

Abstract
Nephronophthisis (NPHP), an autosomal recessive cystic kidney disease, leads to chronic renal failure in children. The genes mutated in NPHP1 and NPHP4 have been identified, and a gene locus associated with infantile nephronophthisis (NPHP2) was mapped. The kidney phenotype of NPHP2 combines clinical features of NPHP and polycystic kidney disease (PKD). Here, we identify inversin (INVS) as the gene mutated in NPHP2 with and without situs inversus. We show molecular interaction of inversin with nephrocystin, the product of the gene mutated in NPHP1 and interaction of nephrocystin with β-tubulin, a main component of primary cilia. We show that nephrocystin, inversin and β-tubulin colocalize to primary cilia of renal tubular cells. Furthermore, we produce a PKD-like renal cystic phenotype and randomization of heart looping by knockdown of invs expression in zebrafish. The interaction and colocalization in cilia of inversin, nephrocystin and β-tubulin connect pathogenetic aspects of NPHP to PKD, to primary cilia function and to left-right axis determination.
NPHP, an autosomal recessive cystic kidney disease, is the most frequent genetic cause for end-stage renal failure in children and young adults1–3. Causative mutations in two genes (NPHP1 and NPHP4) have been identified by positional cloning4–7. There is considerable interest in identifying genes associated with NPHP because its most prominent feature is development of renal interstitial fibrosis8, which in chronic renal disease of all origin represents the pathogenic event correlated most strongly to loss of renal function9. As little was known about the pathogenesis of NPHP, positional cloning was used to identify a new gene, NPHP1, mutations in which cause NPHP1 (OMIM 256100; refs. 4,5). It encodes a novel docking protein, nephrocystin10–13, that interacts with components of cell-cell and cell-matrix signaling, such as focal adhesion kinase 2, tensin, p130Cas and filamin, and with nephrocystin-4 or nephroretinin, the product of NPHP4, mutations in which cause NPHP4 (OMIM 606966; refs. 6,7). Identification of the genes NPHP1 and NPHP4, which are conserved in evolution including in the nematode Caenorhabditis elegans, offered new insights into mechanisms of cell-cell and cell-matrix signaling6,14.
Two additional geneloci were mapped for NPHP. The locus NPHP3 associated with adolescent NPHP localizes to human chromosome 3q22 (ref. 15), and NPHP2 associated with infantile NPHP resides on chromosome 9q21–q22 (ref. 16). The kidney phenotype of NPHP2 combines features of NPHP, including tubular basement membrane disruption and renal interstitial fibrosis, with features of PKD17, including enlarged kidneys and widespread cyst development. We located the human gene INVS in the NPHP2 critical genetic interval16. In the inv/inv mouse model of insertional mutagenesis, a deletion of exons 3–11 of Invs encoding inversin causes a phenotype of cyst formation in enlarged kidneys, situs inversus and pancreatic islet cell dysplasia18,19. Histology of infantile NPHP2 and of the inv/inv mouse identified features resembling NPHP, namely interstitial fibrosis, mild interstitial cell infiltration, tubular cell atrophy, tubular cysts and periglomerular fibrosis. In addition, human NPHP2 and mouse inv/inv phenotypes showed features reminiscent of autosomal dominant PKD, such as kidney enlargement, absence of the tubular basement membrane irregularity characteristic of NPHP and presence of cysts also outside the medullary region. Because of the phenotypic similarities, we examined INVS as a candidate gene by mutational analysis in individuals with NPHP2.
Here we show that recessive mutations in INVS cause NPHP2 with and without situs inversus and that inversin interacts with nephrocystin. We show that both proteins localize to primary cilia of renal tubular cells, the same subcellular compartment that has been recently identified as central to the pathogenesis of PKD20,21.
Results
Mutations in INVS cause NPHP2
We carried out mutational analysis of 16 exons of INVS in genomic DNA from nine affected individuals from seven different families with early onset of NPHP. We included one individual (from family A7) from the initial description17 of infantile NPHP (individual 5) and two affected siblings (VII-1 and VII-3 in family A12) from the Bedouin kindred16 in which the NPHP2 locus was first mapped (Table 1). We detected nine distinct recessive mutations in INVS (Table 1 and Fig. 1). In six individuals, both mutated alleles were detected. In individual A10, only one heterozygous mutation was found. The second mutation might be positioned in a region not covered by exon PCR or might represent a heterozygous deletion.
Table 1.
Clinical characteristics and INVS mutations detected in families with NPHP2
| Family (individual) | Ethnic origin | Nucleotide alteration(s)a | Alteration(s) in coding sequence | Exon, segregationb | Parental consanguinity | Renal cysts | Renal biopsy | Age at ESRDc | Situs inversus (other symptoms)d |
|---|---|---|---|---|---|---|---|---|---|
| A6 | France | C2695T | R899X | 13, hete | − | − | + | <2 y | − |
| 1453delC | Q485fsX509 | 9, hete | |||||||
| A8 | Turkey | C1807T | R603X | 12, home | + | − | + | 14 mo | + (VSDf) |
| A9 | France | C1186T | R396X | 8, hete | − | + | + | <2 y | − |
| C1445G | P482R | 9, het P | |||||||
| A10g | France | 2908delG | E970fsX971 | 14, het M | − | + | + | 12 mo | − |
| A12 (VII-1, VII-3) | Israel | C2719T | R907X | 13, hom M,P | + | + | (+, +) | (30 mo, 30 mo) | −, − (HT, HT) |
| 868 (II-1, II-2) | USA | C2719T | R907X | 13, het M | − | −h | (+, +) | (5 y, 4 y) | −, − (HT, HT) |
| 2747insA | K916fsX1002 | 13, het P | |||||||
| A7 | Portugal | T1478C | L493S | 10, home | + | ND | + | 5 y | − (HT) |
All mutations were absent from at least 100 healthy control subjects.
M, maternal; P, paternal; het, heterozygous; hom, homozygous mutation inherited from both parents; ND, no data available.
ESRD, end-stage renal disease; mo, months.
HT, arterial hypertension.
Parent(s) not available for mutational analysis.
VSD, cardiac ventricular septal defect.
Only one mutation was detected in this individual.
Hyperechogenicity noted as sign of incipient microcysts.
Figure 1.
