

Unipolar depression is ranked as number one before all other somatic and psychiatric illnesses. In spite of its prevalence, the underlying causes of depression are still unclear [1]. This disorder is a significant health problem and 10-20% of all adults suffer from this disease[1]. Different classes of antidepressants have been developed over the past decades. Antidepressants are traditionally thought to elicit their therapeutic effects by increasing extraneuronal concentrations of serotonin and norepinephrine [2]. Earlier tricyclic antidepressants displayed variable norepinephrine and serotonin uptake inhibitory activity, while exhibiting considerable side effects in addition to antidepressant effects due to non-specific interactions in the central nervous system (CNS) [3] Subsequent development of selective serotonin reuptake inhibitors (SSRIs) and serotonin/norepinephrine reuptake inhibitors (SNRIs) are second generation antidepressants which provided drugs with much more selective interaction at serotonin and norepinephrine systems [4-7]. By virtue of their interaction with the SERT and NET only, these drugs had appreciable less unwanted side effects than tricyclic antidepressants [8-9]. An SNRI such as Venlafaxine exhibits antidepressant activity in clinical trials and displays somewhat greater response and remission rates compared to SSRIs [10]. However, a significant unmet need for more improved therapy still exists, as large numbers of depressed people are still refractory to the current existing therapies. In addition, a significant number of people suffer from relapse after treatment with current therapies [11].
Dopaminergic activity has not been included in the current pharmacotherapy of depression even though there are ample evidences pointing towards an important involvement of dopaminergic neurotransmission in depression [12-13]. Inclusion of dopamine activity should act to reduce anhedonia which is associated with a deficit in dopaminergic transmission and is a central component of the depressed state of mind. A successful adjunct therapy approach involving use of dopamine transporter blocker bupropion and SSRI was found to be more efficacious in patients refractory to SSRI.[14-15] Furthermore, the D3 dopamine receptor preferring drug pramipexole was effective in both unipolar and bipolar depression.[16]
This prompted the design of triple uptake inhibitors (TUIs). However, from drug development point of view it is difficult to optimize three uptake inhibition activities in a single molecule. In recent years a number of TUIs, e.g. DOV 216,303, PRC200-SS, JNJ-7925476 and GSK-372,475 have been developed (Figure 1) and have been characterized in animal depression models.[17-18] DOV 216,303 was studied in a human clinical trial for pharmacokinetic properties and efficacy.[18-19] In severe to major depressive disorder, DOV 216,303 was found to be as efficacious as Citalopram.[18]
Figure 1.

Molecular structures of known TUIs and pyran molecule-based TUIs.
In our effort to develop triple uptake inhibitors, we embarked on developing molecules based on asymmetric pyran derivatives. CNS-active pyran compounds are relatively rare. In our recent reports, we have demonstrated the development of asymmetric 3,6-disubstituted and 2,4,5-trisubstituted pyran derivatives targeting monoamine transporter systems.[20-22] Compounds with various profiles were developed. Thus, a number of TUIs were identified as well as compounds with an SSRI, SNRI and dopamine/norepinephrine reuptake inhibitor (DNRI) profile. Two of our lead TUIs 1a (D-142) and 1b (D-161) (Figure 1, Table 1) were shown to be efficacious in animal models of depression.[23-24]
Table 1.
Affinity of drugs at DAT, SERT, and NET in rat brain.
