

Abstract
Background
Triatoma dimidiata is among the main vectors of Chagas disease in Latin America. However, and despite important advances, there is no consensus about the taxonomic status of phenotypically divergent T. dimidiata populations, which in most recent papers are regarded as subspecies.
Methodology and Findings
A total of 126 cyt b sequences (621 bp long) were produced for specimens from across the species range. Forty-seven selected specimens representing the main cyt b clades observed (after a preliminary phylogenetic analysis) were also sequenced for an ND4 fragment (554 bp long) and concatenated with their respective cyt b sequences to produce a combined data set totalling 1175 bp/individual. Bayesian and Maximum-Likelihood phylogenetic analyses of both data sets (cyt b, and cyt b+ND4) disclosed four strongly divergent (all pairwise Kimura 2-parameter distances >0.08), monophyletic groups: Group I occurs from Southern Mexico through Central America into Colombia, with Ecuadorian specimens resembling Nicaraguan material; Group II includes samples from Western-Southwestern Mexico; Group III comprises specimens from the Yucatán peninsula; and Group IV consists of sylvatic samples from Belize. The closely-related, yet formally recognized species T. hegneri from the island of Cozumel falls within the divergence range of the T. dimidiata populations studied.
Conclusions
We propose that Groups I–IV, as well as T. hegneri, should be regarded as separate species. In the Petén of Guatemala, representatives of Groups I, II, and III occur in sympatry; the absence of haplotypes with intermediate genetic distances, as shown by multimodal mismatch distribution plots, clearly indicates that reproductive barriers actively promote within-group cohesion. Some sylvatic specimens from Belize belong to a different species – likely the basal lineage of the T. dimidiata complex, originated ∼8.25 Mya. The evidence presented here strongly supports the proposition that T. dimidiata is a complex of five cryptic species (Groups I–IV plus T. hegneri) that play different roles as vectors of Chagas disease in the region.
Introduction
Chagas disease (American Trypanosomiasis) is one of the most important parasitic diseases in Latin America with about 8–10 million people infected, 10–12 thousand deaths per year and ∼25 million at risk of infection [1], [2]. Humans acquire the disease when they come into contact with Trypanosoma cruzi-infected faeces of blood-sucking bugs of the subfamily Triatominae (Hemiptera: Reduviidae). As the most effective mechanism to prevent Chagas disease transmission relies on vector control strategies, substantial effort has been devoted to the study of the ecology, population structure and evolution of triatomine bugs (for review see [3]). Triatoma dimidiata, T. infestans, and Rhodnius prolixus are the main vectors of Chagas disease. Vector control programs have achieved remarkable success towards the elimination of R. prolixus and T. infestans in several regions of Central and South America, respectively [4]; T. dimidiata is currently the primary target of control efforts across its range [5], which spans from southern Mexico through Central America into Colombia, Peru, and Ecuador [6].
T. dimidiata occupies a wide variety of domestic and peridomestic environments, in both rural areas [7], [8] and urban settings [9], [10]. In the wild, it is also very versatile, and colonies have been found in a wide variety of ecotopes (e.g., rocky outcrops, trees, caves, and palm trees [11], [12], [13]).
Throughout its geographic distribution, T. dimidiata exhibits high phenotypic variability, which has caused considerable taxonomic controversy since the species description in 1811. A number of chromatic, morphometric, and antennal phenotype variants have been recognized; while some of them were regarded as geographic populations, others were formally described as subspecies or species (reviewed by [14]).
The first genetic evidence suggesting the existence of undescribed cryptic species within the T. dimidiata taxon was reported in 2001 [15]. Based on nucleotide sequence analyses of the ribosomal DNA second internal transcribed spacer (ITS-2), substantial differences were observed between one population from Yucatán (Mexico) and those from other localities in Mexico and Central America. Chromosome C-banding patterns, genome size [16], and mitochondrial cyt b sequence analyses [17] later corroborated these findings and extended the distribution of this new species into the forests of Petén, Guatemala. Bargues et al. [18] have proposed that all other T. dimidiata populations (including the closely related species T. hegneri) although also genetically distinct, but with distance values markedly lower than those for the particular population from Yucatán, should be regarded as subspecies. Genetic groups based on the subspecific criteria adopted by Usinger [19] were proposed: Group 1A (specimens from Chiapas in southern Mexico, Guatemala, Honduras, El Salvador, Nicaragua, Costa Rica, and Ecuador), which would correspond to T. dimidiata dimidiata; Group 1B (specimens from Panama and Colombia), corresponding to T. dimidiata capitata; and Group 2 (samples from central and northwestern Mexico, Guatemala and Belize), corresponding to T. dimidiata maculipennis. Results based on cuticular hydrocarbon patterns support the existence of these “three subspecies” and suggest the existence of yet a fourth subspecies and a full species within the taxon T. dimidiata [20].
In summary, phenotypic and genetic studies have indicated that T. dimidiata is a complex of sibling or near-sibling taxa, although the precise number of species and subspecies and their relationship with each other remain uncertain. To further our knowledge on the taxonomy and population subdivision of this important Chagas disease vector, we present new data based on two mitochondrial gene fragments, cytochrome b (cyt b) and nicotinamide adenine dinucleotide dehydrogenase 4 (ND4).
Materials and Methods
Insect Sampling and DNA Isolation
A total of 126 T. dimidiata specimens were collected from 32 localities across the species geographic range (Table 1 and Figure 1). Ten additional specimens of five closely related Triatoma species (T. hegneri, T. flavida, T. phyllosoma, T. pallidipennis, and T. nitida) were also sampled (Table 1). Insects were collected between 1995 and 2004 from domestic, peridomestic, and sylvatic habitats and identified following the Lent and Wygodzinsky [6] taxonomic key. Whenever necessary home/property owners gave consent for traps to be placed. One leg of each individual was stored in 95% ethanol until the DNA purification step. Extractions were performed using the Wizard Genomic DNA extraction kit (Promega, Madison, Wisconsin) following the manufacturer recommendations.
Table 1. Triatoma samples used in the study.
