

Abstract
DNA fragments containing mispaired and modified bases, bulges, lesions and specific sequences have altered conformation. Methods for separating complex samples of DNA fragments based on conformation but independent of length have many applications, including (i) separation of mismatched or unmatched DNA fragments from those perfectly matched; (ii) simultaneous, diagnostic, mismatch scanning of multiple fragments; (iii) isolation of damaged DNA fragments from undamaged fragments; and (iv) estimation of reannealing efficiency of complex DNA samples. We developed a two-dimensional conformation-dependent electrophoresis (2D-CDE) method for separating DNA fragments based on length and conformation in the first dimension and only on length in the second dimension. Differences in migration velocity due to conformation were minimized during second dimension electrophoresis by introducing an intercalator. To test the method, we constructed 298 bp DNA fragments containing cytosine bulges ranging from 1 to 5 nt. Bulge-containing DNA fragments had reduced migration velocity in the first dimension due to altered conformation. After 2D-CDE, bulge-containing DNA fragments had migrated in front of an arc comprising heterogeneous fragments with regular conformation. This simple and robust method could be used in both analytical and preparative applications involving complex DNA samples.
INTRODUCTION
Unusual secondary structures in linear DNA fragments often change the conformation of the entire molecule. Such changes in conformation frequently result in curvature of DNA fragments. Curvature has been detected, for example, in DNA fragments containing mispaired bases (1), insertion/deletion bulges (2), UV lesions (3,4), base adducts (5), base methylation (6), A-tracts (7), GGCC sequences (8), cross-links (9), DNA hairpins or cruciforms (10), slipped-strand DNA (11), nicks (12) and nucleotide gaps (13).
Polyacrylamide gel electrophoresis (PAGE) allows separation of DNA fragments based on both length and conformation, while agarose gel electrophoresis separates DNA fragments essentially only according to length. Migration velocity of DNA fragments in PAGE decreases as the square of the degree of curvature in a DNA fragment (14). This quantitative relationship between curvature and migration velocity of DNA fragments in PAGE has been used experimentally to measure curving of DNA (15,16).
Some methods to screen for mutations or polymorphisms are based on conformation-dependent separation in PAGE. In heteroduplex analysis (HA), the migration velocity of mismatched DNA fragments (altered conformation) and perfectly matched DNA fragments (normal conformation) are compared (17). Conformation-sensitive gel electrophoresis (CSGE) is a variation of heteroduplex analysis where mildly denaturing solvents are used to enhance the tendency of single-base mismatches to produce conformational changes (1).
Use of one-dimensional techniques such as HA and CSGE is limited to situations where DNA fragments to be tested are of known length. In addition, only simple DNA samples containing one or, at most, only a few different DNA fragments can be tested in each lane. If the sample contained many DNA fragments of different lengths, it would be impossible to identify which fragments show conformation-dependent migration since individual bands would not resolve.
Methods for conformation-dependent separation of DNA fragments from a complex sample of DNA fragments of different lengths are of great interest. They could be used for (i) physical separation of mismatched and perfectly matched DNA fragments to allow isolation of either class; (ii) simultaneous mismatch scanning of multiple fragments; (iii) isolation of damaged DNA fragments from undamaged fragments; and (iv) estimation of reannealing efficiency of complex DNA samples. Two two-dimensional gel electrophoresis methods allow length-independent separation of curved DNA fragments containing A-tracts (18–20). One of the methods combines agarose electrophoresis and PAGE using different migration behavior of curved DNA fragments in these two matrices. The other method uses the temperature dependence of an A-tract curvature. Apparently, these two methods have not been used for conformation-dependent separation of curved DNA fragments containing other structures.
Interaction between several intercalators and DNA fragments curved due to A-tracts can suppress altered migration of the fragments in PAGE (21–23). Electron microscopy study further confirmed that the effect of the intercalator on the gel migration was linked to uncurving of DNA fragments (24).
Here we describe a two-dimensional conformation- dependent electrophoresis (2D-CDE). By exploiting interactions between intercalators and curved DNA, we achieved separation of DNA fragments based on differences in conformation but independent of their length. 2D-CDE utilized the same gel matrix in both dimensions, thus eliminating troublesome transfer between two different matrixes. Migration of DNA fragments with normal conformation was primarily determined by their relative lengths in both dimensions, resulting in an arc of DNA fragments lying diagonally through the gel. DNA fragments with altered conformation migrated at a relatively slower rate in the first dimension compared with the second dimension. They were therefore displaced in front of the arc after the second dimension electrophoresis, allowing their easy identification and isolation. Using 2D-CDE, we were able to separate DNA fragments containing two different unusual secondary structures, i.e. bulges and A-tracts, from a complex mixture of heterogeneous DNA fragments.
MATERIALS AND METHODS
Oligonucleotides
HPLC-purified oligonucleotides (Table 1) were purchased from TAG Copenhagen and stored in 100 µl of H2O at –20°C.