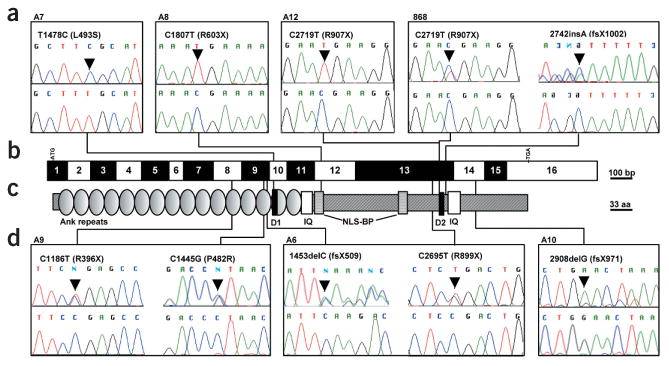
Mutations in INVS in individuals with NPHP2. (a,d) Mutations in INVS (nucleotide exchange and amino acid exchange) are shown together with sequence traces for mutated sequence (top) and sequence from healthy controls (bottom). Family numbers are given above boxes. If only one mutation is shown, it occurred in the homozygous state, except in individual A10, in whom only one mutation in the heterozygous state was detected. In individual 868, the 2742insA mutation is shown in the flipped version of the reverse strand. (b) Exon structure of INVS. Lines indicate relative positions and connect to mutations detected in INVS. Open and filled boxes represent INVS exons drawn relative to scale bar. Positions of start codon (ATG) at nucleotide +1 and of stop codon (TGA) are indicated. (c) Representation of protein motifs is drawn to scale parallel to exon structure. Lines connect to point mutations detected, as shown in boxes (a,d). aa, amino acid residues; Ank, ankyrin/swi6 motif; D1, D box1 (Apc2-binding23); D2, D box2; IQ, calmodulin binding domains.
Seven of the mutations were truncating (four non-sense, three frameshift). Two were missense mutations leading to non-conservative exchanges of amino acids (P482R, L493S). Both missense mutations changed amino acids conserved in evolution in mouse and zebrafish. L493S changes a highly conserved residue in the first D box (Fig. 1a,c). None of the mutations were found in 100 healthy control individuals (200 chromosomes). Truncating mutations differed in their consequence on conceptual translation of the inversin protein. Inversin contains multiple domains and protein-binding motifs, such as 16 N-terminal ankyrin/swi6 repeats, 2 IQ domains (one of which is calmodulin-binding22), two D boxes (the first of which is Apc2-binding23) and a bipartite nuclear localization signal (NLS-BP). Truncating mutations R899X, Q485fsX509, R603X, R396X, E970fsX971, R907X and K916fsX1002 deleted to a varying degree the succession of such domains as distal ankyrin/swi6 repeats, IQ domains, D boxes and NLS-BP (Fig. 1). Thus, we identified recessive mutations in INVS as causing NPHP2 with and without situs inversus.
Inversin coprecipitates with nephrocystin
We tested whether inversin participates in direct molecular interaction with nephrocystin, the product of NPHP1, mutations in which cause NPHP1. Nephrocystin specifically coprecipitated with inversin, (Fig. 2a) and vice versa (Fig. 2b,c), but not with control protein. To confirm interaction among endogenous proteins derived from mouse kidney and testis, we generated an inversin-specific antiserum. We used a maltose binding protein (MBP)–inversin fusion protein that contained amino acids 561–716 of mouse inversin to immunize rabbits. We then used a glutathione S-transferase (GST)–inversin agarose column to affinity-purify the inversin-specific antiserum. The affinity-purified rabbit antiserum specifically recognized inversin expressed in HEK293T cells and recombinant GST–inversin but not two other control GST fusion proteins (Fig. 2d). Western-blot analysis of immunoprecipitates from mouse kidney confirmed the interaction of inversin and nephrocystin (Fig. 2e), indicating that this interaction occurs in vivo.
Figure 2.
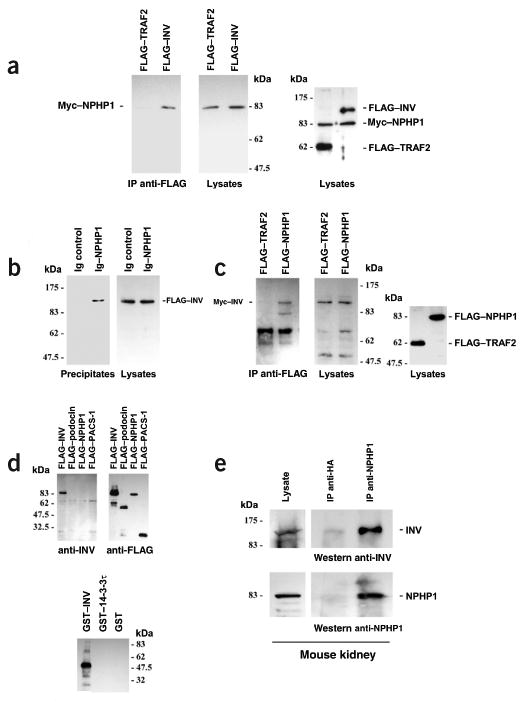
Nephrocystin associates with inversin in HEK293T cells and mouse tissue. (a) Myc-tagged nephrocystin (Myc–NPHP1) was coexpressed with N-terminally FLAG-tagged full-length inversin (FLAG–INV) or FLAG-tagged TRAF2 (FLAG–TRAF2) protein as a negative control. After immunoprecipitation with anti-FLAG antibody, coprecipitating nephrocystin was detected with nephrocystin-specific antiserum (left panel). We controlled for protein expression levels in cellular lysates by immunoblotting using a nephrocystin antibody (middle panel) or FLAG-specific and nephrocystin-specific antibodies (right panel). Molecular weight markers are shown in kDa. (b) Full-length nephrocystin was fused to the CH2 and CH3 domains of human IgG1 and precipitated with protein G sepharose beads. FLAG-tagged inversin specifically coprecipitated with nephrocystin but not with control protein (CH2 and CH3 domains of human IgG1 without nephrocystin fusion) as shown with FLAG-specific antibody. (c) FLAG-tagged nephrocystin or FLAG-tagged TRAF2 protein as a negative control was coexpressed with N-terminally Myc-tagged full-length inversin (Myc–INV). After immunoprecipitation with anti-FLAG antibody, coprecipitating inversin was detected with inversin-specific antiserum (left and middle panels). Right panel shows lysate control developed with FLAG-specific antibody. (d) A rabbit antiserum to an MBP-inversin fusion protein (amino acids 561–716 of mouse inversin) specifically recognized inversin (amino acids 1–716) expressed in HEK293T cells (left panel) but not the FLAG-tagged control proteins podocin (FLAG–podocin), nephrocystin (FLAG-NPHP1) or PACS-1 (FLAG–PACS–1, amino acids 85–280; left panel). It also specifically recognized recombinant GST-inversin (amino acids 561–716) but not two other control GST fusion proteins (lower panel). (e) To show endogenous nephrocystin-inversin interaction in vivo in mouse kidney, half of mouse kidney tissue lysates was immunoprecipitated with a control antibody to hemagglutinin (anti-HA), and the other half was precipitated with anti-nephrocystin antisera. Immobilized inversin was detected with the inversin-specific antisera (right upper panel). Precipitation of endogenous nephrocystin was confirmed by reprobing the blot for nephrocystin (right lower panel). Left panels show lysate control.
