


Abstract
The existence of sequences in the human genome which can be a target for triplex formation, and accordingly are candidates for anti-gene therapies, has been studied by using bioinformatics tools. It was found that the population of triplex-forming oligonucleotide target sequences (TTS) is much more abundant than that expected from simple random models. The population of TTS is large in all the genome, without major differences between chromosomes. A wide analysis along annotated regions of the genome allows us to demonstrate that the largest relative concentration of TTS is found in regulatory regions, especially in promoter zones, which suggests a tremendous potentiality for triplex strategy in the control of gene expression. The dependence of the stability and selectivity of the triplexes on the length of the TTS is also analysed using knowledge-based rules.
INTRODUCTION
The sequencing of the human genome (1,2) has opened the way for the design of new pharmacological therapies based on the inhibition of the synthesis of pathological proteins. The blocking of the synthesis of pathological proteins can be performed at least by two different mechanisms: (i) by inhibition of the translation of mRNA and (ii) by inhibition of the transcription of the corresponding gene. The first approach defines the ‘anti-sense’ strategy (in either its pure anti-sense and its RNA-interference versions), where oligonucleotides are used to specifically bind the target, the mRNA, blocking then the corresponding protein synthesis (3,4). The second approach defines the ‘anti-gene’ strategy that consists of blocking the transcription of specific genes by formation of a triple helix (5–9) at the target DNA duplex.
DNA triplexes were theoretically suggested by Pauling and Corey in 1953 (10), and probed experimentally by Rich and co-workers 4 years later (11). They are formed when a polypurine-rich DNA duplex binds a single-stranded polynucleotide [triplex-forming oligonucleotide (TFO)], through specific major groove interactions (reviewed in 5). Two types of triplexes have been described, based on the orientation of the third strand with respect to the central polypurine Watson–Crick strand: (i) parallel triplexes and (ii) anti-parallel triplexes. The first can be formed following three different motives: d(T·A-T), d(C·G-C)+ (where protonated cytosine is needed in the third strand) and d(C·G-G), where Hoogsteen hydrogen bonds stabilize the interaction between the Watson–Crick (the first two bases of the triad) duplex and the third strand (Fig. 1). The second type of triplex is formed by three triads: d(T·A-A), d(C·G-G) and d(T·A-T), where the third strand makes reverse Hoogsteen pairs with the Watson–Crick duplex (Fig. 1). In general, under normal laboratory conditions, parallel triplexes are expected to be more stable than anti-parallel triplexes (12,13). However, their pH dependence [due to the presence of d(C·G-C)+ triads] might limit their physiological stability.
Figure 1.

Schematic representation of Hoogsteen and reverse Hoogsteen-based triads in parallel and anti-parallel triplexes.
The presence of a third strand introduces severe restrictions in the flexibility of the DNA, changing its ability to recognize specific proteins along the major groove (14,15), and accordingly, altering all the mechanisms controlling DNA function. This, and the specificity of the recognition process between the TFO and the duplex DNA explains the large number of potential applications of triplexes in the biomedical and biotechnological scenario (5–9). Thus, triplexes have been used to construct artificial restriction enzymes or to direct nuclease cutting in certain regions of the genome (6,16–17). Triplexes bound to cleaving agents have been successfully used to induce recombination in both episomal and chromosomal DNA in mammalian cells (18). It also has been reported by different authors that triplexes complexed with psoralen can be used to induce specific mutations in the genome (19–21). Furthermore, recent studies by Glazer’s group (22,23) have shown that even when no chemical mutagen is added, triplex formation induces a dramatic increase in the rate of mutagenesis in the target duplex, probably as a consequence of the inability of the NER system to repair triplexes. These findings open the possibility to use triplexes as an alternative for knocking down/out specific genes (8,20).
