98). The mathematically calculated dose required to achieve a 98% infection rate is 3.9 IU (3.912, to three decimal places), using Eq. (8) in Materials and Methods. The stock of EIAVΔS2 had a titer of 9.9×104 PERT-predicted TU/ml (based on a standard GFP EIAV vector preparation of known biological titer). To empirically determine the number of infectious units associated with this arbitrary titer, the EIAVΔS2 stock was diluted to 12.5 PERT-predicted TU/ml and then 4-fold serially diluted from 5 to 0.02 PERT-predicted TU/0.4 ml of medium. Eight replicate 0.4-ml volumes of this dilution series were used to transduce 92BR cells in the 24-well scale format. After 24 hr of incubation the medium in the wells was replaced and cultures were incubated for a further 9 days, at which point supernatants were sampled for PERT analysis. Three of the five dilutions of viral inoculation resulted in fewer than eight of eight replicate wells being infected (i.e., between one of eight and seven of eight infected), and were used to calculate the MID as follows: data from the 0.313, 0.078, and 0.02 PERT-predicted TU/well dilutions were used to determine that 0.61 PERT-predicted TU is equivalent to 3.9 IU of EIAVΔS2 (see Table 1 for details). Therefore, it was calculated that the stock of EIAVΔS2 had an infectious titer of 6.35×105 IU/ml.
Table 1.
Determination of the MIDp=0.98 of EIAVΔS2, Using Poisson Distribution Mathematics and Empirical Data*
| Input amount of EIAVΔS2 (PERT-predicted TU): a | Observed number of negative replicates: b | p [negative]: c | Calculated IU present: d=−ln (c)* | Correction factor: e=3.912/d | Calculated input of EIAVΔS2 for p=0.98 (PERT-predicted TU): x=a×e | |
|---|---|---|---|---|---|---|
| Empirical data | 0.313 | 1/8 | 0.125 | 2.080 | 1.88 | 0.589 |
| 0.078 | 5/8 | 0.625 | 0.470 | 8.32 | 0.649 | |
| 0.020 | 7/8 | 0.875 | 0.134 | 29.19 | 0.584 | |
| Target | x | 2/100 | 0.02 | 3.912 | 1 | x=0.61** |
EIAVΔS2, EIAV mutated to include two stop codons, so as to stop potential downstream translation initiation within S2; IU, infectious units; p, probability of infection; PERT, product-enhanced reverse transcriptase; TU, transducing units.
The target dose of virus that enables a 98% infection rate is 3.912 IU according to Eq. (8) (see Materials and Methods), which considers the number of negative outcomes likely at this dose, in this case 2/100 or p=0.02. Three sets of replicate 92BR cultures inoculated with a 4-fold serially diluted stock of EIAVΔS2 (a) resulted in observed negative outcomes presented as a fraction of the experimental set (b); this can also be expressed as a probability of noninfection (c). Using Eq. (8), the number of infectious units present in the inoculums enabling the observed infection rate is calculated (d). The correction factor (e) required to achieve a dose of 3.912 IU allows the calculation of the target dose (x) of virus in arbitrary units (in this case PERT-predicted TU).
Average calculated value (x) to 2 dp.
Testing clinical lot FDP, which is concentrated during processing, rather than crude harvest material, allows more flexibility in setting culture medium volume in the test article assay. This volume was previously optimized in the MLV/HEK293 assay in order to maximize test article contact with the amplification cell line without causing vector-associated toxicity; typically 5% of the FDP (test article) is inoculated into 10 T75 flasks, each containing 4 ml of medium, that is, a volume-to-area ratio of 0.053 ml/cm2 (Miskin et al., 2006). We found that incubation of particularly high-titer test article with 92BR cells in this format led to slowing of cell growth (data not shown) and decided to double the flask size while maintaining the volume-to-area ratio, that is, 8 ml per T150 flask. This effective reduction of the test article concentration facilitated efficient growth of 92BR cells at typical growth rates (data not shown). Given that EIAVΔS2 could be detected in 0.4 ml of medium in 24-well plate format (i.e., ∼0.21 ml/cm2), the use of the 0.053-ml/cm2 format was well within the capability of the assay to detect the positive control. We tested several intermediate formats in order to evaluate the robustness of detection of a single infectious unit of EIAVΔS2 in larger volumes. To summarize, inoculation of 1 infectious unit of EIAVΔS2 at 0.053-, 0.106-, 0.133-, and 0.160-ml/cm2 formats in quadruplet produced 2/4, 2/4, 4/4, and 3/4 positive flasks, respectively. Not only did this further demonstrate the capacity of the assay to detect replication-competent virus in volumes greater than that intended for the actual assay, taking all 16 flasks together, the infection rate of 11/16 was in close agreement with the expected rate with a single infectious unit of virus (i.e., 10/16 positive flasks; see Supplementary Table S1, panel III).