β-tubulin is a nephrocystin interaction partner
To identify additional proteins that interact with nephrocystin in cellular lysates of HEK293T cells transfected with FLAG-tagged nephrocystin or FLAG-tagged control proteins (RGS3 or GFP), we carried out immunoprecipitation using an antibody to FLAG. We identified differentially coprecipitating proteins by colloidal Coomassie Blue and Silver staining. Two-dimensional (2D) electrophoresis of coimmuno-precipitates of FLAG-tagged nephrocystin expressed in HEK293T cells identified at least five different nephrocystin-binding proteins (data not shown). We identified (3-tubulin as the most prominent spot using mass spectrometry and western-blot analysis (Fig. 3a). Analyses of immunoprecipitates of FLAG-tagged proteins (GFP, TNF receptor–associated factor 2 (TRAF2) and nephrocystin) expressed in HEK293T cells indicated that β-tubulin specifically associated with nephrocystin (Fig. 3b,c). We localized the interaction to a C-terminal region between amino acid residues 237 and 670 of nephrocystin (Fig. 3c). We reproduced this finding for endogenous proteins from ciliated tubular kidney cells, indicating that this interaction occurs in vivo (Fig. 3d). We confirmed the in vivo interaction in mouse kidney tissue (data not shown). These data suggest that nephrocystin exists in a protein complex containing β-tubulin in vivo (Fig. 3).
Figure 3.
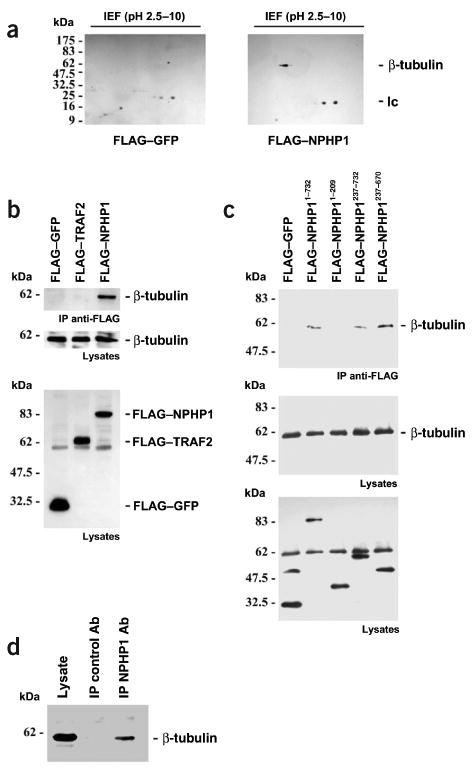
Molecular interaction of nephrocystin with β-tubulin. (a) To identify nephrocystin-interacting proteins, HEK 293T cells were transfected with the FLAG-tagged control protein GFP or FLAG-tagged nephrocystin. Specific association of β-tubulin with nephrocystin was confirmed by immunoblotting of 2D gels using anti-tubulin antibody. lc, immunoglobulin light chain. (b) Several FLAG-tagged nephrocystin truncations were generated to analyze the interaction of nephrocystin with β-tubulin. Endogenous β-tubulin precipitated with transfected full-length nephrocystin but not with the control proteins GFP or TRAF2 (upper panel). Middle panel, expression of native β-tubulin in lysates. Lower panel shows a reprobe of the membrane depicted in middle panel with anti-FLAG antibody (β-tubulin is still detected below the 62 kDa marker), confirming comparable expression levels of the FLAG-tagged proteins. (c) The interaction was mapped to a region of nephrocystin involving amino acids 237–670 (c, upper panel). Expression levels of β-tubulin are shown (c, bottom panel). The blot was reprobed with anti-FLAG antibody to confirm expression of the FLAG-tagged proteins in the lysates (c, lower panel). (d) Coprecipitation of endogenous β-tubulin with native nephrocystin in ciliated mCcd-K1 cells. mCcd-K1 cells were grown for 7 d until cilia were recognized (data not shown). Cell lysates were precleared extensively and immunoprecipitated with a control antibody (Ab) or nephrocystin-specific antiserum and separated by 10% SDS–PAGE. We detected endogenous β-tubulin in the nephrocystin-containing precipitates using a β-tubulin-specific antibody, indicating that the nephrocystin–β-tubulin interaction occurs in vivo.
Nephrocystin and inversin colocalize with β-tubulin to cilia
In kidney cells, β-tubulin is a principal component of primary cilia. Recent evidence has suggested an association between structural or functional defects in the primary apical cilium of kidney epithelia and PKD20,21. We therefore investigated whether nephrocystin is a component of primary cilia as suggested by proteomics data24. We evaluated the subcellular localization of endogenous nephrocystin in Madin–Darby canine kidney (MDCK) cells grown on cell culture inserts for at least 5–7 d after confluence to allow epithelial polarization and cilia formation. Using a highly specific antiserum raised against human nephrocystin (ref. 13; Supplementary Fig. 1 online), we detected specific endogenous nephrocystin expression in primary cilia of MDCK cells. We found that nephrocystin localized in primary cilia and colocalized with β-tubulin-4, a known constituent of the ciliary axoneme (Fig. 4a,b). With more extensive permeabilization of MDCK cells, nephrocystin was also detected in a perinuclear compartment and in vesicle-like structures in close proximity to the bases of developing cilia, possibly representing centrosomes (data not shown).
Figure 4.

Nephrocystin and inversin localize to primary cilia in renal tubular epithelial cells. (a) Colocalization of nephrocystin and β-tubulin-4 in primary cilia of MDCK cells. Two examples of cilia are shown in the upper and lower panels. Wild-type MDCK cells (clone II) were grown on coverslips at 100% confluence and cultivated for 7 d before the experiment to allow full polarization and cilia formation. Localization of nephrocystin was determined by immunofluorescence using nephrocystin-specific antibody with confocal images captured at the level of the apical membrane. Cells were costained with rabbit antibody to nephrocystin (left panels) and mouse antibody to β-tubulin-4 (middle panels) followed by the respective secondary antibodies. (b) Specific localization of nephrocystin in primary cilia was confirmed by the use of blocking recombinant nephrocystin protein. (c) Inversin localizes to primary cilia in MDCK cells. Localization of endogenous inversin was determined by immunofluorescence using inversin-specific antibody with confocal images captured at the level of the apical membrane. Cells were costained with mouse antibody to β-tubulin-4 and rabbit antibody to inversin followed by the respective secondary antibodies (lower panel). In additional stainings, the antibody to β-tubulin-4 was omitted to reduce potential spectral overlap between the inversin and β-tubulin-4 signals (upper panel). (d) Partial colocalization of nephrocystin and inversin in primary cilia. Localization of nephrocystin was determined by immunofluorescence using nephrocystin-specific antibody with confocal images captured at the level of the apical membrane. Cells were costained with goat antibody to inversin (left panel) and rabbit antibody to nephrocystin (middle panel) followed by the respective secondary antibodies. Partial colocalization is shown in the right panel.