| Compound | DAT uptake, Ki, nM, [3H]DA[a] |
SERT uptake, Ki, nM, [3H]-5- HT[a] |
NET uptake, Ki, nM [3H]DA[a] |
|---|---|---|---|
| GBR 12909 | 9.14 ± 1.94 | 132 ± 47 | 38.5 ± 4.9 |
| Reboxetine | > 10,000 | 503 ± 61 | 0.694 ± 0.217 |
|
|
|||
| Fluoxetine | 1092 ± 98 | 12.2 ± 2.4 | 158 ± 58 |
|
|
|||
| 1a | 59.3 ± 13.7 | 14.7 ± 2.1 | 29.3 ± 7.9 |
|
|
|||
| 1b | 42.0 ± 3.3 | 29.1 ± 3.5 | 30.5 ± 7.8 |
|
|
|||
| 1c | 62.4 ± 5.6 | 16.1 ± 1.6 | 12.6 ± 3.7 |
|
|
|||
| 2a | 20 ± 14 | 270 ± 32 | 658 ± 86 |
|
|
|||
| 2b | 62.1± 4.2 | 155 ± 53 | 68.0 ± 15.0 |
|
|
|||
| 2c | 152 ± 11 | 1.05 ± 0.24 | 41.3 ± 21.4 |
|
|
|||
| 2d | 344 ± 74 | 409 ± 52 | 121 ± 8.1 |
|
|
|||
| 2e | 87.7 ± 3.5 | 52.6 ± 13.8 | 8.58 ± 1.29 |
|
|
|||
| 2f | 160 ± 21 | 1.07 ± 0.12 | 15.8 ± 3.7 |
|
|
|||
| 2g | 60.0 ± 8.6 | 79.2 ± 13.8 | 70.3 ± 18.2 |
|
|
|||
| 2h | 11,795 ± 483 | > 100 μm | 3288 ± 996 |
|
|
|||
| 4a | 135 ± 6 | 14.7 ± 3.5 | 22.6 ± 14.9 |
|
|
|||
| 4b | 334 ± 53 | 93.2 ± 17.9 | 244 ± 68 |
|
|
|||
| 4c | 245 ± 47 | 370 ± 42 | 193 ± 43 |
|
|
|||
| 4d | 401 ± 87 | 1.99 ± 0.77 | 57.2 ± 11.9 |
|
|
|||
| 4e | 303 ± 39 | 120 ± 21 | 91.8 ± 16 |
|
|
|||
| 4f | 1393 ± 268 | 2008 ± 513 | 4097 ± 661 |
|
|
|||
| 23a | 32.5 ± 4.3 | 69.1 ± 19.8 | 8.48 ± 1.73 |
|
|
|||
| 23b | 28.8 ± 2.7 | 334 ± 126 | 13.4 ± 5.6 |
|
|
|||
| 23c | 73.0 ± 8.6 | 187 ± 27 | 50.5 ± 18.0 |
|
|
|||
| 23d | 11.2 ± 2.8 | 3.07 ± 0.32 | 6.19 ± 2.48 |
|
|
|||
| 31a | 38.4 ± 2.6 | 58.4 ± 4.0 | 4.20 ± 2.39 |
|
|
|||
| 31b | 424 ± 46 | 72.8 ± 8.7 | 89.1 ± 32.3 |
For uptake by DAT, SERT and NET, [3H]DA, [3H]-5-HT and [3H]DA accumulation was measured. Results are average ± SEM of three to eight independent experiments assayed in triplicate.
Reductive amination of the amines 1 (disubstituted pyran) and 3 (trisubstituted pyran) with various aldehydes in the presence of NaCNBH3 afforded the target compounds 2a-2f, 4a-4e (Scheme 1 and Scheme 2 respectively) in appreciable yields. Nucleophilic displacement of corresponding halides by the amines 1 and 3 in the presence of a weak base (K2CO3 or Et3N) provided compounds 2g-2h and 4f (Scheme 1 and Scheme 2, respectively).
Scheme 1.

Synthesis of 2a-2h: Reagents and conditions: (a) RCHO, NaCNBH3, AcOH, 1,2-dichloroethane, rt, overnight (for compounds 2a-2f), (b) 4-(bromomethyl)benzenesulfonamide, K2CO3, DMT, rt, overnight (for compound 2g) (c) p-Toluenesulfonyl chloride, Et3N, DCM, rt, overnight (for compound 2h)
Scheme 2.

Synthesis of 4a-4f: Reagents and conditions: (a) RCHO, NaCNBH3, AcOH, 1,2-dichloroethane, rt, overnight (for compounds 4a-4e), (b) 4-(bromomethyl)benzenesulfonamide, K2CO3, DMT, rt, overnight (for compound 4f).