Pilot test article ET-RCL assay and testing of the MIDp=0.98 of EIAVΔS2
To confirm that the mathematically determined dose of 3.9 IU was capable of infecting 92BR cultures at a rate close to 98%, we conducted a pilot test article RCL assay, as outlined in Fig. 6A. It was impractical to test a 98% infection rate directly (i.e., to set up 100×T150 flasks), and so an indirect method was chosen. This was achieved by dividing the 3.9 IU of EIAVΔS2 across 10 T150 flasks and comparing the observed versus predicted number of infected flasks. Inclusion of 0.5% test article in each of the 10 flasks additionally allowed evaluation of any potential inhibitory effect of vector on the EIAVΔS2 infection rate. The probability of infection of 0.39 IU of virus per test flask was 32.3% [using Eq. (2)], and therefore the predicted number of positively infected flasks was 3/10. Positive control and spike control flasks were each inoculated with the MIDp=0.98 of EIAVΔS2, the latter also coincubated with 0.5% test article to model the assay inhibition control. Two operators each performed single control flasks and 5 of the 10 test flasks (A–J). The results, displayed in Fig. 7A and B, show that 2/10 flasks (flasks E and F) became infected by the end of the amplification phase, as did both sets of positive and spike controls. This was in close agreement with the predicted 3/10 infection rate, which on average one would expect to occur 26.4% of the time (Supplementary Table S1, panel II). Because a 2/10 infection rate is expected to occur 20.7% of the time with this dose, it was impossible to conclude whether the test article had slightly impacted virus infection or if the observed infection rate was due to chance alone. Nevertheless, these data demonstrate that use of Poisson distribution mathematics enables accurate virus dose setting, and therefore the MIDp=0.98 of EIAVΔS2 was suitably confirmed.
FIG. 6.

ET-RCL assay formats and acceptance criteria. (A) The pilot RCL assay format used to validate the virus dose set for positive and spike controls. The mathematically determined dose of EIAVΔS2 achieving a 98% infection rate (3.9 IU) was divided across 10× test article-containing flasks, leading to a predicted infection rate of ∼32% that is, 3/10 flasks. (B) The final ET-RCL assay format for testing FDP or EOPCs, including the 10× spike flask, which provides an additional measure of potential inhibition in test flasks. The acceptance level determining assay pass/failure is a PERT assay threshold of 37 cycles. Ct, threshold cycle; MID, minimal infectious dose.
FIG. 7.

Pilot FDP ET-RCL assay results. The pilot assay for testing the MIDp=0.98 of EIAVΔS2 (refer to Fig. 6A for a description of the assay format) was run by two operators; the results are displayed in (A) and (B). Positive control and spike control flasks each received the MIDp=0.98, with the latter also receiving 0.5% FDP (1.8×107 RNA-predicted TU test article vector). The 10 test flasks (A–J) each received 0.5% FDP, and the MIDp=0.98 (3.91 IU) was divided across all flasks (each flask receiving 0.39 IU). Flasks were passaged for ∼5 weeks as an amplification phase and prepassaged samples were taken for PERT analysis. Negative control flasks were included as a baseline for RT-negative samples. Each data marker represents accumulation of RT in cultures from the previous passage point, thus representing steady state RT levels; lines between data points are therefore displayed for clarity purposes.
Testing the MIDp=0.98 of EIAVΔS2 infection rate in EOPC/92BR cocultures
To evaluate the potential impact of EOPC/92BR cocultivation on the MIDp=0.98 infection rate we set up quadruplicate 24-well–scale cocultures containing 92BR cells with either HEK293T cells (modeling positive or negative controls) or product-specific EOPCs (modeling spike control). These cultures were inoculated with 0.1 ×, 1 ×, or 10× MIDp=0.98 of EIAVΔS2 as outlined in Table 2, with negative control wells receiving no virus to control for cross-contamination of the 24-well plates. The three different input amounts of EIAVΔS2 enabled evaluation of potential inhibition of EOPCs on virus infection at minor (0.1 ×), moderate (1 ×), and severe (10 ×) levels. Each coculture was subjected to a fully scaled-down RCL assay with a final end-point PERT analysis taken for each condition. None of the EOPC cultures showed evidence of inhibition of virus infection when comparing the predicted outcomes (Table 2). All EOPC/92BR cocultures that received 0.1× MIDp=0.98 (i.e., 0.39 IU; p[infection]=0.32; see Supplementary Table S1, panel I) of EIAVΔS2 yielded 1/4 positive wells except StarGen subrun 2 EOPCs, in which 3/4 positive wells were detected. When considering all results from wells that received 0.1× MIDp=0.98 EIAVΔS2, 7/20 wells were productively infected, which was in close agreement with the predicted probable outcome of 6/20 (see Supplementary Table S1, panel IV). All other cocultures receiving greater inoculums became infected as expected. These data therefore demonstrated that the coculture phase of the EOPC ET-RCL assay did not affect the EIAVΔS2 infection rate.
Table 2.
