

Abstract
The first annual workshop for preclinical and clinical development of radiosensitizers took place at the National Cancer Institute on August 8–9, 2012. Radiotherapy is one of the most commonly applied and effective oncologic treatments for solid tumors. It is well recognized that improved clinical efficacy of radiotherapy would make a substantive impact in clinical practice and patient outcomes. Advances in genomic technologies and high-throughput drug discovery platforms have brought a revolution in cancer treatment by providing molecularly targeted agents for various cancers. Development of predictive biomarkers directed toward specific subsets of cancers has ushered in a new era of personalized therapeutics. The field of radiation oncology stands to gain substantial benefit from these advances given the concerted effort to integrate this progress into radiation therapy. This workshop brought together expert clinicians and scientists working in various disease sites to identify the exciting opportunities and expected challenges in the development of molecularly targeted agents in combination with radiation therapy.
The overarching goal of radiation therapy is to achieve the most optimal therapeutic index. Most innovations in the past half-century have been focused on ways of delivering treatment so that normal organ toxicity can be minimized while a therapeutic dose can be delivered. Advances in computational technologies and imaging innovations have led to marked improvements in radiation planning and therapeutic delivery. The incorporation of the computed tomography scan for treatment planning has brought treatment delivery from two-dimensional to three-dimensional planning. Consequently, the advances in both treatment planning and delivery have reduced toxicity, allowed escalation of dose to the tumor, and consequently improved clinical outcomes (1–4). Although there continue to be new advances in how treatment is planned and delivered, advances in the field of radiation oncology have gradually met a plateau where the therapeutic index cannot be further improved because of physical limitations and dose-limiting structures. For additional progress to be made, technological innovations must be complemented by biological innovations, such as development of novel radiosensitizing agents and biology-driven patient selection. In this commentary, we summarize the collective scientific output from the first annual workshop for preclinical and clinical development of radiosensitizers at the National Cancer Institute on August 8–9, 2012, and provide recommendations for the rational and accelerated development of radiation-based combination therapy (see Table 1).
Table 1.
Summary of workshop recommendations for the accelerated development of novel targeted radiation enhancers
| Determination of agent activity | |
| 1.1 | Agents of interest should enhance radiation effects through either synergistic or additive mechanisms but, if not, should at least have single-agent or combination activity with chemotherapy. |
| 1.2 | Preclinical testing is crucial to provide evidence that the agent of interest has radiation enhancement effect; At least two cell lines from the same disease site should be required, whereas in vivo animal testing should be used to further demonstrate radiation enhancement effect whenever a suitable animal model for the cancer type exists. |
| 1.3 | When generation of in vitro or in vivo preclinical data is not feasible, strong justification must be provided for the inability to perform these studies before proposing a clinical study. |
| Prioritizing agent development | |
| 2.1 | The development of radiation-enhancing agents should be prioritized when biomarker-based patient selection is available. |
| 2.2 | Agents without validated predictive biomarkers available for clinical testing could be brought into clinical testing but with the mandate that there are clear plans for concurrent preclinical research and clinical development of predictive biomarkers from pretreatment tissue specimens. |
| 2.3 | Concurrent development of predictive biomarkers should be a priority during the preclinical and early clinical phases of testing followed by subsequent clinical validation. Clinical studies must mandate pretreatment tumor biopsy and/or serum collection, with strong consideration given to acquisition of serial tissue collection during early therapy and at the time of recurrence. |
| 2.4 | Understanding the proper sequencing of combining targeted agents with radiation therapy will be important before carrying out large clinical trials. |
| Safety | |
| 3.1 | Phase I studies are critical to determine the safety of combining a new agent with radiation (or chemoradiation), but this should only be a site-specific and not a disease-specific requirement. For instance, clinical testing of a novel agent with radiation for rectal cancer need not be repeated for prostate cancer, cervical cancer, or other pelvic tumors. |
| 3.2 | A minimal 30-day observational period after completion of radiotherapy should be used to gauge the acute toxicities of a novel treatment. |
| 3.3 | Late toxicity should be monitored in early-phase clinical trials even when acute toxicity is used as a primary safety endpoint. |
| Clinical trial designs | |
| 4.1 | Alternative innovative phase I designs (eg, time-to-event continuous reassessment method) should be considered to improve study efficiency and cumulative safety analysis. |
| 4.2 | An efficient way to rapidly test novel agents in combination with radiation is through a modular clinical trial platform, in which several agents/combinations are individually tested in parallel noncomparative arms. |
