

Abstract

We describe here details of our investigations into Pd-catalyzed and thermal aza-Claisen–carbocyclizations of N-allyl ynamides to prepare a variety of α,β-unsaturated cyclopentenimines. The nature of the ynamide electron withdrawing group and β-substituent plays critical roles in the success of this tandem cascade. With N-sulfonyl ynamides, the use of palladium catalysis is required, as facile 1,3-sulfonyl shifts dominate under thermal conditions. However, since no analogous 1,3-phosphoryl shift is operational, N-phosphoryl ynamides could be used to prepare similar cyclopentenimines under thermal conditions through zwitter ionic intermediates that undergo N-promoted H-shifts. Alternatively, by employing ynamides bearing tethered carbon nucleophiles, the zwitter ionic intermediates could be intercepted giving rise rapidly to more complex fused bi- and tricyclic scaffolds.
INTRODUCTION
We have been involved with the chemistry of ynamides for the last 17 years, and this burgeoning field has attracted an immense amount of attention from the synthetic community in recent decade.1–3 In our own effort during the past several years, we have been heavily interested in using Pd-catalyzed and thermal aza-Claisen4,5 rearrangements as a means of activating N-allyl ynamides towards (1) nucleophilic additions of amines6 and alcohols,7 (2) inter-8 and intramolecular9 [2 + 2] cycloaddition reactions, (3) skeletal rearrangements yielding nitriles,6c and (4) carbocyclizations10 and cationic polyene cascades.
We previously reported the initial discovery of a tandem cascade of Pd-catalyzed aza-Claisen rearrangement and carbocyclization of TIPS-terminated N-allyl ynamide 1 to cyclopentenimine6b 3 upon exposure to 5 mol% Pd(PPh3)4 [Scheme 1]. Interestingly, our only success in achieving this transformation was using TIPS-terminated ynamides; alkyl and aryl-terminated ynamides underwent facile 1,3-sulfonyl shifts to generate quaternary nitriles 5.6b–6c Furthermore, we demonstrated that the pathway leading to cyclopenteimine 3 involved tautomeric Pd-π-allyl ynamide and ketenimine complexes 2a and 2b, as treatment of the isolable TIPS-ketenimine11 4 to either Pd(0) or thermal conditions gave no cyclopentenimine 3. We wish to disclose here details of this Pd-catalyzed or thermal aza-Claisen–carbocyclization sequence using N-allyl ynamides and the impact of the ynamide electron withdrawing group and β-substituents on this tandem cascade.
Scheme 1.

Initial Discovery of a Pd-Catalyzed Carbocyclization.
RESULTS AND DISCUSSION
While attempting to render the 1,3-sulfonyl shift leading to nitrile formation diastereoselective, we uncovered a fascinating reaction dichotomy with γ-branched ynamides. Upon heating ynamide 6 in toluene at 110 °C, nitrile 7 was isolated in near quantitative yield, albeit with modest diastereoselectivity6c [Scheme 2]. Alternatively, treatment of 6 with 5 mol % Pd(PPh3)4 gave cyclopentenimine 8 in 69% yield and nitrile 7 was not found. This represented the first time we had observed a carbocyclization of a non-silyl terminated N-sulfonyl ynamide.
Scheme 2.

1,3-Sulfonyl Shift versus Carbocyclization Dichotomy.
We were intrigued by the possibility of using γ-branched ynamides as carbocyclization precursors and wanted to further probe the substrate scope. Unfortunately, the preparation of γ-branched ynamide 6 was problematic [Scheme 3]. It involved initial silylation of 3-phenyl-propargyl alcohol 9 followed by bromination to afford 10. The ensuing standard 15 mol% CuSO4·5H2O and 30 mol% 1,10-phenanthroline catalyzed amidative cross-coupling gave 6 in only 12% yield, likely due to the steric bulk of the alkynyl bromide. We later discovered that using 20 mol% CuTC and 40 mol% DMEDA could improve the yield of the cross coupling to 66%. Still, assessing the reaction scope using this protocol would have been a lengthy endeavor.
Scheme 3.

Preparation of γ-Branched Ynamides
Alternatively, inspired by a report by Saá,12 we found that ynamide 11, which was readily accessible in gram quantities, could be lithiated using LHMDS and added to aldehydes such as pivalaldehyde to directly prepare γ-hydroxy ynamide 12a in excellent yield [Scheme 3]. The alcohol could then be functionalized as desired to afford ynamides such as 13a. This allowed us to expediently and fully explore the scope of cyclopentenimine formation.
We soon discovered that the substrate scope for the Pd-catalyzed carbocyclization was quite broad. As shown in Table 1, a variety of functionalized N-allyl-γ-branched ynamides could be employed in cyclopentenimine synthesis using 5 mol % Pd(PPh3)4 in toluene at 70 °C. The reaction nicely tolerated t-Bu, n-hex, c -hex, and even a spirocyclic cyclohexane at the γ-position. The tolerance for functionality on the alcohol moiety was equally general, including methyl, silyl, benzyl, and even trityl protecting groups. Also, both N-Ts and N-Ns containing ynamides underwent the desired carbocyclization in comparable yields. Notably, this methodology provided a means of preparing α,β-unsaturated cyclopentenimines analogous to the α,β-unsaturated enones from a Baylis-Hillman13 type reaction.
Table 1.
Synthesis of α,β-Unsaturated Cyclopentemines from N-Allyl Ynamides
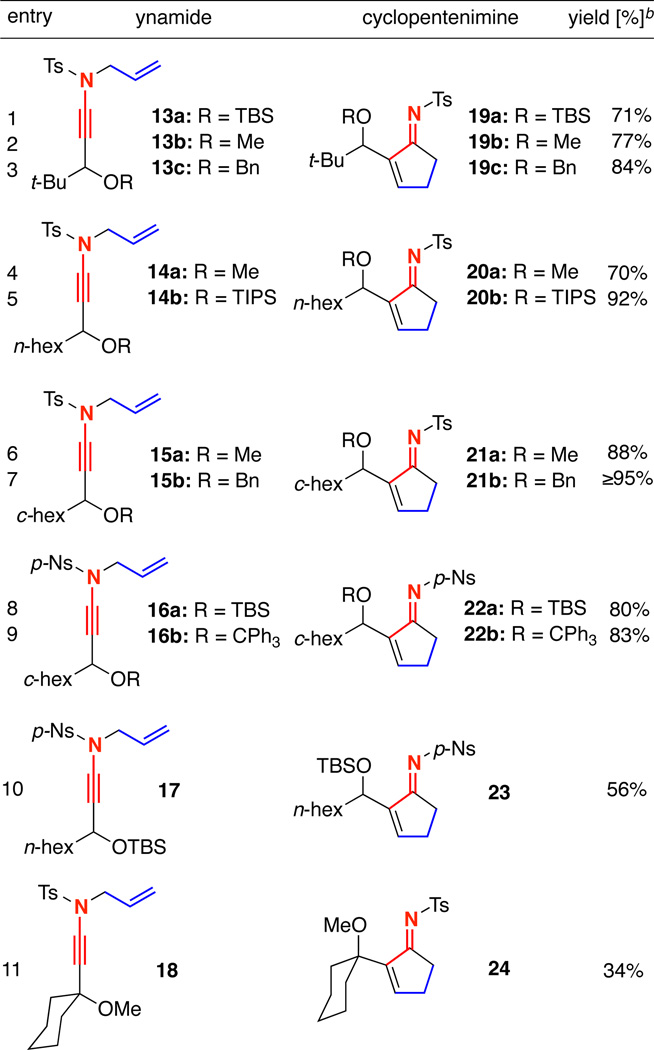 |
Reaction conditions: 5.0 mol% Pd(PPh3)4, toluene [conc = 0.04 M], 70 °C, 1 h.
Isolated yields.
While exploring a potential Staudinger-type [2 + 2] cycloaddition of N-phosphoryl N-allyl ynamides to give azetidinimines 26,14 we discovered another case of cyclopentenimine formation [Scheme 4]. When a mixture of N-phosphoryl ynamide 25 and N-benzylidineaniline was subjected to 5 mol% Pd2(dba)3 and 10 mol% xantphos, conditions which favor reductive elimination of the intermediate Pd-π-allyl ketenimine in our hands, the major product by 1H NMR using phenanthrene as an internal standard was cyclopentenimine 27. This was the first time we had observed the carbocyclization of a phenyl-terminated ynamide. In the analogous phenyl-terminated N-sulfonyl ynamide, the 1,3-sulfonyl shift leading to nitriles dominated the carbocyclization pathway. However, because there was no competing 1,3-phosphoryl shift, heating of N-phosphoryl ynamide 25 to 125 °C in toluene led to cyclopentenimine 27 in an isolated 50% yield.
Scheme 4.

An Unexpected Carbocyclization of an N-Phosphoryl Ynamide.
We soon found that the yield of cyclopentenimine 27 could be increased to 87% yield using 5 mol% Pd(PPh3)4 in toluene at 70 °C [Scheme 5]. Ynamides 30b and 30c bearing terminal n-hexyl and TBS-ether moieties were also tolerated using both thermal and Pd-catalyzed conditions, though the yields were slightly diminished. Interestingly, the Pd-catalyzed carbocyclization of γ-branched ynamide 30d was sluggish, giving <10% yield of the desired cyclopentenimine. The yield could be improved to 69% by heating to 135 °C in toluene. The difference in reactivity of γ-branched N-sulfonyl and N-phosphoryl ynamides under Pd-catalyzed conditions was the first indication that the carbocyclization pathways may be different.
Scheme 5.

Pd-Catalyzed Versus Thermal Carbocyclizations of N-Phosphoryl Ynamides.
Furthermore, treatment of TIPS-terminated N-phosphoryl ynamide 30e with 5 mol% Pd(PPh3)4 led to no discernable formation of cyclopentenimine 31e; only ketenimine 32 resulting from reductive elimination of the intermediate Pd-π-allyl complex was isolated [Scheme 6]. Also, heating of 32 to 185–190 °C for two days did not result in any formation of 31e.
Scheme 6.

Mechanistic Studies Employing an Isolable TIPS-Ketenimine
Collectively, the experimental evidence indicated that the carbocyclization mechanisms for N-sulfonyl and N-phosphoryl ynamides, even under Pd-catalysis, were unique [Scheme 7]. We propose that the carbocyclization of N-sulfonyl ynamides 33 occurs through Pd-π-allyl ketenimines 34b via a Rautenstrauch rearrangement.15–16 In contrast, it seems that the carbocyclization of N-phosphoryl ynamides 36 occurs through ketenimine 37 under both palladium catalyzed and thermal conditions due to the lack of a competing 1,3-phosphoryl shift. Following the carbocyclization17–18 that generates zwitter ionic intermediate 38, a N-promoted 1,2-H shift gives cyclopentenimine 39.
Scheme 7.

Unique Carbocyclization Pathways for N-Sulfonyl and N-Phosphoryl Ynamides.
We were especially excited by the discovery that N-phosphoryl ynamides participated in the carbocyclization without the need for palladium catalysis, thereby avoiding any potential scrambling6b of the allyl moiety through the Pd-π-allyl intermediates. In suitably substituted N-phosphoryl ynamides, we demonstrated that N-promoted Meerwein-Wagner alkyl shifts could compete with the 1,2-H shift leading to fused or spiro bicyclic products.10 In addition to exploiting the zwitter ionic intermediates via N-promoted ring expansions and contractions, we envisioned the possibility of intercepting19 the intermediates with tethered nucleophiles to prepare a variety of bicyclic scaffolds such as 43a–43c [Scheme 8].
Scheme 8.

Potential Carbocyclization Cascades of N-Phosphoryl Ynamides.
Our initial attempts at trapping the zwitter ionic intermediates employed tethered carbon nucleophiles, thereby eliminating any undesired side reactions such as amidine6 or imidate7 formation. When ynamide 44 featuring a tethered benzene ring was heated to 135 °C in toluene, only cyclopentenimine 48 resulting from a 1,2-H shift through 47 was isolated [Scheme 9]. To allow the nucleophilic addition to occur, it was clear that we needed to slow the 1,2-H shift. By introducing a methyl at R1 and tethering a more electron-rich m-methoxy benzene, ynamide 45 cleanly underwent the desired thermal aza-Claisen–Friedel-Craft electrophilic aromatic substitution reaction sequence to give 50 in 85% yield as a mixture of cis and trans isomers at the ring juncture.10
Scheme 9.

Carbocyclization Cascade vs. 1,2-H Shift.
In addition, we had previously demonstrated that geranyl-tethered ynamides such as 51 could be used in thermal carbocyclization cascades to prepare a readily separable mixture of cis-fused bicycle 52 as a 9:1 mixture of alkene isomers and tricycle 53 [Scheme 10].10
Scheme 10.

Carbocyclization Cascade of Geranyl-Tethered Ynamides.
Working from our success thus far in developing carbocyclization cascades20–21 of geranyl-tethered ynamides, we wondered if we could extend the cyclization to include farnesyl-tethered ynamides. The ynamide synthesis is straightforward and outlined in Scheme 11. Following a literature procedure,22 farnesol amine 55 could be prepared from farnesol 54 in 97% over two steps. Then, phosphorylation of farnesol amine gave 56, which could be subjected to our optimized amidative cross-coupling conditions to yield TIPS-terminated ynamide 57 in 65% yield. TBAF-mediated desilylation gave the terminally unsubstituted ynamide 58, which could then be lithiated and quenched with MeI to prepare the desired farnesyl-tethered ynamide 59.
Scheme 11.

Synthesis of a Farnesyl Tethered Ynamide.
With ynamide 59 in hand, we were able to investigate the outcome of the intended polyene carbocyclization cascade. As described in Scheme 12, our hypothesis was that a facile 1,2-H shift could occur to allow equilibration between carbocationic intermediates 61a and 61b, which may be followed by a third cyclization to afford tricycle 64. Unfortunately, when ynamide 59 was heated to 135 °C in toluene, only bicycle 62 resulting from elimination of 61a and tricycle 63 resulting from a formal [4 + 2] cycloaddition could be found.