Evaluation of Effect of EOPC/92BR Cocultivation on the EIAVΔS2 MIDp=0.98 Infection Ratea
| |
|
|
EIAVΔS2 dose/observed infection rate |
||
|---|---|---|---|---|---|
| Coculture ID | Cell type 1 | Cell type 2 | 0.1× MIDp=0.98(p=0.32) | 1× MIDp=0.98(p=0.98) | 10× MIDp=0.98(p=1.00) |
| Positive | 92BR | HEK293T | 1/4 | 4/4 | 4/4 |
| Positive-92BR | 92BR | — | 1/4 | 4/4 | 4/4 |
| StarGen subrun 1 | 92BR | G145/STAR/EPCB001 | 1/4 | 4/4 | 4/4 |
| StarGen subrun 2 | 92BR | G145/STAR/EPCB002 | 3/4 | 4/4 | 4/4 |
| RetinoStat IH48 | 92BR | 140510RT048 (IH48) | 1/4 | 4/4 | 4/4 |
| Negative | 92BR | HEK293T | 0/4 | 0/4 | 0/4 |
| Predicted outcome: 4 flasks: | 1/4b | 4/4 | 4/4 | ||
| 20 flasks: | 6/20b | 20/20 | 20/20 | ||
EIAVΔS2, EIAV mutated to include two stop codons, so as to stop potential downstream translation initiation within S2; ID, identification; MID, minimal infectious dose; p, probability of infection.
Four replicate cocultures containing 92BR cells and EOPCs from difference sources were inoculated with 0.1 ×, 1 ×, or 10× the MIDp=0.98 of EIAVΔS2, resulting in predicted infection rates of ∼32, 98, and 100%, respectively, to test for potential minor, moderate, and severe virus inhibition effects.
See Supplementary Table S1, panels I and IV; MIDp=0.32.
ET-RCL assay performance and modifications
After experimental confirmation of the format of the test article and EOPC ET-RCL assays, evaluation of EIAVΔS2-derived RT activity in the PERT assay (i.e., linearity, range, accuracy, specificity, precision, and robustness) was carried out (data not shown). This work enabled a defined approach for PERT assay “cutoff” between RT-positive and RT-negative samples; RCL assay threshold cycle (Ct) acceptance criteria for PERT end-point outcomes are depicted in Fig. 6B. A number of assays were conducted to GMP standards and the results, along with pilot and troubleshooting runs, are presented in Supplementary Table S2. To date, no equine-tropic RCL has been detected in clinical lots as judged by the ET-RCL assay. An EOPC ET-RCL assay for StarGen (RCL-CC#006) failed once because of lack of RT detection in the spike control (possibly due to an early but contained bacterial/fungal contamination in this flask), and the same control failed in the repeat assay (RCL-CC#009). A third, amended assay of this material included three 1× MIDp=0.98 spike flasks and a 10× MIDp=0.98 spike flask to test whether EOPCs from this subrun of StarGen contained an inhibitor of EIAVΔS2 replication (RCL-CC#014). The 10-fold increase in virus was essentially an arbitrarily set dose but one that still adequately models RCL concentration (see Discussion) and represents a low MOI. However, all three 1× MIDp=0.98 spike flasks and the 10× MIDp=0.98 spike flask became productively infected, and therefore we could not exclude the possibility that the spike failure in RCL-CC#009 was due to chance alone. A total of 22/23 positive controls have passed (the only failed positive control to date was in RCL-CC#017). Because 1 failure in 23 is not highly improbable using this dose (p=0.295; using similar mathematics outlined in Supplementary Table S1), we again concluded that these data were consistent with the 98% infection rate applied to this size of data set. The success/failure of positive and spiked control flasks to amplify virus over the course of future RCL assays will continue to be scrutinized in order to gain further insight and precision regarding viral infection rates.
There was evidence of the presence of an inhibitor of EIAVΔS2 infection for UshStat FDP. The first 1× MIDp=0.98 spike control failure occurred in RCL-CC#005, followed by two of two 1× MIDp=0.98 spike control failures in RCL-CC#007. To test whether UshStat-transduced 92BR cells were capable of being infected by EIAVΔS2, end-of-assay cells (EOACs) from the negative or failed spiked cultures of RCL#005 were infected with the 1× MIDp=0.98 of EIAVΔS2 (RCL-CC#005a). These EOACs were indeed capable of being infected at this dose, excluding the possibility that UshStat-transduced 92BRs were somehow less permissive. The infection resulting from the additional 10× MIDp=0.98 spike control performed in RCL-CC#007 demonstrated that the strength of the putative inhibitor of replication in UshStat FDP was only modest. As a consequence of these spike failures, we amended both the FDP and EOPC ET-RCL assay format to routinely include the 10× MIDp=0.98 spike flask (see Fig. 6B) to assist troubleshooting and data interpretation.
Discussion
Replication-competent lentivirus testing of lentivirus-derived gene therapy vector preparations and EOPCs is an important aspect of safety testing. Here we have contributed to a small but growing body of evidence that RCL formation from contemporary lentiviral vector systems is unlikely (Escarpe et al., 2003; Miskin et al., 2006; Pauwels et al., 2009; Cornetta et al., 2010). This work supports the view that these vectors are sufficiently engineered to be safe for human gene therapy applications. Indeed, no RCLs have been detected in our clinical lots, EOPCs, or from published data (including transduced therapeutic cells) to date (Food and Drug Administration, 2010). Here we report the first RCL assay that has been developed to test the theoretical possibility that nonprimate lentiviral vector production might lead to the formation of RCLs with slow replication kinetics that may not be detected in human cell-based RCL assays. In addition, this paper sets out in detail the mathematical rationale for positive control virus dose setting and monitoring of assay control failures, using Poisson distribution-related mathematics.
