


Abstract
Translesion synthesis is an important mechanism in response to unrepaired DNA lesions during replication. The DNA polymerase ζ (Polζ) mutagenesis pathway is a major error-prone translesion synthesis mechanism requiring Polζ and Rev1. In addition to its dCMP transferase, a non-catalytic function of Rev1 is suspected in cellular response to certain types of DNA lesions. However, it is not well understood about the non-catalytic function of Rev1 in translesion synthesis. We have analyzed the role of Rev1 in translesion synthesis of an acetylaminofluorene (AAF)-dG DNA adduct. Purified yeast Rev1 was essentially unresponsive to a template AAF-dG DNA adduct, in contrast to its efficient C insertion opposite a template 1,N6-ethenoadenine adduct. Purified yeast Polζ was very inefficient in the bypass of the AAF-dG adduct. Combining Rev1 and Polζ, however, led to a synergistic effect on translesion synthesis. Rev1 protein enhanced Polζ-catalyzed nucleotide insertion opposite the AAF-dG adduct and strongly stimulated Polζ-catalyzed extension from opposite the lesion. Rev1 also stimulated the deficient synthesis by Polζ at the very end of undamaged DNA templates. Deleting the C-terminal 205 aa of Rev1 did not affect its dCMP transferase activity, but abolished its stimulatory activity on Polζ-catalyzed extension from opposite the AAF-dG adduct. These results suggest that translesion synthesis of AAF-dG adducts by Polζ is stimulated by Rev1 protein in yeast. Consistent with the in vitro results, both Polζ and Rev1 were found to be equally important for error-prone translesion synthesis across from AAF-dG DNA adducts in yeast cells.
INTRODUCTION
Translesion synthesis is an important mechanism of damage tolerance in response to unrepaired DNA lesions during replication. Translesion synthesis leads to replication of the damaged regions of the genome. It requires a specialized DNA polymerase (Pol) to directly copy the damaged site on the DNA template. Since DNA damage can change the coding property of the bases, alter DNA structure or render the site non-coding, an incorrect nucleotide is often inserted opposite the lesion during translesion synthesis. This is referred to as error-prone translesion synthesis. It is also possible that the correct nucleotide is inserted opposite the lesion, resulting in error-free translesion synthesis.
The major translesion synthesis polymerases are Polζ and the Y family DNA polymerases in eukaryotes. In addition, Polµ has also been shown to possess robust translesion synthesis activity in vitro (1,2). Whether Polµ contributes significantly to translesion synthesis as a damage tolerance mechanism in vivo, however, remains to be determined. In humans, the Y family of DNA polymerases consists of Polη, Polι, Polκ and REV1 (3). In the yeast Saccharomyces cerevisiae, Polι and Polκ are not found. Genetic studies in yeast have indicated that Polζ (the Rev3–Rev7 complex) operates in a biochemical pathway that involves additional proteins including Rev1 (reviewed in refs 4,5), which is also referred to as the Polζ mutagenesis pathway. Unlike other members in the Y family of DNA polymerases, Rev1 is a dCMP transferase, rather than a typical DNA polymerase. Rev1 is able to insert a C opposite a template G and several types of DNA lesions, including apurinic/apyrimidinic (AP) site, 8-oxoguanine, (+)-trans-anti-benzo[a]pyrene-N2-dG, (–)-trans-anti-benzo[a]pyrene-N2-dG and 1,N6-ethenoadenine (6–9). At least in the case of cellular response to AP sites in DNA, the Rev1 dCMP transferase directly participates in nucleotide insertion opposite the lesion during translesion synthesis in vivo (10).
Error-prone translesion synthesis is largely responsible for DNA damage-induced mutagenesis. In yeast, the vast majority of mutations induced by UV radiation, acetylaminofluorene (AAF), AP sites and benzo[a]pyrene diol epoxide are generated by the Polζ mutagenesis pathway (10–14). The Polζ mutagenesis pathway also exists in humans (7,15–17) and is shown to be the major mechanism for UV- and benzo[a]pyrene diol epoxide-induced mutagenesis in cultured human cells (15,17,18). Thus, it appears that the Polζ pathway constitutes a major mechanism of error-prone translesion synthesis and DNA damage-induced mutagenesis in eukaryotes. Although Rev1 is required for the Polζ mutagenesis pathway, very little is known about the role of Rev1 in the mutagenesis process. The dCMP transferase of Rev1 is unable to respond to a template TT dimer and a template TT (6–4) photoproduct in vitro (6,8,10), two major DNA lesions of UV radiation. Yet Rev1 is required for UV-induced mutagenesis (10,19,20). Thus, it has been proposed that Rev1 may play another non-catalytic function in the Polζ mutagenesis pathway (10,20). However, it is not well understood about the non-catalytic function of the Rev1 protein in translesion synthesis.