The synthesis of the intermediate (R)-epoxide 10 is depicted in Scheme 3 and Scheme 4. Commercially available bis(4-fluorophenyl)methane (5) was converted to the corresponding ketone 6 via oxidation in the presence of KMnO4. Wittig reaction of ketone 6 with (methoxymethyl)triphenylphosphonium bromide in the presence of sodium amide under the Schlosser conditions afforded the desired vinyl ether 7 in excellent yield.[25] Compound 7 was converted to the corresponding aldehyde 8 in the presence of glacial acetic acid and concentrated sulfuric acid.[26] Without further purification, the aldehyde 8 was immediately utilized to obtain the racemic epoxide 9 via the Corey-Chakovsky method.[27] The racemate 9 was then subjected to the kinetic hydrolytic resolution in the presence of Jacobsen’s catalyst to obtain the (R)-epoxide 10 and diol 11 in 48% yield (over 99% ee). Selective protection of the primary hydroxyl group of the diol 11 in the presence of TBDMSCl and catalytic imidazole afforded compound 12. Mesylation of compound 12 followed by the deprotection of TBDMS group by TBAF furnished compound 14. Intramolecular SN2 displacement of the OMs group by the primary alcohol group of compound 14 in anhydrous methanol, catalyzed by anhydrous K2CO3 afforded the desired (R)-epoxide 10 in excellent yield.
Scheme 3.

Synthesis of 10 and 11: Reagents and Conditions: a) KMnO4, CH3CN, 0 °C-rt, 99%. b) MeOCH2PPh3 +Br−, anhyd. THF, anhyd. NaNH2, 0 °C-rt, 97%. c) Conc. H2SO4, glacial AcOH, rt, 30 min, 97%. d) trimethylsulfoxonium iodide, anhyd. DMSO, NaH, rt, 1 h, then 60 °C, 2 h 95%. e) (R,R)-(−)-N,N/-bis(3,5-di-tert-butylsalicylidine)-1,2-cyclohexane diaminocobalt (Jacobsen’s catalyst), H2O, THF, over 99% ee and 48% yield for both 10 and 11.
Scheme 4.

Synthesis of 10: Reagents and Conditions: a) TBDMSCl, imidazole, DCM, 0 °C-rt, 1 h. b) CH3SO2Cl, Et3N, DCM, 0 °C-rt, 2 h. c) TBAF, THF, 0 °C-rt, 1 h. d) K2CO3, CH3OH, 0 °C-rt, 5 h.
The synthesis of the key amine intermediate 21 (disubstituted pyran) is described in Scheme 5. The δ,ε-unsaturated alcohol 15 was obtained by the regioselctive ring opening of the (R)-epoxide 10 in the presence of allylmagnesium chloride and catalytic amount of copper(I) iodide. Compound 15 was converted to the corresponding vinyl ether 16 via trans-vinylation with ethyl vinyl ether using catalytic amount of mercury (II) trifluoroacetate. Compound 16 was immediately subjected to ring-closing metathesis in the presence of Grubb’s catalyst (1st genetaion) to afford the cyclic olefin 17. Compound 17 was then subjected to hydroborylation in the presence of 9-BBN in anhydrous THF, followed by oxidation to obtain an inseparable mixture of distereomers exclusively in favor of the trans-isomer 18a. We have established the stereochemistry of the product 18a in our recent and earlier publications.[20-22] The diastereomeric mixture was mesylated with methanesulfonyl chloride and separated by gradient column chromatography to afford 19a as the major isomer in 77% yield. The azide 20 was obtained by nucleophilic SN2 substitution of 19a with sodium azide in anhydrous DMF. Hydrogenation of compound 20 in the presence of 10% Pd/C in methanol provided quantitative yield of the key intermediate cis-amine 21. Compounds 22, 23b-d were obtained by reductive amination with corresponding aldehydes. Compound 23a was obtained by hydrogenation of compound 22 in the presence of 10% Pd/C in methanol for 1 h.
Scheme 5.

Synthesis of 23a-23d: Reagents and conditions: (a) allylmagnesium chloride, anhyd. diethylether, −78 oC to rt, overnight. (b) ethylvinyl ether, Hg(OCOCF3)2, rt, 4 h. (c) 1st generation Grubb’s catalyst, anhyd. benzene, reflux, 2 h. (d) (i) 9-BBN, anhyd. THF, rt, overnight; (ii) 10% NaOH, 30% H2O2, 50 °C, 1 h. (e) CH3SO2Cl, Et3N, anhyd. DCM, rt, 2 h. (f) NaN3, anhyd. DMF, 80 °C, overnight. (g) H2, Pd/C, MeOH, 50 psi, 2 h. (h) RCHO, NaCNBH3, 1,2-dichloroethane, AcOH rt, overnight. (i) H2, Pd/H2, MeOH, 50 psi, 1 h.