All EIAV sequences have been removed from our vector system, except for approximately 800 cis-acting nucleotides that include the packaging signal and the self-inactivating (SIN) LTRs of the vector required for polyadenylation, reverse transcription, and integration of the vector genome. The packaging signal has been modified to remove all Gag translation start codons, the GagPol-encoding gene has been codon-optimized (except for the transframe slip region), and accessory gene sequences have been entirely removed, with the exception of the tat1 exon, which forms part of the packaging signal (see Fig. 2). It is theoretically possible that RCLs formed from this vector system may retain only partial sequences of the vector genome that confer Tat and/or Rev independence, resulting in attenuated RCLs. In modeling replication kinetics of these putative “slow-growing” ET-RCLs in the highly EIAV-permissive equine 92BR cell line, there was no evidence that EIAVΔtat or EIAVΔrev is capable of cell-spreading infection. Although EIAVΔtat was incapable of expressing the tat open reading frame, there is potential for a theoretical RCL to express the tat1 exon as this sequence forms part of the packaging signal. However, assuming the occurrence of specific recombination events, only the first 29 amino acids of Tat could be expressed from tat1; the first 49 amino acids are required for the activation domain of Tat, but the cysteine-rich, core, basic, and trans-activation response region (TAR)-binding domains necessary for protein function (Derse and Newbold, 1993) would be entirely lacking from such an RCL.
VLP expression from pEIAVΔtat-transfected cells could be rescued by Tat in trans in the first 20 hr of the pseudo-infection, whereas pEIAVΔrev required both Rev and Tat to complement VLP production, demonstrating that these mutants could be functionally restored in the short term. Rev is required for the early–late switch from solely multiply spliced mRNA to later include singly spliced or unspliced message during infection (Martarano et al., 1994). Rev downregulates its own expression through the exonic splicing enhancer (Belshan et al., 2000), leading to a balance between Tat-expressing (three exonic) and Rev-expressing (four exonic) mRNAs. We theorize that the unregulated coexpression of Rev with proviral DNA (i.e., by pRev-CO in trans) had a negative effect on virion production because of preferential transport of unspliced viral mRNA before multiple splicing events could occur; because Tat is encoded by multiply spliced mRNA, the Tat feedback loop may have been inhibited, leading to low LTR activity and consequently low virion production. Only when tat was also provided in trans by pTat-CO was pRev-CO able to fully complement the deficiency of EIAVΔrev, suggesting that LTR activity was limiting. The similar observations from cotransfection of pEIAV with pRev-CO±pTat-CO support this theory.
EIAVΔS2 was selected as a positive control for the ET-RCL assay after testing of EIAV accessory gene mutants. Transfection of mammalian cells by plasmid DNA results in multiple copy number (typically in excess of 104) per transfected cell (Carapuca et al., 2007). Therefore, the pseudo-infection time course experiment modeled a “worst case” scenario in terms of RCL production and a “best case” scenario for initiation of infection with the attenuated EIAV provirus DNAs. This led to VLP production levels at 20–40 hr posttransfection that were atypical of an infection of 92BR cells with EIAV at an MOI of 0.1, in which a particle titer of approximately 103 PERT-predicted TU/ml was reached only on day 6 postinfection (Fig. 2). We conclude that this atypical scenario best explains the recovery of VLP titers of tat and rev mutants at 40 hr posttransfection, and the generation of a small amount of integration-proficient mutant virus, which led to a minority of stable VLP-expressing cells in these cultures. It is unlikely that steady state RT levels in pEIAVΔtat- or pEIAVΔrev-transfected culture medium reflected 100% infected cultures resulting from cell-spreading infection. First, viral RNA could not be detected within EIAVΔtat or pEIAVΔrev VLPs on day 34 posttransfection. Second, even when assuming that all virions produced at 40 hr posttransfection contained viral RNA, the maximal possible MOI in cultures was 0.08 (6 ml of 1×104 PERT-predicted TU/ml represents a total of 3.8×105 IU in a vessel containing ∼5×106 cells); in this scenario one would expect steady state RT levels to have increased by at least 10-fold by the end of the time course, as in fact occurred for pEIAV- and pEIAVΔS2-transfected cultures. Instead, RT levels declined from this initial burst of RT expression from pEIAVΔtat- or pEIAVΔrev-transfected cells. It is therefore highly unlikely that a productive infection could be established from limiting quantities of the tat or rev mutants, as required for an RCL assay positive control modeling RCL detection. Given the options, EIAVΔS2 was the only viable accessory gene knockout mutant warranting further development toward the ET-RCL assay.