Most of the biomedical impact of triplex technology is related to the well known ability of triplexes to inhibit mRNA synthesis in target genes, both in vitro (6,7,24,25) and in vivo (6–8,26–28). Some of the genes whose expression can be inhibited by triplex formation include genes associated with different diseases including cancer (6), suggesting that TFOs could generate, in the near future, a new generation of drugs. The inhibition of DNA transcription by triplexes can occur by means of the inhibition of mRNA elongation (29,30). However, the greatest transcription-inhibitory activity of triplexes is found when the target duplex is in the regulatory region of the gene (8).
Despite the promising results found, triplex technology still presents some shortcomings (5) mostly related to: (i) their reduced stability, (ii) sequence restrictions due to the need of polypurine tracks in the triplex target sequence, (iii) susceptibility to nucleases and (iv) problems to deliver TFOs in the cellular nucleus. A large amount of chemical, biochemical and biotechnological research is now focussed on trying to solve these practical problems of triplex technology (6,7,31,32). However, a very basic question is still unanswered: what is the triplex-forming potential of the human genome? In this paper, we perform a very extensive analysis of the human genome in order to determine how many triplex-forming oligonucleotide target sequences (TTS) exist, their location and their potential as targets for anti-gene therapy.
MATERIALS AND METHODS
Genome information
Sequence information of the human genome was taken from the UCSC database (version hg12; June 28, 2002) (http://genome.ucsc.edu/goldenPath/28jun2002) developed by the International Human Genome Mapping Consortium (1). The definition of genes, exons, coding regions, repetitive regions and conserved human–mouse regions were also taken from the UCSC Genome Browser Database (http://genome.ucsc.edu/cgi-bin/hgGateway; 33). Annotation of the genes considered in this study was obtained from the refGene (refSeq) collection, after removing redundant or overlapping genes. Promoter regions were selected as those located 100 bp upstream of the beginning of the gene. A more diffuse upstream regulatory region is selected as that located 1900 bases upstream of the promoter region. Putative downstream regulatory regions are defined 2000 bases downstream of the end of the gene. The best human/mouse conserved regions were those listed in the chrN_blatzBestMouse list in the UCSC database (http://genome.ucsc.edu/goldenPath/28jun2002). Only highly conserved blocks larger than 100 bases were considered. Repeated sequences were those obtained using the RepeatMasker software and listed in the chrN_rmsk database (http://genome.ucsc.edu/goldenPath/28jun2002; http://www.geospiza.com/products/tools/repeatmasker.htm; 34), and were used without further manipulation. Single nucleotide polymorphisms (SNPs) were mapped combining SNP databases in UCSC (snpNih and snpTsc) and the dbSNP database of genetic variation (35).
Definition of TTS
Possible TTS were defined as polypurine tracks of any size. No mismatching in the triplexes was allowed, which means that a strict triplex definition was used. In order to determine whether or not the population of TTS is that expected from a random distribution (36), we developed a simple, but flexible random model, which assuming a binomial behaviour of the TTS distribution, allows the calculation of the expected number of TTS of a given length (in a given genome). This considers that the expected number (P) of TTS of length n in a given genome of length m can be expressed as shown in equation 1, where qn is the probability that a nucleotide belongs to a TTS of length n. The factor 2 appears because the TTS can happen in either DNA chains. The probability factor qn is computed from the average number of TTS of size (n) in the random model using equations 2 and 3. The key parameter <i> is determined assuming that the number of TTS in the random model follows a binomial distribution (see equation 3):
P = 2 × m × qn1
qn = n (<i> / m)2
<i> ≈ (m – n – 1) × α2 × (1 – α)n–13