Scheme 6 describes the synthesis of the other key intermediate amine 30 (trisubstituted pyran). The (R)-epoxide 10 was treated with vinylmagnesium chloride and catalytic amount of copper(I) iodide in anhydrous THF to obtain the homoallylic alcohol 24 in a regioselective fashion. O-Allylation of compound 24 followed by ring-closing metathesis in the presence of Grubb’s catalyst (1st generation) afforded the cyclic olefin 26. Regioselective bromohydrin formation of the olefin 26 in the presence of N-bromoacetamide provided compound 27 which was converted to the trans-epoxide 28 in the presence of 20% NaOH in dioxane. The stereochemistry of trans-epoxide was comprehensively determined in our recent publication.[28] The trans-epoxide 28 was regioselectively converted to the corresponding azide 29 which upon hydrogenation in the presence of 10% Pd/C in methanol afforded the key intermediate amine 30 in a quantitative yield. Reductive amination of the amine 30 with various aldehydes provided the target compounds 31a-b.
Scheme 6.

Synthesis of 31a-31b: Reagents and conditions: (a) vinylmagnesium bromide, CuI, anhyd. THF, −78 °C-rt, 24 h, 93%. b) NaH, allyl bromide, anhyd. DMF, 1.5 h, 0 °C-rt, 95%. c) 1st generation Grubb’s catalyst, anhyd. benzene, reflux, 2 h, 96%. d) N-bromoacetamide, Dioxane-H2O, 0 °C-rt, 4 h, 82%. e) 20% NaOH, Dioxane, 0 °C-rt, 30 min, 90%. f) NaN3, anhyd. DMF, 80 °C, 24 h, 96%. g) Pd/C, MeOH, H2, 30 psi, 98%. h) RCHO, NaCNBH3, 1,2-dichloroethane, AcOH, overnight.
Expanding on SAR studies with asymmetric pyran derivatives, we report here on structural modifications of both di- and trisubstituted pyran derivatives. One of our main goals is to develop suitable TUIs and to further understand the effect of structural modifications on the activity profile for the three monoamine transporters. Our recent communication explored a number of disubstituted pyran derivatives, producing a lead TUI exhibiting balanced activity at all three monoamine transporters and exhibited significant effectiveness in reducing immobility in the forced swim test (FST).[21] In this regard, FST is a well recognized animal model for preclinical screening of potential antidepressant.[21-29] In addition, in our recent report we were able to demonstrate potent in vivo activity of a tri-substituted pyran derivative D-142 in both FST and mouse tail suspension tests.[24]
In our current study, we have further expanded our SAR studies with 3,6-disubstituted compounds. Compounds 2a-h were synthesized and characterized to evaluate their uptake inhibition profile. Several different N-benzyl substitutions were introduced to evaluate their effect on profile of uptake inhibition. To follow up on our previous SAR studies, several different phenyl-alkoxy related derivatives were synthesized. Thus, compounds 2a, 2c, 2e and 2f were made. Out of these compounds, 2c showed exceptional uptake inhibition potency at SERT and moderate to weak potency at NET and DAT (Ki; 152, 1.05 and 47.3 nM for DAT, SERT and NET, respectively), i.e. exhibited an SSRI profile. In this context, compound 2c was 11.6 times more potent than fluoxetine for its interaction with SERT. Thus, compound 2c might qualify as one of the most potent SERT-selective inhibitors known to date. Compound 2f similarly exhibited very high uptake inhibitory potency at SERT, but also had a good affinity for NET (Ki; 160, 1.07 and 15.8 nM for DAT, SERT and NET, respectively) and thus, exhibited a SNRI type profile. Compound 2a, however, did not display appreciable inhibitory activity at either SERT or NET but was potent at DAT. Dioxy compound 2e was potent at NET and moderately potent at both SERT and DAT (Ki; 87.7, 52.6 and 8.58 nM for DAT, SERT and NET, respectively), exhibiting a profile similar to that of a TUI. The precursor to compound 2e, derivative 2d did not show much activity for any one of the three transporters. We next synthesized several sulfonamide derivatives to explore their activity. Compound 2b showed moderate potency at DAT and NET but weaker potency at SERT. Compound 2g showed comparable potency at all three transporters (Ki; 60, 79 and 70 nM for DAT, SERT and NET, respectively) and thus, was a balanced TUI.