Notably, we never found staining of nephrocystin in the entire cilia but rather in a more punctuated pattern, whereas staining with the antibody to β-tubulin-4 almost always extended over the full length of cilia. Punctate staining for nephrocystin might represent predominant expression in varicosities of cilia. On average, 10 of 11 cilia stained positive for nephrocystin (Supplementary Fig. 2 online), whereas all cilia were positive for β-tubulin-4, suggesting that targeting of nephrocystin to cilia may be distinctively regulated. We observed the same nephrocystin localization when using antibody to nephrocystin and rabbit antiserum conjugated with Alexa488 in the absence of anti-serum to β-tubulin-4. We detected no immunofluorescence when secondary antibodies were used alone (data not shown). Because nephrocystin was expressed in primary cilia of MDCK cells and because individuals with nephrocystin defects (NPHP1) and those with inversin defects (NPHP2) share similar renal cystic phenotypes, we anticipated and showed by coimmunofluorescence of inversin with β-tubulin-4 and acetylated tubulin that inversin was expressed in primary apical cilia of MDCK cells (Fig. 4c and Supplementary Fig. 3 online). There was a marked partial colocalization of endogenous nephrocystin and endogenous inversin occurring primarily in varicosities of cilia in MDCK cells (Fig. 4d), as has recently been shown for inversin alone23. These data suggest that inversin and nephrocystin participate in a common pathway and provide a basis for the understanding of the similarities between phenotypes associated with mutations in NPHP1 and NPHP2.
Disruption of zebrafish invs causes renal cyst formation
Injection of translation-blocking morpholino oligonucleotides (atgMO) directed against the AUG initiation codon of the zebrafish invs mRNA caused a marked ventral axis curvature (Fig. 5a,b) and the appearance of severe pronephric cysts by 2.5 d post-fertilization (d.p.f.; Fig. 5d,e). We also observed randomization of heart looping in embryos injected with atgMO: of 60 embryos examined, 21 showed normal heart looping, 29 were reversed and 10 had midline hearts. In 42 embryos injected with the splice-blocking morpholino oligonucleotides (spMO), 22 had normal heart looping, 12 were reversed and 8 had midline hearts (data not shown).
Figure 5.
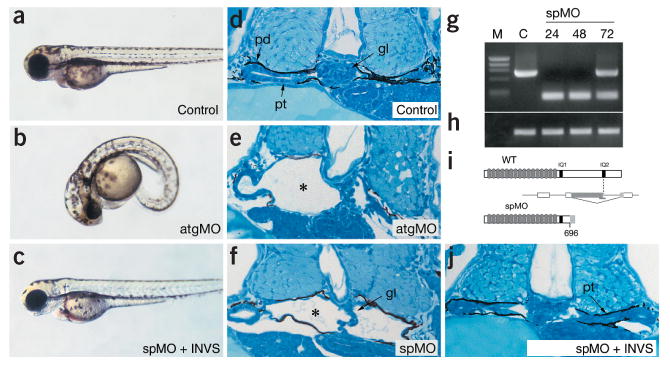
Disruption of zebrafish invs function results in renal cyst formation. (a) Embryos injected with a control, non-specific oligonucleotide have normal morphology. (b) Embryos injected with atgMO and spMO (not shown) have a pronounced ventral axis curvature at 3 d.p.f. (combined totals for atgMO and spMO: 432 of 479 injected embryos; 90%). (c) Coinjection of 100 pg mouse Invs mRNA with spMO completely rescued axis curvature defects (combined totals for atgMO and spMO: 363 of 381 mRNA+MO injected embryos were rescued; 95%). (d) Histological section of a 2.5-d.p.f. control embryo pronephros showing the midline glomerulus (Gl), pronephric tubule (Pt) and pronephric duct (Pd). (e) atgMO-injected 3-d.p.f. embryo showing cystic dilatation of pronephric tubules and glomerulus (indicated with an asterisk) lined with squamous epithelium. (f) spMO similarly causes cystic maldevelopment of the pronephric tubules (marked with an asterisk). (g–i) Molecular analysis of morpholino targeted invs splicing defects: RT–PCR analysis of invs expression in 24-h.p.f. control injected embryos generates a 746-bp invs fragment encoding the C-terminal domain (g; lane C, nucleotides 2,233–2,979 of GenBank AF465261; lane M, ΦX174 markers). spMO-injected embryos (g; lanes spMO; 24, 48 and 72 h.p.f.) analyzed with the same RT–PCR primers generate a 189-bp RT–PCR product representing a C-terminal invs deletion allele. Some recovery of wild-type (WT) mRNA is observed at 72 h.p.f. (h) RT–PCR of ACTB mRNA on the same RNA samples as in g shows no effect of morpholino injection at any time point. (i) Diagram of effect of spMO on invs mRNA processing. Preventing normal splicing in the IQ2 domain recruits a cryptic splice donor in upstream invs coding sequence; the resulting out-of-frame fusion generates a C-terminally truncated invs mRNA at amino acid 696 with an altered 21 amino acid C terminus. (j) Rescue of normal morphology by coinjected spMO and mouse Invs mRNA shows a normal pronephric duct structure (Pt). When injected alone, mouse Invs mRNA had no effect.
To further refine the domains of the inversin protein that are functionally important in vivo for normal epithelial structure, we generated aberrantly spliced invs mRNA in embryos by blocking the splice-donor site (nucleotide 2,921 of the zebrafish invs cDNA) corresponding to the human exon 14 donor. We chose this site because aberrant splicing here would be expected to disrupt the C-terminal IQ2 domain. Similar to the invs knockdown, C-terminal truncation of inversin resulted in ventrally curved embryos with marked renal cysts in the pronephric tubules (Fig. 5f). Morpholino-directed aberrant splicing resulted in the use of an upstream cryptic splice donor in the invs coding sequence (Fig. 5g–i), a frameshift at amino acid 696 and the introduction of a stop codon after 21 amino acids. The predicted protein would be a C-terminal deletion lacking the IQ2 domain. Suppression of wild-type mRNA was complete up to 48 h post fertilization (h.p.f.) as judged by RT–PCR (Fig. 5g).
To confirm that the effects of the morpholino-oligonucleotides were specific to the invs mRNA, we did rescue experiments using coinjected full-length mouse Invs mRNA. Injection of mouse Invs mRNA (100 pg) completely rescued the axis curvature (Fig. 5c) and restored the normal pronephric tubule structure (Fig. 5j). RT–PCR analysis of rescued embryos (data not shown) confirmed that splicing of the endogenous invs mRNA was still disrupted and that the rescued phenotype was probably due to expression of mouse Invs. These results show specificity of the morpholino effects and underscore the conserved function of inversin, particularly the C-terminal domain, in widely divergent vertebrate species. The localization of inversin to cilia prompted us to examine cilia structure in the embryos injected with morpholino oligonucleotides. Neither inversin protein knock-down nor aberrant invs mRNA splicing caused any obvious change in pronephric duct cilia structure as judged by immunofluorescence using a monoclonal antibody to acetylated tubulin (data not shown). This result is consistent with previous reports showing no structural change in tracheal cilia in the inv/inv mutant mouse23,25,26.