Combining equations 2 and 3 we obtained equation 4 which provides us an approximated expression for P in a random model. Note that equation 4 is in fact an approximate solution, but provides a TTS distribution very similar to that obtained in numerical simulations of 50 randomly generated genomes with the size of the human genome (see Results). In an ideal binomial model α should be equal to 1/2 in equations 3 and 4. However, since transitions Pur–Pyr, Pyr–Pur and Pur–Pur are not equally probable in our genome, other values of α might be more suitable. In fact, the fitting to the human genome shows that the most realistic value for α is 0.44 (the value used in the paper):
P ≈ 2 × n × (m – n – 1) × α2 × (1 – α)n–14
Prediction of triplex stability
The stability of the triplex measured in terms of the melting temperature depends on many factors, such as sequence, concentration of the TFO, length of the triplex, presence of modified nucleotides in the TFO or pH (for the most stable parallel triplexes). A rough prediction of the melting temperature of triplexes in the genome, created using the parallel motif, was determined using Roberts and Crothers empirical equations (37) (equations 5–7). The stability of the corresponding anti-parallel triplexes is more difficult to determine and depends on the concentration of divalent cations, and on the possible existence of alternative structures (like the G-DNA). However, recent experiments by Eritja and co-workers (38) suggest that, in general, even for the worst cases, anti-parallel triplexes are only a few degrees less stable than the corresponding parallel triplexes at pH 4.5:
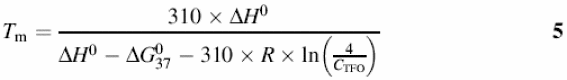
where CTFO is the concentration of target + triplex-forming oligonucleotides. The enthalpy (ΔH0) and Gibbs free energy (ΔG0) are evaluated (in kcal/mol) using equations 6 and 7:
ΔH0 = –4.9(CC) – 8.9(TC + CT) – 7.4(TT)6
where XX means the number of dinucleotides of this particular type in the TFO:
ΔG037 = –3.00(C) – 0.65(T) + 1.65(CC) + 6.0 + (C)(pH – 5.0)[1.26 – 0.08(CC)]7
where (X) means the number of nucleotides of type X in the TFO.
For discussion purposes we have considered three different TFO concentrations (µM and nM), assuming very dilute concentration for the TTS. As a reference, state of the art delivery methodologies allow the delivery of up to 20– 70 µmol of TFO in the interior of the nuclei (31). Two different pH values were considered, physiological (7.0) and acidic (4.5). The use of the latter provides insight into the stability of triplexes formed with TFOs containing modified nucleotides. Triplexes with melting temperature above 50°C were considered as stable. This high temperature implies that we are using a conservative threshold of triplex stability, and probably more triplexes than those detected here can be stable under physiological conditions.
To determine the differential stability of triplexes in the human genome we compared our calculations with two background models. The first (labelled Random) assumes equal populations of A and G and equal possibility for Pur/Pur, Pur/Pyr and Pyr/Pur transitions, the second one (labelled Random H.G) was generated with the restrictions necessary to maintain the ratio A/G and the transition probabilities at the values found in the human genome. In both cases, 50 random genomes with the same length as the human genome were generated.
All the software developed here for the localization and analysis of TTS is available as C-programs upon request to the authors.
RESULTS AND DISCUSSION
The amount of nucleotides appearing in TTS in the human genome is several times larger (Fig. 2) than that expected from a random distribution (see Materials and Methods), and the difference increases as does the size of the tracks. Thus, TTS longer than 20 nt are very rare in random systems, whereas TTS longer than 30 nt are commonly found in the human genome. The existence of such a large density of TTS (or nucleotides in TTS with respect to total nucleotides) could be due in principle to two different phenomena: (i) massive duplication of a small number of TTS (this will imply a large amount of TTS in repetitive DNA) and (ii) the existence of a subtle biological effect related to these sequences. We have investigated both possibilities.
Figure 2.

Number of TTS tracks of different lengths found in the human genome and in random models. The profile found in human genome is compared with that found in analytical or numerical random models (see text for details).