To better understand Rev1 functions, we have analyzed the role of this protein in translesion synthesis of an AAF-dG DNA adduct. Whereas the dCMP transferase of purified yeast Rev1 was essentially unresponsive to a template AAF-dG DNA adduct, this protein strongly stimulated the Polζ-catalyzed translesion synthesis of AAF-dG DNA adducts. These results suggest a non-catalytic function of Rev1 in the bypass of AAF-dG adducts. Consistent with the in vitro results, we found that both Polζ and Rev1 are equally important for error-prone translesion synthesis across from AAF-dG DNA adducts in yeast cells.
MATERIALS AND METHODS
Materials
A mouse monoclonal antibody against the His6 tag and the Escherichia coli expression vector pQE80 were purchased from Qiagen (Valencia, CA). The plasmid vector pQE80C was constructed by inserting a 90mer duplex DNA encoding a calmodulin binding peptide (21) into the BamHI site of pQE80. The coding sequence of the duplex DNA is 5′-GATCCAAGCGACGATGGAAAAAGAATTTCATAGCCGTCTCAGCAGCCAACCGCTTTAAGAAAATCTCATCCTCCGGGGCACTTAGATCTA-3′. The E.coli strain BL21 (DE3 Gold) was obtained from Invitrogen (Carlsbad, CA). Calmodulin affinity resin was purchased from Stratagene (La Jolla, CA). Alkaline phosphatase conjugated anti-mouse IgG was from Sigma Chemicals (St Louis, MO). All oligonucleotides were synthesized by Operon (Alameda, CA). Yeast Polζ (the Rev3–Rev7 complex) and human REV1 were purified to near homogeneity as described previously (8,22). N-Acetoxy-N-2-acetylaminofluorene (AAAF, the activated form of AAF) was obtained from the Midwest Research Institute (Kansas City, MO). A 30mer DNA template containing a site-specific AAF-dG adduct was prepared as described previously (23). Its sequence is 5′-CCTTCTTCATTCGAACATACTTCTTCTTCC-3′, where the modified guanine is underlined. A 36mer template containing a site-specific tetrahydrofuran (AP site analogue) was synthesized by Operon. Its sequence is 5′-GAAGGGATCCTTAAGACTXTAACCGGTCTTCGCGCG-3′, where X designates the AP site.
Yeast strains
Yeast strains used are BY4741Δrad14 (proficient in mutagenesis) (MATa his3 leu2 met15 ura3 rad14::HIS3) and its isogenic BY4741Δrad14Δrev1 (rev1 deletion mutant), and BY4741Δrad14Δrev3 (rev3 deletion mutant). BY4741 was purchased from ATCC (Manassas, VA). BY4741Δrev3 (lacking Polζ) was constructed as described previously (14). BY4741Δrev1 (lacking Rev1) was constructed by transforming BY4741 cells with a linearized rev1 deletion plasmid construct. The rev1 deletion clone was confirmed by a functional assay demonstrating reduced UV-resistance and loss of UV-induced mutagenesis. The rev1 deletion strain was further tested for complementation of UV resistance and UV-induced mutagenesis by a plasmid carrying the wild-type REV1 gene. Finally, the RAD14 gene of BY4741, BY4741Δrev1 and BY4741Δrev3 was deleted by transforming the respective strains with a rad14 deletion plasmid construct and the rad14 deletion phenotype confirmed as described previously (14).
Purification of yeast Rev1
The yeast REV1 gene was obtained by PCR using S.cerevisae DNA as the template and the primers 5′-GAAGATCTATGGGTGAACATGGTGGTCTTG-3′ and 5′-ACGCGTCGACAGGTATTGTT-GCAATGC-3′. The 3-kb REV1 DNA fragment was then cloned into the BglII and SalI sites of the vector pQE80C, yielding pQE80C-yREV1. Escherichia coli strain BL21 (DE3 Gold) containing pQE80C-yREV1 was grown at 37°C to an OD600 of 0.8. Rev1 expression was induced by addition of IPTG to a final concentration of 1 mM and growth for 14 h at 16°C. Cells were collected by centrifugation and resuspended in an extraction buffer containing 50 mM Tris–HCl pH 7.5, 10% sucrose, 20% glycerol, 5 mM β-mercaptoethanol and protease inhibitors (24). Cells were then homogenized by a sonicator, using four pulses of 30 s each with a 2 min pause on ice between pulses. After centrifugation at 18 000 g for 20 min, the clarified sample was loaded onto a 1 ml HiTrap chelating column charged with NiSO4 (Amersham Pharmacia Biotech, Piscataway, NJ), followed by washing the column sequentially with 15 ml of Ni buffer A (50 mM Tris–HCl pH 7.5, 0.5 M NaCl, 10% glycerol, 5 mM β-mercaptoethanol and protease inhibitors) containing 10 mM imidazole and 25 ml of Ni buffer A containing 35 mM imidazole. Bound proteins were eluted with a linear gradient (25 ml) of 35–500 mM imidazole in Ni buffer A. An equal volume of calmodulin binding buffer (50 mM Tris–HCl pH 8.0, 0.3 M NaCl, 4 mM CaCl2 and 10 mM β-mercaptoethanol) was added to the pooled Rev1 fractions. After centrifugation at 15 000 g for 20 min, the sample was loaded onto a 2 ml calmodulin affinity column. The column was washed with 15 ml of the calmodulin column buffer (50 mM Tris–HCl pH 8.0, 0.3 M NaCl and 10 mM β-mercaptoethanol). Finally, the column was eluted with three EGTA step-gradients in calmodulin column buffer containing 1, 2 and 4 mM EGTA, respectively.