Scheme 12.

Attempted Carbocyclization Cascade of Farnesol-Tethered Ynamide 59.
CONCLUSION
We have described here a fascinating study of Pd-catalyzed and thermal carbocyclizations of N-sulfonyl and N-phosphoryl ynamides to prepare α,β-unsaturated cyclopentenimines. In the case of N-sulfonyl ynamides, the use of palladium catalysis is necessary, as the carbocyclization occurs through Pd-π-allyl complexes via a Rautenstrach rearrangement; with N-phosphoryl ynamides, thermal conditions may be employed to yield cyclopentenimines via N-promoted 1,2-H shifts through zwitter ionic intermediates. We also demonstrated the ability to intercept the zwitter ionic intermediates with tethered alkenyl and aryl nucleophiles to prepare a variety of fused bicyclic and tricyclic scaffolds.
EXPERIMENTAL SECTION
All reactions were performed in flame-dried glassware under a nitrogen atmosphere. Solvents were distilled prior to use. Chromatographic separations were performed using 60 Å SiO2. 1H and 13C NMR spectra were obtained on spectrometers using CDCl3 with TMS or residual solvent as standard unless otherwise noted. Infrared spectra were obtained on FTIR and relative intensities are expressed qualitatively as s (strong), m (medium), and w (weak). TLC analysis was performed using 254 nm polyester-backed plates (60 Å, 250 µm) and visualized using UV and a suitable chemical stain. Low-resolution mass spectra were obtained using LC/MSD and APCI.
General Procedure for the Preparation of γ-Hydroxy-Ynamides
To a flame-dried 50-mL RB-flask was added ynamide 11 (470.0 mg, 2.00 mmol) and THF (20 mL). The solution was cooled to −78 °C [− 50 °C works equally well] and LHMDS (3.0 mL, 3.0 mmol, 1 M in THF) was added dropwise over ~1 min. The reaction mixture was stirred at −78 °C [or − 50 °C] for 1 h to ensure complete lithiation of the ynamide and then pivalyl aldehyde was added dropwise over ~1 min. After 20 min, the cooling bath was removed and the reaction was allowed to come to rt. The reaction was monitered by TLC and when progress appeared to be complete after ~2 h, the mixture was diluted with EtOAc (20 mL) and quenched with water (15 mL). The phases were separated and the aqueous phase extracted with EtOAc (20 mL) once. The organic phases were combined, washed with brine, dried over Na2SO4, and concentrated by rotary evaporation. The crude residue was purified by flash silica gel column chromatography (isocratic eluent: 4:1 hexanes/EtOAc + 2% NEt3 [to buffer the column]) to afford the γ-hydroxy ynamide 12a (656.0 mg, 2.00 mmol, ≥95%) as a colorless oil.
12a
(656.0 mg, ≥95%); Rf= 0.27 [3:1 hexanes:EtOAc]; colorless oil; 1H NMR (500 MHz, CDCl3) δ 0.95 (s, 9H), 2.45 (s, 3H), 3.96 (dq, 2H, J = 1.5, 6.5 Hz), 4.11 (brs, 1H), 5.21 (dq, 1H, J = 1.5, 10.0 Hz), 5.24 (dq, 1H, J = 1.5, 17.0 Hz), 5.72 (ddt, J = 6.5, 10.0, 17.0 Hz), 7.34 (d, 2H, J = 8.0 Hz), 7.79 (d, 2H, J = 8.0 Hz); 13C NMR (100 MHz, CDCl3) δ 21.8, 25.6, 36.4, 54.3, 71.0, 71.7, 78.1, 120.2, 127.9, 129.9, 131.1, 134.9, 144.9; IR (film) cm−1 3519brm, 2956m, 2869w, 2244m, 1597m, 1478m, 1462m, 1362s; mass spectrum (ESI): m/e (% relative intensity) 336 (100) (M–H2O+MeOH+H)+; HRMS (QTOF MS ESI) m/e calcd for C17H23NO3SNa [M+Na]+: 344.1291, found 344.1307.
12b
(413.2 mg, 79%); (Rf= 0.28 [3:1 hexanes:EtOAc]; pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 0.90 (t, 3H, J = 6.8 Hz), 1.23–1.42 (m, 8H), 1.59–1.73 (m, 2H), 1.87 (brs, 1H), 2.45 (s, 3H), 3.95 (ddt, 1H, J = 1.2, 6.4, 14.4 Hz), 4.01 (ddt, 1H, J = 1.2, 6.4, 14.4 Hz), 4.48 (t, 1H, J = 6.8 Hz), 5.22 (dq, 1H, J = 1.6, 10.0 Hz), 5.26 (dq, 1H, J = 1.6, 16.4 Hz), 5.77 (ddt, 1H, J = 6.4, 10.0, 16.4 Hz), 7.35 (d, 2H, J = 8.4 Hz), 7.80 (d, 2H, J = 8.4 Hz); 13C NMR (100 MHz, CDCl3) δ 14.2, 21.8, 22.7, 25.3, 29.1, 31.9, 38.0, 54.3, 62.8, 72.4, 78.2, 120.2, 127.9, 129.9, 131.0, 134.8, 144.8; IR (film) cm−1 3376brm, 2925m, 2857m, 2243m, 1597m, 1494w, 1466m, 1419m, 1364s; mass spectrum (ESI): m/e (% relative intensity) 364 (100) (M–H2O+MeOH+H)+; HRMS (QTOF MS ESI) m/e calcd for C19H27NO3SNa 372.1604, found 372.1612.
12c
(211.0 mg, 81%); Rf= 0.21 [3:1 hexanes/EtOAc]; colorless oil; 1H NMR (400 MHz, CDCl3) δ 0.97–1.33 (m, 6H), 1.64–1.83 (m, 5H), 2.45 (s, 3H), 3.94 (ddt, 1H, J = 1.2, 5.6, 14.0 Hz), 3.99 (ddt, 1H, J = 1.2, 5.6, 14.0 Hz), 4.26 (d, 1H, J = 4.0 Hz), 5.21 (dq, 1H, J = 1.4, 7.2 Hz), 5.25 (dq, 1H, J = 1.4, 14.0 Hz), 5.73 (ddt, 1H, J = 6.4, 10.4, 16.8 Hz), 7.34 (d, 2H, J = 8.4 Hz), 7.79 (d, 2H, J = 8.4 Hz); 13C NMR (100 MHz, CDCl3) δ 21.8, 26.0, 26.1, 26.5, 28.2, 28.8, 44.5, 54.3, 67.5, 71.4, 78.9, 120.2, 127.9, 129.9, 131.1, 134.8, 144.9; IR (film) cm−1 2925m, 2853m, 2244m, 1597w, 1364s; mass spectrum (ESI): m/e (% relative intensity) 362 (100) (M–H2O+MeOH+H)+. HRMS (TOF MS ESI) m/e calcd for C19H-26NO3S [M+H]+ 348.1628, found 348.1639.
12d
(184.0 mg, 65%); Rf = 0.30 [3:1 hexanes/EtOAc]; orange oil; 1H NMR (400 MHz, CDCl3) δ 0.97–1.30 (m, 4H), 1.50–1.57 (m, 2H), 1.66–1.84 (m, 5H), 4.02 (ddt, 1H, J = 1.2, 6.4, 14.4 Hz), 4.07 (ddt, 1H, J = 1.2, 6.4, 14.4 Hz), 4.28 (t, 1H, J = 5.6 Hz), 5.25 (dq, 1H, J = 1.2, 10.4 Hz), 5.28 (dq, 1H, J = 1.2, 16.8 Hz), 5.72 (ddt, 1H, J = 6.4, 10.4, 16.8 Hz), 8.10 (d, 2H, J = 9.2 Hz), 8.40 (d, 2H, J = 9.2 Hz); 13C NMR (100 MHz, CDCl3) δ 25.9, 26.0, 26.5, 28.2, 28.7, 44.4, 54.7, 67.3, 72.1, 77.6, 120.9, 124.5, 129.1, 130.3, 143.2, 150.8; IR (film) cm−1 3401brm, 2928m, 2854m, 2247m, 1532s, 1372s; mass spectrum (APCI): m/e (% relative intensity) 393 (100) (M–H2O+MeOH+H)+.
12e
(55.4 mg, 58%); Rf = 0.25 [3:1 hexanes/EtOAc]; pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 0.89 (t, 3H, J = 7.2 Hz), 1.22–1.44 (m, 8H), 1.61–1.76 (m, 2H), 1.80 (brs, 1H), 4.01 (ddt, 1H, J = 1.2, 6.4, 14.8 Hz), 4.05 (ddt, 1H, J = 1.2, 6.4, 14.8 Hz), 4.49 (t, 1H, J = 6.4 Hz), 5.24 (dq, 1H, J = 1.2, 10.4 Hz), 5.27 (dq, 1H, J = 1.2, 16.8 Hz), 5.72 (ddt, 1H, J = 6.4, 10.4, 16.8 Hz), 8.10 (d, 2H, J = 9.2 Hz), 8.41 (d, 2H, 9.2 Hz); 13C NMR (100 MHz, CDCl3) δ 14.3, 22.8, 25.4, 29.1, 32.0, 38.1, 54.8, 62.7, 73.3, 77.0, 121.0, 124.6, 129.2, 130.3, 143.2, 150.9; IR (film) cm−1 3379brm, 2928m, 2858m, 2246m, 1607s, 1348s; mass spectrum (APCI): m/e (% relative intensity) 395 (100) (M–H2O+MeOH+H)+; HRMS (QTOF MS ESI) m/e calcd for C18H24N2O5SNa 403.1298, found 403.1292.
12f
(507.9 mg, ≥95%); Rf = 0.38 [4:1 hexanes/EtOAc]; colorless oil; 1H NMR (400 MHz, CDCl3) δ 1.06–1.16 (m, 1H), 1.30–1.59 (m, 7H), 1.80 (d, 2H, J = 12.4 Hz), 2.36 (s, 3H), 3.10 (s, 1H), 3.86 (d, 2H, J = 6.4 Hz), 5.09–5.17 (m, 2H), 5.58–5.68 (m, 1H), 7.27 (d, 2H, J = 8.0 Hz), 7.72 (d, 2H, J = 8.4 Hz); 13C NMR (100 MHz, CDCl3) δ 21.3, 23.0, 24.9, 39.7, 53.8, 68.5, 74.1, 77.2, 119.7, 127.5, 129.5, 130.6, 134.2, 144.5; IR (film) cm−1 3496w, 3399w, 2933s, 2857m, 2242m, 1597w, 1447w, 1362s; mass spectrum (APCI): m/e (% relative intensity) 334 (24) (M+H)+.
General Procedure for the Silylation of γ-Hydroxy-Ynamides
To a flame-dried screw-cap vial was added ynamide 12a (64.8 mg, 0.20 mmol) and CH2Cl2 (0.57 mL, 0.35 M in ynamide). The solution was cooled to 0 °C and then imidazole (34.0 mg, 0.50 mmol) and TBSCl (37.5 mg, 0.25 mmol) were added. The reaction mixture was allowed to warm to rt. The reaction was monitered by TLC and when the starting material was consumed after ~45 min, the mixture was diluted with CH2Cl2 (3 mL) and quenched with water (3 mL). The phases were separated and the aqueous phase extracted with CH2Cl2 (3 × 5 mL). The organic phases were combined, washed with brine, dried over Na2SO4, and concentrated by rotary evaporation. The crude residue was purified by flash silica gel column chromatography (isocratic eluent: 15:1 hexanes/EtOAc + 2% NEt3 [to buffer the column]) to afford ynamide 13a (78.0 mg, 0.18 mmol, 89%).
13a
(78.0 mg, 89%); Rf= 0.48 [8:1 hexanes:EtOAc]; pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 0.02 (s, 3H), 0.04 (s, 3H), 0.86 (s, 9H), 0.91 (s, 9H), 2.44 (s, 3H), 3.97 (d, 2H, J = 6.4 Hz), 4.03 (s, 1H), 5.21 (dd, 1H, J = 1.2, 10.0 Hz), 5.25 (dd, 1H, J = 1.2, 16.8 Hz), 5.77 (ddt, 1H, J = 6.4, 10.0, 16.8 Hz), 7.33 (d, 2H, J = 8.0 Hz), 7.80 (d, 2H, J = 8.0 Hz); 13C NMR (100 MHz, CDCl3) δ −5.2, −4.3, 18.3, 21.7, 25.7, 25.9, 37.0, 54.4, 71.5, 71.9, 77.7, 120.0, 127.9, 129.8, 131.3, 135.2, 144.6; IR (film) cm−1 2954m, 2929m, 2856m, 2243w, 1598w, 1462m, 1367s, 1292w, 1250m; mass spectrum (ESI): m/e (% relative intensity) 336 (100) (M–OTBS+MeOH+H)+.
General Procedure for the Alkylation of γ-Hydroxy-Ynamides
To a flame-dried screw-cap vial was added ynamide 12a (96.0 mg, 0.30 mmol) and THF (1.0 mL, 0.3 M in ynamide). The solution was cooled to 0 °C and NaH (16.0 mg, 0.39 mmol, 60% dispersion in mineral oil) was added carefully. The reaction mixture was stirred at 0 °C for 30 min to ensure complete deprotonation of the alcohol and then MeI (24.0 µL, 0.78 mmol) was added dropwise over ~1 min. After 30 min, the cooling bath was removed and the reaction was allowed to come to rt. The reaction was monitered by TLC and when progress appeared to be complete after ~2 h, the mixture was diluted with EtOAc (3 mL) and quenched with water (3 mL). The phases were separated and the aqueous phase extracted with EtOAc (10 mL) once. The organic phases were combined, washed with brine, dried over Na2SO4, and concentrated by rotary evaporation. The crude residue was purified by flash silica gel column chromatography (isocratic eluent: 15:1 hexanes/EtOAc + 2% NEt3 [to buffer the column]) to afford ynamide 13b (86.0 mg, 0.256 mmol, 86%). Note: for displacement of alkyl bromides, DMF was used as the solvent. During workup, the organic phase was washed with brine 5 times to remove DMF.