| 4.3 | The economic benefits and a platform-based clinical trial design should be recognized and prioritized. |
| Regulatory | |
| 5.1 | Both the US Food and Drug Administration (FDA) and the pharmaceutical industry should acknowledge that combination trials with radiation can help to dramatically improve cure rates of nonmetastatic patients, representing a new pathway for expediting the drug approval process. The FDA should issue an advisory clarifying requirements for approval of an agent in combination with radiotherapy or with a previously established chemotherapy and radiation therapy combination. |
| 5.2 | The Cancer Therapy Evaluation Program (CTEP) should consistently issue mass solicitations for drug combination trials that have specific calls for proposals in combination with radiation and simultaneously facilitate development of compounds as radiosensitizers within the CTEP portfolio. |
| 5.3 | The development of novel radiation sensitizers should be financially supported consistent with other priorities in cancer research. |
Challenges and Opportunities in Developing Molecularly Targeted Therapies in Combination With Radiation
The choice of agents to combine with radiation is an important consideration. New agents that are developed in combination with radiotherapy should have: 1) a single-agent activity, and/or 2) a radiation-enhancing effect, either as a direct radiation sensitizer or as a synthetically lethal combination when administered with radiation. The definition of radiation enhancement is complex because it may involve both a direct radiation-sensitizing effect and indirect effects (5). We recognize that there are specific challenges in developing radiosensitizing agents. Before embarking on large-scale clinical trials, adequate preclinical data should be acquired to support such hypotheses.
Preclinical Testing and Model Systems
Mechanisms of action must be explored using preclinical experi ments. When embarking on preclinical studies evaluating the combination of molecular targeted therapies and radiation, the first step is to determine the appropriate system (cell culture or animal models) in which to evaluate the combination. In vitro clonogenic survival assays or apoptotic assays may be needed to demonstrate preclinical activity of agents that may have direct cytotoxic effects on the cells themselves (eg, tyrosine kinase inhibitors blocking tumor EGFR mutations) to illustrate the additive effect of the agent over radiation alone. Agents that act upon the tumor microenvironment to influence tumor growth (eg, antiangiogenic agents, anti-HGF antibodies) may require in vivo assays to establish their activity in combination with radiation.
A spectrum of in vitro and in vivo models may be used for testing the preclinical efficacy of combining radiation therapy with molecularly targeted agents. In vitro models could include the use of cell lines in tissue culture or in three-dimensional cultures. In vivo model systems include cell line-derived xenografts (6), patient tumor-derived xenografts, and genetically engineered mouse models that form tumors spontaneously by conditionally expressing a gene of interest (7). However, no matter how intricate these systems are, none fully reflects the degree of diversity and complexity seen in human cancers. For certain disease sites (eg, salivary gland tumors) or agents (eg, immunomodulators such as anti–CTLA-4 antibodies), there are simply no appropriate preclinical models.
Although inadequate to address all the nuances and complexities of human physiology and cancer biology, preclinical studies remain a valuable tool to confirm mechanisms of action and radioenhancement properties when conducted appropriately. We feel that at a minimum, there should be in vitro data from at least two cell lines derived from the disease of interest (eg, lung cancer cell lines for a lung cancer proposal) when they are available, and data should not be extrapolated from unrelated cell lines. Because it is important to study host-tumor effects and to confirm findings in a more complex system, in vivo animal studies are generally recommended; therefore, whenever established animal model systems are available, they should be used to support clinical trial proposals. However, because no model system perfectly recapitulates the human condition and each system has its own inherent limitations, there is no panel consensus on specific recommendations about the best use of the in vivo system. At a minimum, subcutaneous flank or intramuscular thigh models may be sufficient to test in vivo efficacy to verify cell culture results. When there is not a standard or established model system that can be applied to the specific cancer type, in vivo preclinical validation may not be required for a clinical trial proposal. In these situations, data from other disease sites with the same agent/radiation combinations could justify exploratory studies in the disease site of interest.
Selection of Candidate Agents of Interest
Given the number of candidate agents in the pipeline or in early clinical testing, it will be nearly impossible to test every combination with radiation. Many novel agents have mechanisms of action that are scientifically poised to serve as radiation enhancers. However, a comprehensive listing of the data to support this statement is beyond the scope of this article. Although these agents target various molecular pathways in a diversity of disease sites, we believe many of the potentially radiation-sensitizing drugs of interest can be broadly placed into two groups based on the maturity of translational and clinical data: agents with established biomarkers of response or agents with biomarkers in development. Specific examples of these two groups of agents are listed in Table 2.