Discussion
NPHP is the most frequent genetic cause for end-stage renal failure in children and young adults. Its pathomechanism, however, has been obscure for decades. Here, we combined approaches of disease gene identification by positional cloning, protein-protein interaction, studies of subcellular localization and studies of phenotypic evolutionary conservation and applied them to the study of disease mechanisms in NPHP2. We show that recessive mutations in INVS cause NPHP2 in humans and that there is molecular interaction between the NPHP2 product inversin and the NPHP1 product nephrocystin, which is also known to interact with the NPHP4 product nephrocystin-4 (ref. 7) and with the NPHP3 product nephrocystin-3 (ref. 27). In addition, we show that nephrocystin and inversin localize to primary cilia of renal tubular cells, thereby connecting the pathogenesis of NPHP to the same subcellular compartment that has been recently identified as central to the pathogenesis of PKD. We also show evolutionary conservation of the PKD-like prominent renal cystic phenotype of human NPHP2 and of inv/inv mice by the finding of large renal cysts resulting from invs knockdown in zebrafish. These findings point to a role for inversin at the crossroads between pathogenic mechanisms of nephronophthisis and of PKD.
Recent findings have connected the pathogenesis of human PKD to proteins that exert their function in motile or immotile cilia of renal distal tubular cells and other organs. These proteins have a role in ciliogenesis, cilia maintenance and intraflagellar transport20 and, most importantly, mediate mechanosensation of cilia and cell cycle control28,21. These proteins include polaris, polycystin-1 and polycystin-2 (with their respective homologs in C. elegans osm-5 (refs. 29,30), lov-1 (ref. 31) and pkd-2 and cystin32,33. Very recently, polycystin-1 and polycystin-2 have been shown to function in a shared pathway of mechanotransduction exerted by the primary cilia of renal epithelia. Inability of these cells to sense mechanical cues may be causative for the defect in tissue morphogenesis that leads to the PKD phenotype21. Whereas in PKD one network of pathogenic factors is clustered around proteins of primary cilia, another functional cluster involves mechanisms of cell-cell signaling34.
Similarly, nephrocystin has been described as a novel docking protein10–13 that interacts with components of cell-cell and cell-matrix signaling, such as focal adhesion kinase 2, tensin, p130Cas, filamin and nephrocystin-4 or nephroretinin7. These observations suggest that proteins associated with renal cystic disease may have multiple functions depending on their localization in different cell compartments and their association with distinct protein assemblies. The finding that inversin may have an important function in widely divergent subcellular localizations has recently been emphasized35: in renal proximal tubular cells, a 125-kDa isoform of inversin was present in cell-cell contacts in a complex with N-cadherin and the catenins, whereas a 90-kDa inversin isoform localized to the cell nucleus in a complex with β-catenin, which is known to shuttle between cell-cell contacts and the nucleus. More recently, it has been shown that inversin interacts through its D1 box motif with the anaphase-promoting complex protein (Apc2) in a dynamic expression pattern throughout the cell cycle23. In early prophase, inversin localized at the centrosomes, whereas in metaphase and anaphase, inversin localized to the poles of the mitotic spindle, and in cytokinesis, to the midbody between cells, where microtubule overlap occurs. The link between inversin, polycystin-1 and polycystin-2, the cell cycle and calcium signaling was suggested to lie in a function of primary cilia as an environmental sensor for the centrosome, thus regulating the cell cycle23. The relationship between genes involved in renal cystic diseases and their functional expression in primary cilia is of interest in the light of the finding that taxol, a compound that promotes microtubule assembly, inhibited PKD progression in cpk mice36,37.
In individuals with NPHP2, in whom we detected recessive mutations in INVS, the renal phenotype was similar to that described for inv/inv mice18,19. In one of the nine individuals with mutations in INVS, there was complete situs inversus. This finding is compatible with the recent finding that not all inv/inv mice present with situs inversus, but rather that 10% of mice have situs solitus38. The same report described an additional phenotype in inv/inv mice with cardiac malformations of the right ventricular outflow tract or the ventricular septum38. A ventricular septal defect was also present in the only individual (A8) from our cohort who presented with situs inversus (Table 1). This individual was the only one who carried on both alleles a mutation that truncated all protein domains C-terminal to and including the putative first NLS-BP domain (Fig. 1a–c). All other individuals carried at least one missense allele or one allele that truncated inversin further downstream (Fig. 1). The homozygous truncating mutation in individual A8 would be analogous in severity to the molecular defect of the inv/inv mouse, in which exons 3–11 were homozygously deleted and in which situs inversus was present in 90% of mice18,19. Thus, we confirmed in humans the role of inversin for left-right axis specification that has been described in mice18,19.
Recently, it has become apparent that products of other genes associated with renal cystic disease (in addition to inversin) are important in left-right axis determination of the body plan20,39. The gene PKD2, mutations in which cause autosomal dominant PKD and which encodes the calcium release channel polycystin-2, had been shown in a Pkd2−/− mouse model to represent a master gene regulating left-right axis determination, acting upstream of Nodal, Ebaf, Leftb and Pitx2 (ref. 40). In addition to the malformations described previously41,42, homozygous mutant embryos showed right pulmonary isomerism, randomization of embryonic turning, heart looping and abdominal situs inversus. In Pkd2–/– mice, Leftb and Nodal were not expressed in the left lateral plate mesoderm, and Ebaf was absent from the floor-plate40. Pitx2 was bilaterally expressed in the posterior lateral plate mesoderm but not in the anterior lateral plate mesoderm, suggesting that polycystin-2 acts downstream or in parallel to Shh and upstream of the nodal cascade. Inversin has been shown to be expressed in presomite-stage mouse embryos before the expression of Nodal and Ebaf18. In the orpk mice, mutation of the gene TgN737Rpw, encoding polaris, causes a wide spectrum of phenotypes, including random left-right axis specification, PKD, liver and pancreatic defects, hydrocephalus, skeletal patterning abnormalities and studded monocilia on renal distal tubular cells43. In mouse, node monocilia are required upstream of the nodal cascade40. These findings indicate a strong link between ciliary function and development of laterality.
The locus containing Invs on mouse chromosome 4 has been described as a strong modifier locus for multiple renal cystic mouse models, such as the pcy, cpk44 and jck45 mouse models. In the pcy mouse, this locus (MOP1) modifies 36.7% of the variance for the renal cystic pcy phenotype44. The identification of INVS as causing one form of human NPHP (NPHP2) is very interesting in the light of the fact that pcy is the ortholog of NPHP3, recessive mutations of which cause an adolescent form of human NPHP27,46. NPHP3 was recently identified27, and it was shown that its product interacts with products of other genes associated with NPHP, in analogy to the inversin-nephrocystin interaction identified here and in analogy to the interaction of nephrocystin with nephrocystin-4 (ref. 7).