Table 1. Oligonucleotides and primers used in the study.
| Name | Description | Sequence 5′ to 3′ and location | 5′ modification |
|---|---|---|---|
| Forward-LEP | Forward primer for PCR product used for amplification of left end part of tripartite DNA fragmentsa | TAACGGATTCACCACTCCAAG (1298–1319)b | None or Cy5 |
| Reverse-LEP | Reverse primer for PCR product used for amplification of left end part of tripartite DNA fragments | CGATACGGGTTACTGATGATG (1842–1823) | None |
| Forward-REP | Forward primer for PCR product used for amplification of right end part of tripartite DNA fragments | CGACTACGCGATCATGGCGA (339–359) | None |
| Reverse-REP | Reverse primer for PCR product used for amplification of right end part of tripartite DNA fragments | TCATCAGCCCGAAGTGGCGA (495–475) | None |
| TMP | Top middle part | TCGGCAGAGACTTCAGATACTACTCTGTCCAGGAGAGCC | Phosphate |
| BMP | Bottom middle part. Complementary to TMP | CTCCTGGACAGAGTAGTATCTGAAGTCTCTG | Phosphate |
| BMP 1C | BMP containing one extra cytosine at position 16 | CTCCTGGACAGAGTACGTATCTGAAGTCTCTGc | Phosphate |
| BMP 2C | BMP containing two extra cytosines at position 16 | CTCCTGGACAGAGTACCGTATCTGAAGTCTCTG | Phosphate |
| BMP 3C | BMP containing three extra cytosines at position 16 | CTCCTGGACAGAGTACCCGTATCTGAAGTCTCTG | Phosphate |
| BMP 4C | BMP containing four extra cytosines at position 16 | CTCCTGGACAGAGTACCCCGTATCTGAAGTCTCTG | Phosphate |
| BMP 5C | BMP containing five extra cytosines at position 16 | CTCCTGGACAGAGTACCCCCGTATCTGAAGTCTCTG | Phosphate |
| [Forward 1-Cy5] | Forward primer for formation of a series of perfectly matched DNA fragments | GGTGAAAACCTCTGACACATGCAG (2092–2115) | Cy5 |
| [Reverse 1-Cy5 155] | Reverse primer for formation of 155 bp perfectly matched DNA fragment | TACACTCCGCTATCGCTACGTGAC (2246–2223) | Cy5 |
| [Reverse 1-Cy5 346] | Reverse primer for formation of 346 bp perfectly matched DNA fragment | CGTATTACCGCCTTTGAGTGAGC (2437–2415) | Cy5 |
| [Reverse 1-Cy5 543] | Reverse primer for formation of 543 bp perfectly matched DNA fragment | GGGGAAACGCCTGGTATCTTTATAG (2634–2610) | Cy5 |
| [Reverse 1-Cy5 857] | Reverse primer for formation of 857 bp perfectly matched DNA fragment | CCACTTCAAGAACTCTGTAGCACCG (2948–2924) | Cy5 |
Construction of tripartite DNA fragments
We constructed six different 298 bp DNA fragments using the method outlined in Figure 1. One DNA fragment was perfectly matched. Five fragments contained one defined cytosine bulge, 1–5 nt in size.
Figure 1.

Construction of DNA fragments containing an unmatched bulge. Two complementary synthetic oligonucleotides were designed to contain a specific mismatch or bulge after annealing to generate the middle part (MP). Two products, one containing an AvaI site and the other an Eco24I site, were PCR amplified from pBR322. The PCR products were digested with the corresponding enzymes, generating fragments with asymmetrical overhangs. Due to the overhang asymmetry, only a 298 bp tripartite DNA fragment (LEP–MP–REP) could be formed in a tripartite ligation (38). MP = middle part; LEP = left end part; REP = right end part. Gray tags represent overhangs.
Formation of right and left end parts. Two DNA fragments were PCR amplified from plasmid pBR322 (GenBank accession no. J01749). A 545 bp PCR product was amplified using forward and reverse left end part (F-LEP and R-LEP) primers. A 154 bp PCR product was amplified using forward and reverse right end part (F-REP and R-REP) primers. Each reaction (100 µl) contained 1 ng of pBR322, 0.2 µM of each primer, 200 µM of each dNTP, 7 U of KlenTaq 1 polymerase (AB peptides), 0.05 U of PfuI polymerase (Stratagene) and 1× RDA buffer (25). PCR conditions for both reactions were a hot start at 84°C by adding dNTPs, initial denaturation at 94°C for 5 min, 94°C for 1 min, 58°C for 1 min, and 68°C for 2 min for 25 cycles, and an extension step at 68°C for 5 min. PCR products were purified using a GFX PCR and Gel band purification kit (Amersham Biosciences). DNA was eluted in 2.5 mM Tris–HCl pH 8.0. Seven purified PCRs were pooled and concentrated to 175 µl using SpeedVac plus (Savant). Concentrations of PCR and ligation products were quantified with a DynaQuant 200 fluorometer (Amersham Bioscience).
The 545 bp PCR product was digested for 2 h at 37°C with 2 U/µg AvaI (New England Biolabs), and a 127 bp fragment (LEP) was gel isolated without any exposure to ethidium bromide (EtBr) or UV light using a GFX purification kit and EtBr-stained reference lane.
The 154 bp PCR product was digested for 2 h at 37°C with 10 U/µg Eco24I (MBI Fermentas). The 132 bp DNA fragment (REP) was isolated using GFX with a PCR purification protocol and two washing steps to ensure removal of the 22 bp DNA fragment.
Formation of middle parts. Double-stranded middle parts (MPs) were formed by annealing 100 pmol of each of the six different bottom middle parts (BMPs) (Table 1) and 120 pmol of the top middle part (TMP) in 20 µl of 0.3× SSC (1× 150 mM NaCl and 15 mM Na-citrate pH 7.0). The reaction was kept at 85°C for 2 min and then allowed to cool to room temperature at the rate of 1.2°C per min.
Tripartite ligation
LEP, each of the six different MPs and REP (2–10 pmol) were ligated in 50 µl for 16 h at 16°C using 400 U of T4 DNA ligase (New England Biolabs). Products were either gel isolated to remove shorter bipartite products or GFX purified.
