

Abstract
Cervical cancer is second most common cancer in woman worldwide. Cervical cancer caused by human papillomavirus (HPV) oncogene. Inhibition of histone deacetylase (HDAC) activity has been known as a potential strategy for cancer therapy. SAHA is an HDAC inhibitor that has been used in cancer therapy but still has side effects. SAHA modification proposed to minimize side effects. Triazole attachment on the chain of SAHA has been known to enhance the inhibition ability of SAHA and less toxic. In this study, it will be carried out with molecular dynamic simulations of SAHA modifications consisting ligand 1a, 2a and, 2c to interact with six HDAC in hydrated conditions. To all six HDAC Class II, performed docking with SAHA and a modified inhibitor. The docking results were then carried out molecular dynamics simulations to determine the inhibitor affinities in hydrated conditions. The molecular dynamic simulations results show better affinities of ligand 2c with HDAC 4, 6, and 7 than SAHA itself, and good affinity was also shown by ligand 2a and 1c on HDAC 5 and 9. The results of this study can be a reference to obtain better inhibitors.
Keywords: Cervical cancer, HPV, HDAC, Triazole, SAHA, Modified inhibitor, Docking, Dynamic
Background
Cervical cancer is caused by infection of cervical cells by the Human Papilloma Virus (HPV) [1]. In Indonesia, there are 13 new reported cervical cancer cases per 100,000 population of women aged 14-44 years with a mortality rate of 77% [2]. One of the possible treatment of cervical cancer is by inhibiting the process of virus replication in the cell. Histone Deacetylase or HDAC (EC 3.5.1), which exist on HPV, catalyze the role of histone deacetylation process in eukaryotes. Deacetylation is the release of acetyl groups from histone tails [3]. Inhibition of HDAC activity is a potential strategy for cancer therapy [4] and HDAC inhibition by a specific inhibitor induces growth arrest, differentiation, and apoptosis (death) of cancer cells [3]. The main weakness of HDAC inhibitors, such as SAHA, is having a fairly high toxic property [5]. Therefore, modification of the SAHA cluster is required to eliminate the toxic properties and minimize side effects [6]. One way is to replace or add to the triazole group in the chains of SAHA [7]. Triazole is a group of compounds that act as non-classical bioisostere the amide group [8] and can replace it on the side chain of SAHA without significant loss of activity [9]. Some modifications on the triazole compounds on the chain actually has the inhibition ability better than SAHA and have passed the pharmacological parameters according to Lipinski's Rule of Five [10]. Moreover, some other groups have already conducted studies on modified SAHA and cancer, albeit with different emphasize [11].
This study would be carried out by molecular dynamics simulations of the modified compound SAHA to determine the interaction of these compounds with hydrated HDAC on the condition that describes by the dynamic conditions in the wet laboratory. The compounds from the simulation results will be compared with the HDAC SAHA as standard. Finally, the results of these interactions are evaluated to obtain modified ligands or compounds that are potentially being developed as a drug.
Methodology
Preparation of the 3D structure of Homo sapiens Class II HDAC:
Prepare a class II HDAC sequence data (4, 5, 6, 7, 9, and 10) that have been available in the NCBI (http://www.ncbi.nih.gov) and RCSB (http://www.rcsb.org/pdb/) based on the alignment of previous studies [10]. The entire sequence data of HDAC class II is modeled by SWISS model tools (http://swissmodel.expasy.org/).
File Preparation Class II HDAC Homo sapiens:
Polar hydrogen was added into each of these HDAC. The addition of polar hydrogen serves to provide partial/Gasteiger charges to the enzyme. Then, Zn charge on class II HDAC Homo sapiens should be changed from 0 to +2 by using a python script [12]. Then the class II HDAC Homo sapiens was set as macromolecular docking process.
File Preparation Class II HDAC Inhibitor Homo sapiens:
The best modified inhibitor or ligand performed on previous research [10] was computed with software AutoDockTools [13]. The amount of torsion on the ligand is then governed by the rotatable bonds that existed in the ligand (Figure 1).
Figure 1.
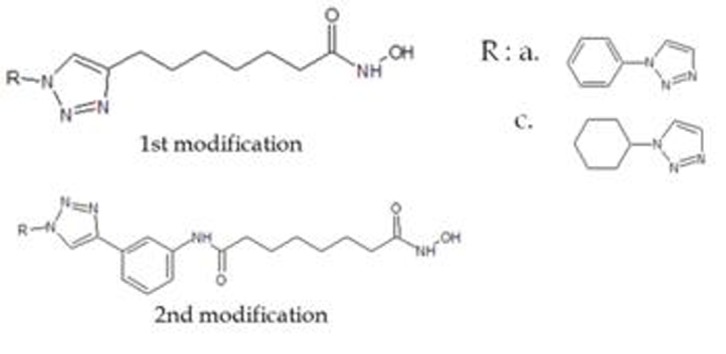
Triazole-modified SAHA chain based on previous research [10]
Grid Setup Box:
Determination of grid box size in this study was carried out base on the docking results of previous research which is also uses the class II HDAC Homo sapiens as a target inhibitor [6]. The utilized dimensional grid box was 50 × 60 × 50 with a grid spacing of 0.375 Å.