| No. | Country | Map No. | Capture Location | Ecotope1 | Sequencecodes | Cytb GenBank No. | ND4 GenBank No. | ||
| T. dimidiata | |||||||||
| 1 | Mexico | 1 | San Luis Potosí | Axtla de Terrazas, Temalacaco | D | MxSa1 | JN585833 | AF454685 | |
| 2 | San Antonio, Xolol | D | MxSa2 | JN585834 | |||||
| 3 | D | MxSa3 | AY062149 | ||||||
| 4 | 2 | Hidalgo | Acomul | D | MxHi3 | AY062151 | AF454686 | ||
| 5 | Canali | D | MxHi4 | AY062151 | JN620155 | ||||
| 6 | D | MxHi5 | AY062151 | JN620155 | |||||
| 7 | 3 | Veracruz | Mesa de Tlanchinol | D | MxVe1 | AY062150 | AF454685 | ||
| 8 | D | MxVe2 | AY062149 | AF454684 | |||||
| 9 | D | MxVe3 | AY062149 | ||||||
| 10 | La Luz | D | MxVe4 | JN585835 | JN620155 | ||||
| 11 | 4 | Oaxaca | Cañada Atotonilco, Los S R Nopala | P | MxOx1 | JN585836 | |||
| 12 | P | MxOx2 | JN585836 | ||||||
| 13 | San Juan Comaltepec | P | MxOx3 | JN585837 | JN620156 | ||||
| 14 | San Juan Juquila | P | MxOx4 | AY062149 | |||||
| 15 | P | MxOx5 | JN585838 | ||||||
| 16 | ND | MxOx6 | JN585837 | ||||||
| 17 | P | MxOx7 | JN585836 | ||||||
| 18 | 5 | Chiapas | Palenque | S | MxCh1 | JN585839 | JN620157 | ||
| 19 | Tapachula, Manacal | P | MxCh2 | JN585840 | JN620158 | ||||
| 20 | P | MxCh3 | JN585841 | JN620159 | |||||
| 21 | P | MxCh4 | JN585842 | JN620159 | |||||
| 22 | Tuxtla Chico, Medio Monte | P | MxCh5 | JN585840 | JN620158 | ||||
| 23 | 6 | Tabasco | El Rosario | D | MxTa | JN585843 | |||
| 24 | 7 | Yucatán | Yaxkukul, Rancho San Antonio | S | MxYu1 | AY062162 | AF454697 | ||
| 25 | S | MxYu2 | AY062160 | AF454695 | |||||
| 26 | S | MxYu3 | AY062163 | AF454698 | |||||
| 27 | S | MxYu4 | JN585844 | ||||||
| 28 | S | MxYu5 | AY062158 | AF454693 | |||||
| 29 | S | MxYu6 | AY062159 | AF454694 | |||||
| 30 | S | MxYu7 | AY062161 | AF454696 | |||||
| 31 | S | MxYu8 | AY062164 | AF454699 | |||||
| 32 | Yucatán | Carretera Paraíso-Progreso | S | MxYu9 | AY062158 | ||||
| 33 | S | MxYu10 | JN585845 | JN620160 | |||||
| 34 | Merida | D | F58MxY | FJ197158* | |||||
| 35 | D | F59MxY | FJ197159* | ||||||
| 36 | Guatemala | 8 | Petén | Yaxhá | S | GuPe1 | JN585846 | ||
| 37 | S | GuPe2 | JN585847 | ||||||
| 38 | S (chultun) | GuPe3 | JN585848 | ||||||
| 39 | S | GuPe4 | JN585839 | ||||||
| 40 | S | GuPe5 | JN585849 | ||||||
| 41 | S (palm) | GuPe6 | JN585850 | ||||||
| 42 | S (palm) | GuPe7 | JN585851 | ||||||
| 43 | S | GuPe8 | JN585852 | ||||||
| 44 | S | GuPe9 | JN585853 | ||||||
| 45 | S (chultun) | GuPe10 | JN585854 | ||||||
| 46 | S (palm) | GuPe11 | JN585855 | ||||||
| 47 | S (palm) | GuPe12 | JN585856 | JN620161 | |||||
| 48 | S | GuPe13 | JN585857 | JN620162 | |||||
| 49 | S | GuPe14 | JN585858 | ||||||
| 50 | 9 | Alta Verapaz | Cahabón | D | GuVe1 | JN585859 | |||
| 51 | D | GuVe2 | JN585860 | ||||||
| 52 | Lanquin | S (cave) | GuVe3 | JN585861 | JN620163 | ||||
| 53 | S (cave) | GuVe4 | JN585859 | ||||||
| 54 | S (cave) | GuVe5 | JN585859 | ||||||
| 55 | S (cave) | GuVe6 | JN585859 | JN620164 | |||||
| 56 | Tamahú | D | GuVe7 | JN585862 | |||||
| 57 | Tucurú | D | GuVe8 | JN585863 | |||||
| 58 | ND | GuVe9 | JN585864 | JN620165 | |||||
| 59 | . | ND | GuVe10 | JN585865 | JN620166 | ||||
| 60 | Tamahú | ND | GuVe11 | JN585866 | JN620165 | ||||
| 61 | San Marcos Lachuá | P | Gua1 | JN585867 | |||||
| 62 | P | Gua2 | JN585868 | ||||||
| 63 | P | Gua3 | JN585869 | JN620167 | |||||
| 64 | P | Gua4 | JN585870 | JN620168 | |||||
| 65 | 10 | Chiquimula | Tuticopote | ND | Gua7 | JN585871 | |||
| 66 | ND | Gua8 | JN585871 | ||||||
| 67 | San Juan Ermita, Chanco | D | GuCh | JN585872 | |||||
| 68 | 11 | Izabal | Los Amates, Hacienda el Manacal | S (palm) | GuIz1 | JN585873 | |||
| 69 | S (palm) | GuIz2 | JN585874 | ||||||
| 70 | 12 | Quiché | Olopa | ND | Gua5 | JN585871 | |||
| 71 | Tituque | ND | Gua6 | JN585871 | |||||
| 72 | Canillá | D | GuQu1 | JN585875 | |||||
| 73 | San Andrés Sajcabajá, Xepalzac | D | GuQu2 | JN585876 | |||||
| 74 | P | GuQu3 | JN585877 | ||||||
| 75 | 13 | Alta Verapaz | San Marcos Lachuá | ND | GUA1B | JN585878 | JN620169 | ||
| 76 | ND | GUA3B | AY062157 | AF454692 | |||||
| 77 | 14 | Zacapa | Río Hondo, El Cajón | P | GuZa | JN585875 | |||
| 78 | 15 | Santa Rosa | Amberes | D | GuSa1 | JN585879 | |||
| 79 | S.Rosa de Lima | Laguna de Pereira | D | GuSa2 | JN585880 | ||||
| 80 | 16 | Jutiapa | Conguaco, Laguna Seca | ND | GuJu1 | JN585881 | |||
| 81 | Quesada, El Tule | D | GuJu2 | JN585881 | |||||
| 82 | San José Acatempa | D | GuJu3 | JN585874 | |||||
| 83 | D | GuJu4 | JN585881 | ||||||
| 84 | D | GuJu5 | JN585881 | ||||||
| 85 | San José Acatempa, Tunillas | P | GuJu6 | JN585882 | |||||
| 86 | P | GuJu7 | JN585881 | ||||||
| 87 | Belize | 17 | Cayo | Río Frio | S (cave) | Bz1 | JN585883 | JN620170 | |
| 88 | S (cave) | Bz2 | JN585884 | ||||||
| 89 | Calla Creek | D | 56BzCa | FJ197156* | |||||
| 90 | 18 | Toledo | San Pedro Columbia | D | 54BzTo | FJ197154* | |||
| 91 | Santa Teresa | D | 55BzTo | FJ197155* | |||||
| 92 | Honduras | 19 | Carrizalón | D | HoCa1 | JN585885 | |||
| 93 | D | HoCa2 | JN585886 | JN620171 | |||||
| 94 | D | HoCa3 | JN585887 | ||||||
| 95 | 20 | Yoro | Yorito, Los Planes | ND | HoYo1 | AY062153 | AF454688 | ||
| 96 | ND | HoYo2 | AY062153 | AF436860 | |||||
| 97 | 21 | Tegucigalpa. | Colonia Nueva Suyapa | ND | HoTe1 | AY062152 | AF454687 | ||
| 98 | ND | HoTe2 | AY062153 | AF436860 | |||||