Formation of Cy5-labeled perfectly matched DNA fragments
DNA fragments (155, 357, 543 and 857 bp) were amplified from pBR322 using a forward Cy5 primer and one of the four reverse Cy5 primers (Table 1). Reactions were identical to the end part PCR described above, except that annealing was at 56°C.
Migration velocity
Migration velocity was evaluated in PAGE, MDE and CSGE in 7 × 8 × 0.1 cm gels using the Mini-Protean II system (Bio-Rad) or in 18 × 24 × 0.1 cm gels using the Hoefer SE 660 system (Amersham Biosciences). Gels were stained for 10 min in electrophoresis buffer containing 1 µg/ml EtBr.
Polyacrylamide gels (6–15%) were made from a 29:1 acrylamide:bisacrylamide mixture (Amersham Bioscience) in 0.5× TBE. Electrophoresis was carried out at room temperature in 0.5× TBE at 20 mA until bromophenol blue (BB) migrated to the lower edge of the gel.
MDE gels (FMC BioProducs) were made from a 0.75× MDE solution and 0.6× TBE. Brief electrophoresis was performed at room temperature in 0.6× TBE at 20 mA for 5 min to load samples into the gel, followed by incubation for 10 min in 0.6× TBE with or without 1 µg/ml of EtBr. After incubation, gels were run in a Pharmacia Multiphor system (Amersham Bioscience) at 5, 20 or 35°C in the same direction as before at 5 mA in 0.6× TBE until BB migrated to the edge.
CSGE gels (10%) were prepared as previously described (26). Electrophoresis was carried out at 35°C in a 0.5× TTE buffer at 10 W until BB migrated two-thirds of the gel.
The migration velocities of DNA fragments were compared using the Conformation factor (C-factor) defined as the apparent migration velocity of perfectly matched DNA fragments divided by the apparent migration velocity of bulge-containing DNA fragments of the same length.
2D-CDE
First dimension. PAGE, MDE and CSGE gels were prepared as described above. A mixture of DNA fragments in ficoll (for CSGE) or glycerol gel-loading buffer was loaded in the second lane of each gel. The sample volume was 10–20 µl and the amount of DNA was 100–1000 ng. Vertical electrophoresis was then carried out and gels subsequently incubated with shaking for 10 min in 100 ml of the appropriated electrophoresis buffer containing 1 µg/ml EtBr.
Second dimension. Electrophoresis was carried out in a Multiphor system perpendicular to the first dimension at 5 W and 20°C using paper electrode wicks until BB migrated to the edge of the gel. The same buffer system was used as for the first dimension except that it contained 1 µg/ml of EtBr.
Fluorescent-labeled DNA fragments were analyzed using the fluorescence-scanning mode of a Typhoon 8610 variable mode imager (Amersham Biosciences).
Two-dimensional temperature-dependent electrophoresis
The same gel and buffer were used as previously described (19). The first dimension was carried out as described above for PAGE in the small gel format. The second dimension was carried out as described for 2D-CDE but without EtBr and at constant temperatures of 35, 50, 60 or 65°C.
Separation and enrichment of DNA fragments containing unmatched bulge from a complex mixture of perfectly matched DNA fragments
λ Phage DNA (dam–, dcm–) was purchased from MBI Fermentas. λ Phage DNA (10 µg) was digested for 60 min with 30 U of NdeII (Roche) at 37°C.
Digested λ phage was labeled by extension of overhangs [10 mg of DNA, 5 U of Klenow fragment (Fermentas), 100 mM unlabeled dNTPs (Fermentas), 100 mM Cy-labeled dCTP (Amersham Biosciences)] for 15 min at 30°C. The reaction was quenched by addition of 1 µl of 0.5 M EDTA. Labeling reactions were purified using a GFX PCR and Gel band purification kit.
Cy5 NdeII-digested λ phage DNA (800 ng) was mixed with a Cy5 tripartite DNA fragment containing 5C bulge (5 ng) and 3C bulge (5 ng). The DNA pool was separated using 2D-CDE in a 9% polyacrylamide gel (7 × 8 × 0.1 cm) and fluorescence scanned as described in ‘2D-CDE’. Using ImageQuant 5.1 (Amersham Biosciences), a multi-segmented string was aligned along the length of the arc of perfectly matched DNA. The width of the string was chosen to match the width of the DNA in the arc. Copies of the string were moved horizontally in steps of one-string width. A real size picture of the gel showing the parallel strings was placed underneath and the gel sliced, using the string boundaries as reference. Each slice was crushed in an Eppendorf tube, 400 µl of 0.05× TE was added, and DNA was eluted overnight at 4°C on a shaker (600 r.p.m.). The collected supernatant was reduced to ∼20 µl using Savant Speedvac and analyzed on a 2% agarose gel.
RESULTS
Construction of 298 bp DNA fragments each containing a specific cytosine bulge near its center
To develop a method for conformation-dependent separation, it was necessary to construct long DNA fragments (∼300 bp) with specific secondary structures that affect the conformation of the entire molecule. A semi-synthetic strategy for construction of tripartite DNA fragments (298 bp) was designed (Fig. 1). We constructed five fragments containing one defined cytosine bulge at the center of the fragment with bulge size ranging from 1 to 5 nt (1C–5C). An identical perfectly matched DNA fragment was prepared as a control. We evaluated purity and length of all fragments with agarose electrophoresis (Fig. 2A). As expected, all DNA fragments migrated at the same rate independently of the bulge size.
Figure 2.