To construct the REV1ΔC gene coding for a truncated Rev1 lacking the C-terminal 205 aa, PCR was performed using the plasmid pQE80C-yREV1 as the template and two primers, 5′-GAAGATCTATGGGTGAACATGGTGGTCTTG-3′ and 5′-ACGCGTCGACTAATAATTTCTTTTCTCGAACTCGTTG. The resulting 2.3 kb DNA fragment was cloned into the BglII and SalI sites of the vector pQE80C, yielding pQE80C-yREV1ΔC. Expression and purification of the C-terminal truncated Rev1ΔC (94 kDa) were similarly carried out as described above for the full-length Rev1 protein.
DNA polymerase assays
Identical conditions were used for dCMP transferase assays and DNA polymerase assays. A standard DNA polymerase reaction mixture (10 µl) contained 25 mM KH2PO4 (pH 7.0), 5 mM MgCl2, 5 mM dithiothreitol, 100 µg/ml bovine serum albumin, 10% glycerol, 50 µM of dNTPs (dATP, dCTP, dTTP and dGTP individually or together as indicated), 50 fmol of an indicated DNA substrate containing a 32P-labeled primer, and purified Polζ, Rev1 or both as indicated. After incubation at 30°C for 30 min, reactions were terminated with 7 µl of a stop solution (20 mM EDTA, 95% formamide, 0.05% bromophenol blue and 0.05% xylene cyanol). The reaction products were resolved on a 20% denaturing polyacrylamide gel and visualized by autoradiography. DNA synthesis products were quantitated by scanning densitometry using the SigmaGel software (Sigma, St Louis, MO) for analysis.
Preparation of AAF-damaged plasmid DNA
Plasmid pCLU (50 µg) was incubated with 10 µM AAAF in a 500-µl reaction mixture containing 20% ethanol and TE buffer (10 mM Tris–HCl, pH 7.5, 1 mM EDTA) for 3 h in the dark at 37°C. The modified DNA was then purified by 5–20% sucrose gradient centrifugation at 28 000 r.p.m. for 17 h at 4°C in a Beckman SW41Ti rotor. Fractions of 0.5 ml each were collected from the bottom of the gradient and 5-µl aliquots were analyzed by electrophoresis on a 1% agarose gel to locate the DNA. Fractions containing supercoiled DNA were pooled, precipitated in ethanol and dissolved in TE buffer.
In vivo mutagenesis assay in yeast
A plasmid-based mutagenesis assay in yeast cells was performed as we reported recently (14). Briefly, damaged or undamaged plasmid pCLU DNA (2 µg) was transformed into yeast cells. Immediately after transformation, cells were collected by centrifugation (20 s at 5000 r.p.m.) in a microcentrifuge, and resuspended in 1 ml of sterile water. An aliquot of 999 µl cell suspension was plated onto three YNB minimal agar plates lacking leucine but supplemented with 5 mM 5-fluoroorotic acid (5-FOA), 150 µM methionine and 380 µM uracil to score for colonies containing ura3 mutant pCLU. Another aliquot of 1 µl cell suspension was diluted and plated onto YNB minimal agar plates lacking leucine to score for colonies containing replicated pCLU (plasmid survival). Yeast colonies were counted after incubation at 30°C for 3–4 days. Mutation frequency was calculated by dividing the number of the ura3 mutant colonies by the number of colonies containing replicated pCLU.