13b
(86.0 mg, 86%); Rf= 0.16 [15:1 hexanes/EtOAc]; colorless oil; 1H NMR (500 MHz, CDCl3) δ 0.94 (s, 9H), 2.45 (s, 3H), 3.31 (s, 3H), 3.63 (s, 1H), 3.94 (ddt, 1H, J = 1.0, 6.5, 14.5 Hz), 4.01 (ddt, 1H, J = 1.0, 6.5, 14.5 Hz), 5.21 (dq, 1H, J = 1.0, 10.0 Hz), 5.24 (dq, 1H, J = 1.0, 17.0 Hz), 5.74 (ddt, 1H, J = 6.5, 10.0, 17.0 Hz), 7.33 (d, 2H, J = 8.0 Hz), 7.79 (d, 2H, J = 8.0 Hz); 13C NMR (125 MHz, CDCl3) δ 21.9, 26.1, 36.0, 54.5, 57.3, 68.9, 79.5, 81.1, 120.2, 128.0, 129.9, 131.3, 135.0, 144.9; IR (film) cm−1 2956m, 2926m, 2820w, 2240m, 1597w, 1366s; mass spectrum (APCI): m/e (% relative intensity) 336 (100) (M+H)+; HRMS (QTOF MS ESI) m/e calcd for C18H25NO3SNa [M+Na]+: 358.1447, found 358.1447.
Ynamide 13c was prepared from 12a following the general procedure for alkylation
13c
(98.0 mg, 79%); Rf= 0.17 [15:1 hexanes/EtOAc]; colorless oil; 1H NMR (500 MHz, CDCl3) δ 0.96 (s, 9H), 2.41 (s, 3H), 3.78 (s, 1H), 3.95 (ddt, 1H, J = 1.0, 6.5, 14.5 Hz), 4.01 (ddt, 1H, J = 1.0, 6.5, 14.5 Hz), 4.38 (d, 1H, J = 12.0 Hz), 4.68 (d, 1H, J = 12.0 Hz), 5.22 (dq, 1H, J = 1.0, 10.0 Hz), 5.25 (dq, 1H, J = 1.0, 17.0 Hz), 5.74 (ddt, 1H, J = 6.5, 10.0, 17.0 Hz), 7.24–7.34 (m, 5H), 7.28 (d, 2H, J = 8.0 Hz), 7.79 (d, 2H, J = 8.0 Hz); 13C NMR (125 MHz, CDCl3) δ 21.8, 26.2, 36.0, 54.5, 69.1, 70.7, 77.9, 79.6, 120.3, 127.6, 127.9, 128.0, 128.4, 129.9, 131.3, 135.0, 138.7, 144.9; IR (film) cm−1 2956m, 2867m, 2241m, 1597w, 1495w, 1365s; mass spectrum (APCI): m/e (% relative intensity) 336 (100) (M– HOBn+MeOH+H)+, 412 (20) (M+H)+; HRMS (QTOF MS ESI) m/e calcd for C24H29NO3SNa [M+Na]+: 434.1760, found 434.1759.
Ynamide 14a was prepared from 12b following the general procedure for alkylation
14a
(70.0 mg, 96%); Rf= 0.41 [4:1 hexanes/EtOAc]; colorless oil; 1H NMR (400 MHz, CDCl3) δ 0.88 (t, 3H, J = 6.8 Hz), 1.21–1.34 (m, 6H), 1.34–1.43 (m, 2H), 1.58–1.74 (m, 2H), 2.45 (s, 3H), 3.30 (s, 3H), 3.93 (ddt, 1H, J = 1.2, 6.4, 14.4 Hz), 4.01 (ddt, 1H, J = 1.2, 6.4, 14.4 Hz), 4.04 (t, 1H, J = 6.4 Hz), 5.20 (dq, 1H, J = 1.2, 10.0 Hz), 5.24 (dq, 1H, J = 1.2, 16.4 Hz), 5.73 (ddt, 1H, J = 6.4, 10.0, 16.4 Hz), 7.34 (d, 2H, J = 8.4 Hz), 7.79 (d, 2H, J = 8.4 Hz); 13C NMR (100 MHz, CDCl3) δ 14.3, 21.9, 22.8, 25.5, 29.2, 32.0, 36.0, 54.4, 56.3, 70.1, 71.7, 79.1, 120.2, 128.0, 129.9, 131.1, 134.9, 144.9; IR (film) cm−1 2926m, 2858m, 2240m, 1465w, 1367s; mass spectrum (APCI): m/e (% relative intensity) 364 (100) (M+H)+.
Ynamide 14b was prepared from 12b following the general procedure for silylation
14b
(101.0 mg, ≥95%); Rf = 0.56 [4:1 hexanes/EtOAc]; colorless oil; 1H NMR (500 MHz, CDCl3) δ 0.89 (t, 3H, J = 7.0 Hz), 1.03 (d, 18H, J = 5.0 Hz), 1.05–1.07 (m, 3H), 1.24–1.34 (m, 7H), 1.34–1.47 (m, 3H), 1.66 (td, 2H, J = 6.0, 8.0 Hz), 2.44 (s, 3H), 3.94 (ddt, 1H, J = 1.5, 5.5, 14.0 Hz), 3.98 (ddt, 1H, J = 1.5, 5.5, 14.0 Hz), 4.56 (t, 1H, J = 6.0 Hz), 5.18 (dq, 1H, J = 1.0, 10.0 Hz), 5.22 (dq, 1H, J = 1.0, 17.0 Hz), 5.73 (ddt, 1H, J = 6.5, 10.0, 17.0 Hz), 7.31 (d, 2H, J = 8.0 Hz), 7.77 (d, 2H, J = 8.0 Hz); 13C NMR (125 MHz, CDCl3) δ 12.5, 14.4, 18.0, 18.3, 18.3, 21.9, 22.9, 25.2, 29.3, 32.1, 39.4, 54.5, 63.5, 73.0, 119.9, 128.0, 129.9, 131.4, 135.2, 144.7; IR (film) cm−1 2941s, 2864s, 2242m, 1597w, 1463m, 1369s; mass spectrum (APCI): m/e (% relative intensity) 364 (100) (M–HOTIPS+MeOH+H)+.
Ynamide 15a was prepared from 12c following the general procedure for alkylation
15a
(60.0 mg, 79%); Rf = 0.11 [15:1 hexanes/EtOAc]; colorless oil; 1H NMR (500 MHz, CDCl3) δ 1.03–1.26 (m, 5H), 1.58 (tdt, 1H, J = 3.0, 6.0, 12.0 Hz), 1.65 (d, 1H, J = 12.0 Hz, 1H), 1.74 (t, 4H, J = 14.0 Hz), 2.45 (s, 3H), 3.29 (s, 3H), 3.83 (d, 1H, J = 6.0 Hz), 3.94 (dd, 1H, J = 6.5, 14.5 Hz), 4.02 (dd, J = 6.5, 14.5 Hz), 5.22 (dq, 1H, J = 1.0, 10.0 Hz), 5.25 (dq, 1H, J = 1.0, 17.0 Hz), 5.74 (ddt, 1H, J = 6.5, 10.0, 17.0 Hz), 7.33 (d, 2H, J = 8.0 Hz), 7.79 (d, 2H, J = 8.0 Hz); 13C NMR (125 MHz, CDCl3) δ 21.9, 26.2, 26.2, 26.7, 28.6, 29.2, 43.0, 54.5, 56.6, 69.1, 76.7, 79.7, 120.2, 128.0, 129.9, 131.2, 134.9, 144.9; IR (film) cm−1 2924m, 2853m, 2239m, 1449m, 1367s; mass spectrum (APCI): m/e (% relative intensity) 362 (M+H)+.
Ynamide 15b was prepared from 12c following the general procedure for alkylation
15b
(72.0 mg, 63%); Rf= 0.49 [3:1 hexanes/EtOAc]; white solid; mp = 62–64° C; 1H NMR (400 MHz, CDCl3) δ 0.80–1.28 (m, 5H), 1.56–1.84 (m, 6H), 2.42 (s, 3H), 3.95 (ddt, 1H, J = 1.2, 6.4, 14.4 Hz), 3.96 (d, 1H, J = 6.2 Hz), 4.03 (ddt, 1H, J = 1.2, 6.4, 14.4 Hz), 4.53 (ABq, 2H, ΔvAB = 33.6 Hz, JAB = 11.8 Hz), 5.23 (dq, 1H, J = 1.2, 10.0 Hz), 5.26 (dq, 1H, J = 1.2, 17.2 Hz), 5.76 (ddt, 1H, J = 6.4, 10.0, 17.2 Hz), 7.24–7.35 (m, 7H), 7.80 (d, 2H, J = 8.8 Hz); 13C NMR (100 MHz, CDCl3) δ 21.8, 26.1, 26.1, 26.6, 28.7, 29.2, 43.0, 54.4, 69.3, 70.3, 73.9, 79.7, 120.2, 127.6, 127.9, 128.0, 128.4, 129.8, 131.2, 134.8, 138.4, 144.8; IR (film) cm−1 2925m, 2852m, 2243m, 1597w, 1451m, 1363s; mass spectrum (APCI): m/e (% relative intensity) 362 (100) (M–BnOH+MeOH+H)+.
Ynamide 16a was prepared from 12d following the general procedure for silylation
16a
(81.0 mg, 82%); Rf= 0.57 [3:1 hexanes/EtOAc]; white solid; mp = 61–62° C; 1H NMR (400 MHz, CDCl3) δ 0.01 (s, 3H), 0.03 (s, 3H), 0.85 (s, 9H), 0.90–1.30 (m, 6H), 1.45 (dtt, 1H, J = 2.8, 6.2, 18.0 Hz), 1.61–1.79 (m, 4H), 4.00 (ddt, 1H, J = 1.2, 6.2, 14.8 Hz), 4.04 (ddt, 1H, J = 1.2, 6.2, 14.8 Hz), 4.18 (d, 1H, J = 6.0 Hz), 5.22 (dq, 1H, J = 1.2, 10.4 Hz), 5.25 (dq, 1H, J = 1.2, 16.8 Hz), 5.70 (ddt, 1H, J = 6.4, 10.0, 16.8 Hz), 8.08 (d, 2H, J = 9.2 Hz), 8.37 (d, 2H, J = 9.2 Hz); 13C NMR (100 MHz, CDCl3) δ −5.0, −4.4, 18.3, 25.8, 26.1, 26.1, 26.6, 28.7, 28.7, 45.3, 54.8, 67.8, 72.8, 76.5, 120.8, 124.4, 129.2, 130.5, 143.5, 150.7; IR (film) cm−1 2929m, 2855m, 2248m, 1535s, 1376m, 1349m; mass spectrum (APCI): m/e (% relative intensity) 393 (100) (M–HOTBS+MeOH+H)+; HRMS (QTOF MS ESI) m/e calcd for C24H-36N2O5SSiNa [M+Na]+: 515.2006, found 515.1992.
Ynamide 16b was prepared from 12d following the general procedure for silylation, but with Ph3C-Cl (1.3 equiv), NEt3 (2 equiv), and DMAP (0.1 equiv) in CH2Cl2
16b
(89.0 mg, 55%); Rf = 0.51 [3:1 hexanes/EtOAc]; yellow foam; 1H NMR (500 MHz, CDCl3) δ 1.09–1.19 (m, 6H), 1.43 (d, 1H, J = 10.0 Hz), 1.63 (t, 2H, J = 8.0 Hz), 1.72 (d, 1H, J = 6.5 Hz), 1.86 (d, 1H, J = 10.0 Hz), 3.70 (dd, 1H, J = 6.5, 15.0 Hz), 3.79 (d, 1H, J = 3.5 Hz), 3.88 (dd, 1H, J = 6.5, 15.0 Hz), 5.16 (d, 1H, J = 16.5 Hz), 5.18 (d, 1H, J = 10.0 Hz), 5.53 (ddt, 1H, J = 6.5, 10.0, 16.5 Hz), 7.11– 7.31 (m, 9H), 7.47 (d, 6H, J = 8.0 Hz), 8.00 (d, 2H, J = 9.0 Hz), 8.34 (d, 2H, J = 9.0 Hz); 13C NMR (125 MHz, CDCl3) δ 26.3, 26.3, 26.8, 28.0, 29.3, 43.6, 54.6, 69.3, 71.5, 78.3, 87.5, 120.3, 124.5, 126.9, 127.2, 127.5, 127.8, 128.2, 128.2, 128.5, 128.6, 129.2, 129.2, 129.7, 130.8, 143.7, 144.9, 147.1, 150.7; IR (film) cm−1 3059w, 3031w, 2930m, 2853m, 2246m, 1717m, 1605m, 1531s, 1491m, 1447m, 1348s; mass spectrum (APCI): m/e (% relative intensity) 293 (100) (p-Ns-allylamine+H)+, 393 (10) (M– HOCPh3+MeOH+H)+.