The density of TTS (defined as the number of nucleotides in TTS related to the total number of nucleotides, i.e. the probability of a nucleotide being part of a TTS) for different chromosomes is similar (data not shown), the largest density of TTS is found in the 19 chromosome and the lowest in the Y chromosome. The analysis of the base composition shows, in general, a larger population of adenines than guanines. For example, for TTS of 15 nt in length there are ∼60% adenines, and for TTS of 30 nt or more the percentage of adenines increase to almost 80%. It is worth noting that adenines constitute ∼60% of the purines in our genome, which means that in long tracks, adenines are over-represented in TTS. We should note that the larger population of adenines in TTS is favourable from the point of view of triplex formation, since Watson–Crick guanines are targeted (Fig. 1) either by Hoogsteen cytosines (leading to a strong pH dependence of the triplex) or by reverse Hoogsteen guanines [leading then to a strong competition in the TFO between single-stranded (that are needed for triplex formation) and tetraplexes] (5).
To analyse whether or not TTS are located in regions of importance for anti-gene therapy, we divided (see Materials and Methods) the human genome into repeated regions (∼50% of the genome), genes (the 10 000 of the RefGene collection), best human–mouse conserved regions (∼5% of the genome) and general regulatory regions (2000 bases up and downstream of the 10 000 selected genes). The profile of TTS found for the genes (Fig. 3) reproduces very well the corresponding profile found for the total human genome. On the contrary, the highly conserved human–mouse region shows a lower ability to make triplex than the average genome, which can be partly explained by the fact that a good portion of highly conserved regions are protein-coding regions, where, as noted below, a low quantity of TTS is found. Interestingly, repeated and regulatory regions exhibit larger concentrations of long TTS than the average for the genome (Fig. 3).
Figure 3.

Probability (in %) of a nucleotide being part of TTS of different lengths in different regions of the human genome.
In order to analyse in more detail the presence of TTS in regions of special relevance, we divided the gene region into: (i) exons, (ii) coding sequences (i.e. the exons after removing UTR), (iii) introns, (iv) promoter regions (100 nt upstream of the beginning of the gene), (v) regulatory region upstream (1900 nt) promoters and (vi) downstream (2000 nt) regulatory regions. Very interestingly (Fig. 3), only two lines appear below that of the global human genome: the exon and the coding regions. This is likely to reflect the compositional bias in the nucleic acid sequence associated with the coded polypeptide. The relative number of nucleotides in TTS is larger in all regulatory regions than in the whole genome (Fig. 4). Interestingly, the very short promoter region contains a very large concentration of nucleotides in TTS (Fig. 4), indicating that the crucial region of control of gene expression can be easily targeted for triplex formation. The existence of this large concentration of TTS in a key region of the genome strongly suggests some subtle biological function for this type of sequence, which might be related to some structural properties which can favour interaction of DNA with control proteins. In any case, for the purpose of this paper we must emphasize that the existence of a large density of TTS in the promoter region provides a tremendous opportunity for the use of triplex-based approaches in anti-gene therapies. In fact, many genes of possible therapeutic impact show extremely large TTS (>40 nt) in the promoter region. Examples are SCA1 (the gene causing type 1 spinocerebellar ataxia), ATP6V1B1 (the gene encoding for ATPase related to sensorineural deafness), PAWR (a key gene in apoptotsis), HIPK3 (a kinase involved in multidrug-resistant cells), SOX10 (mutations in this gene leads to Waardenburg–Hirschsprung disease), NOVA1 (an onconeural antigen related to breast and small cell lung cancer), and many other examples that will be discussed in more detail in a further communication.
Figure 4.

Probability (in %) of a nucleotide being part of TTS of different lengths in different parts of the gene region.