RESULTS
The dCMP transferase of purified yeast Rev1 is essentially unresponsive to a template AAF-dG adduct
To understand the role of Rev1 in translesion synthesis of AAF-dG DNA adducts, we first determined whether the dCMP transferase of yeast Rev1 is able to insert a C opposite the lesion in vitro. To avoid potential contamination by yeast proteins, we purified yeast Rev1 from E.coli cells following its expression from a plasmid construct. The Rev1 preparation contained two bands of ∼120 and 90 kDa, respectively (Fig. 1A). Western blot analysis using a monoclonal antibody against the N-terminal His6 tag confirmed that both bands are yeast Rev1 protein (Fig. 1B). While the larger band is consistent with the calculated molecular mass of yeast Rev1 (112 kDa), thus representing the full-length protein, the smaller band is probably a C-terminal truncated product of Rev1. The eukaryotic Y family DNA polymerases, including Rev1, are relatively unstable and protein truncation is a common feature among them. To determine whether the truncated Rev1 protein retains activity, we deleted 205 aa from its C-terminus, yielding Rev1ΔC (94 kDa). Rev1ΔC was similarly expressed in E.coli and purified (Fig. 1C), yielding a protein of approximately the size of the ∼90 kDa band in Figure 1A. As shown in Figure 1D, both Rev1 and Rev1ΔC preparations were similarly active in dCMP transferase in response to a template AP site. Thus, the C-terminal truncated protein of ∼90 kDa retains the Rev1 dCMP transferase activity.
Figure 1.
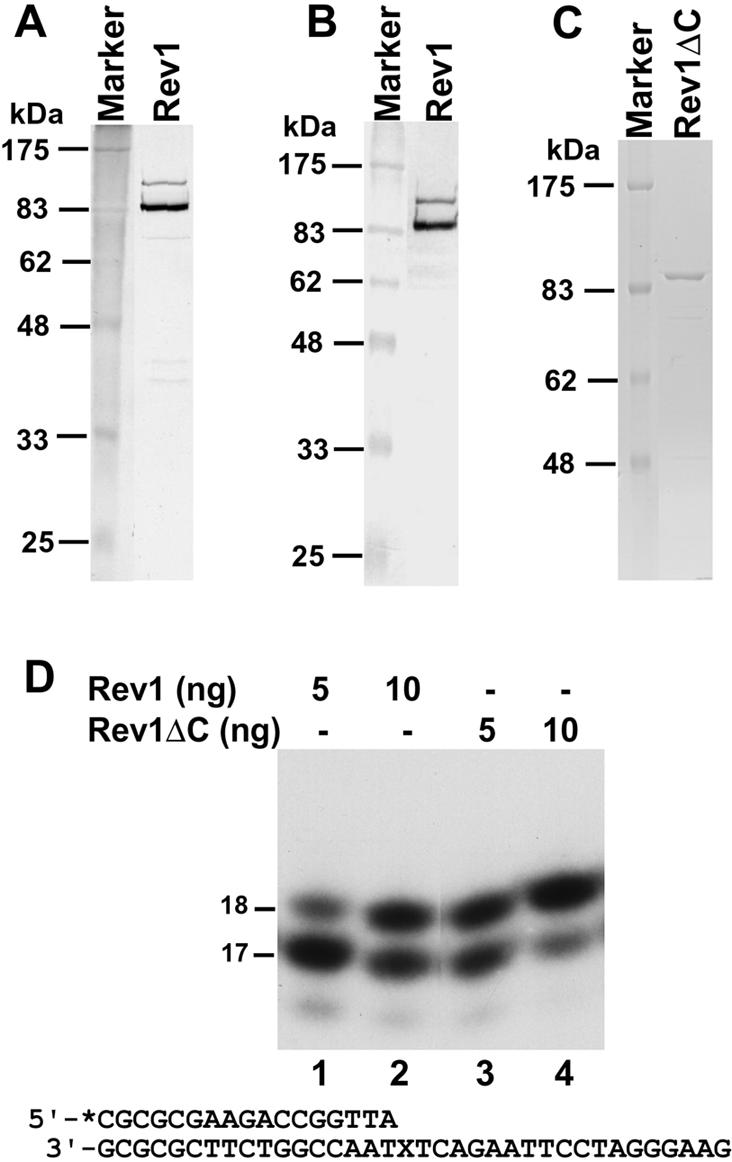
Analyses of purified yeast Rev1. (A) The purified yeast Rev1 protein (300 ng) was analyzed by electrophoresis on a 10% SDS–polyacrylamide gel and visualized by silver staining of the gel. Protein size markers are indicated on the left. (B) The purified yeast Rev1 protein (300 ng) was analyzed by a western blot using a mouse monoclonal antibody against the N-terminal His6 tag. (C) The purified Rev1ΔC protein (300 ng) lacking the C-terminal 205 aa of yeast Rev1 was analyzed by 10% SDS–polyacrylamide gel and visualized by staining with Coomassie blue. The identity of the protein band that migrated at the ∼90 kDa position on the gel was confirmed by western blot analysis using a mouse monoclonal antibody against the N-terminal His6 tag. (D) A 5′-32P-labeled 17mer primer was annealed to a damaged 36mer template, terminating right before the AP site as shown at the bottom. Then, DNA polymerase assays were performed using 5 or 10 ng of purified yeast Rev1 or Rev1ΔC protein as indicated. DNA size markers in nucleotides are indicated on the left. X, the AP site.