Methods
Affected individuals
We obtained blood samples and pedigrees after obtaining informed consent from individuals with NPHP and their parents. Experiments on humans were approved by the University of Michigan Institutional Review Board. In all individuals, diagnosis of infantile NPHP (NPHP2) was based on the following criteria: (i) individual entered end-stage renal disease in the first 5 y of life, (ii) kidneys were enlarged and (iii) renal biopsy identified interstitial fibrosis with only mild interstitial infiltrate, tubular atrophy or lack of differentiation, tubular cysts and glomerula that were surrounded by fibrosis. Renal histology findings were compatible with typical findings of NPHP. Histology in NPHP2 was distinct from non-infantile variants of NPHP by absence of tubular basement membrane irregularity, presence of focal segmental glomerulosclerosis and presence of cysts also outside the medullary region in the renal cortex. In all families, diagnosis of NPHP2 was confirmed by renal biopsy. Some individuals had spontaneously regressive hepatomegaly. None of the individuals had ocular abnormalities, such as retinitis pigmentosa or coloboma, that have been described later in life in non-infantile variants of NPHP. One individual (A8) had complete situs inversus and cardiac ventricular septal defect.
Zebrafish
The Subcommittee on Research Animal Care, Massachusetts General Hospital approved the zebrafish studies.
Mutational analysis
We developed primers flanking 16 exons of INVS from the genomic sequence (primer sequences available on request). We carried out direct sequencing using the dideoxy chain termination method on an ABI capillary sequencer and evaluated sequences with Sequencher software.
Plasmids
FLAG-tagged versions of nephrocystin have been described13. J. Goodship (University of Newcastle, UK) provided mouse Invs cDNA. We generated FLAG-tagged Invs plasmids by PCR and standard cloning techniques. We used site-directed mutagenesis to insert mutations into Invs and NPHP1. Mutations were verified by automated sequencing.
Generation and purification of an INV-specific antiserum
We used a recombinant, gel-purified fragment of mouse inversin (amino acids 561–716) fused to MBP (MBP–inversin) to immunize rabbits (Cocalico Biologicals) following a standard immunization protocol. For protein A purification, we loaded the rabbit antiserum onto a commercially available protein A column (Pierce, Perbio Science) and eluted it from the column with 0.1 M glycine-HCl (pH 3.0). For affinity purification of the rabbit antiserum, we coupled recombinant GST–inversin to AminoLink Plus (Pierce, Perbio Science). We eluted immobilized inversin-specific antiserum from the column with 0.1 M glycine-HCl (pH 3.0) and dialyzed it against phosphate-buffered saline. All recombinant proteins were expressed in Escherichia coli BL21 following standard protocols.
Coimmunoprecipitation
We carried out coimmunoprecipitations as described47. Briefly, we transiently transfected HEK293T cells by the calcium phosphate method. After incubation for 24 h, we washed cells twice and lysed them in 1% Triton X-100 lysis buffer. After centrifugation at 15,000g (15 min, 4 °C) and ultracentrifugation at 100,000g (30 min, 4 °C), we precleared cell lysates containing equal amounts of total protein with protein G–sepharose, incubated them for 1 h at 4 °C with the appropriate antibody and then incubated them with 40 µl of protein G–sepharose beads for approximately 2 h. We washed the beads extensively with lysis buffer and resolved bound proteins by 10% SDS–PAGE. For immunoprecipitation of endogenous proteins, we perfused mouse kidneys in situ with ice-cold phosphate-buffered saline before homogenizing them in a lysis buffer containing 20 mM Tris/HCl, pH 7.5, 1% Triton X-100, 25 mM NaF, 12.5 mM Na4P2O7, 0.1 mM EDTA, 50 mM NaCl, 2 mM Na3VO4 and protease inhibitors. After centrifugation to remove cellular debris, we subjected the supernatant to ultracentrifugation (100,000g) for 30 min followed by extensive preclearing with protein G–sepharose. We then carried out immunoprecipitation with control antibody or nephrocystin antiserum as recently described13.
2D gel analysis
We immunoprecipitated equal amounts of protein from cells transfected with nephrocystin or control plasmid with anti-FLAG M2 beads (Sigma). We separated the precipitates on a 2D gel (BioRad), first dimension pH 3–10, second dimension 8–16% SDS–PAGE. After 2D electrophoresis, we either stained proteins with colloidal Coomassie Blue stain or blotted them onto PVDF membranes, blocked them with 5% bovine serum albumin and stained them with antibodies as indicated. For protein identification, we reduced, alkylated, digested in-gel and then identified the gel-separated proteins by mass spectrometry. Briefly, we analyzed aliquots (10%) of the generated peptide mixtures by MALDI mass spectrometry (Bruker Daltonik) to yield a peptide mass map. We used the list of mono-isotopic peptide masses obtained to query non-redundant protein sequence databases. Proteins were identified by correlating the measured peptide masses with theoretical digests of all proteins present in the database.
Western-blot analysis
We carried out western-blot analysis as described48. The antibodies were from Santa Cruz (antibody to β-tubulin-4 and antibody to hemagglutinin pAb), Sigma (antibody to FLAG M2) or Roche (antibody to hemagglutinin mAb). Equal protein loading was confirmed by amidoblack staining of the membranes.
Immunofluorescence staining of MDCK cells and mCcd-K1 cells
We seeded MDCK cells (strain II) or mCcd-K1 cells on coverslips at 100% confluence and cultivated them for 7 d. After washing with ice-cold phosphate-buffered saline, we fixed cells using 4% paraformaldehyde, pH 7.5 and 0.05% Triton X-100 for 15 min at room temperature. We washed cells three times with phosphate-buffered saline and carried out double immunofluorescence staining sequentially with the antibodies as indicated. After washing, we incubated the filters with the secondary antibody, Alexa 488-labeled anti-rabbit and Cy3-labeled anti-mouse IgG, washed them again and mounted them in a commercially available antifade kit (Molecular Probes). Confocal images were taken using a Zeiss laser scan microscope equipped with a 100X oil immersion objective. The appropriate controls were done without the first or second primary antibodies.