Repetitive regions represent ∼49% of the human genome (1,2), and are clearly those where sequencing is more difficult, and where a larger portion of gaps in the genome sequence exists. Our analysis shows that, as a whole, repetitive regions have a high density of TTS, just after the TTS-rich promoter region, a result that is not unexpected considering that a part of the repetitive DNA is defined by polypurine tracks. In order to study more precisely the distribution of TTS in repetitive regions, we analysed TTS density in different classes of repetitive DNAs: (i) long interspersed elements (LINE), (ii) small interspersed elements (SINE), (iii) long terminal repeat (LTR), (iv) transposons and (v) unclassified repeated DNA (low complexity, simple repeated, satellites and others). LTR and LINE regions show a density of TTS similar to that of the ‘no repeated’ part of the genome (Fig. 5). DNA transposons show a TTS density lower than the no-repeated part of the genome, denoting their origins as coding sequences (see discussion above). SINEs present a density of TTS larger than that of the non-repetitive part of genome, and quite interestingly, a TTS versus length of the track (in the sequence-length considered) profile quite different to the exponential decay found in the other cases (Fig. 5). This suggests that at least a part of SINEs was generated by massive duplication of a number of different short/medium sequences of DNA, biasing the distribution of possible TTS. Finally, unclassified repeated sequences also show a non-exponential decay with length. This is due to the fact that simple repeated sequences (obtained by massive replication of small DNA fragments) and low complexity DNA [rich in polypurine sequences (1,2)] are incorporated within this family of repetitive DNA.
Figure 5.

Probability (in %) of a nucleotide being part of TTS of different lengths in different parts of the repetitive DNA.
The next step in our analysis was to follow the presence of SNPs in TTS sequences. SNPs are the major source of genetic variability in humans (39). Thus, knowing the impact of SNPs in TTS may be of biomedical interest for the purpose of designing individual-adapted therapies. Results in Figure 6 show that the probability of a position in the human genome to be part of a TTS is largely increased if it exhibits polymorphism. No obvious reason was found for this interesting behaviour other than mutations will be made more easily, or worse repaired, in TTS, or that transient triplexes are formed in polypurine tracks leading to an increase in the mutagenic rate (22,23). However, for biomedical and biotechnological purposes, what is clear is that this finding reinforces the interest of anti-gene strategies in individual-directed therapies, as taking into account the structural variability of the protein in the process of drug design is much more complex than designing oligonucleotides with varying sequences.
Figure 6.

Probability (in %) of a nucleotide being part of TTS containing SNPs (results for both snpNih and snpTsc databases are included). The base line corresponding to the whole human genome is displayed for comparison.
Previous analysis shows that TTS are more abundant in the genome than expected, and that they are particularly frequent in regions of special relevance for gene expression, suggesting a priori that anti-gene strategies may be very promising. However, to assess the real biomedical and biotechnological impact of triplex strategies two questions must be answered: (i) what is the selectivity TFO (i.e. what is the degree of uniqueness of a given TTS in the genome) and (ii) how stable are the triplexes formed? To answer the first question we computed how many of all the possible combinations of TTS of a given length are present in the human genome, and in the gene region (genes + regulatory regions). As shown in Figure 7, the human genome samples all possible sequence space in TTS shorter than 17, and no selectivity is possible for the complementary TFOs. As noted in Figure 7, the percentage of TTS sampled in the human genome decreases below 50% for TTS longer than 21 nt, and TTS are almost unique for lengths >26 nt, making it possible to design 100% specific TFOs. If only the gene region is considered in the analysis, the minimum length of the TTS needed to define specific TFOs is smaller [19 (one secondary interaction is expected on average) and 24 (no secondary interaction expected)]. In summary, selectivity can be reached with relatively small oligonucleotides which can be easily introduced inside the cell (6,7,31,32).
Figure 7.

Coverage of the space of sequences of TTS in the human genome and in the gene region.
In order to determine the stability of triplexes under physiological conditions we follow Crother’s empirical rules (see Materials and Methods), which allow us to determine the melting temperature of parallel triplexes based on the triplex sequence, concentration of oligonucleotides (i.e. the amount of TFO that can be internalized at the nuclei), the triplex length and the pH (acidic pH makes anti-parallel triplexes more stable). The stability of parallel triplexes largely depends on the pH. Thus, at acidic pH (4.5), ∼3% of the human genome is found in TTS susceptible to form stable triplexes (melting temperature >50°C) at micromolar concentrations of TFO. When the pH is raised to 7.0 only 0.2% of the human genome can form stable triplexes. For anti-parallel triplexes, no pH dependence is expected, and on the basis of recent experiments by Eritja’s group (38) the range of stabilities are expected to be those corresponding to parallel triplexes at moderately acid pH (∼5–6).