Ynamide 17 was prepared from 12e following the general procedure for silylation
17
(88.4 mg, 90%); Rf = 0.36 [8:1 hexanes/EtOAc]; colorless oil; 1H NMR (500 MHz, CDCl3) δ 0.04 (s, 3H), 0.05 (s, 3H), 0.87 (s, 9H), 1.22–1.41 (m, 10H), 1.57–1.69 (m, 3H), 4.01 (ddt, 1H, J = 1.0, 6.5, 14.5 Hz), 4.05 (ddt, 1H, J = 1.0, 6.5, 14.5 Hz), 4.44 (t, 1H, J = 6.5 Hz), 5.23 (dq, 1H, 1.0, 10.0 Hz), 5.26 (dq, 1H, J = 1.0, 17.0 Hz), 5.71 (ddt, 1H, J = 6.5, 10.0, 17.0 Hz), 8.09 (d, 2H, J = 9.0 Hz), 8.38 (d, 1H, J = 9.0 Hz); 13C NMR (125 MHz, CDCl3) δ −4.8, −4.3, 14.3, 18.4, 22.8, 25.5, 26.0, 29.2, 32.1, 39.0, 54.9, 63.3, 73.8, 76.1, 120.9, 124.5, 129.2, 130.5, 143.5, 150.8; IR (film) cm−1 2954m, 2928m, 2857m, 2245m, 1606w, 1534s, 1377m, 1348s; mass spectrum (APCI): m/e (% relative intensity) 395 (100) (M–HOTBS+MeOH+H)+; HRMS (QTOF MS ESI) m/e calcd for C24H38N2O5SSiNa [M+Na]+: 517.2163, found 517.2158.
Ynamide 18 was prepared from 2f following the general procedure for alkylation
18
(596.6 mg, ≥95%); Rf = 0.38 [6:1 hexanes/EtOAc]; white solid; mp = 50–51 °C; 1H NMR (400 MHz, CDCl3) δ 1.23–1.28 (m, 1H), 1.39–1.54 (m, 5H), 1.59–1.63 (m, 2H), 1.84–1.88 (m, 2H), 2.45 (s, 3H), 3.26 (s, 3H), 3.97 (dt, 2H, J = 6.4, 1.2 Hz), 5.19–5.26 (m, 2H), 5.68–5.77 (m, 1H), 7.34 (d, 2H, J = 8.0 Hz), 7.79 (d, 2H, J = 8.4 Hz); 13C NMR (100 MHz, CDCl3) δ 21.6, 22.8, 25.4, 36.8, 50.5, 54.1, 71.4, 74.3, 79.3, 119.9, 127.8, 129.6, 130.9, 134.5, 144.6; IR (film) cm−1 2934s, 2857w, 2360w, 2341w, 2240m, 1447m, 1366s; mass spectrum (APCI): m/e (% relative intensity) 348 (43) (M+H)+; HRMS (QTOF MS ESI) calcd for C19H25NO3SNa [M+Na]+: 370.1447, found 370.1450.
General Procedure for the Carbocyclization of γ-Branched-N-Allyl Ynamides
To a flame-dried screw-cap vial containing 4 Å M.S. was added ynamide 6 (50.0 mg, 0.100 mmol), Pd(PPh3)4 (5.8 mg, 0.005 mmol), and toluene (2.5 mL, 0.04 M in ynamide). The vial was sealed under nitrogen and heated to 70 °C for 1 h, at which time TLC analysis showed consumption of the starting material. The reaction mixture was cooled to rt, filtered through Celite™ and purified by flash silica gel column chromatography (isocratic eluent: 8:1 hexanes/EtOAc) to afford cyclopentenimine 8 (34.5 mg, 0.069 mmol) in 69% yield.
8
(34.5 mg, 69%); Rf = 0.11 [8:1 hexanes/EtOAc]; colorless oil; 1H NMR (500 MHz, CDCl3) δ 0.91 (d, 9H, J = 7.0 Hz), 0.94 (d, 9H, J = 7.0 Hz), 1.01 (sept, 3H, J = 7.0 Hz), 2.44 (s, 3H), 2.63 (ddt, 1H, J = 2.5, 6.0, 20.5 Hz), 2.73 (ddt, 1H, J = 2.5, 6.0, 20.5 Hz), 3.06 (ddd, 1H, J = 1.5, 6.0, 20.5 Hz), 3.20 (ddd, 1H, J = 1.5, 6.0, 20.5 Hz), 5.57 (s, 1H), 7.15 (dd, 2H, J = 2.0, 5.0 Hz), 7.29 (d, 2H, J = 8.0 Hz), 7.30–7.32 (m, 3H), 7.57 (td, 1H, J = 1.5, 2.5 Hz), 7.75 (d, 2H, J = 8.0 Hz); 13C NMR (125 MHz, CDCl3) δ 12.3, 18.1, 18.2, 21.8, 30.9, 33.2, 70.0, 126.7, 127.2, 127.4, 128.0, 129.5, 138.5, 143.1, 143.5, 151.4, 157.2, 189.4; IR (film) cm−1 2942m, 2865m, 1591s, 1461m, 1317s; mass spectrum (APCI): m/e (% relative intensity) 498 (100) (M+H)+.
19a
(54.1 mg, 71%); Rf= 0.26 [8:1 hexanes:EtOAc]; yellow oil; 1H NMR (400 MHz, CDCl3) δ −0.24 (s, 3H), −0.04 (s, 3H), 0.78 (s, 9H), 0.84 (s, 9H), 2.43 (s, 3H), 2.70–2.73 (m, 2H), 3.11 (ddd, 1H, J = 2.4, 5.6, 20.4 Hz), 3.29 (ddd, 1H, J = 2.4, 5.6, 20.4 Hz), 4.27 (s, 1H), 7.31 (d, 2H, J = 8.0 Hz), 7.38 (t, 1H, J = 2.4 Hz), 7.84 (d, 2H, J = 8.0 Hz); 13C NMR (100 MHz, CDCl3) δ −5.2, −4.5, 18.1, 21.7, 25.7, 25.9, 30.9, 32.4, 36.3, 72.9, 127.1, 129.4, 138.4, 143.4, 149.5, 160.4, 190.9; IR (film) cm−1 2954m, 2928m, 2857w, 1587s, 1471m, 1438m, 1389m, 1304m; mass spectrum (ESI): m/e (% relative intensity) 436 (100) (M+H)+; HRMS (QTOF MS ESI) calcd for C23H37NO3SSiNa [M+Na]+: 458.2156, found 458.2165.
19b
(49.4 mg, 77%); Rf = 0.25 [4:1 hexanes/EtOAc]; white solid, mp = 113–114 °C; 1H NMR (500 MHz, CDCl3) δ 0.79 (s, 9H), 2.43 (s, 3H), 2.78 (dq, 2H, J = 3.0, 5.0 Hz), 3.12 (dddd, 1H, J = 1.0, 3.0, 5.0, 21.0 Hz), 3.14 (s, 3H), 3.35 (dddd, 1H, J = 1.0, 3.0, 5.0, 21.0 Hz), 3.82 (s, 1H), 7.31 (d, 2H, J = 8.5 Hz), 7.35 (t, 1H, J = 3.0 Hz, 1H), 7.84 (d, 2H, J = 8.5 Hz); 13C NMR (125 MHz, CDCl3) δ 21.8, 25.9, 31.1, 32.7, 35.8, 57.8, 82.6, 127.2, 129.6, 138.5, 143.6, 146.3, 160.1, 192.2; IR (film) cm−1 2955m, 2870m, 1586s, 1315s; mass spectrum (APCI): m/e (% relative intensity) 336 (100) (M+H)+; HRMS (QTOF MS ESI) calcd for C18H26NO3S [M+H]+: 336.1628, found 336.1637.
19c
(65.9 mg, 84%); Rf = 0.31 [4:1 hexanes/EtOAc]; pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 0.83 (s, 9H), 2.44 (s, 3H), 2.67–2.78 (m, 2H), 3.11 (ddd, 1H, J = 2.8, 5.6, 20.4 Hz), 3.31 (ddd, 1H, J = 2.8, 5.6, 20.4 Hz), 4.05 (s, 1H), 7.22–7.29 (m, 5H), 4.29 (ABq, 2H, ΔvAB = 18.5 Hz, JAB = 11.8 Hz), 7.31 (d, 2H, J = 8.0 Hz), 7.38 (td, 1H, J = 0.8, 2.8 Hz), 7.84 (d, 2H, J = 8.0 Hz); 13C NMR (125 MHz, CDCl3) δ 21.8, 26.0, 31.1, 32.6, 35.9, 71.8, 80.2, 127.2, 127.6, 127.8, 128.4, 129.6, 138.5, 138.9, 143.6, 146.5, 160.5, 191.8; IR (film) cm−1 2955m, 2867m, 1584s, 1364m, 1314s; mass spectrum (APCI): m/e (% relative intensity) 412 (100) (M+H)+; HRMS (QTOF MS ESI) calcd for C24H29NO3SNa [M+Na]+: 434.1760, found 434.1779.
20a
(48.3 mg, 70%); Rf = 0.17 [4:1 hexanes/EtOAc]; pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 0.85 (t, 3H, J = 7.2 Hz), 1.18–1.26 (m, 8H), 1.43–1.63 (m, 2H), 2.43 (s, 3H), 2.72–2.76 (m, 2H), 3.20 (ddd, 1H, J = 2.8, 5.2, 20.4 Hz), 3.25 (s, 3H), 3.27 (ddd, 1H, J = 2.8, 5.2, 20.4 Hz), 4.03 (dddt, 1H, J = 1.8, 2.8, 4.4, 5.8 Hz), 7.31 (d, 2H, J = 8.0 Hz), 7.33 (td, 1H, J = 1.2, 2.8 Hz), 7.86 (d, 2H, J = 8.0 Hz); 13C NMR (100 MHz, CDCl3) δ 14.3, 21.8, 22.8, 25.2, 29.3, 31.0, 31.9, 33.1, 34.8, 57.6, 76.2, 127.2, 129.6, 138.5, 143.6, 148.1, 157.6, 191.0; IR (film) cm−1 2924m, 2864m, 1587s, 1464m, 1381m, 1316s; mass spectrum (APCI): m/e (% relative intensity) 364 (100) (M+H)+; HRMS (QTOF MS ESI) calcd for C20H29NO3SNa [M+Na]+: 386.1760, found 386.1758.
20b
(92.0 mg, 92%); Rf = 0.43 [4:1 hexanes/EtOAc]; colorless oil; 1H NMR (500 MHz, CDCl3) δ 0.97 (d, 9H, J = 5.5 Hz), 0.99 (d, 9H, J = 4.5 Hz), 1.05 (s, 3H), 1.53–1.67 (m, 2H), 2.43 (s, 3H), 2.66–2.74 (m, 2H), 3.17 (dt, 1H, J = 4.0, 20.5 Hz), 3.23 (dt, 1H, J = 4.0, 20.5 Hz), 4.70 (td, 1H, J = 1.0, 5.0 Hz), 7.30 (d, 2H, J = 8.5 Hz), 7.42 (td, 1H, J = 1.0, 2.5 Hz), 7.83 (d, 2H, J = 8.5 Hz); 13C NMR (125 MHz, CDCl3) δ 12.5, 14.3, 17.9, 18.3, 18.3, 21.8, 22.9, 23.8, 29.6, 30.8, 32.0, 33.2, 36.9, 67.5, 127.2, 129.6, 138.6, 143.5, 151.0, 158.6, 190.2; IR (film) cm−1 2925m, 2865m, 1587s, 1463m, 1382m, 1317m; mass spectrum (APCI): m/e (% relative intensity) 506 (100) (M+H)+; HRMS (QTOF MS ESI) calcd for C28H47NO3SSiNa [M+Na]+: 528.2938, found 528.2949.
21a
(52.1 mg, 88%); Rf = 0.24 [3:1 hexanes/EtOAc]; pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 0.87–0.97 (m, 1H), 1.00–1.18 (m, 4H), 1.43 (d, 1H, J = 12.0 Hz), 1.50 (tdt, J = 3.2, 5.6, 11.2 Hz), 1.55– 1.73 (m, 4H), 2.44 (s, 3H), 2.75–2.78 (m, 2H), 3.17 (ddd, 1H, J = 3.2, 4.8, 20.4 Hz), 3.19 (s, 3H), 3.29 (ddd, 1H, J = 3.2, 4.8, 20.4 Hz), 3.88 (dd, 1H, J = 0.8, 5.6 Hz), 7.32 (d, 2H, J = 8.0 Hz), 7.85 (d, 2H, J =8.0 Hz); 13C NMR (100 MHz, CDCl3) δ 21.8, 26.3, 26.4, 26.6, 28.1, 29.2, 31.1, 33.0, 42.6, 57.9, 80.2, 127.2, 129.6, 138.5, 143.6, 146.8, 158.7, 191.5; IR (film) cm−1 2925m, 2851m, 1586s, 1449m, 1314s; mass spectrum (APCI): m/e (% relative intensity) 362 (100) (M+H)+; HRMS (QTOF MS ESI) calcd for C20H27NO3SNa [M+Na]+: 384.1604, found 384.1615.
21b
(88.5 mg, >95%); Rf = 0.31 [3:1 hexanes/EtOAc]; pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 0.80–1.18 (m, 5H), 1.41 (d, 1H, J = 10.0 Hz), 1.49–1.72 (m, 4H), 1.74 (d, 1H, J = 12.8 Hz), 2.44 (s, 3H), 2.70–2.76 (m, 2H), 3.17 (ddd, 1H, J = 3.6, 4.4, 20.4 Hz), 3.27 (ddd, 1H, J = 3.6, 4.4, 20.4 Hz), 4.09 (d, 1H, J = 5.2 Hz), 4.32 (ABq, 2H, ΔvAB = 20.2 Hz, JAB = 11.7 Hz), 7.22–7.29 (m, 5H), 7.31 (d, 2H, J = 8.4 Hz), 7.36 (t, 1H, J = 2.7 Hz), 7.85 (d, 2H, J = 8.4 Hz); 13C NMR (100 MHz, CDCl3) δ 21.8, 26.2, 26.4, 26.6, 28.3, 29.4, 31.1, 32.9, 42.8, 71.9, 77.9, 127.2, 127.7, 127.9, 128.5, 129.6, 138.5, 138.7, 143.6, 147.3, 159.2, 191.3; IR (film) cm−1 2925m, 2852m, 1585s, 1451m, 1314s, 1302s; mass spectrum (APCI): m/e (% relative intensity) 438 (100) (M+H)+; HRMS (QTOF MS ESI) calcd for C26H31NO3SNa [M+Na]+: 460.1917, found 460.1919.