We used FDA guidelines on RCL testing to determine the sensitivity and robustness of the assay (Food and Drug Administration, 2006). These guidelines recommend that an RCL assay must be sufficiently sensitive to detect an RCL with 95% confidence or greater. The theoretical minimal volume of crude harvest required for testing is therefore 300 ml, assuming an RCL concentration of no greater than 0.01 RCL/ml [Eq. (3)]. Once the maximal volume in which a single infectious unit of positive control virus can be detected has been empirically determined, the minimal number of replicate test vessels may be calculated. Because we currently test 5% of FDP in the ET-RCL assay, this equates to ∼3.5 liters of crude harvest material, giving essentially 100% confidence of detecting an RCL based on the above theoretical RCL concentration. Put in other terms [using Eq. (4)], the current testing scale effectively allows a 95% detection rate of 1 RCL IU in more than 1 liter of crude harvest material. Because of this high confidence level, and being mindful that the assay should be demonstrably sensitive, we decided to set the detection rate for the EIAVΔS2 positive control at 98% per flask, because there was no reason to set the positive control failure rate higher than one might expect for a putative RCL. This has advantages over the use of the median tissue culture infective dose (TCID50) as a test dose [theoretically 0.693 IU, using Eq. (8); see Wulff et al., 2012, for practical considerations], which requires at least five positive control replicates in order to achieve a ≥95% success rate in the assay. This has further consequences for increased test article volume when considering spike controls in testing crude vector material (see below).
The use of Poisson distribution mathematics to determine positive control virus dose proved to be highly accurate, in agreement with others (Grigorov et al., 2011). When considering data from across all dose testing experiments, the observed infection rates agreed closely with that of the mathematically predicted rates (total observed infection rate of virus across all experimental settings was 20/46 versus the predicted rate of 19/46). Evaluation of virus detection in terms of volume alone makes little sense unless vessel area is also considered (excess of permissive cells is assumed). For example, the ability to detect a single infectious unit of virus in 10 ml of medium within a T150 flask does not necessarily translate in testing the same inoculum volume in a T25 flask. Ultimately, this is likely due to local concentration of virus above the cell monolayer, which must be greater when using smaller volume-to-area ratios. Virus incubation time must also be a factor, but we have not observed any differences in virus infection rate when comparing 6- and 24-hr incubation times (data not shown). We have yet to find the maximal volume-to-area ratio in which a single infectious unit of EIAVΔS2 can be detected but have demonstrated that it is at least 0.21 ml/cm2, which is four times greater than that used in the ET-RCL assay format. This will give scope to dilute the clinical lot FDP further without losing sensitivity should there be cytotoxicity associated with these highly concentrated cultures, which is also a concern for RCL assay design in general (Cornetta et al., 2010). In the current assay format, a maximal vector MOI of 5 (based on DNA integration titer) or 25 (based on RNA-predicted titer) is inoculated per test or spike flask; more than 10 test flasks would be required should the clinical lot FDP titer exceed these limits.
We initially tested single positive control and spike control flasks, but on observing potential EIAVΔS2 inhibition in the FDP ET-RCL assay, we modified our GMP assays to included the 10× MIDp=0.98 dose. The dose of 39 IU of virus per spike flask still appropriately models detection of RCL in crude harvest material at 0.01 RCL/ml (i.e., 3500 ml×0.01=35 IU) that is represented in 5% FDP. We have yet to observe a failure in the 10× spike control, and given that this increased dose still represents a low MOI (∼8×10–6), the degree of EIAVΔS2 inhibition at 1× MIDp=0.98 observed to date must be modest.
Although the ET-RCL assay has further demonstrated the safety of EIAV-based vectors for use in treating patients, we believe that the most clinically relevant assay is one that uses human cells to amplify putative RCLs. Given that the vector system lacks all EIAV accessory genes, of which tat and rev have now been demonstrably shown to be necessary for virus replication in equine cells, it is difficult to envisage how the EIAV vector-manufacturing process could generate an RCL that could be detected only in the ET-RCL assay and not the MLV/HEK293-based assay. In addition, the use of EIAVΔS2 as a positive control models replication of an ET-RCL expressing the EIAV envelope, which is lacking from both the vector system and HEK293T production cells. A putative RCL is most likely to encode the VSV-G envelope, which is pantropic. This furthers the rationale of using amphotropic MLV as a surrogate positive control because of the broad tropism of MLV. We currently hold a conservative position regarding EIAV engineering by withholding from constructing an EIAV genome in which the natural envelope is replaced with VSV-G, and so the use of MLV as a surrogate RCL positive control in the HEK293 cell-based RCL assay remains the most logical approach to ensure the absence of RCL from vector and EOPCs, as stated previously (Miskin et al., 2006).
The mathematical principles used in this work outline a useful tool for monitoring observed versus probable failure rates amongst the controls within accumulating data sets from RCL assay runs. We recommend performing statistical analysis of failure rates on segregated data sets, that is, positive control-only and spike control-only. Ultimately, product-specific spike control data should be considered in isolation once sufficiently large data sets can be established. This provides an option to set a “threshold-of-failure rate” (TFR), above which the operators should begin investigation into why improbable rates of failure are being observed. This approach would lead the investigator to retitrate the positive control virus stock (when a value above the TFR is observed for the positive control-only data set) or to attempt to characterize potential inhibitor(s) within the test article or EOPCs (when a value above the TFR is observed for the spike control-only data set).