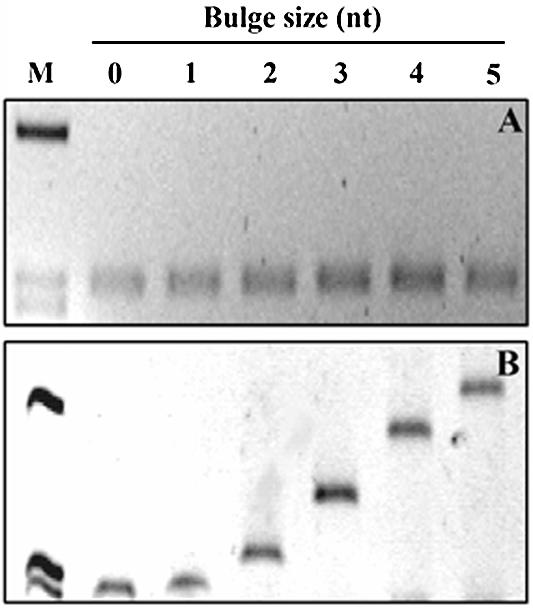
Migration of tripartite DNA fragments by agarose electrophoresis and PAGE. Six tripartite DNA fragments either perfectly matched or containing a cytosine bulge ranging in size from 1 to 5 nt (1C–5C) were analyzed. (A) Electrophoresis in 2% agarose. All products migrated consistently with the length of the tripartite products. (B) Electrophoresis in 8% polyacrylamide. Migration velocity of different fragments depended on the size of the bulge. M = φX174 DNA/digested with HaeIII.
Migration velocity in PAGE of tripartite DNA fragments containing an unmatched bulge
DNA fragments containing a bulge have reduced migration velocity in PAGE compared with perfectly matched DNA fragments of the same length, and the reduction is dependent on the bulge size (2). Migration velocities of tripartite DNA fragments were evaluated in PAGE to validate formation of specific cytosine bulges (Fig. 2B). The DNA fragment containing the 1C bulge migrated slightly more slowly than the perfectly matched fragment. We identified a strong inverse correlation between bulge size in fragments containing 2–5 cytosine bulges and migration velocity in gels (Fig. 2B and Table 2). To enhance the migration differences between perfectly matched DNA fragments and DNA fragments containing bulges, we used an MDE™ gel matrix. Higher C-factors were obtained in MDE™ gels for DNA fragments containing large bulges (Table 2).
Table 2. Inverse correlation between bulge size from 1 to 5 nt in DNA fragments and migration velocity.
| Size of bulge | C-factor in 8% PAGE | C-factor in 0.75× MDE |
|---|---|---|
| 0 | 1.0 | 1.0 |
| 1 | 1.0 | 1.0 |
| 2 | 1.1 | 1.2 |
| 3 | 1.4 | 1.5 |
| 4 | 1.7 | 2.2 |
| 5 | 2.0 | 2.6 |
Conformation factors (C-factors) were calculated as the apparent migration of a perfectly matched DNA fragment divided by the apparent migration of a bulge-containing DNA fragment of equal length.
To define the effect of temperature and intercalators on altered migration, three DNA fragments (perfectly matched, 2C and 4C bulge) were analyzed in an MDE gel at three constant temperatures (4, 20 or 35°C), both with and without EtBr. Calculated C-factors revealed that the altered migration velocity of bulge-containing DNA fragments was almost independent of temperature (Table 3). In contrast, the effect of a bulge on migration velocity was strongly reduced by addition of the intercalator. In the presence of EtBr, bulge-containing DNA fragments migrated more consistently based on their length and with much less dependence on the size of the bulge. The difference in migration velocity, whether EtBr was present or not, was most pronounced for the fragment containing the larger 4C bulge.
Table 3. Effect of temperature and EtBr intercalator on C-factors of bulge-containing DNA fragments.
| Size of bulge in DNA fragment | 4°C – EtBr | 4°C + EtBr | 20°C – EtBr | 20°C + EtBr | 35°C – EtBr | 35°C + EtBr |
|---|---|---|---|---|---|---|
| 0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| 2 | 1.1 | 1.0 | 1.3 | 1.0 | 1.1 | 1.0 |
| 4 | 1.8 | 1.1 | 2.4 | 1.2 | 2.4 | 1.4 |
These results allowed us to define conditions where DNA fragments either showed conformation-dependent migration, or not, in a single gel matrix. Gels without EtBr allowed separation according to both length and conformation of each DNA fragment. Gels containing EtBr allowed separation mainly according to length. By combining these two conditions in 2D gel electrophoresis, one should be able to achieve conformation-dependent separation of DNA fragments containing an unusual conformation from a complex sample of heterogeneous DNA fragments.
2D-CDE
We tested this hypothesis using a mixture of DNA fragments with normal or altered conformation in a small MDE™ gel. The mixture included an HaeIII digest of φX174 DNA (11 perfectly matched DNA fragments) and a 298 bp DNA fragment containing a 3C bulge. One DNA fragment (281 bp) from the φX174 digest is known to show sequence-induced altered migration at 20°C (27) and should serve as an internal control for conformation-dependent separation. Two DNA fragments clearly migrated in front of the arc after 2D-CDE (Fig. 3). The slower migrating band was the 3C DNA fragment and the faster migrating band was the 281 bp DNA fragment from HaeIII-digested φX174. By increasing the time of second dimension electrophoresis, further separation was achieved (Fig. 3B–D).
Figure 3.