In our next exploration, selected tri-substituted pyran derivatives corresponding to their already characterized disubstituted compounds were synthesized. Thus, compounds 4a-f were synthesized and characterized. Compound 4a which is a trisubstituted derivative of 2e, showed an interesting profile of triple uptake inhibition (Ki; 135, 14.7 and 25.9 nM for DAT, SERT and NET). Compound 4a exhibited higher inhibition potency at SERT compared to its corresponding disubstituted version 2e (Ki; 14.7 vs. 52.6 nM for SERT for 4a and 2e, respectively). Compounds 4b and 4c were in general weakly active at all three transporters, indicating low tolerance of hydroxyl substitution on the aromatic ring. Similar to its disubstituted counterpart 2c, compound 4d exhibited high uptake inhibition activity for SERT and was an SSRI (Ki; 401, 1.99 and 57.2 nM for DAT, SERT and NET, respectively). Thus, it seems that N-benzyl aryl substitutions carrying tetrahydrofuran or methoxy moieties (in particular tetrahydrofuran) conveye high affinity for SERT. Compounds 4e and 4f were not active at the three transporters targets.
In our next goal, we wanted to evaluate the effect of introduction of fluorine atoms in the aromatic rings of pyran derivatives on uptake inhibitory potency. Another rationale behind introducing fluorine is to increase possible metabolic stability in the lead compounds which will have an impact in improving their pharmacokinetical properties. A fluorinated version of a previous lead molecule, 23a exhibited a dopamine and norepinephrine reuptake inhibitory profile (DNRI) with more potent inhibition of dopamine and norepinephrine transporters compared to SERT (Ki; 32.5, 69 and 8.5 nM, for DAT, SERT and NET, respectively). The unsubstituted version of 23a, compound 1c (Table 1), was more potent at SERT and less potent at DAT compared to 23a.20 Compound 23b which is a fluorinated version of D-161, exhibited a DNRI-type profile (Ki; 28.8, 334 and 13.4 nM for DAT, SERT and NET, respectively). In this regard, it is important to mention that there are only few compounds which are known to exhibit a DNRI-type profile. One well known example of this is bupropion, used as an antidepressant agent in the clinic.[15, 30] Disubstituted fluorinated indole compound 23c exhibited good potency at DAT and NET but was weaker at SERT. Compound 23d, which is a fluorinated version of 2c, exhibited high uptake inhibition potency at all three transporters (Ki; 11, 3.1 and 6.2 nM for DAT, SERT and NET, respectively). Thus, compound 23d is one of the most potent TUIs that we have developed so far.
Followed by synthesis of disubstituted fluorinated compounds, two trisubstituted fluorinated derivatives 31a and 31b were made. Compound 31a exhibited a triple uptake inhibitory profile with highest uptake inhibition shown for NET and DAT (Ki; 4.2 and 38.4 nM, respectively for NET and DAT), exhibiting a profile similar to a DNRI. Compound 31b was modestly potent at SERT and weak at DAT and NET transporters. Overall, it seems that fluorinated versions of compounds showed propensity for higher activity at DAT and NET giving rise to a more DNRI-type profile. We have incorporated the current SAR results along with our earlier findings into the pharmacophore model we presented previously (ref 20). This updated pharmacophore model is shown in Figure 2.
Figure 2.
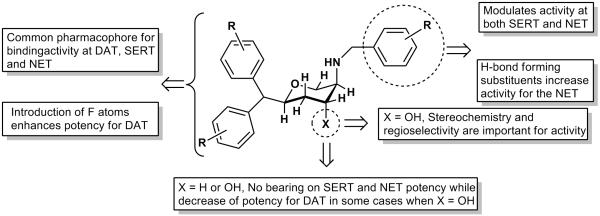
An updated interaction model of pyran derivatives with monoamine transporters molecules.