| 99 | Barrio El Bosque | ND | HoTe3 | AY062154 | JN620172 | ||||
| 100 | ND | HoTe4 | AY062156 | AF454691 | |||||
| 101 | Colonia San Miguel | ND | HoTe5 | AY062154 | AF454689 | ||||
| 102 | ND | HoTe6 | AY062155 | AF454690 | |||||
| 103 | El Salvador | 22 | Santa Ana | Monte Largo | D | SaSa1 | JN585888 | ||
| 104 | D | SaSa2 | JN585889 | ||||||
| 105 | D | SaSa3 | JN585890 | ||||||
| 106 | 23 | La Unión | El Farito | D | SaLa1 | JN585891 | |||
| 107 | Amapolita | D | SaLa2 | JN58589 | |||||
| 108 | El Farito | D | SaLa3 | JN585836 | JN620173 | ||||
| 109 | Nicaragua | 24 | Masaya | Masatepe | D | Nic | JN585892 | ||
| 110 | Costa Rica | 25 | Heredia | Santo Tomás de Santo Domingo | P | CR1 | JN585893 | ||
| 111 | P | CR2 | JN585894 | ||||||
| 112 | P | CR3 | JN585894 | ||||||
| 113 | San José | P | CR4 | JN585895 | |||||
| 114 | P | CR5 | JN585895 | JN620174 | |||||
| 115 | P | CR6 | JN585894 | ||||||
| 116 | Panama | 26 | Veraguas | Santa Fe, El Macho | D | Pan1 | JN585896 | ||
| 117 | D | Pan2 | JN585897 | ||||||
| 118 | D | Pan3 | JN585898 | JN620175 | |||||
| 119 | El Pantano | S (palm) | Pan4 | JN585899 | |||||
| 120 | S (palm) | Pan5 | JN585900 | ||||||
| 121 | Colombia | 27 | Sucre | San Onofre | S | CoSu | JN585901 | ||
| 122 | 28 | Bolívar | San Fernando | S (palm) | CoBo1 | JN585902 | |||
| 123 | La Margarita | S (palm) | CoBo2 | JN585903 | |||||
| 124 | 29 | Magdalena, Santa Marta | Cacahualito | S (palm) | CoMa1 | JN585904 | |||
| 125 | Las Tinajas | S (palm) | CoMa2 | JN585905 | |||||
| 126 | 30 | Santander | San Joaquin | S (rock pile) | CoSa1 | JN585906 | |||
| 127 | Del Carmen | S (rock pile) | CoSa2 | JN585907 | |||||
| 128 | 31 | Boyacá | Boavita | D | CoBy1 | JN585908 | JN620176 | ||
| 129 | D | CoBy2 | JN585909 | ||||||
| 130 | Ecuador | 32 | Manabi | El Cade | ND | Ec3 | JN585910 | JN620177 | |
| 131 | P | Ec5 | JN585910 | ||||||
| T. hegneri | |||||||||
| 132 | Mexico | 33 | Cozumel Island | Quintana Roo, Rancho Exekalihche | P | HgC207 | JN585830 | JN620178 | |
| 133 | P | HgCz85 | JN585831 | ||||||
| 134 | P | HgCz88 | JN585831 | ||||||
| 135 | P | Heg29 | JN585832 | ||||||
| T. flavida | |||||||||
| 136 | Cuba | Pinar del Río | Península Guanacahibes | S (cave) | fl4715 | JX848648 | JX848653 | ||
| T. phyllosoma | |||||||||
| 137 | Mexico | Oaxaca | P | Phy446-2 | JX844671 | JX848649 | |||
| 138 | Phy446-7 | JX844672 | JX848650 | ||||||
| T. pallidipennis | |||||||||
| 139 | Mexico | Morelos | Chalcatzingo | D | Palli465 | JX848645 | |||
| T. nitida | |||||||||
| 140 | Guatemala | ND | nitN30n | JX848646 | JX848651 | ||||
| 141 | Guatemala | ND | nitN27 | JX848647 | JX848652 | ||||
D = domestic, P = peridomestic, S = sylvatic, ND = no data.
indicates cyt b haplotypes from Dorn et al. [17].
Figure 1. Sampling sites of representatives of the Triatoma dimidiata species complex. Numbers in the circles correspond to numbers of sampled locations listed in Table 1.

PCR Amplification and DNA Sequencing
A fragment of the cyt b mitochondrial gene was amplified from each specimen using primers 7432F (5′ GGACGWGGWATTTATTATGGATC 3′), and 7433R (5′ GCWCCAATTCAR GTTARTAA3′) [21]. The ND4 mitochondrial gene was also amplified and sequenced for a subset of specimens using primers ND4deg01F (5′ GGSGCYTCAACATGAGCCYT 3′), and ND4b02R (5′ TAATTCGTTGTCATGGTAATG 3′) [22]. When the DNA of the sample was of poor quality, a nested PCR was performed with the primers ND4deg (5′ TCAACATGA GCCCTTGGAAG 3′), and ND4neR (5′ TAATTCGTACTCATGGTAATG 3′) [22]. An average of 1–3 µL of purified DNA was amplified in a 50 µL reaction: 5 µL 10× buffer (Promega), 4 µL dNTPs (2.5 mM each), 2 µL MgCl2 (25 mM), 0.5 µL Taq DNA polymerase (Promega), and 2 µL of each primer (10 pmol/µL). Amplification conditions were: 94°C for 5 min, followed by 35 cycles of 94°C for 30 sec, 47°C for 30 sec, 72°C for 1 min, and a final elongation step at 72°C for 10 min. Purification of PCR products was performed with MultiScreen PCR purification plates (Millipore) following the manufacturer recommendations. Direct sequencing of both forward and reverse sequences was performed on an ABI 3130 (Applied Biosystems) automated sequencer.
Sequence Variation and Phylogenetic Analyses
Standard genetic diversity indices such as both nucleotide (π) and haplotype (h) diversities, and number of variable and parsimony informative sites were estimated using DNASP 5.10 [23]. Tajima’s D [24], as implemented in ARLEQUIN 3.11 [25], were used to test for mutation-drift equilibrium deviation in the overall sample.
The strategy employed to infer the phylogenetic relationships among T. dimidiata populations was to first generate a tree based on all cyt b sequences available, identify the main clades present, and then select at least one representative specimen of each clade to be further sequenced for the ND4 gene fragment to be used in a cyt b+ND4 combined analysis. Other Triatoma species (T. flavida, T. phyllosoma, T. pallidipennis, and T. nitida) were used as outgroups in the phylogenetic analyses. Best-fitting substitution models for each dataset were determined with JMODELTEST 0.1 [26], [27] based on Akaike’s information criterion (AIC [28]), which led to the selection of the Hasegawa-Kishino-Yano (HKY) model [29] with a proportion of invariable sites (+I), and gamma-distributed rate heterogeneity among the remaining sites (+G).