Morpholino antisense experiments in zebrafish
We directed antisense morpholino oligonucleotides (Gene Tools) against the zebrafish inversin (invs) translation initiation codon (atgMO) and also against the splice-donor site of the exon corresponding to nucleotides 2,259–2,921 of the invs cDNA (spMO). Primer sequences for atgMPO, spMO and the control oligonucleotide (human β-globin pre mRNA) are available on request. We derived the genomic sequence for invs from assembly of whole-genome shotgun sequence at the Sanger Center and confirmed it locally using phred, phrap and the 11/30/02 wgs trace repository data. We injected oligonucleotides into 1–2-cell–stage embryos in a solution of 0.25 mM morpholino oligonucleotide, 200 mM KCl and 0.1% phenol red. Final cytoplasmic oligonucleotide concentration was approximately 100 nM. mRNA rescue experiments were carried out by coinjecting 100 pg of in vitro transcribed human INVS mRNA with the morpholino oligonucleotide. For histological analysis, we fixed 3-d.p.f. embryos, embedded them in glycolmethacrylate (JB-4; Polysciences) and stained them with methylene blue and azure II as previously described49. We assessed heart looping visually at 27–29 h.p.f. We analyzed cilia structure in morpholino-injected zebrafish embryos using the monoclonal antibody to acetylated tubulin (Clone 6-11B-1; Sigma) on whole-mount preparations of 24-h.p.f. embryos. The oligonucleotides to zebrafish invs sequence used for RT-PCR (2233F) and zebrafish invs (2979R) are available on request.
Supplementary Material
Acknowledgments
We thank the affected individuals and their families for participation, J. Robillard for discussion, R.H. Lyons and A. Imm for large-scale sequencing and P. Cochat for contribution of clinical data on individuals A8 and A10. F.H., G.W. and T.B. are supported by Sonderforschungsbereich 592 of the German Research Foundation and by DFG grants to T.B. and G.W.; I.A.D. and T.O. are supported by grants from the US National Institutes of Health to I.A.D.
Footnotes
GenBank accession numbers. INVS cDNA, NM_014425; Invs cDNA, NM_010569; invs cDNA, AF465261; INVS in chromosome 9 genome contig, NT_008470.
URLs. Additional information is available at http://danio.mgh.harvard.edu/blast/blast.html.
Note: Supplementary information is available on the Nature Genetics website.
Competing Interests Statement: The authors declare that they have no competing financial interests.
References
- 1.Smith C, Graham J. Congenital medullary cysts of the kidneys with severe refractory anemia. Am J Dis Child. 1945;69:369–377. [Google Scholar]
- 2.Fanconi G, Hanhart E, Albertini A. Die familiäre juvenile Nephronophthise. Hel Pediatr Acta. 1951;6:1–49. [PubMed] [Google Scholar]
- 3.Hildebrandt F. Juvenile nephronophthisis. In: Barratt TM, Avner ED, Harmon WE, editors. Pediatric nephrology. Williams & Wilkins; Baltimore: 1999. [Google Scholar]
- 4.Hildebrandt F, et al. A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat Genet. 1997;17:149–153. doi: 10.1038/ng1097-149. [DOI] [PubMed] [Google Scholar]
- 5.Saunier S, et al. A novel gene that encodes a protein with a putative src homology 3 domain is a candidate gene for familial juvenile nephronophthisis. Hum Mol Genet. 1997;6:2317–2323. doi: 10.1093/hmg/6.13.2317. [DOI] [PubMed] [Google Scholar]
- 6.Otto E, et al. A gene mutated in nephronophthisis and retinitis pigmentosa encodes a novel protein, nephroretinin, conserved in evolution. Am J Hum Genet. 2002;71:1167–1171. doi: 10.1086/344395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mollet G, et al. The gene mutated in juvenile nephronophthisis type 4 encodes a novel protein that interacts with nephrocystin. Nat Genet. 2002;32:300–305. doi: 10.1038/ng996. [DOI] [PubMed] [Google Scholar]
- 8.Waldherr R, Lennert T, Weber HP, Fodisch HJ, Scharer K. The nephronophthisis complex. A clinicopathologic study in children. Virchows Arch. 1982;394:235–254. doi: 10.1007/BF00430668. Pathol Anat. [DOI] [PubMed] [Google Scholar]
- 9.Zeisberg M, Strutz F, Muller GA. Renal fibrosis: an update. Curr Opin Nephrol Hypertens. 2001;10:315–320. doi: 10.1097/00041552-200105000-00004. [DOI] [PubMed] [Google Scholar]
- 10.Otto E, et al. Nephrocystin: gene expression and sequence conservation between human, mouse, and Caenorhabditis elegans. J Am Soc Nephrol. 2000;11:270–282. doi: 10.1681/ASN.V112270. [DOI] [PubMed] [Google Scholar]
- 11.Donaldson JC, et al. Crk-associated substrate p130(Cas) interacts with nephrocystin and both proteins localize to cell-cell contacts of polarized epithelial cells. Exp Cell Res. 2000;256:168–178. doi: 10.1006/excr.2000.4822. [DOI] [PubMed] [Google Scholar]
- 12.Donaldson JC, Dise RS, Ritchie MD, Hanks SK. Nephrocystin-conserved domains involved in targeting to epithelial cell-cell junctions, interaction with filamins, and establishing cell polarity. J Biol Chem. 2002;277:29028–29035. doi: 10.1074/jbc.M111697200. [DOI] [PubMed] [Google Scholar]
- 13.Benzing T, et al. Nephrocystin interacts with Pyk2, p130(Cas), and tensin and triggers phosphorylation of Pyk2. Proc Natl Acad Sci USA. 2001;98:9784–9789. doi: 10.1073/pnas.171269898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hildebrandt F, Otto E. Molecular genetics of nephronophthisis and medullary cystic kidney disease. J Am Soc Nephrol. 2000;11:1753–1761. doi: 10.1681/ASN.V1191753. [DOI] [PubMed] [Google Scholar]
- 15.Omran H, et al. Identification of a new gene locus for adolescent nephronophthisis, on chromosome 3q22 in a large Venezuelan pedigree. Am J Hum Genet. 2000;66:118–127. doi: 10.1086/302705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haider NB, Carmi R, Shalev H, Sheffield VC, Landau D. A Bedouin kindred with infantile nephronophthisis demonstrates linkage to chromosome 9 by homozygosity mapping. Am J Hum Genet. 1998;63:1404–1410. doi: 10.1086/302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gagnadoux MF, Bacri JL, Broyer M, Habib R. Infantile chronic tubulo-interstitial nephritis with cortical microcysts: variant of nephronophthisis or new disease entity? Pediatr Nephrol. 1989;3:50–55. doi: 10.1007/BF00859626. [DOI] [PubMed] [Google Scholar]
- 18.Mochizuki T, et al. Cloning of inv, a gene that controls left/right asymmetry and kidney development. Nature. 1998;395:177–181. doi: 10.1038/26006. [DOI] [PubMed] [Google Scholar]