Finally, we carried out an in vivo study with one of our lead TUIs, 2g. Based on physiochemical properties calculated by using Molecular Operating Environment (MOE) 2011.10 version program, of three lead TUIs (Table 2), this compound was chosen for in vivo rat FST experiment over 4a and 23d as it was expected to provide more favorable bioavailability. Based on our previous experience with transporter inhibitors with comparable affinities, a dose of 10 mg/kg (ip) was selected to evaluate the effect on immobility and was compared with vehicle. Imipramine was used as a positive control. Compound 2g was able to reduce the immobility significantly compared to vehicle (Figure 3). The in vivo activity of 2g was similar to structurally different PRC200-SS (Figure 1) under the same ip drug administration conditions.17 With respect to other, structurally different, compounds, DOV 21,947 and DOV 216,303 (Figure 1), no direct correlation could be made as the DOV compounds were administered orally.[31-32] In regards to our own results, it appears compound 2g is marginally more efficacious in reducing immobility compared to D-142 (Ref. 24) but was similar to D-391 (compound 10f in ref. 21).[21, 24] We plan to carry out a broad spectrum screening of compound 2g in the near future to assess its interaction with CNS receptors. Based on core pyran structural similarity with our previous lead molecules, it is expected that similar selectivity for monoamine transporters would be exhibited by compound 2g as found for our disubstituted and trisubstituted pyran derivatives D-161, D-391 and D-142.[21, 23, 24]
Table 2.
Physicochemical parameters of lead TUIs[a]
| Compound | Mol. Wt. |
Number of H- bond acceptor |
Number of H- bond donor |
Number of rotatable bonds |
TPSA | Log P |
|---|---|---|---|---|---|---|
| 2g | 421.55 | 4 | 2 | 7 | 81.42 | 4.32 |
| 4a | 417.50 | 4 | 1 | 6 | 39.72 | 5.60 |
|
|
||||||
| 23d | 435.51 | 3 | 1 | 6 | 30.49 | 6.10 |
Physicochemical parameters were calculated with molecular modelling program MOE 2011.10 version.
Figure 3.

Effect of sub-chronic administration of vehicle (Veh), 2g, and imipramine (Imp) on the duration of immobility in the forced swimming test in rats. One way ANOVA analysis demonstrates significant effect among treatments: F (3,95) = 8.93 (P< 0.0042). Dunnett’s analysis showed that the effect of 2g at a dose (10 mg/kg) on immobility was statistically significant different compared to vehicle (P< 0.01). The effect of reference imipramine (15 mg/kg) on immobility was also statistically significantly different (P< 0.05) from vehicle. Asterisks indicate a statistically significant difference toward control group that received saline i.p. **P < 0.01. Each treatment group contained six to eight rats.
In our goal to determine whether the efficacy of 2g originated, in part or in full, from locomotor activation, we carried out locomotor activity studies with the 10 mg/kg dose of 2g as applied in the FST. The results indicate that 10 mg/kg of 2g was ineffective in producing locomotor activity as tested under same experimental protocol applied in rat FST (Figure 4a). Additionally, determination of total locomotor activity for a period of one and a half an hour post administration of drugs indicated no significant difference of activity between 2g and vehicle (Figure 4b). Thus, the reduction of immobility by 2g in these animal models was not due to increased locomotor activity.
Figure 4.

Effects of drugs, 2g and GBR 12909, on locomotor activity (horizontal activity, HACTV). Rats were injected (i.p.) with either vehicle or 2g followed by measurement of locomotor activity for one and a half an hour post administration of drugs. GBR 12909 is shown for comparison from our previous work (ref). a) This time frame represents measurement of locomotor activity for ½ h after post 1 h of administration of drugs and mimicked the condition of rat FST test. One way ANOVA analysis demonstrates non significant effect between control and the dose of 2g but significant effect between control and GBR 12909 : F (3,95) = 6.73 (P< 0.05). b) This time frame represents total locomotor activity for one and a half an hour post administration of drugs. One way ANOVA analysis demonstrates non significant effect between control and the dose of 2g but significant effect between control and GBR 12909 : F (3,95) = 24.21 (P< 0.01). Asterisks indicate a statistically significant difference toward control group that received saline i.p. **P < 0.01. Each treatment group contains three to four rats.