Separation by 2D-CDE of a bulge-containing DNA (298 bp) and a curved DNA fragment (281 bp) due to an A-tract from perfectly matched HaeIII fragments of RF φX174. (A) First dimension electrophoresis (20°C in 0.75× MDE matrix) to separate DNA fragments according to both their length and conformation. (B–D) Second dimension electrophoresis perpendicular to the first dimension was carried out after addition of EtBr to separate mainly according to the length of the DNA fragments. Interim time points of 30 min (B), 45 min (C) and 60 min (D). Two DNA fragments were displaced in front of the perfectly matched DNA fragments. The DNA fragment labeled 1 contained a 3C bulge. The DNA fragment labeled 2 was a 281 bp DNA fragment from the HaeIII digest of the RF φX174 containing an A-tract.
We also ran two-dimensional electrophoresis on identical DNA mixtures where the only difference between dimensions was temperature (20°C in the first and 35, 50, 60 or 65°C in the second dimension). No separation was detected for the 3C bulge DNA fragment, but the 281 bp A-tract DNA fragment migrated in front of the arc under temperatures up to 60°C in the second dimension (data not shown). This result further confirmed the temperature dependency of the curvature associated with A-tracts.
2D-CDE was also performed in the large gel format using 10% PAGE. This system allowed separation of 2C–5C DNA fragments from perfectly matched DNA fragments (data not shown).
To further increase the resolution of 2D-CDE, we used CSGE matrix and buffers in a large gel format (1). Using this approach, we were able to separate tripartite and dipartite DNA fragments containing 1C–5C bulges from a mixture of perfectly matched DNA fragments (Fig. 4).
Figure 4.

DNA fragments containing 1C–5C bulges separated from perfectly matched DNA by 2D-CDE using a CSGE gel matrix. DNA fragments containing 1C–5C bulges either at their center in the tripartite 298 bp DNA fragment (labeled T-1C to T-5C) or 15 bp from their end in bipartite 162 or 168 bp DNA fragments (MP–REP or LEP–MP) (labeled D-1C to D-5C) were mixed with perfectly matched Cy5 PCR products (155, 346, 543 and 857 bp) and analyzed. Perfectly matched DNA fragments formed an arc with unmatched bulge-containing DNA fragments migrating in front of the arc. DNA fragments containing bulges either at the center or close to the end essentially aligned in a vertical ladder of DNA bands, consistent with the fact that all bulge-containing fragments migrate at a similar rate in the second dimension.
We adapted 2D-CDE for using fluorescent detection to increase the sensitivity and versatility of the method. This required a washing step after 2D-CDE to reduce background fluorescent detection from the intercalator. By using double-dye fluorescent detection we were able to visualize the separation of perfectly matched DNA fragments and 3C–5C DNA fragments (Fig. 5).
Figure 5.

Two-color fluorescent image of 2D-CDE in 7 × 8 cm, 10% PAGE. Nine perfectly matched DNA fragments 200–1000 bp (100 bp Fluorescein Molecular Ruler, Bio-Rad) aligned diagonally through the gel (labeled green). DNA fragments containing a 0C–5C bulge at the center were labeled with Cy5 (red). Separation was achieved between perfectly matched DNA and DNA containing bulges in the size range between 3 and 5 nt.
Isolation of DNA fragments containing bulges from complex DNA samples
We also tested whether DNA fragments containing bulges can be efficiently separated from λ phage DNA. Digested λ phage DNA was mixed with 3C and 5C DNA fragment in an equimolar concentration. The mixture was separated using 2D-CDE in a small gel format (Fig. 6). The bulge-containing DNA fragments were displaced in front of the arc of digested λ phage DNA fragments. Quantitative analysis of the gel revealed several hundred-fold enrichment for both 3C and 5C DNA fragments (Fig. 7). To further demonstrate the potential of the system, we gel-isolated DNA from the arc and DNA displaced in front of the arc. These samples were analysed with agarose gel electrophoresis (Fig. 8). As expected, no DNA was detected in the segment excised from behind the arc, λ DNA was detected in the arc, and only bulge-containing DNA fragments in the gel segments excised from in front of the arc. These results showed the efficiency of the method for preparatory work.
Figure 6.

2D-CDE separation of 298 bp DNA fragments containing a 3C or 5C bulge from NdeII-digested λ phage DNA in 7 × 8 cm, 9% PAGE. All fragments were Cy5 labeled. (A) No physical separation was achieved after the first dimension. (B) Second dimension electrophoresis resulted in displacement of the 3C and 5C DNA fragments in front of the arc of perfectly matched DNA.
Figure 7.

Enrichment of the 3C and 5C bulge-containing DNA fragments from λ phage DNA. A multi-segmented arc-wide line was aligned on top of the arc in the gel and horizontally displaced copies of it in front of the arc through the bulge-containing DNA fragments. The density for the Cy5 channel was measured in all the lines and the density in fluorescent units was plotted as a function of length of the lines in pixels. The dotted line in the graph represents the density of perfectly matched DNA in the arc. The red line represents the first multi-segmented line placed one arc width in front of the arc (Arc+1). The green line represents the second multi-segmented line placed two arc widths in front of the arc (Arc+2). The bulge-containing DNA fragment was enriched several hundred fold after 2D-CDE.
Figure 8.