Table 2.
Select examples of novel agents based on the currently available biomarkers for patient selection
| Agents with established biomarkers | ||
| Drug | Targets | Biomarker |
| Erlotinib | EGFR | EGFR mutations |
| Crizotinib | ALK/ROS1 fusion genes | EML4/ALK and ROS1 fusion |
| Vemurafenib, dabrafenib | BRAF | BRAF mutations |
| MK-1775 | Wee-1 | TP53 mutations |
| MK-8776 | Chk-1 | TP53 mutations |
| Nutlin-3 | MDM2 | TP53 WT |
| ARQ-197, XL-184 | c-MET | c-MET mutations |
| Trametinib, salumetinib | MEK1/2 | K-ras mutations |
| BEZ235 | PI3K/mTOR/DNA-PK | K-ras/PIK3CA/PTEN mutations |
| AZD1480 | JAK2 | TEL-JAK2 fusion |
| Trastuzumab | Her-2 | Her-2 overexpression |
| Tamoxifen, aromatase inhibitors | ER | ER expression |
| Agents with promising biomarkers in development | ||
| Drug | Targets | Potential Biomarker |
| Bevacizumab, aflibercept | VEGF | VEGF-A |
| Vorinostat | HDAC | — |
| Azacytidine | DNA, DNMTs | — |
| ABT-888 | PARP 1 or 2 | — |
| Cisplatin | DNA | ERCC1 |
| 5-FU | Thymidylate synthase | — |
| Gemcitabine | Ribonucleotide reductase | hENT1 |
| Bortezomib | 26S proteasome | — |
| RO4929097 | Notch | — |
| BMS 833923 | Smoothened | — |
| Fostamatinib | SYK | — |
| Cetuximab | EGFR | Kras mutation is a negative prognostic factor |
| Ipilimumab | CTLA-4 | CTLA-4 expression |
| BMS-936558 | PD-1 | PD-1 expression |
Agents With Established Biomarkers of Clinical Benefit
Agents in this group have some clinical or preclinical evidence to support specific biomarkers that are predictive for clinical benefit. These biomarkers are mostly tumor-specific biomarkers, which, when present, can be used to select optimal patients for specific treatment. These agents are mostly inhibitors targeting activated tyrosine kinases, serine/threonine kinases, cell cycle regulators, and/or hormonal receptors. For most of these compounds, there is compelling clinical evidence that the response to therapy is predicted by the presence of specific biomarkers reflecting a clear on-target effect [EGFR mutations (8), ALK or ROS1 translocation (9,10), Her-2 overexpression (11), estrogen receptor expression (12)], whereas some other agents in early development lack such clinical association, having only preclinical data [chk1/wee1-TP53 mutation(13)]. Many of these agents demonstrate considerable activity in appropriately selected patient populations but appear relatively ineffective when used in an unselected manner (Figure 1A). To accelerate development, we recommend the prioritization of radiosensitizer development in compounds or classes for which biomarker-based patient selection is currently available.
Figure 1.
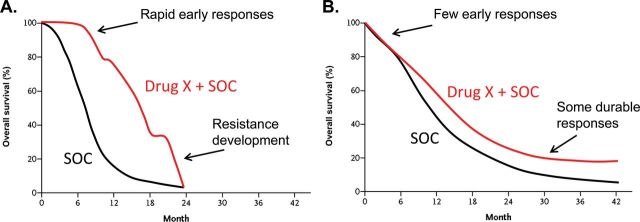
Hypothetical examples of Kaplan–Meier overall survival curves comparing drug developments in a selected patient population based on biomarkers vs unselected patient population. A) Like many drug trials directed at specific populations of patients based on biomarker selection, the initial part of the curve separates early in the targeted agent group (Drug X) compared with the standard of care (SOC) arm, but eventually resistance develops in the majority of these patients. B) For a trial testing the efficacy of Drug X in an unselected group of patients, practically no real separation is seen in the curves between the experimental (Drug X + SOC) and SOC arms early on; the curves eventually split, and a statistically significant separation occurs later in the trial, with some patients having durable responses.