To examine the response of yeast Rev1 to an AAF-dG adduct, we annealed a 32P-labeled 17mer primer to the damaged template with the primer 3′ end terminating right before the lesion (Fig. 2A), and performed standard dCMP transferase assays. As shown in Figure 2A, purified yeast Rev1 was unable to effectively catalyze nucleotide insertion opposite the AAF-dG adduct. In contrast, it efficiently inserted a C opposite a template 1,N6-ethenoadenine adduct (Fig. 2B). These results show that the yeast Rev1 dCMP transferase is essentially unresponsive to a template AAF-dG adduct.
Figure 2.

Response of the yeast Rev1 dCMP transferase to AAF-dG and 1,N6-ethenoadenine DNA adducts. DNA polymerase assays were performed with 5 ng (45 fmol) of purified yeast Rev1 in the presence of all four dNTPs (N4), or a single deoxyribonucleotide triphosphate, dATP (A), dCTP (C), dGTP (G), or dTTP (T). DNA size markers in nucleotides are indicated on the left. Quantitation of extended primers is shown below the gel. (A) Assays with DNA templates containing a site-specific AAF-dG adduct. A 17mer primer was labeled with 32P at its 5′ end and annealed to the damaged template with the primer 3′ end terminating right before the lesion as shown on the top. (B) Assays with DNA templates containing a site- specific 1,N6-ethenoadenine adduct. A 20mer primer was labeled with 32P at its 5′ end and annealed to the damaged template with the primer 3′ end terminating right before the lesion as shown on the top.
Synergistic effect of Polζ and Rev1 combination during bypass of a template AAF-dG adduct in vitro
Since purified yeast Polζ alone is inefficient in performing translesion synthesis across from a template AAF-dG adduct (22), we suspected that accessory factor(s) might be required to stimulate lesion bypass by Polζ in vivo. To test whether Rev1 could play such a role, we performed DNA synthesis reactions with the AAF-damaged template by combining purified yeast Polζ and Rev1 together. In the reaction, a 13mer 32P-labeled primer was annealed to the damaged template, terminating 4 nt before the lesion (Fig. 3). Consistent with our previous finding (22), the AAF-dG adduct strongly blocked DNA synthesis by yeast Polζ right before the lesion, as evidenced by the strong 17mer DNA band (Fig. 3, lane 3). Only very small amounts of the damaged templates were bypassed (longer than 18mer DNA bands), and the 30mer full-length product or near full-length products were not detected (Fig. 3, lane 3). When purified yeast Rev1 protein was added to the reaction, much higher levels of the damaged templates were bypassed and the 30mer full-length product was readily detected (Fig. 3, lane 4). These results show that combination of yeast Polζ and Rev1 has a synergistic effect on the bypass of AAF-dG DNA adducts.
Figure 3.

Effect of Rev1 protein on Polζ-catalyzed bypass of the template AAF-dG adduct. A 5′-32P-labeled 13mer primer was annealed to a damaged 30mer template with the primer 3′ end terminating 4 nt before the AAF-dG adduct as shown on the top. As the control, the 32P-labeled primer was separately annealed to the undamaged template with identical sequence. DNA polymerase assays were then performed with 10 ng (50 fmol) of yeast Polζ in the absence (lanes 1 and 3) or presence (lanes 2 and 4) of purified yeast Rev1 protein (5 ng, 45 fmol). Lanes 1 and 2, undamaged DNA template; lanes 3 and 4, damaged DNA template. DNA size markers in nucleotides are indicated on the left.
We consistently observed that the last template base was inefficiently copied by Polζ even from undamaged templates (22) (Fig. 3, lane 1). Surprisingly, addition of yeast Rev1 protein greatly stimulated DNA synthesis at the end of the undamaged template (Fig. 3, lane 2). The 30mer full-length product migrated at the same position in the absence or presence of Rev1 (Fig. 3, compare lanes 1 and 2). Therefore, the last nucleotide copied at the very end of the template is consistent with Polζ-catalyzed G addition, rather than C addition catalyzed by the Rev1 dCMP transferase. C addition would have resulted in a 30mer product that migrated significantly faster than the 30mer DNA band in lane 1 of Figure 3. These results show that Rev1 stimulates the inefficient DNA synthesis of Polζ at the end of the template.