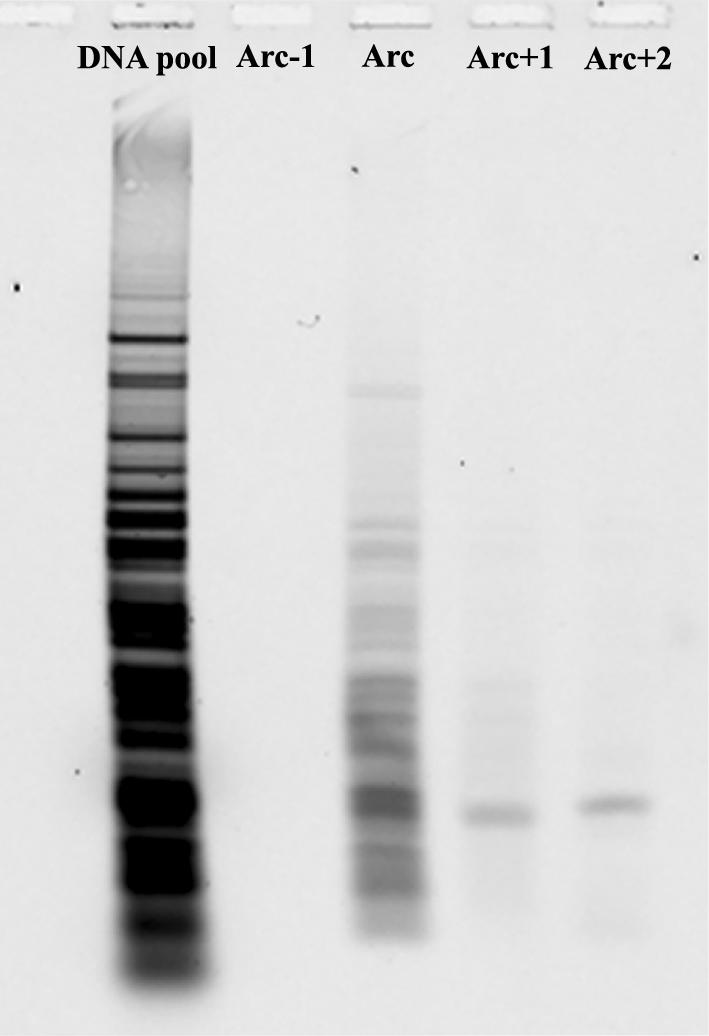
Isolation of DNA fragments after 2D-CDE. After separation by 2D-CDE, the gel was cut, parallel to the arc, into four identical gel slices each one arc in width. One slice was from behind the arc (Arc-1), one containing the arc (Arc) and two parallel in front of the arc (Arc+1 and Arc+2). DNA isolated from each gel slice was separated on agarose and compared with the DNA sample before separation. No DNA was detected behind the arc, only λ DNA was detected in the arc, 3C DNA in Arc+1, and 5C DNA in arc+2.
DISCUSSION
In this paper, we describe a novel physicochemical method, 2D-CDE. The method allows separation of DNA fragments according to their conformation but independent of their length. The method should be a powerful tool for various tasks where length-independent separation and isolation of DNA fragments with altered conformation is needed. It is easy to carry out and requires only limited labor. Due to its simplicity, the method is reproducible and robust. The method does not require a specific instrument. All reagents are inexpensive and commonly used in the biosciences.
In principle, this method can be used to separate DNA fragments that have conformational changes that can be eliminated by addition of an intercalator. We showed that 2D-CDE can be used to separate and enrich DNA fragments containing bulges from complex samples of perfectly matched DNA fragments (∼50–2000 bp). 2D-CDE can also be used to separate DNA fragments containing an A-tract from a complex DNA mixture. The method may also separate DNA fragments containing other local sequences and secondary structures that are known to affect conformation of DNA, including UV lesions, GGCC sequences, cross-links, slipped-strand DNA, nicks and nucleotide gaps.
The resolution of 2D-CDE will vary according to the conditions used. A gel matrix such as the MDE™ allowed better conformation-dependent separation than a regular polyacrylamide gel matrix. In our experience, the highest resolution was achieved using CSGE conditions and a gel matrix in the first dimension. We were able to separate DNA fragments containing one nucleotide bulge using CSGE conditions from a mixture of perfectly matched fragments ranging in length from 155 to 857 bp. We have also detected an increased difference in migration of a DNA fragment containing single base differences, but the increase was not sufficient for two-dimensional separation (data not shown). We do not exclude the possibility of length-independent separation of single base differences with 2D-CDE using a larger gel format and CSGE conditions. Another possibility for detection of single-base mismatches that we are currently pursuing is to combine enzyme treatment of DNA pools with 2D-CDE.
In this paper, we have shown that bulges as large as 5 nt can be separated using the 2D-CDE. For efficient separation of DNA fragment-containing microsatellite repeats from human DNA, it is important to be able to resolve larger bulges (typically 10–20 nt) The largest bulge we have tested to date was 18 nt in a 657 bp DNA fragment. This fragment separated well in 2D-CDE (unpublished results).
To eliminate or decrease differences in conformation between DNA fragments after the first dimension electrophoresis, we exploited interactions between an intercalator and DNA. Insertion of intercalating molecules causes elongation of the DNA duplex and local unwinding of the base pairs. Values for unwinding have been reported for many intercalators. They constitute an indirect measurement of conformation alterations of DNA (28). We reviewed previously reported values of 19 different intercalators where values ranged from 8 to 80° (29,30). We chose to use EtBr, which has an intermediate unwinding value of 26°. EtBr intercalates double-stranded nucleic acids with limited sequence specificity with the exception of poly(dA)–poly(dT) (31). It has been documented that EtBr shows higher affinity for double-stranded DNA-containing bulges than their perfectly matched counterparts (32–34).
Suppression of altered migration by adding EtBr into the gel matrix was achieved for all tested bulge-containing DNA fragments. We believe that intercalators showing unwinding values similar to or higher than EtBr are also potential candidates for various applications of 2D-CDE.