The timely reevaluation of RCR/RCL testing requirements is helpful to the retroviral gene therapy vector field, particularly as lessons are learned from previous vector formats (Pauwels et al., 2009) and as manufacturing scale increases in correlation to clinical trial size. One aspect of RCL testing that may need to be revisited is the rationale for EOPC testing, specifically when cytotoxic genes are expressed. For example, the VSV-G envelope is cytotoxic unless regulated in packaging/producer cell lines (Stewart et al., 2009). Given that transient methods of VSV-G-pseudotyped vector production results in robust expression of VSV-G in production cells (and from producer cell lines), it remains questionable as to whether the remaining EOPCs tested within the cocultivation assay represent viable pools of RCL replication. At present, 12% volumes of EIAV-based vector clinical batches are tested in test article RCL assays under requirements by the FDA (5% test article and 0.5% applied to both spiked controls in each of the human-tropic and ET-RCL assays). Testing of this large proportion of each manufactured lot is highly burdensome, especially on occasions when repeat testing is required. We are currently reevaluating the option to test the theoretical minimal volume of crude harvest vector material outlined above, and to introduce scaled-down spike controls. The mathematical principles outlined and tested in this paper demonstrate that the virus infection rate is independent of test scale when an empirically determined volume-to-area ratio is maintained. Therefore, performing spike controls at reduced scale will model any potential RCL inhibitor that might be in the test article, which will be present at the same concentration in spike and test vessels. If this approach is adopted, it will translate to a significant saving in test article material when testing crude harvest material in multiple replicate spike controls containing ≤1 IU of virus, as required to achieve acceptable assay pass rates (e.g., ≥95%). Finally, entirely new assay formats that minimize scale and handling of these labor-intensive RCL assays are being researched; spinoculation approaches (Forestell et al., 1996) and the use of multilayered tissue culture vessels are currently being evaluated.
Supplementary Material
Acknowledgments
The authors thank Ronald C. Montelaro for the kind gift of the NIH3T3(ELR1/cyc) cell line used in this research, and Pippa Radcliffe for comments in preparation of this manuscript.
Author Disclosure Statement
All authors are employees of Oxford BioMedica; no competing financial interests exist.
References
- Beisel C.E. Edwards J.F. Dunn L.L. Rice N.R. Analysis of multiple mRNAs from pathogenic equine infectious anemia virus (EIAV) in an acutely infected horse reveals a novel protein, Ttm, derived from the carboxy terminus of the EIAV transmembrane protein. J. Virol. 1993;67:832–842. doi: 10.1128/jvi.67.2.832-842.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belshan M. Park G.S. Bilodeau P., et al. Binding of equine infectious anemia virus Rev to an exon splicing enhancer mediates alternative splicing and nuclear export of viral mRNAs. Mol. Cell. Biol. 2000;20:3550–3557. doi: 10.1128/mcb.20.10.3550-3557.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasco L. Baricordi C. Aiuti A. Retroviral integrations in gene therapy trials. Mol. Ther. 2012;20:709–716. doi: 10.1038/mt.2011.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieniasz P.D. Grdina T.A. Bogerd H.P. Cullen B.R. Highly divergent lentiviral Tat proteins activate viral gene expression by a common mechanism. Mol. Cell. Biol. 1999;19:4592–4599. doi: 10.1128/mcb.19.7.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi A. Bartolomae C.C. Cesana D., et al. Lentiviral vector common integration sites in preclinical models and a clinical trial reflect a benign integration bias and not oncogenic selection. Blood. 2011;117:5332–5339. doi: 10.1182/blood-2010-09-306761. [DOI] [PubMed] [Google Scholar]
- Bouillant A.M. Nielsen K. Ruckerbauer G.M., et al. The persistent infection of a canine thymus cell line by equine infectious anaemia virus and preliminary data on the production of viral antigens. J. Virol. Methods. 1986;13:309–321. doi: 10.1016/0166-0934(86)90056-x. [DOI] [PubMed] [Google Scholar]
- Cartier N. Hacein-Bey-Abina S. Bartholomae C.C., et al. Lentiviral hematopoietic cell gene therapy for X-linked adrenoleukodystrophy. Methods Enzymol. 2012;507:187–198. doi: 10.1016/B978-0-12-386509-0.00010-7. [DOI] [PubMed] [Google Scholar]
- Campochiaro P.A. Gene transfer for ocular neovascularization and macular edema. Gene Ther. 2012;19:121–126. doi: 10.1038/gt.2011.164. [DOI] [PubMed] [Google Scholar]
- Carapuca E. Azzoni A.R. Prazeres D.M., et al. Time-course determination of plasmid content in eukaryotic and prokaryotic cells using real-time PCR. Mol. Biotechnol. 2007;37:120–126. doi: 10.1007/s12033-007-0007-3. [DOI] [PubMed] [Google Scholar]