In this report we have shown the development of novel compounds with TUI, DNRI, SNRI, or SSRI profile. Both disubstituted and trisubstituted pyran compounds exhibited such profiles. Compounds 2g, 4a and 23d exhibited a TUI profile. Compound 2g exhibited balanced potency at all three monoamine transporters. Compound 4a was most potent at SERT and NET with moderate potency at DAT. Disubstituted compound 2c and trisubstituted compound 4d exhibited an SSRI-type profile with a nanomolar potency selectively at SERT. Introduction of fluorine atom in general increased activity for the DAT. Compound 23b exhibited DNRI-type profile. An in vivo rat study with balanced TUI 2g indicated significant reduction of immobility in the FST depression model compared to vehicle, indicating potential antidepressant property.
Supplementary Material
Acknowledgements
This work is supported by the National Institute of Mental Health/National Institute of Health (MH084888, AKD). We thank Mr. Gyan Modi for the help with locomotor activity study.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemmedchem.org or from the author.((Please delete if not appropriate))
References
- [1].Millan MJ. Neurotherapeutics. 2009;6:53–77. doi: 10.1016/j.nurt.2008.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Richelson E. J Clin Psychiatry. 2003;64(Suppl 13):5–12. [PubMed] [Google Scholar]
- [3].Baldessarini RH. The pharmacological basis of the therapeutica. McGrawe Hills; New York: 1996. In Goodman and Gilman’s. [Google Scholar]
- [4].Spinks D, Spinks G. Curr Med Chem. 2002;9:799–810. doi: 10.2174/0929867024606795. [DOI] [PubMed] [Google Scholar]
- [5].Wong DT, Bymaster FP. Adv Exp Med Biol. 1995;363:77–95. [PubMed] [Google Scholar]
- [6].Hiemke C, Hartter S. Pharmacol Ther. 2000;85:11–28. doi: 10.1016/s0163-7258(99)00048-0. [DOI] [PubMed] [Google Scholar]
- [7].Owens MJ, Nemeroff CB. Depress Anxiety. 1998;8(Suppl 1):5–12. [PubMed] [Google Scholar]
- [8].Barbey JT, Roose SP. J Clin Psychiatry. 1998;59(Suppl 15):42–48. [PubMed] [Google Scholar]
- [9].Goldstein BJ, Goodnick PJ. J Psychopharmacol. 1998;12:S55–87. doi: 10.1177/0269881198012003041. [DOI] [PubMed] [Google Scholar]
- [10].Thase ME, Entsuah AR, Rudolph RL. Br J Psychiatry. 2001;178:234–241. doi: 10.1192/bjp.178.3.234. [DOI] [PubMed] [Google Scholar]
- [11].Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, Niederehe G, Thase ME, Lavori PW, Lebowitz BD, McGrath PJ, Rosenbaum JF, Sackeim HA, Kupfer DJ, Luther J, Fava M. Am J Psychiatry. 2006;163:1905–1917. doi: 10.1176/ajp.2006.163.11.1905. [DOI] [PubMed] [Google Scholar]
- [12].D’Aquila PS, Collu M, Gessa GL, Serra G. Eur J Pharmacol. 2000;405:365–373. doi: 10.1016/s0014-2999(00)00566-5. [DOI] [PubMed] [Google Scholar]
- [13].Papakostas GI. Eur Neuropsychopharmacol. 2006;16:391–402. doi: 10.1016/j.euroneuro.2005.12.002. [DOI] [PubMed] [Google Scholar]
- [14].Mischoulon D, Nierenberg AA, Kizilbash L, Rosenbaum JF, Fava M. Can J Psychiatry. 2000;45:476–481. doi: 10.1177/070674370004500509. [DOI] [PubMed] [Google Scholar]
- [15].Ascher JA, Cole JO, Colin JN, Feighner JP, Ferris RM, Fibiger HC, Golden RN, Martin P, Potter WZ, Richelson E, et al. J Clin Psychiatry. 1995;56:395–401. [PubMed] [Google Scholar]
- [16].Sporn J, Ghaemi SN, Sambur MR, Rankin MA, Recht J, Sachs GS, Rosenbaum JF, Fava M. Ann Clin Psychiatry. 2000;12:137–140. doi: 10.1023/a:1009060800999. [DOI] [PubMed] [Google Scholar]