The human genome shows a surprisingly good ability to form stable triplexes, much better than that expected from numerical random models (data not shown, but available upon request) at any pH, but the difference is especially large for neutral pH, indicating that triplexes in the human genome are less sensitive to the pH than expected. The larger density of stable triplexes is found in the regulatory regions, in particular in promoter regions, where triplexes are stable even in unfavourable pH conditions (data not shown, but available upon request). In summary, our results strongly suggest that there is a large percentage of the human genome that can lead, under physiological conditions, to stable triplexes. In fact, our calculations strongly suggest that in the design of TFOs the requirement for selectivity is stricter than the requirement for stable triplexes.
CONCLUSIONS
In silico analysis of the human genome allows us to identify regions that can lead to triplex formation when a suitable TFO is available. The regions susceptible to form triplex (TTS) are more common in the human genome than expected by random models, even when these models are adapted to the composition of the human genome. A large density of TTS, which can yield stable and specific triplexes under physiological conditions, are found in promoter regions, opening interesting possibilities for the use of triplexes in the control of gene expression. Also, interestingly, TTS present an unusually large number of SNPs, which suggests that triplex strategies may be of interest in individual-oriented therapies (in fact, several examples are found of SNPs located in large TTS segments located in promoter regions). Overall, our results show the large possibilities of triplex technology in the biomedical and biotechnological scenario.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Roderic Guigó for many helpful discussions. This work has been supported by the Spanish Ministry of Science and Technology (PM99-0046 and BIO2003-06848). R.G. is supported by a fellowship of the IRBB-PCB.
REFERENCES
- 1.International Human Genome Sequencing Consortium (2001) Initial sequencing and analysis of the human genome. Nature, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- 2.Venter J.C., Adams,M.D., Myers,E.W., Li,P.W., Mural,R.J., Sutton,G.G., Smith,H.O., Yandell,M., Evans,C.A., Holt,R.A. et al. (2001) The sequence of the human genome. Science, 291, 1304–1351. [DOI] [PubMed] [Google Scholar]
- 3.Hammond S.M., Caudy,A.A. and Hannon,G.J. (2001) Post-transcriptional gene silencing by double-stranded RNA. Nature Rev. Genet., 2, 110–119. [DOI] [PubMed] [Google Scholar]
- 4.Stein C.A. and Cheng,Y.C. (1993) Antisense oligonucleotides as therapeutic agents–is the bullet really magical? Science, 261, 1004–1012. [DOI] [PubMed] [Google Scholar]
- 5.Robles J., Grandas,A., Pedroso,E., Luque,F.J., Eritja,R. and Orozco,M. (2002) Nucleic acid triple helices: stability effects of nucleobase modifications. Curr. Org. Chem., 6, 1333–1368. [Google Scholar]
- 6.Soyfer V.N. and Potaman,V.N. (1996) Triple-Helical Nucleic Acids. Springer-Verlag: New York. [Google Scholar]
- 7.Giovannangeli C. and Hélène,C. (2000) Triplex technology takes off. Nat. Biotechnol., 18, 1245–1246. [DOI] [PubMed] [Google Scholar]
- 8.Knauert M.P. and Glazer,P.M. (2001) Triplex forming oligonucleotides: sequence-specific tools for gene targeting. Hum. Mol. Genet., 10, 2243–2251. [DOI] [PubMed] [Google Scholar]