Effect of Rev1 on Polζ-catalyzed nucleotide insertion opposite a template AAF-dG adduct
Translesion synthesis consists of two distinct steps: nucleotide insertion opposite the lesion and extension DNA synthesis from opposite the lesion. To understand the role of Rev1 in the synergistic bypass by the Rev1–Polζ combination, we determined whether Rev1 affects nucleotide insertion by Polζ opposite the AAF-dG adduct. Yeast Polζ alone inserted 14 fmol C and 12 fmol G opposite the AAF-dG adduct (Fig. 4, lanes 3 and 4, respectively). In the presence of purified Rev1 protein, C insertion opposite the lesion was increased to 23 fmol (1.6-fold increase), while G insertion was increased to 18 fmol (1.5-fold increase) (Fig. 4, compare lanes 3 with 8, and 4 with 9). These results show that while Rev1 stimulates Polζ-catalyzed nucleotide insertion opposite the template AAF-dG adduct, it does not alter the specificity of nucleotide insertion by Polζ opposite the lesion.
Figure 4.

Effect of Rev1 protein on Polζ-catalyzed nucleotide insertion opposite the AAF-dG adduct. A 17mer primer was labeled with 32P at its 5′ end and annealed to the damaged template with the primer 3′ end terminating right before the AAF-dG adduct as shown on the top. DNA polymerase assays were then performed with 10 ng (50 fmol) of yeast Polζ in the presence of all four dNTPs (N4), or a single deoxyribonucleotide triphosphate, dATP (A), dCTP (C), dGTP (G) or dTTP (T). Lanes 1–5, reactions with Polζ alone; lanes 6–10, reactions with Polζ plus purified yeast Rev1 protein (5 ng, 45 fmol). DNA size markers in nucleotides are indicated on the left.
Effect of Rev1 on Polζ-catalyzed extension DNA synthesis from opposite the AAF-dG adduct
To determine whether Rev1 affects extension DNA synthesis from opposite the AAF-dG adduct, we annealed a 32P-labeled 18mer primer to the damaged template, terminating with an A opposite the lesion (Fig. 5). DNA synthesis assays were then performed with purified yeast Polζ in the absence or presence of purified yeast Rev1 protein. Rev1 alone was unable to perform extension synthesis from opposite the AAF-dG adduct (Fig. 5A, lane 1). Polζ alone extended the primer only by 1 nt from opposite the AAF-dG adduct, and further extension synthesis was strongly blocked by the lesion (Fig. 5A, lane 2). In the presence of Rev1 protein, longer extension synthesis by Polζ was greatly stimulated (Fig. 5A, lanes 3–7). Furthermore, the 30mer full-length extension product was readily detected and its production increased with increasing concentrations of Rev1 in the reaction (Fig. 5A, lanes 3–7). Prior heat inactivation of the Rev1 protein abolished its activation activity (data not shown). Thus, stimulation of Polζ-catalyzed extension synthesis depends on a properly folded Rev1 protein. In contrast, the inefficient extension synthesis by Polη from opposite the AAF-dG adduct was not affected by Rev1 protein (Fig. 5B). Moreover, human REV1 protein had no effect on extension synthesis by the yeast Polζ from opposite the AAF-dG adduct (Fig. 5C).
Figure 5.

Effect of Rev1 protein on Polζ-catalyzed extension synthesis from opposite the AAF-dG adduct. An 18mer 32P-labeled primer was annealed to a damaged 30mer template with the primer 3′ A opposite the AAF-dG adduct as shown on the top. (A) DNA polymerase assays were performed with 16 ng (79 fmol) of yeast Polζ in the absence (lane 2) or presence (lanes 3–7) of 63 ng (562 fmol), 32 ng (286 fmol), 21 ng (188 fmol), 15 ng (134 fmol) and 10 ng (89 fmol), respectively, of purified yeast Rev1 protein. Lane 1, DNA polymerase assay with 63 ng of Rev1 alone. (B) DNA polymerase assays were performed with 12 ng (169 fmol) of yeast Polη in the absence (lane 1) or presence (lanes 2–5) of 26 (232 fmol), 13 (116 fmol), 7 (63 fmol) and 3 ng (27 fmol), respectively, of purified yeast Rev1 protein. (C) DNA polymerase assays were performed with 21 ng (104 fmol) of yeast Polζ in the presence of 13 (94 fmol, lane 1), 26 (188 fmol, lane 2), 39 (283 fmol, lane 3) and 52 ng (377 fmol, lane 4) of purified human REV1 protein. DNA size markers in nucleotides are indicated on the left.