Agents With Biomarkers in Development
There are many other agents that currently do not have validated biomarkers (Table 2), despite having a specific target known to be important for cancer development, growth, invasiveness, or metastatic spread. Although there may be gene signatures (eg, mRNA expression, methylation, microRNA) that may be associated with response from retrospective analysis of clinical samples, these signatures should not be used for patient selection until the completion of prospective validation studies. The disease-free survival results from these studies often differ substantially from agents with established biomarkers (Figure 1B). Of note, the radiosensitization properties of any given class of agents might still be effective in a nonselected patient population if the mechanism of radiation enhancement is not known, but the degree of radiosensitization is profound in preclinical studies. In this case, testing a novel agent with radiation therapy in the absence of a biomarker may be appropriate. Thus this developmental prioritization requires ongoing refinement. Although we recognize that there may be some caveats with the broad grouping of these compounds, we believe it encompasses the majority of agents to be brought forward into clinical testing with radiotherapy.
Strategies to Develop Predictive Biomarkers for Agents of Interest
To bring agents with established biomarkers into clinical testing with radiation, there must be some preclinical evidence that demonstrates evidence for synergy with radiotherapy as previously stated. Early phases of testing should focus on confirming activity and identifying mechanisms of resistance. To improve the likelihood of clinical success, concurrent development of potentially predictive biomarkers should be a priority during the preclinical and early clinical phases of testing followed by subsequent clinical validation. Clinical studies must mandate pretreatment tumor biopsy with strong consideration given to acquisition of additional tissue early during therapy and again at the time of recurrence with appropriate safety and incentives for such a priority. Paired samples of pretreatment and early on-treatment biopsies might provide not only biomarkers of treatment resistance detected upfront but also dynamically changing biomarker profiles suggestive of inducible molecular resistance pathways that might overcome treatment efficacy.
Determining the Sequence of Combining Radiation
It is often assumed that sensitizers must be added concurrently with radiation for synergistic effect. However, this may not always be the case. Depending on the mechanism of action of the targeted drugs, concurrent administration may even have antagonistic effects. For instance, because radiation cytotoxic effects are highly dependent on the cell-cycle phase, with the greatest sensitivity occurring at the G2/M phase, and the greatest resistance in the G1/S phase (14), drugs that cause cell-cycle arrest or prolong cells in the radioresistant phase of the cell cycle may jeopardize the radiation effect. Therefore the appropriate timing and sequencing of the combination must be considered as well. Although Bonner et al. demonstrated survival benefit in head and neck cancers treated with cetuximab and radiation compared with radiation alone (15), adding cetuximab to chemoradiation did not improve survival rates over chemoradiation alone, as shown in RTOG 0522 (16). Several theories have been advanced to explain why addition of cetuximab to chemoradiation did not show survival benefit, including an overlapping mechanism of action between cetuximab and cisplatin as radiation sensitizers. However, the EXTREME trial, which used cetuximab maintenance until disease progression, suggests cetuximab may be best used as a maintenance monotherapy after completing the primary treatment. Additionally, this may be a disease-specific phenomena because the phase II trial in lung cancer using cetuximab and a different combination chemotherapy regimen (carboplatin/paclitaxel) was promising (17) and is currently being tested in RTOG 0617. Therefore, mechanisms of interaction between targeted agents, radiation, and/or chemoradiotherapy should be carefully evaluated in preclinical models and in early-phase clinical trials before definitive studies are undertaken.
Phase I Clinical Safety
As is true for all preclinical models, demonstration of radiation enhancement in the laboratory may not translate to activity in the clinical setting. Thus clinical testing in patients is required to demonstrate both safety and subsequent efficacy. Many of the agents being considered as radiosensitizers are also being tested alone or in combination with standard chemotherapy. Most of these clinical trials are conducted in the advanced disease setting after multiple lines of standard therapeutics. The safety data from these studies, although important to identify unique ancillary testing requirements (eg, electrocardiogram, echocardiography, ophthalmic exams, specific blood tests), are not sufficient to confirm the dose and safety when used in combination with radiation. Thus, for every concept incorporating a new agent with radiotherapy, a phase I study to demonstrate dose and safety must be performed. This could be met by a standard single-arm phase I study or a safety run-in component in a phase I/II trial. Combination therapy at completely different anatomic sites cannot be used to generalize the safety of the combination. However, data obtained where a drug is evaluated in combination with radiotherapy in the same anatomic region (eg, esophageal cancer and lung cancer) could be used. In summary, safety data of phase I trials combining radiation and novel agents should be more organ/location specific rather than cancer specific.