Phylogenies were inferred by Maximum Likelihood (ML) using PHYML 2.4.4 [30]. Bootstrap node support values were estimated from 1000 pseudoreplicates. ML trees were also submitted as user trees to MRBAYES 3.1.2 [31] for a Bayesian analysis. Posterior probabilities of phylogenetic trees were estimated by a 10,000,000-generation Metropolis-coupled Markov chain Monte Carlo (MCMC) simulation (four chains, chain temperature = 0.2) under the HKY+I+G model of substitution, with parameters estimated from the dataset. A majority-rule (50%) consensus tree was constructed following 200,000 burn-in generations to allow likelihood values to reach stationary equilibrium. Identical conditions were used for the cyt b and the combined (cyt b+ND4) datasets.
Population-level Inferences and Divergence Times
Mean intra- and inter-group Kimura 2-parameters genetic distances [32] were estimated in MEGA5 [33], with standard errors estimated by bootstrapping (pseudoreplicates). A median-joining network analysis [34] was performed using NETWORK 4.5.1.6 (http://www.fluxus-engineering.com). The maximum number of mutations between haplotypes within the same network for cyt b was 61. The 95% connection limit was not used because of the high levels of sequence divergence, which would cause an undesired fragmentation of the network.
Principal component analysis (PCA) was used to classify all input sequences at once into one or more groups. Variable sites from the nucleotide sequence dataset were selected and nucleotide bases were coded (A = 1, C = 2, G = 3, T = 4) and combined in an alignment matrix, where each row represents a specimen’s DNA sequence. This alignment matrix was then converted into a genetic distance matrix as implemented in GENALEX 6.4 [35].
DNASP was used to generate distribution plots of pairwise sequence differences. No attempt was made to compare the observed distributions with expected distributions, because all models available in the software for producing expected distributions are suitable only for population-level analysis.
We used cyt b to estimate divergence times as we had more data for this particular gene fragment (both for specimens and haplotypes), and because reliable evolutionary rate estimates are available for this gene [36]. The time to the most recent common ancestor (tMRCA) was estimated for all major genetic groups revealed in the phylogenetic analyses using a Bayesian approach with BEAST 1.6.1 [37]. The analysis was performed using an HKY+I+G model of nucleotide substitution with gamma-distributed rate variation among sites and four rate categories – the substitution model selected using JMODELTEST. We used the suggested divergence rate of 1.1 to 1.8% per Myr [36]. The Yule process was chosen as speciation process for our data set. Results from two independent runs (20,000,000 generations, with the first 2,000,000 discarded as burn-in and parameter values sampled every 1000 generations) were analyzed with TRACER 1.5 [38] to assess convergence and confirm that the combined effective sample sizes for all parameters were >200, ensuring that the MCMC had ran long enough to produce valid estimates for the parameters [39]. The dating process was based on all specimens per group to calculate the distance (time) to the nearest node that determines each group.
Results
Sequence Variation and Phylogenetic Analyses
A total of 126 cyt b (621 bp long) and 47 ND4 sequences (554 bp long) were produced for T. dimidiata. In addition, four 4 cyt b and one ND4 sequences were generated for T. hegneri, along with nine sequences (three cyt b and six ND4) for the outgroup species (Table 1). Five T. dimidiata cyt b sequences previously reported [17] were retrieved from GenBank and also included in the analyses. There was no indication of the presence of pseudogenes or numts among the sequences generated as no indels or stop codons were detected and all sequences appeared to be evolving (i.e. accumulating mutations) as expected for normal mtDNA protein coding genes. Inspection of the T. dimidiata mtDNA sequences revealed the existence of 97 and 36 unique haplotypes for the cyt b and ND4 gene fragments, respectively. Basic statistics are presented in Table 2. Neutrality tests failed to reject the null hypothesis that mtDNA sequences were evolving in a neutral manner in the studied species (Tajima’s D: P>0.70; Table 2).
Table 2. DNA polymorphism and neutrality tests.
| cytb | ND4 | Cytb+ND4 | |
| Length (bp) | 621 | 572 | 1175 |
| Nseq | 131 | 47 | 47 |
| Nhap | 97 | 36 | 43 |
| S | 223 | 95 | 262 |
| P | 188 | 93 | 228 |
| h | 0.9938 | 0.9885 | 0.9950 |
| π | 0.0770 | 0.0554 | 0.0697 |
| Tajima’s D | 0.5544a | 0.7549a | 0.9460a |
Nseq: Number of sequences; Nhap: Number of haplotypes; S: Variable sites; P: Parsimony informative sites; h: Haplotype diversity; π: Nucleotide diversity; Tajima’s D (Tajima 1989);
P>0.70.
Saturation plots for both cyt b and ND4 gene fragments (third codon position transversion and transition substitutions against HKY+G+I distances), show no indication of saturation (results not shown).
ML and Bayesian phylogenetic methods yielded the same tree topologies for both datasets used (cyt b and cyt b+ND4). However, the analysis of the larger cyt b+ND4 combined dataset (1175 bp) led to the resolution of the Group I/Group II/T. hegneri polyphyly that was not discriminated in the cyt b tree (Figure 2a).
Figure 2. ML phylogenetic trees for Triatoma dimidiata species complex based on cyt b (left) and cyt b+ND4 (right) gene fragments.

Bootstrap support values (above 50) are given above branches; Posterior probabilities for the Bayesian analysis are given below branches. Branch color codes indicate each of the four different genetic groups (plus T. hegneri) that comprise the T. dimidiata species complex. The three haplotypes in blue (Nic, Ec3, and Ec5) call attention to the high genetic similarity between specimens from Manabi, in Ecuador, and those from Nicaragua, indicating that T. dimidiata populations from the latter most likely represent the source of insects that were recently introduced into Ecuador.
Both cyt b and cyt b+ND4 tree topologies indicate the existence of four well-defined monophyletic groups: Group I includes samples from Mexico (Chiapas), Guatemala, Honduras, El Salvador, Nicaragua, Costa Rica, Panama, Ecuador, and Colombia; Group II comprises the westernmost samples from Mexico but also includes specimens from Tabasco and Petén; Group III includes specimens from Petén (Guatemala), Yucatán (Mexico), and Belize; and Group IV includes sylvatic samples from Belize. T. hegneri, from the island of Cozumel, Mexico, appears as yet another independent lineage within the range of between-group variability observed (Figures 2 and 3). Mean Kimura 2-parameters pairwise cyt b genetic distances among Groups I–IV plus T. hegneri were very high, ranging from 0.080 to 0.155 (Table 3).
Figure 3. Median-joining haplotype network based on T. dimidiata and T. hegneri cytochrome b haplotypes.

The size of the circles on the network is proportional to haplotype frequency. Small red circles represent missing haplotypes. Network circle colors are the same as in Fig. 1. Circles with dotted red outlines denote the presence of haplotypes from Guatemala, Petén among groups I and II.
Table 3. Mean K2P pairwise genetic distances between the four genetic groups revealed in the study (and Triatoma hegneri), for the cytochrome b gene fragment.
| Group I | Group II | Group III | Group IV | Triatoma hegneri | |
| Group I | 0.045 | ||||
| Group II | 0.088 | 0.021 | |||
| Group III | 0.147 | 0.148 | 0.024 | ||
| Group IV | 0.149 | 0.144 | 0.153 | 0.018 | |
| Triatoma hegneri | 0.080 | 0.089 | 0.136 | 0.155 | 0.043 |
Intergroup distances are in the lower left section; mean intragroup distances are in bold.