High level of stimulation on Polζ-catalyzed extension synthesis required relatively high concentrations of the purified Rev1 protein (Fig. 5A). This raised the possibility that the ∼90 kDa truncated Rev1 in the protein preparation (Fig. 1A) may not be able to stimulate Polζ in extension synthesis. To test this possibility, we performed the extension DNA synthesis assay again using the purified 94 kDa Rev1ΔC. As shown in Figure 6 (lanes 4–7), even at very high concentrations of the Rev1ΔC protein, stimulation on Polζ-catalyzed extension from opposite the AAF-dG adduct was not observed. Thus, whereas deleting the C-terminal 205 aa of Rev1 does not affect its dCMP transferase, this C-terminal truncation abolishes the stimulating activity of Rev1 on Polζ-catalyzed extension from opposite the AAF-dG adduct.
Figure 6.
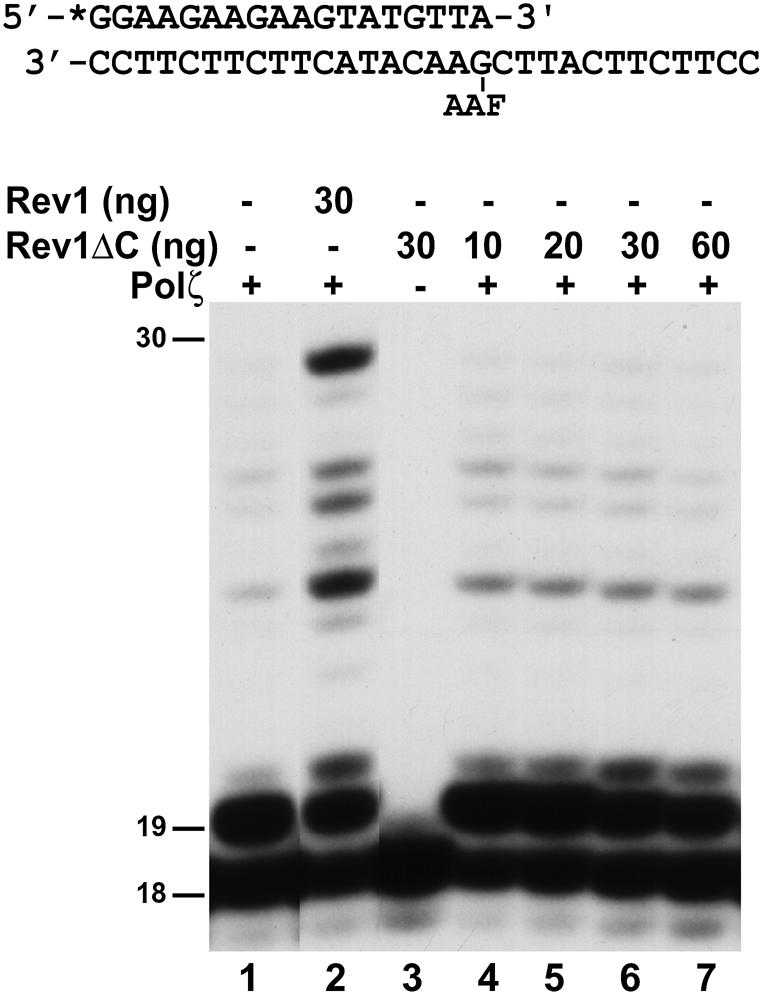
Polζ-catalyzed extension synthesis from opposite the AAF-dG adduct in the presence of the C-terminal truncated Rev1 (Rev1ΔC). An 18mer 32P-labeled primer was annealed to a damaged 30mer template with the primer 3′ A opposite the AAF-dG adduct as shown on the top. DNA polymerase assays were performed with 21 ng (104 fmol) of yeast Polζ in the presence of 30 ng (268 fmol, lane 2) of purified yeast Rev1, or increasing amounts of purified Rev1ΔC as indicated (lanes 4–7). Lane 1, Polζ (21 ng) alone; lane 3, Rev1ΔC (30 ng) alone. DNA size markers in nucleotides are indicated on the left.
To determine whether or not the Rev1 stimulatory effect on Polζ-catalyzed extension synthesis is specific to the A-terminated primer, we performed additional assays using four different DNA substrates containing an 18mer primer terminating with a C, A, T or G, respectively, opposite the AAF-dG adduct (Fig. 7). As shown in Figure 7A–D (compare lane 1 with lane 6), Rev1 stimulated extension synthesis by Polζ from opposite the lesion in every case. With or without Rev1, Polζ predominantly incorporated the correct G opposite the undamaged template C immediately 5′ to the lesion (Figs 6D and 7A, lanes 4 and 9). Thus, the specificity of nucleotide incorporation during extension synthesis by Polζ from opposite the AAF-dG adduct was not affected by Rev1 protein. These results show that Rev1 protein strongly and specifically stimulates Polζ-catalyzed extension DNA synthesis from opposite the AAF-dG adduct, and that Rev1 does not alter the Polζ specificity of nucleotide incorporation during extension synthesis.