- [17].Liang Y, Shaw AM, Boules M, Briody S, Robinson J, Oliveros A, Blazar E, Williams K, Zhang Y, Carlier PR, Richelson E. J Pharmacol Exp Ther. 2008;327:573–583. doi: 10.1124/jpet.108.143610. [DOI] [PubMed] [Google Scholar]
- [18].Skolnick P, Krieter P, Tizzano J, Basile A, Popik P, Czobor P, Lippa A. CNS Drug Rev. 2006;12:123–134. doi: 10.1111/j.1527-3458.2006.00123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Beer B, Stark J, Krieter P, Czobor P, Beer G, Lippa A, Skolnick P. J Clin Pharmacol. 2004;44:1360–1367. doi: 10.1177/0091270004269560. [DOI] [PubMed] [Google Scholar]
- [20].Zhang S, Fernandez F, Hazeldine S, Deschamps J, Zhen J, Reith MEA, Dutta AK. J Med Chem. 2006;49:4239–4247. doi: 10.1021/jm0601699. [DOI] [PubMed] [Google Scholar]
- [21].Gopishetty B, Hazeldine S, Santra S, Johnson M, Modi G, Ali S, Zhen J, Reith M, Dutta AK. J Med Chem. 2011;54:2924–2932. doi: 10.1021/jm200020a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang S, Zhen J, Reith MEA, Dutta AK. J Med Chem. 2005;48:4962–4971. doi: 10.1021/jm049021k. [DOI] [PubMed] [Google Scholar]
- [23].Dutta AK, Ghosh B, Biswas S, Reith MEA. Eur J Pharmacol. 2008;589:73–79. doi: 10.1016/j.ejphar.2008.05.008. [DOI] [PubMed] [Google Scholar]
- [24].Dutta AK, Gopishetty B, Gogoi S, Ali S, Zhen J, Reith MEA. Eur J Pharmacol. 2011;671:39–44. doi: 10.1016/j.ejphar.2011.09.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Schlosser M, Schaub B. Chimica. 1982;36:396. [Google Scholar]
- [26].Clark JA, Clark MS, Gardner DV, Gaster LM, Hadley MS, Miller D, Shah A. Med Chem. 1979;22:1373–9. doi: 10.1021/jm00197a018. [DOI] [PubMed] [Google Scholar]
- [27].Corey EJ, Chaykovsky M. J. Am. Chem. Soc. 1965;87(6):1353–1364. [Google Scholar]
- [28].Gopishetty B, Gogoi S, Dutta AK. Tetrahedron Asymmetry. 22:1081–1086. doi: 10.1016/j.tetasy.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Porsolt RD, Le Pichon M, Jalfre M. Nature. 1977;266:730–732. doi: 10.1038/266730a0. [DOI] [PubMed] [Google Scholar]
- [30].Dwoskin LP, Rauhut AS, King-Pospisil KA, Bardo MT. CNS Drug Rev. 2006;12:178–207. doi: 10.1111/j.1527-3458.2006.00178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Skolnick P, Popik P, Janowsky A, Beer B, Lippa AS. Eur J Pharmacol. 2003;461:99–104. doi: 10.1016/s0014-2999(03)01310-4. [DOI] [PubMed] [Google Scholar]
- [32].Skolnick P, Popik P, Janowsky A, Beer B, Lippa AS. Life Sci. 2003;73:3175–3179. doi: 10.1016/j.lfs.2003.06.007. [DOI] [PubMed] [Google Scholar]
- [33].Cheng Y, Prusoff WH. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- [34].Pacholczyk T, Blakely RD, Amara SG. Nature. 1991;350:350–354. doi: 10.1038/350350a0. [DOI] [PubMed] [Google Scholar]
- [35].Buck KJ, Amara SG. Chimeric Proc Natl Acad Sci U S A. 1994;91:12584–12588. doi: 10.1073/pnas.91.26.12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gu H, Wall SC, Rudnick G. J Biol Chem. 1994;269:7124–7130. [PubMed] [Google Scholar]
- [37].Snyder SH, Coyle JT. J Pharmacol Exp Ther. 1969;165:78–86. [PubMed] [Google Scholar]
- [38].Masserano JM, Venable D, Wyatt RJ. J Pharmacol Exp Ther. 1994;270:133–141. [PubMed] [Google Scholar]
- [39].Williams JM, Steketee JD. J Neurosci Methods. 2004;137:161–165. doi: 10.1016/j.jneumeth.2004.02.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.