Group I is the most widely distributed and genetically variable clade (Figure 4), with within-group divergence reaching values as high as 8.5% (when Colombian haplotypes are compared with Central American haplotypes). Thus, we can suppose that it might harbor more than one species. Observation of the cyt b tree (Figure 2a) towards the lower part of Group I reveals a striking similarity between haplotypes obtained from a specimen collected in Masaya, Nicaragua (Nic) with those obtained from specimens from Manabí, in Ecuador (Ec3 and Ec5), suggesting a very recent common origin.
Figure 4. Map showing the geographic distribution of the four genetically divergent monophyletic groups identified within Triatoma dimidiata (plus T. hegneri) based upon Bayesian and ML phylogenetic analyses of cyt b and ND4 sequences.

Group I occurs from Southern Mexico through Central America and into Colombia and Ecuador; Group II comprises samples from Western and Southwestern Mexico; Group III includes specimens from the Yucatán peninsula (excluding Belize and T. hegneri), Group IV includes sylvatic samples from Belize; and T. hegneri, from the island of Cozumel. The doted blue outline around the Easternmost sites of Group I demarcates the geographic distribution of the T. dimidiata samples from Colombia. The doted black line serves to indicate, based on haplotype similarity, Nicaragua as the most likely source of insects to have colonized Ecuador sometime in the recent past.
Specimens collected from the Lanquín caves, in Alta Verapaz, Guatemala (GuVe3, GuVe4, GuVe5, and GuVe6), are genetically similar to other T. dimidiata Group I insects (Figure 2a).
Population-level Inferences and Divergence Times
With the observation of the magnitude of the inter-population divergence levels of the mtDNA sequences generated, and after unsuccessful attempts to extract meaningful population-level information from the data, we realized that it would be worthless (uninformative) to proceed with regular population-level inferences such as FST or AMOVA, and therefore decided to exclude such analyses from this paper.
The median-joining haplotype network shows that most specimens presented unique haplotypes (Figure 3). Moreover, highly divergent haplotypes were found in Petén, Guatemala, which segregated into different parts of the network. Conversely, certain haplotypes found in geographically distant (Ecuador and Nicaragua) were strikingly similar. Overall, the network displayed the same groups detected in the phylogenetic analyses. The median-joining haplotype network illustrates the intricate genetic structure that characterizes Group I.
PCA based on cyt b sequences alone revealed only three groups, with T. hegneri falling within Group I, while PCA of the combined cyt b+ND4 dataset resolved the same four groups (plus T. hegneri) recovered in the phylogenetic analyses (Figures 5a, 5b).
Figure 5. Principal component plots constructed from genetic similarity matrix based on cyt b sequences (a) and on cyt b and ND4 (b) combined data.
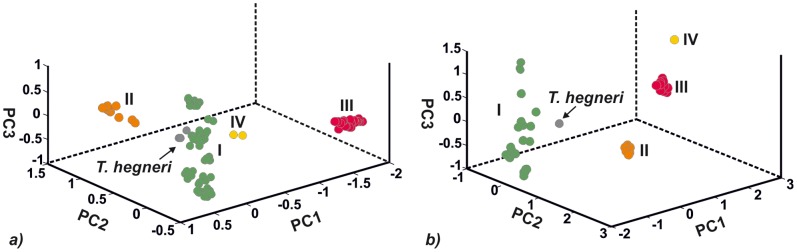
Color codes for different groups of the Triatoma dimidiata complex coincide with colors used in Fig. 2.
Results of within- and between-group comparative analysis of mismatch distributions are depicted in Figure 6. Mismatch distribution within Group III exhibits a unimodal distribution, with most pairwise comparisons revealing small genetic distances. Mismatch distributions within Groups I and II are multimodal and ragged, and contain a larger proportion of comparisons resulting in larger genetic distances. All inter-group mismatch distributions are clearly multimodal and have similar shapes, with most pairwise comparisons revealing large genetic distances; this same pattern is evident when including all T. dimidiata species complex members (Figure 6a).
Figure 6. Mismatch distribution of Triatoma dimidiata species complex based on cyt b nucleotide sequences.

(a) Total distribution. (b, c, d) Mismatch distribution within each group. (f, g, h) Mismatch distribution between different pairs of groups. (e) Mismatch distribution of haplotypes from Petén, Guatemala.
Figure 6e represents the mismatch distribution for individuals from Petén, Guatemala, where representatives of Groups I, II, and III occur in sympatry. The mismatch frequency distribution is multimodal, with visibly separated peaks that reflect the absence of haplotypes with intermediate genetic distances. This clearly suggests the existence of reproductive barriers isolating these groups from one another. These Petén specimens are therefore very likely to belong to different biological species; divergence time estimates suggest that they have been evolving independently for at least about five million years (Table 4).
Table 4. Time of divergence within and among putative species of the Triatoma dimidiata complex (and 95% confidence intervals) calculated using the program Beast.
| Taxa | tMRCA (95%CI) |
| Group I | 2.81 (1.97–3.76) |
| Group II | 1.91 (1.18–2.80) |
| Group III | 0.87 (0.51–1.24) |
| Group IV | 0.69 (0.27–1.13) |
| Triatoma hegneri | 2.64 (1.60–3.89) |
| Group I+ Triatoma hegneri | 3.92 (2.72–5.34) |
| Group I+ Triatoma hegneri +Group II | 4.21 (2.93–5.66) |
| Group I+ Triatoma hegneri +Group II+Group III | 7.10 (4.86–9.64) |
| Triatoma dimidiata species complex | 8.25 (5.75–11.22) |
tMRCA, time to the most recent common ancestor in million years before the present.
Discussion
Since its description by Pierre André Latreille, in 1811 (as Reduvius dimidiatus), the taxonomy of T. dimidiata has been a topic of controversy (reviewed in [14]). Central to the debate was the issue of whether morphologically recognized subspecies should merit specific status. Lent and Wygodzinsky [6] put an end to the dispute by concluding, after the examination of 160 specimens from the species’ distribution range, that the differences observed were “clinal in nature”, and thus, all morphological types should be considered variations within the same species. Our results unmistakably reject this hypothesis.
We report the existence of very high levels of mitochondrial DNA (cyt b and ND4) sequence divergence among populations of T. dimidiata sampled throughout its geographic range. Bayesian and ML phylogenetic analyses indicate the existence of five well defined monophyletic groups, including the formally described species T. hegneri from the island of Cozumel. Group I stretches from Southern Mexico (Chiapas), all the way through Central America into Colombia, with Ecuadorian specimens resembling Nicaraguan material; Group II comprises samples from western and northwestern Mexico, as well as from Petén in Guatemala; Group III includes specimens from the Yucatán peninsula (including Petén, Cozumel and domestic specimens from Belize); and Group IV includes sylvatic samples from Belize (Figures 2 and 4). We will argue that each of these groups merits specific status.