- Cornetta K. Yao J. Jasti A., et al. Replication-competent lentivirus analysis of clinical grade vector products. Mol. Ther. 2010;19:557–566. doi: 10.1038/mt.2010.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covaleda L. Fuller F.J. Payne S.L. EIAV S2 enhances pro-inflammatory cytokine and chemokine response in infected macrophages. Virology. 2010;397:217–223. doi: 10.1016/j.virol.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham T.P. Montelaro R.C. Rushlow K.E. Lentivirus envelope sequences and proviral genomes are stabilized in Escherichia coli when cloned in low-copy-number plasmid vectors. Gene. 1993;124:93–98. doi: 10.1016/0378-1119(93)90766-v. [DOI] [PubMed] [Google Scholar]
- Derse D. Newbold S.H. Mutagenesis of EIAV TAT reveals structural features essential for transcriptional activation and TAR element recognition. Virology. 1993;194:530–536. doi: 10.1006/viro.1993.1291. [DOI] [PubMed] [Google Scholar]
- Derse D. Dorn P.L. Levy L., et al. Characterization of equine infectious anemia virus long terminal repeat. J. Virol. 1987;61:743–747. doi: 10.1128/jvi.61.3.743-747.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiusto D.L. Krishnan A. Li L., et al. RNA-based gene therapy for HIV with lentiviral vector-modified CD34+ cells in patients undergoing transplantation for AIDS-related lymphoma. Sci. Transl. Med. 2010;2:36ra43. doi: 10.1126/scitranslmed.3000931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escarpe P. Zayek N. Chin P., et al. Development of a sensitive assay for detection of replication-competent recombinant lentivirus in large-scale HIV-based vector preparations. Mol. Ther. 2003;8:332–341. doi: 10.1016/s1525-0016(03)00167-9. [DOI] [PubMed] [Google Scholar]
- Fidalgo-Carvalho I. Craigo J.K. Barnes S., et al. Characterization of an equine macrophage cell line: application to studies of EIAV infection. Vet. Microbiol. 2009;136:8–19. doi: 10.1016/j.vetmic.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Food and Drug Administration. Guidance for Industry: Supplemental Guidance on Testing for Replication Competent Retrovirus in Retroviral Vector Based Gene Therapy Products and During Follow-up of Patients in Clinical Trials Using Retroviral Vectors. 2006. http://www.fda.gov/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/cellularandgenetherapy/ucm072961.htm. http://www.fda.gov/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/cellularandgenetherapy/ucm072961.htm [DOI] [PubMed]
- Food and Drug Administration. Food and Drug Administration Cellular, Tissue, and Gene Therapies Advisory Committee Proceedings (November 19, 2010) 2010. http://www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/BloodVaccinesandOtherBiologics/CellularTissueandGeneTherapiesAdvisoryCommittee/ucm230544.htm http://www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/BloodVaccinesandOtherBiologics/CellularTissueandGeneTherapiesAdvisoryCommittee/ucm230544.htm
- Forestell S.P. Dando J.S. Bohnlein E. Rigg R.J. Improved detection of replication-competent retrovirus. J. Virol. Methods. 1996;60:171–178. doi: 10.1016/0166-0934(96)02052-6. [DOI] [PubMed] [Google Scholar]
- Fridell R.A. Partin K.M. Carpenter S. Cullen B.R. Identification of the activation domain of equine infectious anemia virus Rev. J. Virol. 1993;67:7317–7323. doi: 10.1128/jvi.67.12.7317-7323.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galy A. Thrasher A.J. Gene therapy for the Wiskott-Aldrich syndrome. Curr. Opin. Allergy Clin. Immunol. 2010;11:545–550. doi: 10.1097/ACI.0b013e32834c230c. [DOI] [PubMed] [Google Scholar]
- Grigorov B. Rabilloud J. Lawrence P. Gerlier D. Rapid titration of measles and other viruses: Optimization with determination of replication cycle length. PLoS One. 2011;6:e24135. doi: 10.1371/journal.pone.0024135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker C.V. Vink C.A. Wardell T.W., et al. The integration profile of EIAV-based vectors. Mol. Ther. 2006;14:536–545. doi: 10.1016/j.ymthe.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Harris M.E. Gontarek R.R. Derse D. Hope T.J. Differential requirements for alternative splicing and nuclear export functions of equine infectious anemia virus Rev protein. Mol. Cell. Biol. 1998;18:3889–3899. doi: 10.1128/mcb.18.7.3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines R. Maury W. DH82 cells: A macrophage cell line for the replication and study of equine infectious anemia virus. J. Virol. Methods. 2001;95:47–56. doi: 10.1016/s0166-0934(01)00288-9. [DOI] [PubMed] [Google Scholar]
- Jarraya B. Boulet S. Ralph G.S., et al. Dopamine gene therapy for Parkinson's disease in a nonhuman primate without associated dyskinesia. Sci. Transl. Med. 2009;1:2ra4. doi: 10.1126/scitranslmed.3000130. [DOI] [PubMed] [Google Scholar]
- Li F. Puffer B.A. Montelaro R.C. The S2 gene of equine infectious anemia virus is dispensable for viral replication in vitro. J. Virol. 1998;72:8344–8348. doi: 10.1128/jvi.72.10.8344-8348.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F. Leroux C. Craigo J.K., et al. The S2 gene of equine infectious anemia virus is a highly conserved determinant of viral replication and virulence properties in experimentally infected ponies. J. Virol. 2000;74:573–579. doi: 10.1128/jvi.74.1.573-579.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmquist W.A. Barnett D. Becvar C.S. Production of equine infectious anemia antigen in a persistently infected cell line. Arch. Gesamte Virusforsch. 1973;42:361–370. doi: 10.1007/BF01250717. [DOI] [PubMed] [Google Scholar]