22a
(39.6 mg, 80%); Rf= 0.21 [8:1 hexanes/EtOAc]; white solid; mp = 137–140° C [decomp]; 1H NMR (400 MHz, CDCl3) δ−0.15 (s, 3H), −0.02 (s, 3H), 0.86 (s, 9H), 0.98–1.15 (m, 5H), 1.35–1.45 (m, 2H), 1.48 (d, 1H, J = 13.2 Hz), 1.54–1.73 (m, 3H), 2.72–2.82 (m, 2H), 3.23 (ddd, 1H, J = 3.2, 4.4, 20.4 Hz), 3.30 (ddd, 1H, J = 3.2, 4.4, 20.4 Hz), 4.27 (d, 1H, J = 2.9 Hz), 7.44 (td, 1H, J = 1.2, 2.8 Hz), 8.16 (d, 2H, J = 8.8 Hz), 8.38 (d, 2H, J = 8.8 Hz); 13C NMR (100 MHz, CDCl3) δ −4.9, −4.4, 18.3, 25.9, 26.3, 26.5, 26.6, 26.9, 29.9, 31.0, 33.7, 43.4, 71.2, 124.2, 126.5, 128.3, 133.6, 150.0, 161.1, 192.0; IR (film) cm-1 2931m, 2855m, 1557s, 1384s, 1348s; mass spectrum (APCI): m/e (% relative intensity) 493 (100) (M+H)+; HRMS (QTOF MS ESI) calcd for C24H36N2O5SSiNa [M+Na]+: 515.2006, found 515.2013.
22b
(73.4 mg, 83%); Rf = 0.26 [5:1 hexanes/EtOAc]; white solid; mp = 93–95 °C; 1H NMR (400 MHz, CDCl3) δ 0.43 (qd, 1H, J = 3.6, 12.4 Hz), 0.97 (qt, 1H, J = 3.2, 12.8 Hz), 1.12 (qt, 1H, J = 3.2, 12.8 Hz), 1.20–1.34 (m, 2H), 1.37 (d, 1H, J = 12.4 Hz), 1.54 (d, 1H, J = 12.4 Hz), 1.62 (d, 1H, J = 12.4 Hz) 1.78 (d, 1H, J = 12.8 Hz), 1.84 (ddt, 1H, J = 3.6, 12.0, 19.6 Hz), 2.01–2.13 (m, 2H), 2.43 (dd, 1H, J = 6.0, 19.6 Hz), 2.69 (dd, 1H, J = 6.0, 20.4 Hz), 2.87 (dd, 1H, J = 6.0, 20.4 Hz), 4.38 (d, 1H, J = 4.0 Hz), 7.03 (t, 1H, J = 3.6 Hz), 7.17–7.27 (m, 11H), 7.35–7.40 (m, 4H), 8.02 (d, 2H, J = 8.8 Hz), 8.30 (d, 2H, J = 8.8 Hz); 13C NMR (100 MHz, CDCl3) δ 26.3, 26.6, 26.7, 26.8, 29.6, 31.1, 32.9, 44.3, 71.1, 87.3, 124.2, 127.3, 128.1, 128.3, 129.1, 144.6, 146.4, 147.3, 150.1, 162.9, 192.22; IR (film) cm−1 3058w, 3032w, 2929m, 2853m, 1736m, 1576s, 1530s, 1448m, 1349s; mass spectrum (APCI): m/e (% relative intensity) 605 (100) (M+H)+; HRMS (QTOF MS ESI) calcd for C37H36N2O5SNa [M+Na]+: 643.2237, found 643.2262.
23
(27.8 mg, 56%); Rf = 0.23 [6:1 hexanes/EtOAc]; pale yellow oil; 1H NMR (400 MHz, CDCl3) δ −0.08 (s, 3H), 0.00, (s, 3H), 0.83 (t, 3H, J = 7.2 Hz), 0.87 (s, 9H), 1.12–1.29 (m, 8H), 1.41–1.61 (m, 2H), 2.55–2.92 (m, 3H), 3.21–3.35 (m, 2H), 4.44 (tdt, 1H, J = 1.6, 3.2, 5.2 Hz), 7.48 (td, 1H, J = 1.2, 3.2 Hz), 8.16 (d, 2H, J = 8.8 Hz), 8.37 (d, 2H, J = 8.8 Hz); 13C NMR (100 MHz, CDCl3) δ −4.7, −4.5, 14.2, 18.3, 22.8, 24.8, 26.0, 29.3, 31.0, 31.9, 33.9, 36.9, 67.7, 124.3, 128.5, 147.1, 150.3, 151.0, 160.2, 191.8; IR (film) cm−1 2954m, 2928m, 2857m, 1580s, 1531s, 1348s; mass spectrum (APCI): m/e (% relative intensity) 495 (100) (M+H)+; HRMS (QTOF MS ESI) calcd for C24H38N2O5SSiNa [M+Na]+: 517.2173, found 517.2190.
24
(23.8 mg, 34%); Rf = 0.17 [6:1 hexane/EtOAc]; pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 1.42–1.73 (m, 10H), 1.96 (td, 2H, J = 13.6, 4.8 Hz), 2.44 (s, 3H), 2.67–2.70 (m, 2H), 3.14 (s, 3H), 3.25–3.27 (m, 2H), 7.27–7.33 (m, 3H), 7.88 (d, 2H, J = 8.4 Hz); 13C NMR (100 MHz, CDCl3) δ 21.3, 21.5, 25.3, 29.9, 32.1, 33.5, 49.9, 76.5, 126.8, 129.4, 138.7, 143.2, 149.3, 159.2, 190.2; IR (film) cm−1 2931s, 2856m, 1585s, 1448m, 1302s, 1153s; mass spectrum (APCI): m/e (% relative intensity) 348 (64) (M+H)+; HRMS (QTOF MS ESI) calcd for C19H25NO3SNa [M+Na]+: 370.1447, found 370.1452.
Preparations of Ynamides 30b, 30c, and 44
Method A
To a flame-dried screw-cap vial was added (in the following order) the appropriate phosphoramidate (309.0 mg, 1.50 mmol), CuTC (58.0 mg, 0.30 mmol), Cs2CO3 (976.0 mg, 3.00 mmol), dioxane (3.9 mL, 0.4 M in amide), (±)-N,N’-dimethyl-1,2-cyclohexanediamine (94.0 µL, 0.60 mmol), and the alkynyl bromide (369.0 mg, 1.95 mmol) under a nitrogen atmosphere. The vial was evacuated under vacuum and flushed with nitrogen three times, then sealed under nitrogen and heated to 60 °C. When the reaction was judged to be complete by TLC after 48 hours, the mixture was cooled to rt, filtered through Celite™, and purified by flash silica gel column chromatrography (isocratic eluent: 2:1 hexanes/EtOAc + 2% NEt3) to afford ynamide 30b (373.0 mg, 1.19 mmol, 79%).
30b
(373.0 mg, 79%); Rf = 0.36 [1:1 hexanes/EtOAc]; pale yellow solid; mp = 47–49 °C; 1H NMR (400 MHz, CDCl3) δ 0.89 (t, 3H, J = 6.8 Hz), 1.05 (s, 3H), 1.16 (s, 3H), 1.24–1.31 (m, 5H), 1.37 (pent, 2H, J = 7.2 Hz), 1.47 (pent, 2H, J = 7.2 Hz), 2.22 (td, 2H, J = 3.2, 7.2 Hz), 3.87 (ddt, 2H, J = 1.6, 6.2, 9.2 Hz), 4.08 (dd, 2H, J = 11.2, 14.4 Hz), 4.21 (dd, 2H, J = 9.6, 11.2 Hz), 5.24 (dd, 1H, J = 1.6, 10.4 Hz), 5.31 (dq, 1H, J = 1.6, 17.2 Hz), 5.90 (ddt, 1H, J = 6.2, 10.4, 17.2 Hz); 13C NMR (100 MHz, CDCl3) δ 14.2, 18.5, 21.4 (d, J = 11.3 Hz), 22.7, 28.6, 29.3, 31.5, 32.3 (d, J = 6.4 Hz), 53.4 (d, J = 6.3 Hz), 60.5, 65.2, 75.3, 77.9 (d, J = 6.4 Hz), 118.6, 133.0 (d, J = 1.5 Hz); 31P NMR (162 MHz, CDCl3) δ 0.63; IR (film) cm−1 2929m, 2895m, 2855m, 2256m, 1469m, 1367m, 1255s; mass spectrum (APCI): m/e (% relative intensity) 314 (20) (M+H)+, 346 (100) (M+MeOH+H)+.
30a
(231.7 mg, 71%); Rf= 0.23 [1:1 hexanes:EtOAc]; pale yellow solid; mp = 108–109 °C; 1H NMR (400 MHz, CDCl3) δ 1.09 (s, 3H), 1.19 (s, 3H), 4.02 (ddt, 2H, J = 1.6, 6.2, 9.2 Hz), 4.14–4.22 (m, 4H), 5.30 (d, 1H, J = 10.4 Hz), 5.39 (dd, 1H, J = 1.6, 17.2 Hz), 5.97 (ddt, 1H, J = 6.2, 10.4, 17.2 Hz), 7.23–7.30 (m, 3H), 7.31–7.34 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 21.5, 21.5, 32.5, 53.9 (d, J = 6.1 Hz), 66.3 (d, J = 5.7 Hz), 78.3 (d, J = 6.3 Hz), 85.3 (d, J = 4.1 Hz), 119.2, 123.7 (d, J = 2.1 Hz), 127.5, 128.5, 131.2 (d, J = 1.5 Hz), 132.8 (d, J = 1.4 Hz); 31P NMR (162 MHz, CDCl3) δ 1.01; IR (film) cm−1 3081w, 2970w, 2896w, 2244s, 1370m, 1326m, 1254s; mass spectrum (ESI): m/e (% relative intensity) 306 (100) (M+H)+;
30e
(86.3 mg, 57%); Rf= 0.17 [2:1 hexanes:EtOAc]; colorless oil; 1H NMR (400 MHz, CDCl3) δ 1.05 (s, 18H), 1.06 (s, 3H), 1.08 (s, 3H), 1.14 (s, 3H), 3.93 (dd, 2H, J = 6.8, 8.8 Hz), 4.11–4.25 (m, 4H), 5.27 (d, 1H, J = 10.0 Hz), 5.35 (d, 1H, J = 16.8 Hz), 5.96 (ddt, 1H, J = 6.8, 10.0, 16.8 Hz); 13C NMR (100 MHz, CDCl3) δ 11.5, 18.7, 21.2, 21.4, 32.3 (d, J = 6.8 Hz), 53.8 (d, J = 6.1 Hz), 60.4, 63.4 (d, J = 4.6 Hz), 78.3 (d, J = 6.9 Hz), 99.5 (d, J = 3.8 Hz), 119.1, 132.3 (d, J = 1.6 Hz); 31P NMR (162 MHz, CDCl3) δ −2.20; IR (film) cm−1 2942m, 2865m, 2163m, 1740w, 1464m, 1373w, 1313m, 1277s; mass spectrum (APCI): m/e (% relative intensity) 386 (100) (M+H)+.
Method B
To a flame-dried screw-cap vial was added (in the following order) the appropriate phosphoramidate (554.0 mg, 2.70 mmol), CuSO4·5H2O (103.0 mg, 0.41 mmol), 1,10-phenanthroline (145.8 mg, 0.81 mmol), K3PO4 (1.14 g, 5.40 mmol), toluene (6.8 mL, 0.4 M in amide), and the alkynyl bromide (1.02 g, 3.51 mmol) under a nitrogen atmosphere. The vial was evacuated under vacuum and flushed with nitrogen three times, then sealed under nitrogen and heated to 60 °C. When the reaction was judged to be complete by TLC after 48 hours, the mixture was cooled to rt, filtered through Celite™, and purified by flash silica gel column chromatrography (isocratic eluent: 2:1 hexanes/EtOAc + 2% NEt3) to afford ynamide 30c (253.1 mg, 0.61 mmol, 23%).
30c
(253.1 mg, 23%); Rf = 0.51 [3:1 hexanes/EtOAc]; brown solid; mp = 42–43 °C; 1H NMR (400 MHz, CDCl3) δ 0.08 (s, 6H), 0.77 (s, 9H), 0.94 (s, 3H), 1.04 (s, 3H), 1.40–1.50 (m, 4H), 2.12–2.15 (m, 2H), 3.49 (t, 2H, J = 6.0 Hz), 3.74 (t, 2H, J = 7.6 Hz), 3.97 (dd, 2H, J = 12.4, 14.0 Hz), 4.07 (dd, 2H, J = 10.4 Hz), 5.11 (d, 1H, J = 10.4 Hz), 5.19 (dd, 1H, J = 1.6, 17.2 Hz), 5.72–5.82 (m, 1H); 13C NMR (100 MHz, CDCl3) δ −5.6, 17.9, 18.0, 20.9, 21.0, 25.4, 25.7, 31.7, 31.8 (d, J = 6.1 Hz), 53.0 (d, J = 6.9 Hz), 62.3, 64.5 (d, J = 5.4 Hz), 75.1 (d, J = 4.5 Hz), 77,5 (d, J = 6.9 Hz), 118.1, 132.5 (d, J = 1.5 Hz); 31P NMR (162 MHz, CDCl3) δ 0.24; IR (film) cm−1 2929m, 2858m, 2256w, 1472m, 1309w, 1255s; mass spectrum (APCI): m/e (% relative intensity) 416.3 (43) (M+H)+; HRMS (QTOF MS ESI) calcd for C20H38NO4PSiNa [M+Na]+: 438.2200, found 438.2182.