Hypothesis-testing and Taxonomic Implications
The comprehensive study of Bargues et al. [18] on the phylogeography of T. dimidiata based on ITS-2 sequences greatly advanced our knowledge on the taxonomy and evolution of this important vector. Our mtDNA-based results corroborate those of Bargues et al. [18] in the sense that they identify, overall, the same genetic groups present within T. dimidiata s.l. (Figure 7). However, it appears that the ITS-2 region may be too conserved to fully resolve the phylogenetic relationship among those different genetic groups [17]. By adding resolution to this matter the mtDNA gene fragments bring about a few discrepancies. For example, ITS-2 sequence data place samples from Panama together with those from Colombia, in sub-group 1B, and position T. hegneri specimens within group 2 (sensu Bargues et al. 2008 [18]). The mtDNA markers used here seem to be more appropriate for this level of taxonomic investigation. Being less conserved, and thus more informative, they allow for the detection of readily recognizable, well supported monophyletic groups.
Figure 7. This figure shows how the topology recovered for the T. dimidiata species complex based on the phylogenetic analysis of ITS-2 sequence data of Bargues et al. [18] .

(a) compares to the one derived from the mtDNA sequence data (cytb+ND4) presented in this paper (b). Examination of this new figure shows that ITS-2 groups 1, 2, and 3 of Bargues et al. are essentially the same as our mtDNA groups I, II, and III (i.e. they include specimens collected from the same geographic areas). Branch color codes in “b” indicate each of the four different genetic groups (plus T. hegneri) that comprise the T. dimidiata species complex. See Discussion section for more details on the few incongruities between the two topologies and on how these were interpreted and discussed.
The level of sequence divergence between groups I to IV exceeds the value of 8% reported to separate several closely related Triatoma species [40]. The smallest distances observed here resulted from the comparison of Groups I and II (0.088); all other inter-group comparisons gave values that surpass 13%. T. hegneri and Group I cyt b sequences differ by an average of 8% (Table 3).
Three distinct chromatic forms of T. brasiliensis from northeast Brazil analyzed with the same marker (cyt b) showed large genetic distances (d>0.075), which led to their recognition as members of a species complex [40]. Two of these forms were subsequently formally raised to the specific level [41], [42]. Divergence levels of the same magnitude (d>9%), again coupled with chromatic differences, led to the proposition that T. rubida cochimiensis should be considered a full species [34]. Other recent comparisons between valid Triatoma species based on the same marker are T. nitida vs. T. rubida sonoriana/uhleri (d = 10–11%) and T. longipennis vs. T. recurva (d = 11%; [34]). In addition to the very high mtDNA genetic distances among the T. dimidiata groups here described, high values of ITS-2 sequence divergence were also reported for haplotypes belonging to groups I and II (5.62%), which, according to the authors of the study, is suggestive of speciation [18]. These are very convincing arguments in favor of the hypothesis that T. dimidiata is a true species complex.
Group I is the most geographically widespread and genetically variable. Pairwise within-group genetic distances can be as high as 8.5%. The divergent samples from Colombia appear as a monophyletic sister clade with respect to all other specimens in the group (which are predominantly from Central America). Colombian specimens were once described as T. dimidiata capitata on morphological grounds [43], to be later synonymized [6]. Thus, it is fair to speculate that this group might conceal yet another biological species.
Sympatric Occurrence of Different Genetic Groups
Sympatry between Groups II and III is well documented in the Yucatán peninsula [17]. Although there seems to be extensive hybridization, reproductive isolating barriers (RIBs) do exist (such as reduced viability of female hybrids [44]) and appear to prevent the two species from merging into a single entity. This is a compelling argument in favor of the validation of Group III insects as a different species, as previously suggested [16], [18].
Remarkably, in Petén, Guatemala, there is not only overlapping occurrence of Groups II and III as in Yucatán, but also of Group I insects (Figure 4). Mismatch distribution results reveal multimodality caused by the absence of haplotypes with intermediate genetic distances among groups (Figure 6). This is a very significant finding as it points to the probable existence of RIBs for all combinations among these three groups, lending further support to their recognition as different biological species.
Divergence Times and Biogeography
The Isthmus of Tehuantepec is known to represent an important recent geological barrier for a number of sister taxa of birds, mammals, and butterflies [45]. It has been shown to be a phylogeographical barrier to both highland [46] and lowland species [47]. Given the present distribution of the genetic groups revealed by the mtDNA fragments analyzed in this study, it is tempting to speculate that the Isthmus of Tehuantepec orogeny split the original population and caused the allopatric generation of Groups I and II.
The isolation that might have led to the origin of Group III insects from the Yucatán peninsula could be explained by changes in climate and vegetation that took place particularly during the Pleistocene period. Lee [48] suggests that a period of Pleistocene aridity, during which there was a continuous subhumid to xeric habitat, extended from the Pacific side of Mexico across the Isthmus of Tehuantepec to the gulf coast and from there to the Yucatán Peninsula. The increase in humidity together with the introduction of mesophytic vegetation in the area resulted in an isolation of this subhumid environment from the west of Mexico, leading to speciation.
Triatoma Dimidiata in Ecuador
Bargues et al. [18] proposed that Ecuadorian T. dimidiata populations may have derived from recently introduced specimens originally from the Guatemala-Honduras-Nicaragua region, as a result of human migrations. This view was further supported by subsequent molecular analyses [15], [18] and by ecological and biogeographic observations, including the absence of records of wild populations in Ecuador (in contrast with abundant observations elsewhere) and the discontinuous distribution of the species, with Ecuadorian populations isolated from their Colombian relatives by the Central Colombian Massif and the humid Chocó eco-region [10]. The fact that T. dimidiata populations seem to have disappeared from some formerly infested rural areas of Ecuador [49], [50] and appear to persist only in a few urban foci (Abad-Franch F, pers. obs.) also matches the predictions of the artificial introduction hypothesis. As shown in the cyt b tree (highlighted in blue on Figure 2a) and the haplotype network (Figure 3), there is a striking similarity between haplotypes obtained from a specimen collected in Masaya, Nicaragua (Nic) and from Ecuadorian material (Ec3 and Ec5). This genetically pinpoints T. dimidiata populations from Nicaragua as the most likely source of insects to have colonized Ecuador sometime in the recent past.
Lanquín Cave Specimens
Studies based on morphometry [51], RAPD [52], antennal sensilla [53] and cuticular hydrocarbons [20], [54] of cave-dwelling specimens from Lanquín, Alta Verapaz, in Guatemala, revealed great phenotypic divergence from all other T. dimidiata populations analyzed. The differentiation was so remarkable that it was suggested that these insects could represent an incipient species [51], [54]. A different interpretation was put forth by Bargues et al. [18], based on the phylogenetic analysis of the ITS-2 region of the rDNA, that these specimens would have derived from the ancestor which gave rise to the subspecies T. d. dimidiata. Yet another result, also derived from the ITS-2 marker, contradicts the former and depicts Lanquín samples as a separate independent lineage [17]. Our results indicate that the Lanquín cave specimens are no different from other T. dimidiata Group I specimens from Central America (see haplotypes GuVe4, GuVe5, and GuVe6 in the upper portion of Group I, and GuVe3 close to haplotypes GuIz2 and GuJu3 in Figure 2). This suggests that Lanquín cave-dwelling specimens represent a striking case of phenotypic plasticity, most likely related to micro-habitat adaptation, within a single genetic cluster.