- Martarano L. Stephens R. Rice N. Derse D. Equine infectious anemia virus trans-regulatory protein Rev controls viral mRNA stability, accumulation, and alternative splicing. J. Virol. 1994;68:3102–3111. doi: 10.1128/jvi.68.5.3102-3111.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Rendon E. White L.J. Olsen A., et al. New methods to titrate EIAV-based lentiviral vectors. Mol. Ther. 2002;5:566–570. doi: 10.1006/mthe.2002.0576. [DOI] [PubMed] [Google Scholar]
- Miskin J. Chipchase D. Rohll J., et al. A replication competent lentivirus (RCL) assay for equine infectious anaemia virus (EIAV)-based lentiviral vectors. Gene Ther. 2006;13:196–205. doi: 10.1038/sj.gt.3302666. [DOI] [PubMed] [Google Scholar]
- Mitrophanous K. Yoon S. Rohll J., et al. Stable gene transfer to the nervous system using a non-primate lentiviral vector. Gene Ther. 1999;6:1808–1818. doi: 10.1038/sj.gt.3301023. [DOI] [PubMed] [Google Scholar]
- Miyoshi H. Blomer U. Takahashi M., et al. Development of a self-inactivating lentivirus vector. J. Virol. 1998;72:8150–8157. doi: 10.1128/jvi.72.10.8150-8157.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montelaro R.C. Lohrey N. Parekh B., et al. Isolation and comparative biochemical properties of the major internal polypeptides of equine infectious anemia virus. J. Virol. 1982;42:1029–1038. doi: 10.1128/jvi.42.3.1029-1038.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauwels K. Gijsbers R. Toelen J., et al. State-of-the-art lentiviral vectors for research use: Risk assessment and biosafety recommendations. Curr. Gene Ther. 2009;9:459–474. doi: 10.2174/156652309790031120. [DOI] [PubMed] [Google Scholar]
- Payne S.L. Fang F.D. Liu C.P., et al. Antigenic variation and lentivirus persistence: Variations in envelope gene sequences during EIAV infection resemble changes reported for sequential isolates of HIV. Virology. 1987;161:321–331. doi: 10.1016/0042-6822(87)90124-3. [DOI] [PubMed] [Google Scholar]
- Payne S.L. Rausch J. Rushlow K., et al. Characterization of infectious molecular clones of equine infectious anaemia virus. J. Gen. Virol. 1994;75:425–429. doi: 10.1099/0022-1317-75-2-425. [DOI] [PubMed] [Google Scholar]
- Porter D.L. Levine B.L. Kalos M., et al. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rwambo P.M. Issel C.J. Hussain K.A. Montelaro R.C. In vitro isolation of a neutralization escape mutant of equine infectious anemia virus (EIAV) Arch. Virol. 1990;111:275–280. doi: 10.1007/BF01311062. [DOI] [PubMed] [Google Scholar]
- Sadelain M. Riviere I. Wang X., et al. Strategy for a multicenter phase I clinical trial to evaluate globin gene transfer in β-thalassemia. Ann. N. Y. Acad. Sci. 2010;1202:52–58. doi: 10.1111/j.1749-6632.2010.05597.x. [DOI] [PubMed] [Google Scholar]
- Sastry L. Cornetta K. Detection of replication competent retrovirus and lentivirus. Methods Mol. Biol. 2009;506:243–263. doi: 10.1007/978-1-59745-409-4_17. [DOI] [PubMed] [Google Scholar]
- Sastry L. Xu Y. Johnson T., et al. Certification assays for HIV-1-based vectors: Frequent passage of gag sequences without evidence of replication-competent viruses. Mol. Ther. 2003;8:830–839. doi: 10.1016/j.ymthe.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Sastry L. Xu Y. Duffy L., et al. Product-enhanced reverse transcriptase assay for replication-competent retrovirus and lentivirus detection. Hum. Gene. Ther. 2005;16:1227–1236. doi: 10.1089/hum.2005.16.1227. [DOI] [PubMed] [Google Scholar]
- Segall H.I. Yoo E. Sutton R.E. Characterization and detection of artificial replication-competent lentivirus of altered host range. Mol. Ther. 2003;8:118–129. doi: 10.1016/s1525-0016(03)00134-5. [DOI] [PubMed] [Google Scholar]
- Stewart H.J. Leroux-Carlucci M.A. Sion C.J., et al. Development of inducible EIAV-based lentiviral vector packaging and producer cell lines. Gene Ther. 2009;16:805–814. doi: 10.1038/gt.2009.20. [DOI] [PubMed] [Google Scholar]
- Wulff N.H. Tzatzaris M. Young P.J. Monte Carlo simulation of the Spearman–Kaerber TCID50. J. Clin. Bioinforma. 2012;2:5. doi: 10.1186/2043-9113-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S. Green College, University of Oxford; Oxford, UK: 1999. Functional analysis of accessory proteins in equine infectous anaemia virus. Ph.D. thesis. [Google Scholar]
- Zhang B. Montelaro R.C. Replication of equine infectious anemia virus in engineered mouse NIH3T3 cells. J. Virol. 2009;83:2034–2037. doi: 10.1128/JVI.01883-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B. Jin S. Jin J., et al. A tumor necrosis factor receptor family protein serves as a cellular receptor for the macrophage-tropic equine lentivirus. Proc. Natl. Acad. Sci. U.S.A. 2005;102:9918–9923. doi: 10.1073/pnas.0501560102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.