Ynamide 44 was prepared via following Method B
44
(1.20 g, 28%); Rf = 0.51 [1:1 hexanes/EtOAc]; pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 1.08 (s, 3H), 1.16 (s, 3H), 2.40 (q, 2H, J = 7.2 Hz), 2.70 (dt, 2H, J = 8.4, 7.2 Hz), 3.93–3.97 (m, 2H), 4.18 (ABX, 4H, ΔvAB = 25.8 Hz, JAB = 10.8 Hz, JAX = 10.8 Hz, JBX = 13.2 Hz), 5.61–5.69 (m, 1H), 5.84 (dt, 2H, J = 15.2, 6.8 Hz), 7.14–7.18 (m, 4H), 7.23–7.34 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 20.77, 20.80, 31.7 (d, J = 6.1 Hz), 33.7, 35.1, 52.6 (d, J = 6.1 Hz), 65.8 (d, J = 6.1 Hz), 77.7 (d, J = 6.1 Hz), 85.0 (d, J = 4.6 Hz), 124.4 (d, J = 1.6 Hz), 125.5, 126.9, 127.9, 128.0, 128.06, 128.10, 130.5 (d, J = 1.5 Hz), 134.9, 141.2; 31P NMR (162 MHz, CDCl3) δ −1.02; IR (film) cm−1 2966w, 2933m, 2239s, 1371m, 1274s; mass spectrum (APCI): m/e (% relative intensity) 410 (21) (M+H)+. HRMS (QTOF MS ESI) calcd for C24H28NO3PNa [M+Na]+: 432.1699, found 432.1705.
Preparation of Ynamide 30d
To a flame-dried 25-mL RB-flask was added ynamide 30e (493.0 mg, 1.30 mmol) and THF (8.5 mL, 0.15 M in ynamide). The flask was cooled to 0 °C and TBAF (1.65 mL, 1.65 mmol, 1.0 M in THF) was added dropwise. After the addition was complete, the reaction was allowed to warm to rt. The reaction was judged to be complete by TLC analysis after 2 hr and the solvent was removed by rotary evaporation. The crude residue was purified by flash silica gel column chromatography (isocratic eluent: 1:1 hexanes/EtOAc + 2% NEt3) to afford ynamide 30f (290.0 mg, 1.27 mmol, 97%) as a white solid.
30f
(290.0 mg, 97%); Rf = 0.26 [1:1 hexanes/EtOAc]; white solid; mp = 55–57 °C; 1H NMR (400 MHz, CDCl3) δ 1.02 (s, 3H), 1.07 (s, 3H), 2.39 (d, 2H, J = 4.0 Hz), 3.82–3.86 (m, 2H), 4.10 (ABX, 4H, ΔvAB = 25.8 Hz, JAB = 10.8 Hz, JAX = 11.2 Hz, JBX = 10.4 Hz), 5.21 (d, 1H, J = 10.4 Hz), 5.28 (d, 1H, J = 17.2 Hz), 5.84 (ddt, 1H, J = 6.2, 10.4, 17.2 Hz); 13C NMR (100 MHz, CDCl3) δ 21.3, 21.3, 32.3 (d, J = 6.3 Hz), 53.2 (d, J = 5.3 Hz), 54.0 (d, J = 5.6 Hz), 78.2 (d, J = 6.3 Hz), 78.7 (d, J = 4.8 Hz), 119.0, 132.2 (d, J = 1.6 Hz); 31P NMR (162 MHz, CDCl3) δ 0.64; IR (film) cm−1 2972m, 2923m, 2134m, 1473m, 1373m, 1308s, 1276s; mass spectrum (APCI): m/e (% relative intensity) 230 (10) (M+H)+, 261 (100) (M+MeOH+H)+.
Ynamide 30g was prepared from 30f following the general procedure for preparation of γ-hydroxy-ynamides
30g
(206.3 mg, 51%); Rf = 0.25 [3:1 hexanes/EtOAc]; white solid; mp = 45–46 °C; 1H NMR (400 MHz, CDCl3) δ 0.97 (s, 3H), 1.09 (s, 3H), 1.13 (s, 3H), 2.16 (br, 1H), 3.87–3.92 (m, 2H), 4.09–4.21 (m, 5H), 5.26 (dd, 1H, J = 1.2, 10.4 Hz), 5.33 (dd, 1H, J = 1.2, 16.8 Hz), 5.90 (ddt, 1H, J = 6, 10.4, 16.4 Hz); 13C NMR (100 MHz, CDCl3) δ 21.1, 21.2, 25.4, 32.1 (d, J = 6.9 Hz), 36.0, 53.3 (d, J = 5.3 Hz), 65.4 (d, J = 6.1 Hz), 71.5, 77.9 (dd, J = 3.0, 6.1 Hz), 80.9 (d, J = 4.6 Hz), 118.9, 132.4 (d, J = 2.3 Hz); 31P NMR (162 MHz, CDCl3) δ −0.62; IR (film) cm−1 3402brm, 2966s, 2870m, 2246s, 1474m, 1363w, 1273s; mass spectrum (APCI): m/e (% relative intensity) 316.2 (39) (M+H)+.
Ynamide 30d was prepared from 30g following the general procedure for silylation
30d
(218.8 mg, 72%); Rf = 0.25 [3:1 hexanes/EtOAc]; white solid; mp = 45–46 °C; 1H NMR (400 MHz, CDCl3) δ 0.03 (s, 3H), 0.08 (s, 3H), 0.85 (s, 9H), 0.89 (s, 9H), 1.06 (s, 6H), 3.81–3.88 (m, 2H), 3.98 (d, 1H, J = 3.6 Hz), 4.01–4.15 (m, 4H), 5.20 (dq, 1H, J = 1.1, 10.4 Hz), 5.28 (dq, 1H, J = 1.6, 17.2 Hz), 5.85 (ddt, 1H, J = 6.0, 10.4, 17.2 Hz); 13C NMR (100 MHz, CDCl3) δ −5.4, −4.4, 18.1, 21.2, 25.5, 25.7, 32.1 (d, J = 6.1 Hz), 36.6, 53.3 (d, J = 6.1 Hz), 65.6 (d, J = 6.1 Hz), 71.8, 77.7 (dd, J = 6.9, 14.6 Hz), 80.0 (d, J = 5.3 Hz), 118.6, 132.6 (d, J = 1.5 Hz); 31P NMR (162 MHz, CDCl3) δ −0.45; IR (film) cm−1 1255s, 1275s, 2247m, 2858m, 2892m, 2931m, 2957m; mass spectrum (APCI): m/e (% relative intensity) 330 (100) (M-TBS+MeOH+H)+. HRMS (TOF MS ESI) m/e calcd for C21H44N2O4PSi [M+NH4]+ 447.2803, found 447.2820.
General Procedure for the Carbocyclization of N-Phosphoryl Ynamides
Method A
To a flame-dried screw-cap vial containing ~60 mg 4 Å M.S. was added ynamide 30a (61.2 mg, 0.20 mmol), Pd(PPh3)4 (11.6 mg, 0.01 mmol), and toluene (5.0 mL, 0.04 M in ynamide). The vial was sealed under nitrogen and heated to 70 °C for 90 min, at which time TLC analysis showed consumption of the starting material. The reaction mixture was cooled to rt, filtered through Celite™, and purified by flash silica gel column chromatography (isocratic eluent: 1:3 hexanes/EtOAc) to afford cyclopentenimine 31a (53.0 mg, 0.17 mmol) in 87% yield.
Method B
To a flame-dried screw-cap vial containing ~60 mg 4 Å M.S. was added ynamide 30a (61.2 mg, 0.20 mmol) and toluene (5.0 mL, 0.04 M in ynamide). The vial was sealed under nitrogen and heated to 125 °C for 2 h, at which time TLC analysis showed consumption of the starting material. The reaction mixture was cooled to rt, filtered through Celite™, and purified by flash silica gel column chromatography (isocratic eluent: 1:3 hexanes/EtOAc) to afford cyclopentenimine 31a (30.8 mg, 0.10 mmol) in 50% yield.
31a
(Method A: 53.0 mg, 87%); Rf = 0.17 [1:2 hexanes/EtOAc]; pale yellow oil; 1H NMR (500 MHz, CDCl3) δ 0.83 (s, 3H), 1.32 (s, 3H), 2.77 (dd, 2H, J = 3.5, 7.5 Hz), 3.15 (dd, 2H, J = 3.5, 7.5 Hz), 3.89 (dd, 2H, J = 10.5, 19.5 Hz), 4.17 (d, 2H, J = 10.5 Hz), 7.34–7.40 (m, 2H), 7.47 (t, 1H, J = 7.5 Hz), 7.62–7.70 (m, 3H); 13C NMR (125 MHz, CDCl3) δ 21.0, 22.5, 29.8, 32.7 (d, J = 5.8 Hz), 35.6 (d, J = 10.0 Hz), 77.9 (d, J = 7.3 Hz), 128.3 (d, J = 11.1 Hz), 128.5, 128.8 (d, J = 12.3 Hz), 132.4 (d, J = 9.8 Hz), 145.9 (d, J = 28.3 Hz), 159.0, 194.6 (d, J = 2.9 Hz); 31P NMR (162 MHz, CDCl3) δ 0.64; IR (film) cm−1 2962m, 2884m, 1638s, 1593m, 1276s; mass spectrum (APCI): m/e (% relative intensity) 306 (100) (M+H)+; HRMS (QTOF MS ESI) calcd for C16H20NO3PNa [M+Na]+: 328.1073, found 328.1079.
31b
(Method A: 35.2 mg, 56%); Rf = 0.19 [1:1 hexane/EtOAc]; pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 0.87–0.91 (m, 3H), 0.90 (s, 3H), 1.29–1.32 (m, 6H), 1.31 (s, 3H), 1.48–1.54 (m, 2H), 2.22– 2.26 (m, 2H), 2.60–2.62 (m, 2H), 2.91–2.94 (m, 2H), 3.92 (ddt, 2H, J = 1.2, 10.8, 18.8 Hz), 4.18 (dd, 2H, J = 4.0, 10.4 Hz), 7.15–7.17 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 14.0, 20.7, 22.0, 22.5, 25.5, 27.7, 29.1, 29.5, 31.6, 32.4 (d, J = 5.3 Hz), 34.2 (d, J = 10.0 Hz), 77.5 (d, J = 6.9 Hz), 148.0 (d, J = 28.4 Hz), 156.3, 197.1 (d, J = 3.1 Hz); 31P NMR (162 MHz, CDCl3) δ 1.44; IR (film) cm−1 2959m, 2928s, 2857m, 1638s, 1615s, 1470w, 1275s; mass spectrum (APCI): m/e (% relative intensity) 314 (100) (M+H)+.
31c
(Method A: 21.1 mg, 34%); Rf = 0.18 [1:1 hexane/EtOAc]; pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 0.04 (s, 6H), 0.89 (s, 12H), 1.30 (s, 3H), 1.54–1.57 (m, 4H), 2.25 (t, 2H, J = 6.4 Hz), 2.59–2.60 (m, 2H), 2.91–2.94 (m, 2H), 3.62 (t, 2H, J = 6.2 Hz), 3.92 (ddd, 2H, J = 1.2, 9.6, 18.8 Hz), 4.16 (dd, 2H, J = 3.6, 10.4 Hz), 7.15–7.17 (m, 1H); 13C NMR (100 MHz, CDCl3) δ −5.3, 18.3, 20.8, 22.0, 24.0, 25.2, 25.9, 29.5, 32.4 (d, J = 6.1 Hz), 32.6, 34.3 (d, J = 10.0 Hz), 62.9, 77.5 (d, J = 6.8 Hz), 147.8 (d, J = 28.5 Hz), 156.3, 197.0; 31P NMR (162 MHz, CDCl3) δ 1.39; IR (film) cm−1 2954m, 2928m, 2858m, 1615m, 1471w, 1257s; mass spectrum (APCI): m/e (% relative intensity) 416 (100) (M+H)+; HRMS (QTOF MS ESI) calcd for C20H38NO4PSiNa [M+Na]+: 438.2200, found 438.2197.
31d
(41.4 mg, 69%); Rf = 0.14 [1:1 hexanes/EtOAc]; pale yellow oil; 1H NMR (400 MHz, CDCl3) δ −0.18 (s, 3H), 0.84 (s, 3H), 0.85 (s, 9H), 0.86 (s, 9H), 1.32 (s, 3H), 2.57–2.71 (m, 2H), 2.84 (ddt, 1H, J = 2.8, 6.0, 20.0 Hz), 3.04 (ddt, 1H, J = 2.8, 6.0, 20.0 Hz), 3.82–3.93 (m, 2H), 4.14 (ddd, 2H, J = 2.4, 10.8, 21.2 Hz), 4.36 (s, 1H), 7.40 (dd, 1H, J = 2.8, 6.0 Hz); 13C NMR (100 MHz, CDCl3) −5.1, −4.4, 18.2, 20.8, 22.3, 25.9 (d, J = 3.9 Hz), 29.8, 32.5 (d, J = 6.0 Hz), 34.5 (d, J = 10.0 Hz), 36.3, 73.1, 77.5 (d, J = 7.9 Hz), 150.0 (d, J = 27.1 Hz), 160.2, 196.0 (d, J = 2.9 Hz); 31P NMR (162 MHz, CDCl3) δ 0.84; IR (film) cm−1 2957m, 2930m, 2885m, 2857m, 1632m, 1613m, 1472m, 1281m, 1257m; mass spectrum (APCI): m/e (% relative intensity) 430 (100) (M+H)+; HRMS (QTOF MS ESI) calcd for C21H40NO4PSiNa [M+Na]+: 452.2356, found 452.2359.