The effect of temperature on migration velocity was also tested. We found that relative migration of bulge-containing fragments compared with perfectly matched DNA fragment was essentially independent of temperature. The temperature independency of migration for bulge-containing DNA was confirmed further by analyses of a two-dimensional system similar to the one described by Mizuno (19) (unpublished results). This system uses temperature as a variable between dimensions and was reported to be efficient for separation of DNA fragments containing A-tracts from a complex DNA pool. Although in our experiments the temperature was raised up to 60°C in the second dimension, no separation was achieved for bulge-containing DNA. With the same system setup, it was enough to use 35°C in the second dimension to allow separation of DNA fragments containing A-tracts. We suggest that elevated temperature, e.g. 35°C, in the first dimension could be used to avoid 2D-CDE separation of DNA fragments with A-tracts (35).
Genome mismatch scanning (GMS) was developed to circumvent the need to screen markers one at a time to identify genomic regions that are identical by descent (IBD). In theory, the method assays every polymorphic base between two genomes. Enrichment of perfectly matched heterohybrids is a key step in GMS. When GMS was applied successfully to yeast, DNA enrichment was achieved using the Escherichia coli mismatch repair proteins mut H, L and S (36). GMS has not been applied efficiently to the human genome and it has had limited application in genetics. 2D-CDE is a candidate method for such mismatch scanning due to its simplicity and robustness. Further, the current setup of 2D-CDE allows specific enrichment of bulge-containing fragments that are formed between highly polymorphic sequences such as microsatellite repeats. Bulge-containing heteroduplexes are presumably frequently co-isolated with the perfectly matched heterohybrids using the mut HLS system since mut S only interacts with single-base mismatches and bulges up to 4 nt (37). Larger bulges are not recognized.
2D-CDE can be used for various preparative and analytical tasks where separation according to conformation but independent of length can be exploited. An application we are currently pursuing is the separation of polymorphic DNA sequences from the human genome. We are also applying 2D-CDE for separation of DNA containing UV lesions from intact DNA and for insertion/deletion variation scanning in cDNA.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by the Icelandic Research Council, the University of Iceland Research Fund, the Science Fund of the Landspitali-University Hospital and BioCule Inc.
REFERENCES
- 1.Ganguly A., Rock,M.J. and Prockop,D.J. (1993) Conformation-sensitive gel electrophoresis for rapid detection of single-base differences in double-stranded PCR products and DNA fragments: evidence for solvent-induced bends in DNA heteroduplexes. Proc. Natl Acad. Sci. USA, 90, 10325–10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lilley D.M. (1995) Kinking of DNA and RNA by base bulges. Proc. Natl Acad. Sci. USA, 92, 7140–7142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Husain I., Griffith,J. and Sancar,A. (1988) Thymine dimers bend DNA. Proc. Natl Acad. Sci. USA, 85, 2558–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim J.K., Patel,D. and Choi,B.S. (1995) Contrasting structural impacts induced by cis–syn cyclobutane dimer and (6–4) adduct in DNA duplex decamers: implication in mutagenesis and repair activity. Photochem. Photobiol., 62, 44–50. [DOI] [PubMed] [Google Scholar]
- 5.Pietrasanta L.I., Smith,B.L. and MacLeod,M.C. (2000) A novel approach for analyzing the structure of DNA modified by benzo[a]pyrene diol epoxide at single-molecule resolution. Chem. Res. Toxicol., 13, 351–355. [DOI] [PubMed] [Google Scholar]
- 6.Diekmann S. (1987) DNA methylation can enhance or induce DNA curvature. EMBO J., 6, 4213–4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacDonald D., Herbert,K., Zhang,X., Polgruto,T. and Lu,P. (2001) Solution structure of an A-tract DNA bend. J. Mol. Biol., 306, 1081–1098. [DOI] [PubMed] [Google Scholar]
- 8.Wildeson J. and Murphy,C.J. (2000) Intrinsic bending in GGCC tracts as probed by fluorescence resonance energy transfer. Anal. Biochem., 284, 99–106. [DOI] [PubMed] [Google Scholar]
- 9.Hwang G.S., Kim,J.K. and Choi,B.S. (1996) The solution structure of a psoralen cross-linked DNA duplex by NMR and relaxation matrix refinement. Biochem. Biophys. Res. Commun., 219, 191–197. [DOI] [PubMed] [Google Scholar]
- 10.Diekmann S. and Lilley,D.M. (1987) The anomalous gel migration of a stable cruciform: temperature and salt dependence and some comparisons with curved DNA. Nucleic Acids Res., 15, 5765–5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pearson C.E., Wang,Y.H., Griffith,J.D. and Sinden,R.R. (1998) Structural analysis of slipped-strand DNA (S-DNA) formed in (CTG)n·(CAG)n repeats from the myotonic dystrophy locus. Nucleic Acids Res., 26, 816–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LeCam E., Fack,F., Menissier-de Murcia,J., Cognet,J.A., Barbin,A., Sarantoglou,V., Revet,B., Delain,E. and de Murcia,G. (1994) Conformational analysis of a 139 base-pair DNA fragment containing a single-stranded break and its interaction with human poly(ADP-ribose) polymerase. J. Mol. Biol., 235, 1062–1071. [DOI] [PubMed] [Google Scholar]