Docking Simulation:
Autogrid docking performed using AutoDock 4.2 and 4.2 through the terminal. The utilized algorithm is Lamarckian Genetic Algorithm (LGA) and two repetitions was performed by each docking simulation [14].
Molecular Dynamic Simulation:
Partial geometry optimization of complex enzyme-ligand was utilized by forcefield parameters. The stage of energy minimization was proceeded with MMFF94× [15]. The utilized parameters are the NVT ensemble (N: number of atoms; V: volume, T: temperature) with the NPA algorithm [16]. Stages of molecular dynamics simulations are as follows: initialization, equilibration, and production.
Dynamic Process Simulation:
Molecular dynamics simulations was carried out with the enzyme-inhibitor complex at 300 K with 10 ps heating, and then the simulation was executed for 5 ns [15]. The inhibitors used are the modified ligands and standards. The next stage of the process was 20 ps cooling until the temperature reaches 1 K. Results of position, velocity and acceleration are saved in every 0.5 ps. Then the interaction between the enzyme and the ligand for molecular dynamic processes can be observed in the MOE database viewer. Observations of contact residues and hydrogen bonding can be seen in LigX Interaction in MOE 2008.10 software.
Results and Discussion
Preparation of Structure HDAC Class II Homo sapiens:
A captured HDAC class II Homo sapiens sequence was based on the alignments of previous research [10]. 3D structure of the HDAC class II Homo sapiens used a catalytic region that is based on the maintained area (conserved region) [6].
Visualization of the Active Side of HDAC Class II Homo sapiens:
To determine the catalytic side of the HDAC class II Homo sapiens, the 3D structure visualization was made by PyMOL. The visualization results show that the enzyme has the form of a catalytic cofactor Zn2+ binding to three amino acid residues, namely two aspartic acids and one histidine.
Preparation of Class II HDAC Inhibitor Homo sapiens:
The ligands prepared is ligand from the previous modifications result that having the best affinity and low toxicity according to Lipinski's Rule of Five [10]. Each of HDAC II will be paired with SAHA as standard and the best ligand in order to know the interaction differences between the two ligands in the simulation.
Preparation for Enzyme and Ligand Docking:
In the process of preparation, AutoDock will respond to the number of rotatable bonds of the ligand, and the number of rotatable bonds indicate the degree of flexibility of a ligand.
Docking:
Oriented docking is a process that already has a grid box specific parameters in determining the location of the enzyme's active site. In this study an oriented docking process was conducted.
Docking Analysis
Ligand Interactions with Class II HDAC Homo sapiens:
One model out of 100 ligand docking results will be taken based on the most negative bond energy value and polar groups of the ligand is having a strong interaction with the Zn2+ ligand cofactor in the catalytic process. Interactions are selected based on the formation of chelate pentagon rings between O atoms in the cluster of C=O and -OH on the ligand with Zn2+ cofactor.
Free Energy Association (ΔGbinding) and inhibition constants (Ki):
AutoDock 4.2 ΔGbinding calculation results show a negative value for all ligands. This shows that conformation of complex SAHA standard ligand and ligand modification by HDAC 4, HDAC 5, HDAC 6, HDAC 7, 9 HDAC and HDAC 10 are more stable than the individual conformation, because if there is bonding interaction, it would release energy that can be used to lower the activation energy of the catalytic reaction [6].
Visualization Docking Results:
Visualization of the entire results of docking ligand conformations with HDAC class II Homo sapiens has been done by PyMOL. 3D ligand interactions with the HDAC class II Homo sapiens can be seen in Figure 2. The visualization result shows the interaction of the Zn2+ ligands as catalytic side of the HDAC class II Homo sapiens.
Figure 2.

3D interaction of docking simulation results between (A) ligand 2c (B) SAHA with HDAC 4. Between (C) ligand 2a (D) with SAHA HDAC 5. Between (E) ligand 2c (F) with SAHA HDAC 6. between (G) ligand 2c (H) with SAHA HDAC 7. Between (I) the ligand 1c (J) with SAHA HDAC 9. Between (K) ligand 1c (L) with SAHA HDAC 10.
Molecular Dynamic Simulation:
Molecular dynamics simulations of the ligand and the enzyme treated in a flexible state with solvent, in order to study the effects of the present of solvent in the system, then applied to explore the conformation of the receptor protein to improve the process of drug design [17]. The simulation process was carried out on 12 pairs of ligand-enzyme complex of molecular docking results. Minimization process has been done so that the geometry of atoms that do not fit can be returned to produce the lowest potential energy to the system [18].