- 9.van Dongen M.J.P., Doreleijers,J.F., van der Marel,G.A., van Boom,J.H., Hilbers,C.W. and Wijmenga,S.S. (1999) Structure and mechanism of formation of the H-y5 isomer of an intramolecular DNA triple helix. Nature Struct. Biol., 6, 854–859. [DOI] [PubMed] [Google Scholar]
- 10.Pauling L. and Corey,R.B. (1953) A proposed structure for the nucleic acids. Proc. Natl Acad. Sci. USA, 39, 84–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Felsenfeld G., Davis,D.R. and Rich,A. (1957) Formation of a three-stranded polynucleotide molecule. J. Am. Chem. Soc., 79, 2023–2024. [Google Scholar]
- 12.Scaria P.V. and Shafer,R.H. (1996) Calorimetric analysis of triple helices targeted to the d(G3A4G3).d(C3T4C3) duplex. Biochemistry, 35, 10985–10994. [DOI] [PubMed] [Google Scholar]
- 13.Chandler S.P. and Fox,K.R. (1996) Specificity of antiparallel DNA triple helix formation. Biochemistry, 35, 15038–15048. [DOI] [PubMed] [Google Scholar]
- 14.Shields G.A., Laughton,C.A. and Orozco,M. (1997) Molecular dynamics simulations of the d(T·A·T) triple helix. J. Am. Chem. Soc., 119, 7463–7469. [Google Scholar]
- 15.Jiménez E., Vaquero,A., Espinás,M.L., Soliva,R., Orozco,M., Bernués,J. and Azorin,F. (1998) The GAGA factor of Drosophila binds triple-stranded DNA. J. Biol. Chem., 273, 24640–24648. [DOI] [PubMed] [Google Scholar]
- 16.Strobel S.A. and Dervan,P.B. (1992) Triple helix-mediated single-site enzymatic cleavage of megabase genomic DNA. Methods Enzymol., 216, 309–321. [DOI] [PubMed] [Google Scholar]
- 17.Zain R., Marchand,C., Sun,J., Nguyen,C.H., Bisagni,E., Garestier,T. and Hélène,C. (1999) Design of a triple-helix-specific cleaving reagent. Chem. Biol., 6, 771–777. [DOI] [PubMed] [Google Scholar]
- 18.Luo Z., Macris,M.A., Faruqi,A.F. and Glazer,P.M. (2000) High-frequency intrachromosomal gene conversion induced by triplex-forming oligonucleotides microinjected into mouse cells. Proc. Natl Acad. Sci. USA, 97, 9003–9008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Havre P.A., Gunther,E.J., Gasparro,F.P. and Glazer,P.M. (1993) Targeted mutagenesis of DNA using triple helix-forming oligonucleotides linked to psoralen. Proc. Natl Acad. Sci. USA, 90, 7879–7883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Majumdar A., Khorlin,A., Dyatkina,N., Lin,F.L., Powell,J., Liu,J., Fei,Z., Khripine,Y., Watanabe,K.A., George,J., Glazer,P.M. and Seidman,M.M. (1998) Targeted gene knockout mediated by triple helix forming oligonucleotides. Nature Genet., 20, 212–214. [DOI] [PubMed] [Google Scholar]
- 21.Barre F.X., Ait-Si-Ali,S., Giovannangeli,C., Luis,R., Robin,P., Pritchard,L.L., Hélène,C. and Harel-Bellan,A. (2000) Unambiguous demonstration of triple-helix-directed gene modification. Proc. Natl Acad. Sci. USA, 97, 3084–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang G., Seidman,M.M. and Glazer.P.M. (1996) Mutagenesis in mammalian cells induced by triple helix formation and transcription-coupled repair. Science, 271, 802–805. [DOI] [PubMed] [Google Scholar]
- 23.Vasquez K.M., Narayanan,L. and Glazer,P.M. (2000) Specific mutations induced by triplex-forming oligonucleotides in mice. Science, 290, 530–533. [DOI] [PubMed] [Google Scholar]
- 24.Duval-Valentin G., Thuong,N.T. and Hélène,C. (1992) Specific inhibition of transcription by triple helix-forming oligonucleotides. Proc. Natl Acad. Sci. USA, 89, 504–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cooney M., Czernuszewicz,G., Postel,E.H., Flint,S.J. and Hogan,M.E. (1988) Site-specific oligonucleotide binding represses transcription of the human c-myc gene in vitro. Science, 241, 456–459. [DOI] [PubMed] [Google Scholar]