- 13.Roll C., Ketterle,C., Faibis,V., Fazakerley,G.V. and Boulard,Y. (1998) Conformations of nicked and gapped DNA structures by NMR and molecular dynamic simulations in water. Biochemistry, 37, 4059–4070. [DOI] [PubMed] [Google Scholar]
- 14.Crothers D.M., Drak,J., Kahn,J.D. and Levene,S.D. (1992) DNA bending, flexibility and helical repeat by cyclization kinetics. Methods Enzymol., 212, 3–29. [DOI] [PubMed] [Google Scholar]
- 15.Hardwidge P.R., Zimmerman,J.M. and Maher,L.J.,3rd (2000) Design and calibration of a semi-synthetic DNA phasing assay. Nucleic Acids Res., 28, e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ross E.D., Den,R.B., Hardwidge,P.R. and Maher,L.J.,3rd (1999) Improved quantitation of DNA curvature using ligation ladders. Nucleic Acids Res., 27, 4135–4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soto D. and Sukumar,S. (1992) Improved detection of mutations in the p53 gene in human tumors as single-stranded conformation polymorphs and double-stranded heteroduplex DNA. PCR Methods Appl., 2, 96–98. [DOI] [PubMed] [Google Scholar]
- 18.Ohyama T. and Kusakabe,T. (1993) High-efficiency shotgun cloning of curved DNA segments from chromosomal DNA. Anal. Biochem., 212, 287–289. [DOI] [PubMed] [Google Scholar]
- 19.Mizuno T. (1987) Random cloning of bent DNA segments from Escherichia coli chromosome and primary characterization of their structures. Nucleic Acids Res., 15, 6827–6841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson J.N. (1986) Detection, sequence patterns and function of unusual DNA structures. Nucleic Acids Res., 14, 8513–8533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Griffith J., Bleyman,M., Rauch,C.A., Kitchin,P.A. and Englund,P.T. (1986) Visualization of the bent helix in kinetoplast DNA by electron microscopy. Cell, 46, 717–724. [DOI] [PubMed] [Google Scholar]
- 22.Cons B.M. and Fox,K.R. (1990) Effects of sequence selective drugs on the gel mobility of a bent DNA fragment. Biochem. Biophys. Res. Commun., 171, 1064–1070. [DOI] [PubMed] [Google Scholar]
- 23.Nightingale K.P. and Fox,K.R. (1992) Interaction of bleomycin with a bent DNA fragment. Biochem. J., 284, 929–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barcelo F., Muzard,G., Mendoza,R., Revet,B., Roques,B.P. and Le Pecq,J.B. (1991) Removal of DNA curving by DNA ligands: gel electrophoresis study. Biochemistry, 30, 4863–4873. [DOI] [PubMed] [Google Scholar]
- 25.Lisitsyn N. and Wigler,M. (1993) Cloning the differences between two complex genomes. Science, 259, 946–951. [DOI] [PubMed] [Google Scholar]
- 26.Korkko J., Annunen,S., Pihlajamaa,T., Prockop,D.J. and Ala-Kokko,L. (1998) Conformation sensitive gel electrophoresis for simple and accurate detection of mutations: comparison with denaturing gradient gel electrophoresis and nucleotide sequencing. Proc. Natl Acad. Sci. USA, 95, 1681–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wenz H.M. (1994) Capillary electrophoresis as a technique to analyze sequence-induced anomalously migrating DNA fragments. Nucleic Acids Res., 22, 4002–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brukner I., Belmaaza,A. and Chartrand,P. (1997) Differential behavior of curved DNA upon untwisting. Proc. Natl Acad. Sci. USA, 94, 403–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Utsuno K. and Tsuboi,M. (1997) Degree of DNA unwinding caused by the binding of aclacinomycin A. Chem. Pharm. Bull. (Tokyo), 45, 1551–1557. [DOI] [PubMed] [Google Scholar]
- 30.Petersen M. and Jacobsen,J.P. (1998) Solution structure of a DNA complex with the fluorescent bis-intercalator TOTO modified on the benzothiazole ring. Bioconjug. Chem., 9, 331–340. [DOI] [PubMed] [Google Scholar]
- 31.Ren J. and Chaires,J.B. (1999) Sequence and structural selectivity of nucleic acid binding ligands. Biochemistry, 38, 16067–16075. [DOI] [PubMed] [Google Scholar]
- 32.White S.A. and Draper,D.E. (1989) Effects of single-base bulges on intercalator binding to small RNA and DNA hairpins and a ribosomal RNA fragment. Biochemistry, 28, 1892–1897. [DOI] [PubMed] [Google Scholar]
- 33.White S.A. and Draper,D.E. (1987) Single base bulges in small RNA hairpins enhance ethidium binding and promote an allosteric transition. Nucleic Acids Res., 15, 4049–4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nelson J.W. and Tinoco,I.,Jr (1985) Ethidium ion binds more strongly to a DNA double helix with a bulged cytosine than to a regular double helix. Biochemistry, 24, 6416–6421. [DOI] [PubMed] [Google Scholar]
- 35.Jerkovic B. and Bolton,P.H. (2000) The curvature of dA tracts is temperature dependent. Biochemistry, 39, 12121–12127. [DOI] [PubMed] [Google Scholar]
- 36.Nelson S.F., McCusker,J.H., Sander,M.A., Kee,Y., Modrich,P. and Brown,P.O. (1993) Genomic mismatch scanning: a new approach to genetic linkage mapping. Nature Genet., 4, 11–18. [DOI] [PubMed] [Google Scholar]
- 37.Harfe B.D. and Jinks-Robertson,S. (2000) DNA mismatch repair and genetic instability. Annu. Rev. Genet., 34, 359–399. [DOI] [PubMed] [Google Scholar]
- 38.Lee S., Elenbaas,B., Levine,A. and Griffith,J. (1995) p53 and its 14 kDa C-terminal domain recognize primary DNA damage in the form of insertion/deletion mismatches. Cell, 81, 1013–1020. [DOI] [PubMed] [Google Scholar]