The observational period to determine the toxicity of radiation combination trials must balance the time it takes for acute/subacute effects to take place and the practicality of carrying out studies in a timely fashion under time, patient, and logistical constraints. This timing may also differ between the organ at risk and individual susceptibility. Currently, a 3 + 3 design (n = 3 patients to enroll at each dose level, with toxicity to keep below 33%, or n = 1 patient or less to establish maximum tolerated dose [MTD]) is the traditional approach for phase I clinical trial designs. Unfortunately, with the requirement of complete toxicity monitoring of all three patients at each dose level for the time window, this process is inefficient and often stalls trial advancement. The use of alternative statistical designs, such as time-to-event continuous reassessment method could improve the enrollment efficiency and cumulative safety analysis of phase I trials (18). As is true for nonradiotherapy safety reporting, a minimum 30-day observational period after completion of treatment should be used to monitor for toxicities. This period will account for the majority of acute toxicities of therapies (which typically develop within 2 to 3 weeks of the start of radiation) and follows the patient until toxicity recovery (typically 2–3 weeks). A direct relationship exists between the severity of acute side effects and the severity of late toxicities for some normal tissues (19). Therefore establishing the MTD by monitoring acute effects is an imperfect but efficient surrogate for assessing late effects. A limitation to this approach is the potential to miss some late side effects that are clinically relevant (eg, radiation pneumonitis—3–6 months after radiation therapy; fibrosis, radiation proctitis, tracheoesophageal fistula, or memory deficits —months to years after radiation therapy). Although we are not minimizing these effects and realize their potential morbidity, such potential problems will be identified in subsequent lines of investigation, and therefore short-term toxicity assessment should be the major focus of early clinical testing. However, patients should understand the risks of potential late effects during the informed consent process and should be monitored for a sufficient period of time for the development of late effects. Although late effects will not be the primary endpoints of the phase I and II clinical trials, it is important to determine if a new radiation sensitizer has late-term toxicities so that this information is expeditiously captured through the trials and can be considered by the US Food and Drug Administration (FDA) and investigators for later phase trials.
Novel Clinical Trial Designs
The resources needed to design, open, conduct, and complete a clinical trial are substantial. Initiating and opening a series of independent phase I studies with similar design and endpoints would be a poor use of scarce resources. A more efficient way to rapidly test novel agents in combination with radiation is through modular clinical trial platforms. In this regard, several agents can be rapidly tested in parallel noncomparative arms. This allows investigators to generate more clinical data within the same trial infrastructure with consistent eligibility, assessments, endpoints, methodology, and biomarker analyses. By establishing consistent clinical endpoints (eg, landmark survival), agents can be identified with a higher likelihood of success for validation testing in subsequent randomized controlled trials. Several platform-based clinical trials [eg, I-SPY1 trial testing neoadjuvant drug therapy for pathologic response measurements in early-stage breast cancer prior to lumpectomy (20) and various BATTLE platforms for lung cancer trials at MD Anderson Cancer Center (21)] have confirmed the feasibility of such an approach. These have substantial benefits compared with traditional approaches because there is uniformity in eligibility and data collection, efficient enrollment of patients into various subgroup trials under the same enrollment/screening process, and elimination of protocol-dictated downtime that occurs when cohorts are filled by conducting multiple trials in tandem. They also decrease the administrative burden and costs of repetitive institutional review board and other regulatory review through a master protocol.
Source of Agents and Path to Registration
Development of novel therapeutics as radiation enhancers requires the involvement of multiple partners. Traditionally, many of these agents have come from the pharmaceutical industry. However, combining drugs with radiation has not been a priority for industry partners because this is not a common pathway leading to FDA approval. This is particularly unfortunate because drug combinations with radiotherapy are frequently employed in potentially curative therapies. It is therefore important for the FDA to recognize that radiotherapy in combination with a novel drug could be a valid pathway for initial registration and establish clear guidance in this regard. Similarly, the pharmaceutical industry must recognize that combination trials with radiation may represent an opportunity for a first or new indication based upon improvement in cure rates in common solid tumors. The lack of efficacy when a new agent is combined with standard chemotherapy for metastatic disease does not necessarily imply that a clinical benefit might not be gleaned in the locally advanced setting when the novel agent is combined with radiation. The National Cancer Institute (NCI) Clinical Therapy Evaluation Program (CTEP) can serve as a critical intermediary in this regard. Specifically, CTEP should consistently issue mass solicitations for drug combination trials that have specific calls for proposals in combination with radiation and should simultaneously facilitate development of compounds as radiosensitizers within their portfolio, particularly through multiarm clinical trial platforms with multiple industry partners.