48
(71.2 mg, 87%); Rf = 0.41 [1:1 DCM/EtOAc]; pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 0.73 (s, 3H), 1.26 (s, 3H), 2.75 (t, 2H, J = 4.0 Hz), 2.82–2.85 (m, 4H), 3.04–3.07 (m, 2H), 3.77 (ddd, 2H, J = 1.2, 9.6, 19.6 Hz), 4.01 (dd, 2H, J = 1.6, 10.4 Hz), 7.08–7.12 (m, 4H), 7.19–7.35 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 20.5, 22.1, 32.2 (d, J = 6.1 Hz), 32.5, 33.2 (d, J = 1.6 Hz), 33.7, 33.8 (d, J = 10.8 Hz), 77.5 (d, J = 7.7 Hz), 126.3, 127.5, 127.7, 128.1, 128.5, 129.5, 132.4, 140.4, 142.7 (d, J = 29.2 Hz), 173.5, 195.1 (d, J = 1.5 Hz); 31P NMR (162 MHz, CDCl3) δ 0.79; IR (film) cm−1 2963w, 2929w, 1617s, 1592s, 1268s; mass spectrum (APCI): m/e (% relative intensity) 410 (100) (M+H)+; HRMS (QTOF MS ESI) calcd for C24H28NO3PNa [M+Na]+: 432.1699, found 432.1699.
Characterization of N-Phosphoryl-TIPS-Ketenimine 32
32
(41.1 mg, 53%); Rf = 0.17 [4:1 hexanes:EtOAc]; yellow wax; mp = 47–50 °C; 1H NMR (400 MHz, CDCl3) δ 0.86 (s, 3H), 1.13 (d, 18H, J = 6.0 Hz), 1.18 (sept, 3H, J = 6.0 Hz), 1.29 (s, 3H), 2.77 (ddt, 2H, J = 1.2, 6.8, 8.0 Hz), 3.95 (dd, 2H, J = 11.2, 22.0 Hz), 4.18 (d, 2H, J = 10.4 Hz), 5.10 (dd, 1H, J = 1.2, 10.0 Hz), 5.21 (dq, 1H, J = 1.6, 3.2, 17.2 Hz), 5.98 (ddt, 1H, J = 6.4, 10.0, 16.4 Hz); 13C NMR (100 MHz, CDCl3) δ 11.9, 17.9, 18.7, 20.5, 22.2, 29.0 (d, J = 5.4 Hz), 32.4 (d, J = 6.1 Hz), 40.0 (d, J = 15.3); 76.9 (d, J = 31.3 Hz), 116.3 (d, J = 3.8 Hz), 136.1 (d, J = 3.1 Hz), 187.9; 31P NMR (162 MHz, CDCl3) δ −9.05; IR (film) cm−1 2943m, 2890w, 2866m, 1993s, 1464m, 1372w, 1341w, 1301s; mass spectrum (ESI): m/e (% relative intensity) 387 (100) (M+H)+; HRMS (QTOF MS ESI) calcd for C19H36NO3PSiNa [M+Na]+: 408.2094, found 408.2091.
Preparation of Farnesyl-Tethered Ynamide 59
To a flame-dried 25-mL RB-flask was added CH2Cl2 (7.0 mL, 0.5 M in allyl amine), farnesyl amine (752.0 mg, 3.39 mmol), and NEt3 (0.70 mL, 5.09 mmol) under a nitrogen atmosphere. The reaction flask was cooled to 0 °C and 2-chloro-5,5-dimethyl-1,3,2-dioxaphosphorinane 2-oxide (753.0 mg, 4.07 mmol) was added slowly dropwise. Once the addition was complete, the reaction was allowed to warm to rt and stir overnight. After the reaction was judged to be complete by TLC, water (10 mL) was added, the organic layer separated, and the aqueous phase was extracted with CH2Cl2 (3 × 25 mL). The combined organic phase was washed with sat. NaCl and dried over Na2SO4. The crude material was purified by flash silica gel column chromatrography (1:1 EtOAc:CH2Cl2) to afford phosphoramidate 56 (943.0 mg, 2.56 mmol, 75%).
56
(943.0 mg, 75%); Rf= 0.20 [1:1 EtOAc/CH2Cl2]; colorless oil; 1H NMR (400 MHz, CDCl3) δ 0.90 (s, 3H), 1.21 (s, 3H), 1.60 (s, 6H), 1.65 (s, 3H), 1.68 (s, 3H), 2.12-1.94 (m, 8H), 2.65 (brs, 1H), 3.59 (dt, 2H, J = 6.8, 9.2 Hz), 3.81 (ddt, 2H, J = 1.2, 11.2, 19.6 Hz), 4.29 (dd, 2H, J = 4.4, 11.2 Hz), 5.09 (t, 2H, J = 6.8 Hz), 5.25 (tq, 1H, J = 1.6, 6.8 Hz); 13C NMR (100 MHz, CDCl3) δ 16.1, 16.3, 17.8, 20.9, 22.1, 25.8, 26.4, 26.8, 32.0 (d, J = 5.2 Hz), 39.2, 39.6, 39.8, 76.2 (d, J = 5.5 Hz), 122.0 (d, J = 7.3 Hz), 123.8, 124.4, 131.4, 135.5, 139.1; 31P NMR (162 MHz, CDCl3) δ 6.37; IR (film) cm−1 3210brm, 2965m, 2917m, 1447m, 1374m, 1231m; mass spectrum (APCI): m/e (% relative intensity) 370 (100) (M+H)+. HRMS (TOF MS ESI) m/e calcd for C20H37NO3P [M+H]+: 370.2506, found 370.2523.
57
(298.0 mg, 65%); Rf= 0.16 [3:1 hexanes/EtOAc]; colorless oil; 1H NMR (400 MHz, CDCl3) δ 1.04 (s, 18H), 1.03–1.06 (m, 3H), 1.06 (s, 3H), 1.14 (s, 3H), 1.59 (d, 3H, J = 1.2 Hz), 1.60 (d, 3H, J = 0.8 Hz), 1.68 (d, 3H, J = 1.2 Hz), 1.69 (d, 3H, J = 1.2 Hz), 1.94–2.17 (m, 8H), 3.94 (t, 2H, J = 7.6 Hz), 4.15 (ABX, 4H, ΔvAB = 32.8 Hz, JAB = 11.2 Hz, JAX = 10.7 Hz, JBX = 10.9 Hz), 5.06–5.14 (m, 2H), 5.38 (tq, 1H, J = 1.2, 6.8 Hz); 13C NMR (100 MHz, CDCl3) δ 11.6, 16.1, 16.7, 17.8, 18.8, 18.8, 21.3, 21.6, 25.8, 26.5, 26.9, 32.4 (d, J = 6.9 Hz), 39.8, 48.7 (d, J = 5.4 Hz), 63.0 (d, J = 4.5 Hz), 77.4, 78.3 (d, J = 6.8 Hz), 118.8 (d, J = 2.3 Hz), 124.0, 124.5, 131.4, 135.4, 141.4; 31P NMR (162 MHz, CDCl3) δ 1.65; IR (film) cm−1 2923m, 2864m, 2161m, 1463m, 1380m, 1235m; mass spectrum (APCI): m/e (% relative intensity) 370 (70) (M–C≡CTIPS+H)+, 571 (10) (M+H)+.
To a flame-dried 10-mL RB-flask was added ynamide 57 (298.0 mg, 0.54 mmol) and THF (2.2 mL, 0.25 M in ynamide). The flask was cooled to 0 °C and TBAF (0.65 mL, 0.65 mmol, 1.0 M in THF) was added dropwise. After the addition was complete, the reaction was allowed to warm to rt. The reaction was judged to be complete by TLC analysis after 2 h and the solvent was removed by rotary evaporation. The crude residue was purified by flash silica gel column chromatography (isocratic eluent: 2:1 hexanes/EtOAc + 2% NEt3) to afford ynamide 58 (174.0 mg, 0.44 mmol, 82%) as a colorless oil.
58
(174.0 mg, 82%); Rf= 0.29 [2:1 hexanes/EtOAc]; colorless oil; 1H NMR (400 MHz, CDCl3) δ 1.09 (s, 3H), 1.12 (s, 3H), 1.60 (s, 3H), 1.60 (s, 3H), 1.68 (d, 3H, J = 0.8 Hz), 1.71 (d, 3H, J = 0.8 Hz), 1.97 (t, 2H, J = 7.6 Hz), 2.01–2.14 (m, 6H), 2.40 (d, 1H, J = 3.6 Hz), 3.94 (t, 2H, J = 7.6 Hz), 4.15 (ABX, 4H, ΔvAB = 23.5 Hz, JAB = 10.9 Hz, JAX = 11.0 Hz, JBX = 8.8 Hz), 5.07–5.13 (m, 2H), 5.37 (tq, 1H, J = 1.2, 6.8 Hz); 13C NMR (100 MHz, CDCl3) δ 16.2, 16.6, 17.8, 21.3, 21.4, 25.8, 26.5, 26.9, 32.4 (d, J = 6.3 Hz), 39.8, 39.8, 48.2 (d, J = 5.0 Hz), 53.8 (d, J = 5.8 Hz), 77.4, 78.1 (d, J = 6.3 Hz), 118.6 (d, J = 2.3 Hz), 123.9, 124.5, 131.4, 135.4, 141.9; 31P NMR (162 MHz, CDCl3) δ 0.22; IR (film) cm−1 2965m, 2920m, 2133m, 1769w, 1666w, 1448m, 1305s, 1275s; mass spectrum (APCI): m/e (% relative intensity) 394 (100) (M+H)+
To a flame-dried 25-mL RB-flask was added ynamide 58 (174.0 mg, 0.44 mmol) and THF (4.4 mL, 0.1 M in ynamide) under a nitrogen atmosphere. The flask was cooled to −78 °C and LHMDS (0.89 mL, 0.88 mmol, 1 M in THF) was added dropwise. The reaction was kept at −78 °C for 1 h to ensure complete deprotonation and then MeI (83.0 µL, 1.32 mmol) was added dropwise. The reaction was allowed to warm to rt overnight, at which time TLC analysis showed consumption of the starting material. The reaction was diluted with EtOAc (5 mL) and quenched with water (5 mL). The layers were separated and the aqueous layer was extracted with EtOAc (10 mL x 3), then the combined organic layers were washed with brine and dried over Na2SO4. Purification of the crude residue by flash silica gel column chromatography (isocratic eluent: 1:1 hexanes/EtOAc + 2% NEt3) afforded ynamide 59 (147.2 mg, 0.36 mmol, 82%).
59
(147.2 mg, 82%); Rf= 0.16 [2:1 hexanes/EtOAc]; colorless oil; 1H NMR (400 MHz, CDCl3) δ 1.03 (s, 3H), 1.17 (s, 3H), 1.60 (s, 6H), 1.68 (s, 3H), 1.70 (d, 3H, J = 0.8 Hz), 1.85 (d, 1H, J = 3.2 Hz), 1.97 (t, 2H, J = 7.6 Hz), 2.04–2.14 (m, 6H), 3.89 (t, 2H, J = 8.0 Hz), 4.03 (dd, 2H, J = 11.2, 14.8 Hz), 4.20 (dd, 2H, J = 9.2, 10.8 Hz), 5.07–5.14 (m, 2H), 5.36 (tq, 1H, J = 1.2, 6.8 Hz); 13C NMR (100 MHz, CDCl3) δ 3.2 (d, J = 1.9 Hz), 16.1, 16.5, 17.8, 21.3, 21.4, 25.8, 26.5, 26.9, 32.3 (d, J = 6.1 Hz), 39.8 (d, J = 4.4 Hz), 48.2 (d, J = 3.7 Hz), 60.2 (d, J = 5.8 Hz), 74.6 (d, J = 4.6 Hz), 77.6 (d, J = 6.2 Hz), 78.0 (d, J = 3.7 Hz), 119.3 (d, J = 2.2 Hz), 123.9, 124.5, 131.4, 135.3, 141.2; 31P NMR (162 MHz, CDCl3) δ 1.41; IR (film) cm−1 2968m, 2919m, 2262w, 1737m, 1447m, 1373m, 1271m; mass spectrum (APCI): m/e (% relative intensity) 408 (100) (M+H)+; HRMS (QTOF MS ESI) calcd for C23H38NO3PNa [M+Na]+: 430.2482, found 430.2489.
Carbocyclization of Farnesyl-Tethered Ynamide 59
62
(41.7 mg, 43%); Rf= 0.18 [1:2 hexanes/EtOAc]; colorless oil; 1H NMR (400 MHz, CDCl3) all isomers δ 0.98 (s, 9H), 1.08 (s, 3H), 1.09 (s, 3H), 1.11 (s, 3H), 1.13 (s, 3H), 1.14 (s, 3H), 1.19–1.37 (m, 6H), 1.47 (s, 3H), 1.48 (s, 3H), 1.48 (s, 3H), 1.60 (s, 12H), 1.67 (s, 6H), 1.69 (s, 6H), 1.70–1.75 (m, 2H), 1.91–2.11 (m, 16H), 2.17–2.38 (m, 6H), 2.40–2.49 (m, 1H), 2.53–2.60 (m, 1H), 2.62–2.72 (m, 2H), 2.91–3.00 (m, 2H), 3.87 (ddd, 6H, J = 1.5, 11.5, 16.5 Hz), 4.18–4.30 (m, 8H), 4.33 (d, 2H, J = 8.0 Hz), 4.68 (s, 1H), 4.83 (s, 1H), 5.94–5.19 (m, 3H); 31P NMR (162 MHz, CDCl3) all isomers δ 1.56, 1.57, 1.63; IR (film) cm−1 2962m, 2930m, 1737s, 1670m, 1456m, 1373m; mass spectrum (APCI): m/e (% relative intensity) 408 (100) (M+H)+. HRMS (TOF MS ESI) m/e calcd for C23H39NO3P [M+H]+ : 408.2663, found 408.2675.