- 26.Grigoriev M., Praseuth,D., Robin,P., Hemar,A. and Saison-Behmoaras,T. (1992) A triple helix-forming oligonucleotide-intercalator conjugate acts as a transcriptional repressor via inhibition of NF kappa B binding to interleukin-2 receptor alpha-regulatory sequence. J. Biol. Chem., 267, 3389–3395. [PubMed] [Google Scholar]
- 27.Joseph J., Kandala,J.C., Veerapanane,D., Weber,K.T. and Guntaka,R.V. (1997) Antiparallel polypurine phosphorothioate oligonucleotides form stable triplexes with the rat alpha1(I) collagen gene promoter and inhibit transcription in cultured rat fibroblasts. Nucleic Acids Res., 25, 2182–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Postel E.H., Flint,S.J., Kessler,D.J. and Hogan,M.E. (1991) Evidence that a triplex-forming oligodeoxyribonucleotide binds to the c-myc promoter in HeLa cells, thereby reducing c-myc mRNA levels. Proc. Natl Acad. Sci. USA, 88, 8227–8231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Young S.L., Krawczyk,S.H., Matteucci,M.D. and Toole,J.J. (1991) Triple helix formation inhibits transcription elongation in vitro. Proc. Natl Acad. Sci. USA, 88, 10023–10026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Faria M., Wood,C.D., Perrouault,L., Nelson,J.S., Winter,A., White,M.R., Hélène,C. and Giovannangeli,C. (2000) Targeted inhibition of transcription elongation in cells mediated by triplex-forming oligonucleotides. Proc. Natl Acad. Sci. USA, 97, 3862–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ebbinghaus S.W., Vigneswaran,N., Miller,C.R., Chee-Awai,R.A., Mayfield,C.A., Curiel,D.T. and Miller,D.M. (1996) Efficient delivery of triplex forming oligonucleotides to tumor cells by adenovirus-polylysine complexes. Gene Ther., 3, 287–297. [PubMed] [Google Scholar]
- 32.Zendegui J.G., Vasquez,K.M., Tinsley,J.H., Kessler,D.J. and Hogan,M.E. (1992) In vivo stability and kinetics of absorption and disposition of 3′ phosphopropyl amine oligonucleotides. Nucleic Acids Res., 20, 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karolchik D., Baertsch,R., Diekhans,M., Furey,T.S., Hinrichs,A., Lu,Y.T., Roskin,K.M., Schwartz,M., Sugnet,C.W., Thomas,D.J., Weber,R.J. and Kent,W.J. (2003) The UCSC Genome Browser Database. Nucleic Acids Res., 31, 51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith A.F. (1999) Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr. Opin. Genet. Dev., 9, 657–663. [DOI] [PubMed] [Google Scholar]
- 35.Sherry S.-T., Ward,M.-H., Kholodov,J., Baker,L., Pham,L., Smigielski,E.M. and Sirotkin,K. (2001) dbSNP: The NCBI database of genetic variation. Nucleic Acids Res., 29, 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ussery D., Soumpasis,D.M., Brunak,S., Staerfeldt,H.H., Worning,P. and Krog,A. (2002) Bias of purine stretches in sequenced chromosomes. Comput. Chem., 26, 531–541. [DOI] [PubMed] [Google Scholar]
- 37.Roberts R.W. and Crothers,D.M. (1996) Prediction of the stability of DNA triplexes. Proc. Natl Acad. Sci. USA, 93, 4320–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jaumot J., Eritja,R., Tauler,R. and Gargallo,R. (2003) Resolution of parallel and antiparallel oligonucleotide triple helices formation and melting processes by means of multivariate curve resolution. J. Biomol. Struct. Dyn., 21, 267–278. [DOI] [PubMed] [Google Scholar]
- 39.Collins F.S., Brooks,L.D. and Chakravarti,A. (1998) A DNA polymorphism discovery resource for research on human genetic variation. Genome Res., 8, 1229–1231. [DOI] [PubMed] [Google Scholar]