Funding Sources
The development of novel radiosensitizers should be financially supported consistent with other priorities in cancer research. The funding of preclinical studies can occur through established mechanisms including, but not limited to, seed funding by institutional/departmental assets and competitive grants through private or public resources. For some academic centers, U01 and N01 mechanisms provide contractual agreements with CTEP to conduct phase I or phase II studies, respectively. The NCI-sponsored cooperative groups represent an ideal forum to conduct clinical trials necessary to prove activity and efficacy, but not necessarily preclinical or early-phase studies. Given limited resources, studies must be scrutinized for scientific rationale and prioritized based on the scientific quality and clinical need. However, even with clinical study funding sources identified, often biomarker analyses and development cannot be completely supported through these means. By linking correlative biomarker studies with existing grant mechanisms (R01, P01, SPORE), applying for new competitive grants (CPRIT, R01), or establishing scientific collaborations with groups and centers that have the expertise and funding but lack the clinical samples could all be alternative avenues to fund the critical biomarker analyses. Given the financial restrictions in clinical investigation, the economic benefits and thus prioritization of platform-based clinical trial designs should be recognized.
Programmatic Examples of the Opportunities and Challenges
In 2009, leadership at the Molecular Radiation Therapeutics Branch (MRTB) of the NCI identified a need to bring together key stakeholders and experts in an attempt to develop an organized effort to develop novel radiosensitizers. These working groups were comprised of radiation biologists, translational scientists, and clinicians and focused on specific clinical areas of need (Supplementary Table 1, available online). Through government, private, and academic collaboration, these working groups have tackled many of the challenges and have operationalized many of the opportunities outlined in the sections above. We have briefly outlined the efforts of the Breast-to-Brain Metastases working group as an example of ongoing efforts of all working groups.
The Breast-to-Brain Metastases working group was the inaugural group established by the MRTB. The goal of this group, similar to the other groups, is to identify gaps in the preclinical research that would help advance some of the research in bringing targeted agents in combination with whole brain radiotherapy (WBRT) for brain metastases from breast cancer. Brain metastases involve more than 60 000 patients per year, the bulk of whom require WBRT as the standard of care. Breast and non–small cell lung cancers make up the majority of these patients. As systemic disease control improves, control of central nervous system disease becomes ever more important. Intracranial disease control is achieved in less than half of patients treated with WBRT alone, and escalation of WBRT in brain metastases is untenable. Although there are numerous potential agents to combine with WBRT, PARP inhibitors comprise the class receiving the highest attention from the group. Ample preclinical data demonstrate the radiosensitizing effect of PARP inhibitors (22,23). An important early-phase study with ABT-888 (veliparib) combined with WBRT (NCT00649207) is ongoing. Another important key study involves adding a gamma-secretase inhibitor RO4929097 to WBRT for breast cancer patients (NCT01217411).
Although most other agents with activity in breast and lung cancers (eg, VEGF inhibitors, EGFR inhibitors) have been shown in preclinical data to have radiosensitizing effects, they lack phase I safety data with WBRT. To rapidly develop novel radiosensitizers in combination with WBRT, the working group has focused attention on the new collaborative trial between the Radiation Therapy Oncology Group (RTOG) and the Korean Radiation Oncology Group. RTOG 1118 has been designed as a multiarm phase I/II randomized trial that incorporates multiple agents in comparison with WBRT alone. The phase I portion includes broad enrollment eligibility and focuses on safety. The phase II portion of the study limits eligibility to only breast and non–small cell lung cancers patients, with these studies being done in tandem as individual phase I components get completed (with WBRT alone serving as control). Patients are randomized to the various agent experimental arms with stratification for the primary tumor site (lung vs breast). The primary endpoint is intracranial response rate (RR) at 4 months after WBRT. Early stopping rules are incorporated so that if the novel sensitizer does not improve RR by 5% or better, that arm of the trial will be dropped. A 15% improvement in RR makes the drug a potential agent to explore further in definitive randomized controlled trials. The trial will open first with three arms: 1) WBRT alone, 2) WBRT with cilengitide, and 3) WBRT with veliparib. A unique challenge facing development in this clinical arena involves the need to maintain systemic disease control in a heavily pretreated patient population while focusing therapeutic attention on the central nervous system disease.