Samples from Belize
Sylvatic specimens from Belize (Cayo District) represent a different species which constitutes the most basal lineage of the T. dimidiata species complex, as previously suggested based on cuticular hydrocarbon patterns [20]. Divergence time estimates show that this lineage has been evolving independently for approximately 8.25 My (Table 4). These insects are clearly different from the domestic Belize specimens studied by Dorn et al. [17], which belong in Groups I and III (Figure 2). A possible explanation for this incongruence is that the specimens we studied were collected in the Rio Frio Cave. Interestingly, unlike the specimens collected from the Lanquín caves in Guatemala, these insects are quite large and present lighter tegument coloration throughout all developmental stages (Marcet PL, Dotson EM, pers. obs.).
Concluding Remarks
Bargues et al. [18] state, in the Discussion section of their paper, that – “Results of the present study do not support the rise of the abovementioned subspecific taxa to species level for the time being, although it is evident that in the three cases relatively long divergence processes have taken place. Similar genetic studies with other molecular markers may contribute to a more complete assessment of these evolutionary isolation and speciation processes.” We believe we have made an important contribution toward that end. The data presented here unmistakably reject the hypothesis that the intraspecific variation seen in T. dimidiata is clinal. The results further support previous analyses that indicated the existence of clearly recognizable genetic groups within T. dimidiata. We report the finding of very high levels of mitochondrial DNA (cyt b and ND4) sequence divergence among monophyletic populations of this vector which are incompatible with current views that regard most of these populations (with the exception of the Yucatán clade) as subspecies. We alternatively defend the interpretation that all four genetic groups revealed herein merit specific status. All the evidence presented strongly supports the proposition that T. dimidiata is a complex of five species (as it also includes T. hegneri) that play different roles as vectors of Chagas disease, from the apparently strictly sylvatic populations of Group IV in Belize to the heavily synanthropic populations (Groups I and II) in Mesoamerica, Colombia and Ecuador – and with the Yucatán clade (Group III) apparently presenting intermediate behavior.
Acknowledgments
Primers used in this work were synthesized at the CDC core facilities.
Funding Statement
AGP received support from CODI-Universidad de Antioquia (project CPT-0812), and COLCIENCIAS (project #111545921460). This work received support from the Brazilian National Research Council, CNPq. The findings and conclusions in this manuscript are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.World Health Organization (2006) Report of the Scientific Working Group on Chagas disease, Buenos Aires, Argentina. 2005, Geneva. p.7.
- 2.World Health Organization (2012) Fact Sheet Nr 340. August 2012. Chagas disease (American trypanosomiasis). http://wwwwhoint/mediacentre/factsheets/fs340/en/indexhtml.
- 3.Telleria J, Tibayrenc M (2010) American Trypanosomiasis: Chagas Disease One Hundred Years of Research; Telleria JT, M., editor. Burlington, MA, USA: Elsevier. 848 p.
- 4. Dias JC (2007) Southern Cone Initiative for the elimination of domestic populations of Triatoma infestans and the interruption of transfusional Chagas disease. Historical aspects, present situation, and perspectives. Mem Inst Oswaldo Cruz 102 Suppl 111–18. [DOI] [PubMed] [Google Scholar]
- 5.World Health Organization (2002) Control of Chagas Disease: Second report of the WHO Expert Comitee. Geneva: WHO.
- 6. Lent H, Wygodzinsky P (1979) Revision of the Triatominae (Hemiptera, Reduviidae), and their significance as vectors of Chagas’ disease. Bull Am Mus Nat Hist 163: 125–520. [Google Scholar]
- 7. Acevedo F, Godoy E, Schofield CJ (2000) Comparison of intervention strategies for control of Triatoma dimidiata in Nicaragua. Mem Inst Oswaldo Cruz 95: 867–871. [DOI] [PubMed] [Google Scholar]
- 8. Monroy C, Mejia M, Rodas A, Rosales R, Horio M, et al. (1998) Comparison of indoor searches with whole house demolition collections of the vectors of Chagas disease and their indoor distribution. Med Entomol Zool 49: 195–200. [Google Scholar]
- 9. Zeledón R, Rabinovich JE (1981) Chagas' disease: an ecological appraisal with special emphasis on its insect vectors. Annu Rev Entomol 26: 101–133. [DOI] [PubMed] [Google Scholar]
- 10. Abad-Franch F, Paucar A, Carpio C, Cuba CA, Aguilar HM, et al. (2001) Biogeography of Triatominae (Hemiptera: Reduviidae) in Ecuador: implications for the design of control strategies. Mem Inst Oswaldo Cruz 96: 611–620. [DOI] [PubMed] [Google Scholar]
- 11.Zeledón R (1981) El Triatoma dimidiata (Latreille, 1811) y su relación con la Enfermedad de Chagas. Editorial Universidad Estatal a Distancia (EUNED). San Jose, Costa Rica. 146 p.
- 12. Dumonteil E, Gourbière S, Barrera-Pérez M, Rodriguez-Félix E, Ruiz-Piña H, et al. (2002) Geographic distribution of Triatoma dimidiata and transmission dynamics of Trypanosoma cruzi in the Yucatan peninsula of Mexico. Am J Trop Med Hyg 67: 176–183. [DOI] [PubMed] [Google Scholar]
- 13. Monroy MC, Bustamante DM, Rodas AG, Enriquez ME, Rosales RG (2003) Habitats, dispersion and invasion of sylvatic Triatoma dimidiata (Hemiptera: Reduviidae: Triatominae) in Peten, Guatemala. J Med Entomol 40: 800–806. [DOI] [PubMed] [Google Scholar]
- 14. Dorn PL, Monroy C, Curtis A (2007) Triatoma dimidiata (Latreille, 1811): A review of its diversity across its geographic range and the relationship among populations. Infect Genet Evol 7: 343–352. [DOI] [PubMed] [Google Scholar]
- 15. Marcilla A, Bargues MD, Ramsey JM, Magallón-Gastelum E, Salazar-Schettino PM, et al. (2001) The ITS-2 of the nuclear rDNA as a molecular marker for populations, species, and phylogenetic relationships in Triatominae (Hemiptera: Reduviidae), vectors of Chagas disease. Mol Phylogenet Evol 18: 136–142. [DOI] [PubMed] [Google Scholar]
- 16. Panzera F, Ferrandis I, Ramsey J, Ordoñez R, Salazar-Schettino PM, et al. (2006) Chromosomal variation and genome size support existence of cryptic species of Triatoma dimidiata with different epidemiological importance as Chagas disease vectors. Trop Med Int Health 11: 1092–1103. [DOI] [PubMed] [Google Scholar]
- 17. Dorn PL, Calderón C, Melgar S, Moguel B, Solorzano E, et al. (2009) Two distinct Triatoma dimidiata (Latreille, 1811) taxa are found in sympatry in Guatemala and Mexico. PLoS Negl Trop Dis 3: e393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bargues MD, Klisiowicz DR, Gonzalez-Candelas F, Ramsey JM, Monroy C, et al. (2008) Phylogeography and genetic variation of Triatoma dimidiata, the main Chagas disease vector in Central America, and its position within the genus Triatoma . PLoS Negl Trop Dis 2: e233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Usinger RL (1944) The triatominae of North and Central America and the West Indies and their public health significance. Pub Health Bull. 82.