- 19.Morgan D, et al. Inversin, a novel gene in the vertebrate left-right axis pathway, is partially deleted in the inv mouse. Nat Genet. 1998;20:149–156. doi: 10.1038/2450. [DOI] [PubMed] [Google Scholar]
- 20.Igarashi P, Somlo S. Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2002;13:2384–2398. doi: 10.1097/01.asn.0000028643.17901.42. [DOI] [PubMed] [Google Scholar]
- 21.Nauli SM, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 22.Morgan D, et al. The left-right determinant inversin has highly conserved ankyrin repeat and IQ domains and interacts with calmodulin. Hum Genet. 2002;110:377–384. doi: 10.1007/s00439-002-0696-4. [DOI] [PubMed] [Google Scholar]
- 23.Morgan D, et al. Expression analyses and interaction with the anaphase promoting complex protein Apc2 suggest a role for inversin in primary cilia and involvement in the cell cycle. Hum Mol Genet. 2002;11:3345–3350. doi: 10.1093/hmg/11.26.3345. [DOI] [PubMed] [Google Scholar]
- 24.Ostrowski LE, et al. A proteomic analysis of human cilia: identification of novel components. Mol Cell Proteomics. 2002;1:451–465. doi: 10.1074/mcp.m200037-mcp200. [DOI] [PubMed] [Google Scholar]
- 25.Yokoyama T, et al. Reversal of left-right asymmetry: a situs inversus mutation. Science. 1993;260:679–682. doi: 10.1126/science.8480178. [DOI] [PubMed] [Google Scholar]
- 26.Okada Y, et al. Abnormal nodal flow precedes situs inversus in iv and inv mice. Mol Cell. 1999;4:459–468. doi: 10.1016/s1097-2765(00)80197-5. [DOI] [PubMed] [Google Scholar]
- 27.Olbrich H, et al. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapetoretinal degeneration and hepatic fibrosis. Nat Genet. 2003 Jul 20; doi: 10.1038/ng1216. advance online publication. [DOI] [PubMed] [Google Scholar]
- 28.Bhunia AK, et al. PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell. 2002;109:157–168. doi: 10.1016/s0092-8674(02)00716-x. [DOI] [PubMed] [Google Scholar]
- 29.Taulman PD, Haycraft CJ, Balkovetz DF, Yoder BK. Polaris, a protein involved in left-right axis patterning, localizes to basal bodies and cilia. Mol Biol Cell. 2001;12:589–599. doi: 10.1091/mbc.12.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haycraft CJ, Swoboda P, Taulman PD, Thomas JH, Yoder BK. The C. elegans homolog of the murine cystic kidney disease gene Tg737 functions in a ciliogenic pathway and is disrupted in osm-5 mutant worms. Development. 2001;128:1493–1505. doi: 10.1242/dev.128.9.1493. [DOI] [PubMed] [Google Scholar]
- 31.Barr MM, et al. The Caenorhabditis elegans autosomal dominant polycystic kidney disease gene homologs lov-1 and pkd-2 act in the same pathway. Curr Biol. 2001;11:1341–1346. doi: 10.1016/s0960-9822(01)00423-7. [DOI] [PubMed] [Google Scholar]
- 32.Yoder BK, Hou X, Guay-Woodford LM. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol. 2002;13:2508–2516. doi: 10.1097/01.asn.0000029587.47950.25. [DOI] [PubMed] [Google Scholar]
- 33.Hou X, et al. Cystin, a novel cilia-associated protein, is disrupted in the cpk mouse model of polycystic kidney disease. J Clin Invest. 2002;109:533–540. doi: 10.1172/JCI14099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calvet JP. Ciliary signaling goes down the tubes. Nat Genet. 2003;33:113–114. doi: 10.1038/ng1078. [DOI] [PubMed] [Google Scholar]
- 35.Nurnberger J, Bacallao RL, Phillips CL. Inversin forms a complex with catenins and N-cadherin in polarized epithelial cells. Mol Biol Cell. 2002;13:3096–3106. doi: 10.1091/mbc.E02-04-0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woo DD, Miao SY, Pelayo JC, Woolf AS. Taxol inhibits progression of congenital polycystic kidney disease. Nature. 1994;368:750–753. doi: 10.1038/368750a0. [DOI] [PubMed] [Google Scholar]
- 37.Woo DD, Tabancay AP, Jr, Wang CJ. Microtubule active taxanes inhibit polycystic kidney disease progression in cpk mice. Kidney Int. 1997;51:1613–1618. doi: 10.1038/ki.1997.222. [DOI] [PubMed] [Google Scholar]
- 38.McQuinn TC, Miga DE, Mjaatvedt CH, Phelps AL, Wessels A. Cardiopulmonary malformations in the inv/inv mouse. Anat Rec. 2001;263:62–71. doi: 10.1002/ar.1077. [DOI] [PubMed] [Google Scholar]
- 39.Hirokawa N, Noda Y, Okada Y. Kinesin and dynein superfamily proteins in organelle transport and cell division. Curr Opin Cell Biol. 1998;10:60–73. doi: 10.1016/s0955-0674(98)80087-2. [DOI] [PubMed] [Google Scholar]
- 40.Pennekamp P, et al. The ion channel polycystin-2 is required for left-right axis determination in mice. Curr Biol. 2002;12:938–943. doi: 10.1016/s0960-9822(02)00869-2. [DOI] [PubMed] [Google Scholar]
- 41.Wu G, et al. Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell. 1998;93:177–188. doi: 10.1016/s0092-8674(00)81570-6. [DOI] [PubMed] [Google Scholar]
- 42.Wu G, et al. Cardiac defects and renal failure in mice with targeted mutations in Pkd2. Nat Genet. 2000;24:75–78. doi: 10.1038/71724. [DOI] [PubMed] [Google Scholar]
- 43.Moyer JH, et al. Candidate gene associated with a mutation causing recessive polycystic kidney disease in mice. Science. 1994;264:1329–1333. doi: 10.1126/science.8191288. [DOI] [PubMed] [Google Scholar]
- 44.Woo DD, Nguyen DK, Khatibi N, Olsen P. Genetic identification of two major modifier loci of polycystic kidney disease progression in pcy mice. J Clin Invest. 1997;100:1934–1940. doi: 10.1172/JCI119724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuida S, Beier DR. Genetic localization of interacting modifiers affecting severity in a murine model of polycystic kidney disease. Genome Res. 2000;10:49–54. [PMC free article] [PubMed] [Google Scholar]
- 46.Omran H, et al. Human adolescent nephronophthisis: gene locus synteny with polycystic kidney disease in pcy mice. J Am Soc Nephrol. 2001;12:107–113. doi: 10.1681/ASN.V121107. [DOI] [PubMed] [Google Scholar]
- 47.Benzing T, et al. 14-3-3 interacts with regulator of G protein signaling proteins and modulates their activity. J Biol Chem. 2000;275:28167–28172. doi: 10.1074/jbc.M002905200. [DOI] [PubMed] [Google Scholar]
- 48.Benzing T, et al. Upregulation of RGS7 may contribute to tumor necrosis factor-induced changes in central nervous function. Nat Med. 1999;5:913–918. doi: 10.1038/11354. [DOI] [PubMed] [Google Scholar]
- 49.Drummond IA, et al. Early development of the zebrafish pronephros and analysis of mutations affecting pronephric function. Development. 1998;125:4655–4667. doi: 10.1242/dev.125.23.4655. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.