Summary and Near-Term Milestones
In summary, we believe molecularly targeted approaches will best complement the technological advances that are already in place to help accelerate the pace of progress in radiation oncology. With the opportunities and challenges reviewed and recommendations presented, we are poised to meet these challenges to reach our ultimate goal: improve cancer cure rates for our patients. Advances in cancer treatment, including radiotherapy, should optimally start with proper patient selection through innovative preclinical research (Figure 2). Continued progress in biomarker research using various in vitro and in vivo cancer models integrated with various genomic-scale datasets will hopefully accelerate the development of new predictive biomarkers for patient selection and increase the number of agents with clearly validated biomarkers for upfront patient selection in clinical trials. Despite the clinical importance and multitude of potential agents, there are few incentives for industry to develop or test novel radiosensitizers. A systematic and rational approach to the development and testing of novel radiosensitizers is needed, an operational process that the NCI MRTB working groups are beginning to tackle. Through a cooperative group/CTEP/industry partnership, a critical programmatic national approach can be implemented to rapidly identify and test compounds. In the near term, we aim to identify agents with established biomarkers for each working group to move forward through CTEP and industry as solicited or unsolicited concept submissions. There are already several important studies or trial platforms being developed at each of the disease working groups that we hope to see activated within the next year. Finally, we encourage adoption of these workshop recommendations so as to accelerate the development of novel molecularly targeted therapies in combination with radiation therapies.
Figure 2.
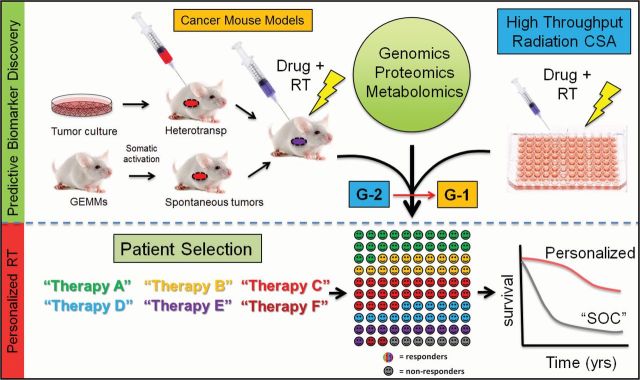
Schematic to accelerate progress in molecular-targeted radiation therapy. To bring upfront patient selection into radiation treatment selection, we need more predictive biomarker discovery and validation, which will increase the number of established biomarkers associated with novel therapeutics and subsequent improvement in patient outcome. CSA = clonogenic survival assays; GEMM = genetically engineered mouse models; G-1 = group 1 drugs (drugs with biomarkers); G-2 = group 2 drugs (drugs without biomarkers); RT = radiation therapy; SOC = standard of care.
Funding
Steven H. Lin: the American Society for Radiation Oncology (ASTRO) Junior Faculty Fellowship Award, the University of Texas MD Anderson Cancer Center, and the National Cancer Institute Cancer Center Support Grant CA016672; Quynh Le: 1R01CA161585-01A1, 1R21DE021167A1-01; Theodore S. Lawrence: R01CA138723, P30CA130810.
Supplementary Material
Minesh Mehta is a consultant for Abbott, Bistol-Meyers-Squibb, Elekta, Genentech, Merck, Novartis, Novelos, Novocure, Viewray; holds stock options in Accuray, Colby, Pharmacyclics, Procertus, Stemina; is on the Board of Directors for Pharmacyclics; is on the Medical Advisory Board for Colby, Stemina, and Procertus; and is a speaker for Merck. All others have no conflicts of interest to disclose.