- 20. Calderón-Fernandez GM, Girotti JR, Juarez MP (2011) Cuticular hydrocarbons of Triatoma dimidiata (Hemiptera: Reduviidae): intraspecific variation and chemotaxonomy. J Med Entomol 48: 262–271. [DOI] [PubMed] [Google Scholar]
- 21. Monteiro FA, Barrett TV, Fitzpatrick S, Cordon-Rosales C, Feliciangeli D, et al. (2003) Molecular phylogeography of the Amazonian Chagas disease vectors Rhodnius prolixus and R. robustus . Mol Ecol 12: 997–1006. [DOI] [PubMed] [Google Scholar]
- 22. Dotson E, Beard CB (2001) Sequence and organization of the mitochondrial genome of the Chagas disease vector, Triatoma dimidiata . Insect Mol Biol 10: 205–215. [DOI] [PubMed] [Google Scholar]
- 23. Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452. [DOI] [PubMed] [Google Scholar]
- 24. Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol Bioinf Online 2005: 1. [PMC free article] [PubMed] [Google Scholar]
- 26. Posada D (2008) jModelTest: phylogenetic model averaging. Mol Biol Evol 25: 1253–1256. [DOI] [PubMed] [Google Scholar]
- 27. Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52: 696–704. [DOI] [PubMed] [Google Scholar]
- 28.Akaike H (1973) Information theory and an extension of the maximum likelihood principle. In: Kiado A, editor. Second International Symposium on Information Theory. Budapest, Hungary. 267–281.
- 29. Hasegawa M, Kishino H, Yano T (1985) Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22: 160–174. [DOI] [PubMed] [Google Scholar]
- 30. Bevan RB, Lang BF, Bryant D (2005) Calculating the evolutionary rates of different genes: a fast, accurate estimator with applications to maximum likelihood phylogenetic analysis. Syst Biol 54: 900–915. [DOI] [PubMed] [Google Scholar]
- 31. Huelsenbeck JP, Ronquist F (2001) MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics 17: 754–755. [DOI] [PubMed] [Google Scholar]
- 32. Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16: 111–120. [DOI] [PubMed] [Google Scholar]
- 33. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, et al. (2011) MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol 28: 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bandelt HJ, Forster P, Rohl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16: 37–48. [DOI] [PubMed] [Google Scholar]
- 35. Peakall R, Smouse P (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6: 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pfeiler E, Bitler BG, Ramsey JM, Palacios-Cardiel C, Markow TA (2006) Genetic variation, population structure, and phylogenetic relationships of Triatoma rubida and T. recurva (Hemiptera: Reduviidae: Triatominae) from the Sonoran Desert, insect vectors of the Chagas' disease parasite Trypanosoma cruzi . Mol Phylogenet Evol 41: 209–221. [DOI] [PubMed] [Google Scholar]
- 37.Drummond AJ, Ho SYH, Rawlence HN, Rambaut A (2007) A rough guide to BEAST 1.4. Available at: http://beast.bio.ed.ac.uk/Main_Page.
- 38.Rambaut A, Drummond AJ (2007) Tracer v1.4, Available from http://beast.bio.ed.ac.uk/Tracer.
- 39. Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7: 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Monteiro FA, Donnelly MJ, Beard CB, Costa J (2004) Nested clade and phylogeographic analyses of the Chagas disease vector Triatoma brasiliensis in Northeast Brazil. Mol Phylogenet Evol 32: 46–56. [DOI] [PubMed] [Google Scholar]
- 41. Costa J, Argolo AM, Felix M (2006) Redescription of Triatoma melanica Neiva & Lent, 1941, new status (Hemiptera: Reduviidae: Triatominae). Zootaxa 1385: 47–58. [Google Scholar]
- 42. Costa J, Felix M (2007) Triatoma juazeirensis sp. nov. from the state of Bahia, Northeastern Brazil (Hemiptera: Reduviidae: Triatominae). Mem Inst Oswaldo Cruz 102: 87–90. [DOI] [PubMed] [Google Scholar]
- 43. Usinger R (1941) Notes and descriptions of neotropical Triatominae (Hemiptera, Reduviidae). Pan-Pacific Entomol 17: 49–57. [Google Scholar]
- 44. Herrera-Aguilar M, Be-Barragán LA, Ramirez-Sierra MJ, Tripet F, Dorn P, et al. (2009) Identification of a large hybrid zone between sympatric sibling species of Triatoma dimidiata in the Yucatan peninsula, Mexico, and its epidemiological importance. Infect Genet Evol 9: 1345–1351. [DOI] [PubMed] [Google Scholar]
- 45. Peterson AT, Sober nJ, Sanchez-Cordero VV (1999) Conservatism of ecological niches in evolutionary time. Science 285: 1265–1267. [DOI] [PubMed] [Google Scholar]
- 46.González C, Ornelas J, Gutiérrez-Rodríguez C (2011) Selection and geographic isolation influence hummingbird speciation: genetic, acoustic and morphological divergence in the wedge-tailed sabrewing (Campylopterus curvipennis). BMC Evol Biol 11. [DOI] [PMC free article] [PubMed]
- 47. Devitt T (2006) Phylogeography of the Western Lyresnake (Trimorphodonbiscutatus): testing aridland biogeographical hypotheses across the Nearctic–Neotropical transition Mol Ecol. 15: 4387–4407. [DOI] [PubMed] [Google Scholar]
- 48.Lee JC (1996) The Amphibians and Reptiles of the Yucatán Peninsula: Cornell Univ. Press, Ithaca, NY.
- 49. Grijalva MJ, Palomeque FS, Villacis AG, Black CL, Arcos-Teran L (2010) Absence of domestic triatomine colonies in an area of the coastal region of Ecuador where Chagas disease is endemic. Mem Inst Oswaldo Cruz 105: 677–681. [DOI] [PubMed] [Google Scholar]
- 50. Grijalva MJ, Villacis AG, Ocana-Mayorga S, Yumiseva CA, Baus EG (2011) Limitations of selective deltamethrin application for triatomine control in central coastal Ecuador. Parasit Vectors 4: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bustamante DM, Monroy C, Menes M, Rodas A, Salazar-Schettino PM, et al. (2004) Metric variation among geographic populations of the Chagas vector Triatoma dimidiata (Hemiptera: Reduviidae: Triatominae) and related species. J Med Entomol 41: 296–301. [DOI] [PubMed] [Google Scholar]
- 52. Calderón CI, Dorn PL, Melgar S, Chavez JJ, Rodas A, et al. (2004) A preliminary assessment of genetic differentiation of Triatoma dimidiata (Hemiptera: Reduviidae) in Guatemala by random amplification of polymorphic DNA-polymerase chain reaction. J Med Entomol 41: 882–887. [DOI] [PubMed] [Google Scholar]
- 53. Catalá S, Sachetto C, Moreno M, Rosales R, Salazar-Schetrino PM, et al. (2005) Antennal phenotype of Triatoma dimidiata populations and its relationship with species of phyllosoma and protracta complexes. J Med Entomol 42: 719–725. [DOI] [PubMed] [Google Scholar]
- 54. Calderón Fernandez G, Juarez MP, Ramsey J, Salazar Schettino PM, Monroy MC, et al. (2005) Cuticular hydrocarbon variability among Triatoma dimidiata (Hemiptera: Reduviidae) populations from Mexico and Guatemala. J Med Entomol 42: 780–788. [PubMed] [Google Scholar]