63
(36.8 mg, 38%); Rf= 0.47 [1:2 hexanes/EtOAc]; colorless oil; 1H NMR (500 MHz, CDCl3) 0.76 (s, 3H), 0.80 (s, 3H), 0.86 (s, 3H), 0.93 (s, 3H), 0.93 (ddd, 1H, J = 6.0, 11.0, 13.5 Hz), 1.13 (ddd, 1H, J = 6.0, 11.0, 13.5 Hz), 1.30 (s, 3H), 1.43 (t, 2H, J = 11.0 Hz), 1.52 (ddt, 1H, J = 3.0, 5.5, 11.5 Hz), 1.56 (s, 3H), 1.63 (s, 3H), 1.65–1.76 (m, 1H), 1.88 (ddd, 2H, J = 6.5, 13.5, 19.0 Hz), 1.92 (s, 1H), 2.00 (s, 1H), 2.69 (t, 2H, J = 3.0 Hz), 3.87 (dddd, 2H, J = 3.0, 11.0, 19.0, 41.0 Hz), 4.16 (ddd, 2H, J = 3.5, 11.0, 43.0 Hz), 4.97–5.00 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 7.3, 16.6, 17.8, 20.2, 20.9, 22.3, 23.7, 25.9, 27.3, 30.0, 32.6 (d, J = 5.7 Hz), 36.1 (d, J = 11.3 Hz), 40.3 (d, J = 6.6 Hz), 47.1, 49.9, 56.9, 64.3 (d, J = 23.1 Hz), 77.8 (d, J = 7.5 Hz), 124.8, 131.4, 206.0 (d, J = 7.4 Hz); 31P NMR (162 MHz, CDCl3) δ 1.71; IR (film) cm−1 2959m, 2931m, 2877m, 1738m, 1689m, 1460m, 1374m; mass spectrum (APCI): m/e (% relative intensity) 408 (100) (M+H)+; HRMS (QTOF MS ESI) calcd for C23H38NO3PNa [M+Na]+: 430.2482, found 430.2486.
Supplementary Material
ACKNOWLEDGEMENT
Authors thank the NIH [GM066055] for financial support. KAD thanks the American Chemical Society for a Division of Medical Chemistry Predoctoral Fellowship.
Footnotes
SUPPORTING INFORMATION AVAILABLE. Experimental procedures, characterization data for all new compounds, and 1H/13C NMR spectra are available free of charge via Internet at http://pubs.acs.org.
Contributor Information
Richard P. Hsung, Email: rhsung@wisc.edu.
Kyle A. DeKorver, Email: kadekorver@dow.com.
REFERENCES
- 1.For current leading reviews on chemistry of ynamides, see: Evano G, Coste A, Jouvin K. Angew. Chem. Int. Ed. 2010;49:2840. doi: 10.1002/anie.200905817. DeKorver KA, Li H, Lohse AG, Hayashi R, Lu Z, Zhang Y, Hsung RP. Chem. Rev. 2010;110:5064. doi: 10.1021/cr100003s. For reviews partially accounting chemistry of ynamides, see: Ackermann L, Potukuchi HK. Org. Biomol. Chem. 2010;8:4503. doi: 10.1039/c0ob00212g. Domínguez G, Perez-Castells J. Chem. Soc. Rev. 2011;40:3430. doi: 10.1039/c1cs15029d. Weding N, Hapke M. Chem. Soc. Rev. 2011;40:4525. doi: 10.1039/c0cs00189a. Madelaine C, Valerio V, Maulide N. Chem. Asian J. 2011;6:2224. doi: 10.1002/asia.201100108. For reviews on syntheses of ynamides, see: Mulder JA, Kurtz KCM, Hsung RP. Synlett. 2003:1379. Dehli JR, Legros J, Bolm C. Chem. Commun. 2005;43:973. doi: 10.1039/b415954c. Tracey MR, Hsung RP, Antoline JA, Kurtz KCM, Shen L, Slafer BW, Zhang Y. In: Science of Synthesis, Houben-Weyl Methods of Molecular Transformations. Weinreb Steve M., editor. Stuttgart, Germany: Georg Thieme Verlag KG; 2005. Chapter 21.4. Evano G, Blanchard N, Toumi M. Chem. Rev. 2008;108:3054. doi: 10.1021/cr8002505. Evano G, Jouvinb K, Coste A. Synthesis. 2013:17.
- 2.For syntheses of ynamides published in 2012, see: Jin X, Yamaguchi K, Mizuno N. Chem. Lett. 2012;41:866. doi: 10.1039/c2cc31159c. Jin X, Yamaguchi K, Mizuno N. Chem. Commun. 2012;48:4974. doi: 10.1039/c2cc31159c. Jouvin K, Heimburger J, Evano G. Chem. Sci. 2012;3:756. Jouvin K, Coste A, Bayle A, Legrand F, Karthikeyan G, Tadiparthi K, Evano G. Organometallics. 2012;31:7933. Tong X, Ni G, Deng X, Xia C. Synlett. 2012;23:2497. Wang M-G, Wu J, Shang Z-C. Synlett. 2012;23:589. Laouiti A, Jouvin K, Bourdreux F, Rammah MM, Rammah MB, Evano G. Synthesis. 2012;44:1491. For syntheses of novel structural analogs of ynamides, see: Yne-Sulfoxyimines: Wang L, Huang H, Priebbenow DL, Pan F-F, Bolm C. Angew. Chem. Int. Ed. 2013;52:3478. doi: 10.1002/anie.201209975. Yne-hydrazides: Beveridge RE, Batey RA. Org. Lett. 2012;14:540. doi: 10.1021/ol2031608. Yne-imines: Laouiti A, Rammah MM, Rammah MB, Marrot J, Couty F, Evano G. Org. Lett. 2012;14:6. doi: 10.1021/ol2032152. Ynimides: Souto JA, Becker P, Iglesias Á, Muñiz K. J. Am. Chem. Soc. 2012;134:15505. doi: 10.1021/ja306211q. Ynimides: Sueda T, Oshima A, Teno N. Org. Lett. 2011;13:3996. doi: 10.1021/ol2014973.Ynimides: Sueda T, Kawada A, Urash Y, Teno N. Org. Lett. 2013;15:1560. doi: 10.1021/ol400338x.Diamino-acetylenes: Petrov AR, Daniliuc CG, Jones PG, Tamm M. Chem.-Eur. J. 2010;16:11804. doi: 10.1002/chem.201002211.Amidinyl-ynamides: Li J, Neuville L. Org. Lett. 2013;15:1752. doi: 10.1021/ol400560m.
- 3.Given the volume, for ynamide chemistry published since September of 2012, see: Karmakar R, Mamidipalli P, Yun SY, Lee D. Org. Lett. 2013;15:1938. doi: 10.1021/ol4005905. Brioche J, Meyer C, Cossy J. Org. Lett. 2013;15:1626. doi: 10.1021/ol400402n. Yun SY, Wang K-P, Lee N-K, Mamidipalli P, Lee D. J. Am. Chem. Soc. 2013;135:4668. doi: 10.1021/ja400477r. Heffernan SJ, Beddoes JM, Mahon MF, Hennessy AJ, Carbery DR. Chem. Commun. 2013;49:2314. doi: 10.1039/c3cc00273j. Kong Y, Jiang K, Cao J, Fu L, Yu L, Lai G, Cui Y, Hu Z, Wang G. Org. Lett. 2013;15:422. doi: 10.1021/ol303474a. Yavari I, Nematpour M, Sodagar E. Synlett. 2013;24:161. Yavari I, Nematpour M. Synlett. 2013;24:165. Bhunia S, Chang C-J, Liu R-S. Org. Lett. 2012;14:5522. doi: 10.1021/ol302621z. Minko Y, Pasco M, Lercher L, Botoshansky M, Marek I. Nature. 2012;490:522. doi: 10.1038/nature11569. Hoye TR, Baire B, Niu D, Willoughby PH, Woods BP. Nature. 2012;490:208. doi: 10.1038/nature11518. Cao J, Kong Y, Deng Y, Lai G, Cui Y, Hu Z, Wang G. Org. Biomol. Chem. 2012;10:9556. doi: 10.1039/c2ob26727f. Greenaway RL, Campbell CD, Chapman HA, Anderson EA. Adv. Synth. Catal. 2012;354:3187. Jiang Z, Lu P, Wang Y. Org. Lett. 2012;14:6266. doi: 10.1021/ol303023y. Gati W, Rammah MM, Rammah MB, Evano G. Beilstein J. Org. Chem. 2012;8:2214. doi: 10.3762/bjoc.8.250. Smith DL, Chidipudi SR, Goundry WR, Lam HW. Org. Lett. 2012;14:4934. doi: 10.1021/ol302259v. Heffernan SJ, Carbery DR. Tetrahedron Lett. 2012;53:5180.
- 4.For reviews on Claisen rearrangements, see: Castro AMM. Chem. Rev. 2004;104:2939. doi: 10.1021/cr020703u. Ito H, Taguchi T. Chem. Soc. Rev. 1999;28:43. Enders D, Knopp M, Schiffers R. Tetrahedron Asym. 1996;7:1847. Wipf Pin. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 5. Oxford: Pergamon Press; 1991. p. 827. Hill RK. In: Asymmetric Synthesis. Morrison JD, editor. New York: Academic Press; 1984.
- 5.For leading reviews on aza-Claisen rearrangments, see: For leading reviews on aza-Claisen rearrangements, see: Majumdar KC, Bhayyacharyya T, Chattopadhyay B, Nandi RK. Synthesis. 2009:2117. Nubbemeyer U. Top Curr. Chem. 2005;244:149.
- 6.(a) Zhang Y, DeKorver KA, Lohse AG, Zhang Y-S, Huang J, Hsung RP. Org. Lett. 2009;11:899. doi: 10.1021/ol802844z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) DeKorver KA, Hsung RP, Lohse AG, Zhang Y. Org. Lett. 2010;12:1840. doi: 10.1021/ol100446p. [DOI] [PMC free article] [PubMed] [Google Scholar]; c DeKorver KA, Johnson WL, Zhang Y, Hsung RP, Dai H, Deng J, Lohse AG, Zhang Y-S. J. Org. Chem. 2011;76:5092. doi: 10.1021/jo200780x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeKorver KA, North TD, Hsung RP. Synlett. 2010:2397. doi: 10.1055/s-0030-1258544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeKorver KA, Walton MC, North TD, Hsung RP. Org. Lett. 2011;13:4862. doi: 10.1021/ol201947b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeKorver KA, Hsung RP, Song W-Z, Wang X-N, Walton MC. Org. Lett. 2012;14:3214. doi: 10.1021/ol3013233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeKorver KA, Wang X-N, Walton MC, Hsung RP. Org. Lett. 2012;14:1768. doi: 10.1021/ol300366e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For reviews on the chemistry of ketenimines, see: Krow GR. Angew. Chem., Int. Ed. 1971;10:435. Gambaryan NP. Usp. Khim. 1976;45:1251. Dondoni A. Heterocycles. 1980;14:1547. Barker MW, McHenry WEIn. In: The Chemistry of Ketenes, Allenes and Related Compounds. Patai S, editor. Chichester, UK: Wiley-Interscience; 1980. pp. 701–720. Part 2. Alajarín M, Vidal A, Tovar F. Targets Heterocycl. Syst. 2000;4:293.
- 12.Rodríguez D, Martínez-Esperón MF, Castedo L, Saá C. Synlett. 2007;12:1963. [Google Scholar]
- 13.For a review, see: Basavaiah D, Jaganmohan Rao A, Satyanarayana T. Chem. Rev. 2003;103:811. doi: 10.1021/cr010043d.
- 14.For a related report, see: Whiting M, Fokin VV. Angew. Chem. Int. Ed. 2006;45:3157. doi: 10.1002/anie.200503936.
- 15.Rautenstrauch V. J. Org. Chem. 1984;49:950. [Google Scholar]
- 16.For a leading reference on Rautenstrauch rearrangements, see: Shi X, Gorin DJ, Toste FD. J. Am. Chem. Soc. 2005;127:5802. doi: 10.1021/ja051689g.
- 17.For a related imino carbocyclization, see: Hanessian S, Tremblay M, Marzi M, Del Valle JR. J. Org. Chem. 2005;70:5070. doi: 10.1021/jo050326w.
- 18.For related carbocyclizations of ketenes, see: Sosa JR, Tudjarian AA, Minehan TG. Org. Lett. 2008;10:5091. doi: 10.1021/ol802147h. Tudjarian AA, Minehan TG. J. Org. Chem. 2011;76:3576. doi: 10.1021/jo200271s.
- 19.For related examples of intercepted Nazarov cyclizations, see: Giese S, Kastrup L, Stiens D, West FG. Angew. Chem., Int. Ed. 2000;39:1970. doi: 10.1002/1521-3773(20000602)39:11<1970::aid-anie1970>3.0.co;2-b. Giese S, West FG. Tetrahedron. 2000;56:10221. Wang Y, Schill BD, Arif AM, West FG. Org. Lett. 2003;5:2747. doi: 10.1021/ol034985b. Huang J, Leboeuf D, Frontier AJ. J. Am. Chem. Soc. 2011;133:6307. doi: 10.1021/ja111504w.
- 20.For reviews on polyene cyclizations, see: Johnson WS. Acc. Chem. Res. 1968;1:1. Johnson WS. Angew. Chem. Int. Ed. 1976;15:9. doi: 10.1002/anie.197600091. For a key account, see: Johnson WS, Telfer SJ, Cheng S, Schubert U. J. Am. Chem. Soc. 1987;109:2517.
- 21.Also see: Stork G, Burgstahler AW. J. Am. Chem. Soc. 1955;77:5068. Eschenmoser A, Ruzicka L, Jeger O, Arigoni D. Helv. Chim. Acta. 1955;38:1890. Stadler PA, Eschenmoser A, Schinz H, Stork G. Helv. Chim. Acta. 1957;40:2191.
- 22.Coppola GM, Prashad M. Syn. Commun. 1993;23:535. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.