Molecular Dynamics Simulation on Temperature 300 K:
Initialization results continued at a temperature of 300 K to determine the initial interaction of the enzyme-ligand complex prior to the temperature of the human body, 310 K. In this study initialization conducted only at 300 K. Nevertheless, the results of interaction simulation in this study can be a reference to proceed at a temperature of 310 K.
Analysis of Results Docking and Molecular Dynamics Simulations:
The data of table 1 shows that the interaction of molecular dynamics simulation results that reinforce the results obtained in the docking process despite the use of different methods. On the results of the energy analysis shows that the ligand docking has modified the Gibbs free energy more negative compared with SAHA. Free energy data is strengthened by the results of residue contacts in the docking and molecular dynamics simulations. Therefore, the modified ligands are feasible to be developed as a drug.
Conclusion
From this study, several conclusions can be drawn. First, the results of SAHA standard ligand docking and modified ligand with HDAC class II Homo sapiens showed that both of ligands, SAHA standard ligand and modified ligand, they still have the same interaction to the HDAC class II Homo sapiens. It occurred at the O atoms of C=O and –OH functional groups that binds to the Zn2+ ions as the catalytic enzyme. The binding free energy (ΔGbinding) and inhibition constants (Ki) analysis results suggest that the modified ligand still have a better affinity than SAHA. The dynamic analysis results show that all modified ligands have good interaction with the active site of HDAC, except ligand 1c with HDAC 10 are no longer bind to the Zn2+ ions. It means that ligand 1c do not meet the requirements to be developed further. The dynamic interactions of modified ligand and SAHA have nearly the same affinity and interaction. However, the docking results of ligand 2c, 2a, and 1c are still better than SAHA. The dynamic results have reinforce the docking results of these ligands. Based on the results of ligand docking and dynamic, ligands that can potentially be developed as a drug is ligand 2c, because it can interact with HDAC 4, 6, and 7 much better than SAHA. However, it is also possible for ligands 2a and 1c could be developed as a drug for one very specific HDAC.
Supplementary material
Acknowledgments
This research is supported by Hibah BOPTN Ditjen Dikti No: 2788/H2.R12/HKP.05.00/2013. Usman Sumo Friend Tambunan, Ridla Bakri, and Djati Kerami supervised this research, while Tirtana Prasetia was working on the technical details. Arli Aditya Parikesit prepared the English manuscript and re-verified the data.
Footnotes
Citation:Tambunan et al, Bioinformation 9(13): 696-700 (2013)
References
- 1.Schiffman MH, et al. JNCI Journal of the National Cancer Institute. 1993;85:958. doi: 10.1093/jnci/85.12.958. [DOI] [PubMed] [Google Scholar]
- 2.WHO, Cervical Cancer Summary Report. 2010 [Google Scholar]
- 3.Subha K, et al. Bioinformation. 2008;3:218. doi: 10.6026/97320630003218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pan LN, et al. Cellular & Molecular Immunology. 2007;4:337. [PubMed] [Google Scholar]
- 5.Rikiishi H, et al. J Biomed Biotechnol. 2011;2011:830260. doi: 10.1155/2011/830260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tambunan US, et al. BMC Bioinformatics. 2010;11:S16. doi: 10.1186/1471-2105-11-S7-S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gryder BE, et al. Future Med Chem. 2012;4:505. doi: 10.4155/fmc.12.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meanwell NA, et al. J Med Chem. 2011;54:2529. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 9.Pirali T, et al. J Comb Chem. 2008;10:624. doi: 10.1021/cc800061c. [DOI] [PubMed] [Google Scholar]
- 10.Tambunan US, et al. BMC Bioinformatics. 2011;12:S23. doi: 10.1186/1471-2105-12-S13-S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi SE, et al. Bioorg Med Chem Lett. 2012;22:7084. doi: 10.1016/j.bmcl.2012.09.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bjerling P, et al. Molecular and Cellular Biology. 2002;22:2170. doi: 10.1128/MCB.22.7.2170-2181.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morris GM, et al. Curr Protoc Bioinformatics. 2008 doi: 10.1002/0471250953.bi0814s24. [DOI] [PubMed] [Google Scholar]
- 14.Morris GM, et al. Journal of Computational Chemistry. 1998;19:1639. [Google Scholar]
- 15.Tambunan USF, et al. OnLine Journal of Biological Sciences. 2011;11:48. [Google Scholar]
- 16.Johnson BC, et al. Antimicrob Agents Chemother. 2012;56:411. doi: 10.1128/AAC.05292-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alonso H, et al. Med Res Rev. 2006;26:531. doi: 10.1002/med.20067. [DOI] [PubMed] [Google Scholar]
- 18.Nurbaiti S, et al. International Journal of Integrative Biology. 2010;9:104. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.