References
- 1. Mañon RR, Jaradat H, Patel R, et al. Potential for radiation therapy technology innovations to permit dose escalation for non-small-cell lung cancer. Clin Lung Cancer. 2005; 7(2):107–113 [DOI] [PubMed] [Google Scholar]
- 2. Liu Y, Li P. Progress in image-guided adaptive radiation therapy. Chinese J Clin Oncol. 2008; 35(22):1314–1316–1320 [Google Scholar]
- 3. Mohan R, Bortfeld T. Proton therapy: clinical gains through current and future treatment programs. Front Radiat Ther Oncol. 2011; 440–464 [DOI] [PubMed]
- 4. Chen AB, Neville BA, Sher DJ, Chen K, Schrag D. Survival outcomes after radiation therapy for stage III non-small-cell lung cancer after adoption of computed tomography-based simulation. J Clin Oncol. 2011; 29(17):2305–2311 [DOI] [PubMed] [Google Scholar]
- 5. Bentzen SM, Harari PM, Bernier J. Exploitable mechanisms for combining drugs with radiation: concepts, achievements and future directions. Nat Clin Pract Oncol. 2007; 4(3):172–180 [DOI] [PubMed] [Google Scholar]
- 6. Pathak AK, Bhutani M, Saintigny P, Mao L. Heterotransplant mouse model cohorts of human malignancies: a novel platform for systematic preclinical efficacy evaluation of drugs (SPEED). Am J Transl Res. 2009; 1(1):16–22 [PMC free article] [PubMed] [Google Scholar]
- 7. Chen Z, Cheng K, Walton Z, et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature. 2012; 483(7391):613–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004; 350(21):2129–2139 [DOI] [PubMed] [Google Scholar]
- 9. Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007; 448(7153):561–566 [DOI] [PubMed] [Google Scholar]
- 10. Yasuda H, De Figueiredo-Pontes LL, Kobayashi S, Costa DB. Preclinical rationale for use of the clinically available multitargeted tyrosine kinase inhibitor crizotinib in ROS1-translocated lung cancer. J Thorac Oncol. 2012; 7(7):1086–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yamauchi H, Stearns V, Hayes DF. When is a tumor marker ready for prime time? A case study of c-erbB-2 as a predictive factor in breast cancer. J Clin Oncol. 2001; 19(8):2334–2356 [DOI] [PubMed] [Google Scholar]
- 12. Abe O, Abe R, Enomoto K, et al. Tamoxifen for early breast cancer: An overview of the randomised trials. Lancet. 1998; 351(9114):1451–1467 [PubMed] [Google Scholar]
- 13. Li J, Wang Y, Sun Y, Lawrence TS. Wild-type TP53 inhibits G2-phase checkpoint abrogation and radiosensitization induced by PD0166285 a WEE1 kinase inhibitor. Radiat Res. 2002; 157(3):322–330 [DOI] [PubMed] [Google Scholar]
- 14. Hall EJ, Giaccia AJ. Radiobiology for the Radiologist. 6th ed Philadelphia, PA: Lippincott Williams and Wilkins Publishing; 2005. [Google Scholar]
- 15. Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006; 354(6):567–578 [DOI] [PubMed] [Google Scholar]
- 16. Ang KK, Zhang QE, Rosenthal DI, et al. A randomized phase III trial (RTOG 0522) of concurrent accelerated radiation plus cisplatin with or without cetuximab for stage III-IV head and neck squamous cell carcinomas (HNC). J Clin Oncol. 2011; 29(Suppl):abstract 5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blumenschein GR, Jr, Paulus R, Curran WJ, et al. Phase II study of cetuximab in combination with chemoradiation in patients with stage IIIA/B non-small-cell lung cancer: RTOG 0324. J Clin Oncol. 2011; 29(17):2312–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ying Kuen C, Chappell R. Sequential designs for phase I clinical trials with late-onset toxicities. Biometrics. 2000; 56(4):1177–1182 [DOI] [PubMed] [Google Scholar]
- 19. Dörr W, Hendry JH. Consequential late effects in normal tissues. Radiother Oncol. 2001; 61(3):223–231 [DOI] [PubMed] [Google Scholar]
- 20. Esserman LJ, Berry DA, Cheang MC, et al. Chemotherapy response and recurrence-free survival in neoadjuvant breast cancer depends on biomarker profiles: results from the I-SPY 1 TRIAL (CALGB 150007/150012; ACRIN 6657). Breast Cancer Res Treat. 2012; 132(3):1049–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim ES, Herbst RS, Wistuba II, et al. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discovery. 2011; 1(1):44–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Albert JM, Cao C, Kwang WK, et al. Inhibition of poly(ADP-ribose) polymerase enhances cell death and improves tumor growth delay in irradiated lung cancer models. Clin Cancer Res. 2007; 13(10):3033–3042 [DOI] [PubMed] [Google Scholar]
- 23. Donawho CK, Luo Y, Penning TD, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007; 13(9):2728–2737 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.