Figure 7.

Specificity of nucleotide incorporation during extension synthesis from opposite the AAF-dG adduct by yeast Polζ with or without Rev1. Four 18mer primers that differed by 1 nt at the 3′ end were labeled with 32P at their 5′ ends and separately annealed to the damaged template with the primer 3′ end opposite the AAF-dG adduct as shown on the top. DNA polymerase assays were then performed with 10 ng (50 fmol) of yeast Polζ in the presence of all four dNTPs (N4), or a single deoxyribonucleotide triphosphate dATP (A), dCTP (C), dGTP (G) or dTTP (T) as indicated. Lanes 1–5, reactions with Polζ alone; lanes 6–10, reactions with Polζ plus purified yeast Rev1 protein (5 ng, 45 fmol). DNA size markers in nucleotides are indicated on the left. (A) C opposite the lesion; (B) A opposite the lesion; (C) T opposite the lesion and (D) G opposite the lesion.
Both Polζ and Rev1 are required for AAF adduct-induced mutagenesis in yeast cells
Since Rev1 is able to strongly stimulate Polζ-catalyzed extension synthesis from mismatched primer end opposite the AAF-dG adduct, it is likely that both Polζ and Rev1 are critical to AAF-induced mutagenesis in cells. To test this hypothesis, we examined the effect of inactivating Polζ and Rev1 on AAF adduct-induced mutagenesis in yeast cells, using a plasmid-based in vivo mutagenesis assay (14). In this assay, the pCLU plasmid was first damaged by 10 µM AAAF in vitro and purified by centrifugation in a linear 5–20% sucrose gradient. Then, the damaged plasmid was transformed into yeast cells. The plasmid contains the yeast LEU2 gene as the selection marker and the yeast URA3 gene as the mutagenesis target. Transformed cells were plated on leucine-lacking minimal medium plates with or without 5-FOA. Plates without 5-FOA allowed cells that had replicated the damaged pCLU plasmid to grow to colonies, thus measuring replication of the damaged plasmid (plasmid ‘survival’). Plates with 5-FOA allowed only those cells that contained inactivating mutations at the plasmid URA3 gene to grow to colonies, thus measuring mutagenesis of the plasmid URA3 gene. The chromosomal URA3 gene had been deleted from all yeast strains used. Once inside cells, AAF adducts on the plasmid DNA are subject to removal by nucleotide excision repair (25–27). In order to effectively detect induced mutagenesis at a relatively low AAAF concentration, we inactivated the nucleotide excision repair pathway by deleting the RAD14 gene from yeast strains used in this study.
Untreated pCLU plasmid DNA was used in the assay as the control for spontaneous mutagenesis. In cells proficient in mutagenesis (rad14), a spontaneous mutation frequency of 3.2 × 14–4 was obtained (Table 1). A similar spontaneous mutation frequency was observed with cells lacking Rev1 (rad14 rev1) (Table 1). In cells lacking Polζ (rad14 rev3), the spontaneous mutation frequency was slightly increased (Table 1). After treatment of the plasmid DNA with 10 µM AAAF, the induced mutation frequency of the plasmid URA3 gene was increased by 10-fold, as compared with the spontaneous mutation frequency (Table 1). Without Rev1 or Polζ, AAF adduct-induced mutation was largely abolished to levels of only 2.1- and 1.6-fold above the spontaneous mutation frequency, respectively (Table 1). These results show that both Polζ and Rev1 are required for AAF adduct-induced mutagenesis in yeast cells. Since the induced mutation frequencies in cells lacking Rev1 or Polζ were reduced to similar levels of 8.2 × 14–4 and 8.9 × 14–4, respectively (Table 1), we conclude that both Rev1 and Polζ are equally important for error-prone translesion synthesis across from AAF-dG DNA adducts in vivo.
Table 1. AAF adduct-induced mutation frequency in various yeast strainsa.
| Strain | Spontaneous (×10–4) | AAF-induced (×10–4) | Ratiob |
|---|---|---|---|
|
rad14 |
3.2 ± 1.0 |
33 ± 10 |
10 |
|
rad14rev1 |
4.0 ± 0.5 |
8.2 ± 3.0 |
2.1 |
| rad14rev3 | 5.7 ± 0.2 | 8.9 ± 2.0 | 1.6 |
aIn vivo mutagenesis assays were performed as described in Materials and Methods. Mutation frequency was calculated by dividing the number of ura3 mutant colonies by the number of colonies containing replicated plasmid. Mutation frequencies with standard deviations are shown. Each result is the average of three independent experiments. rad14, proficient in mutagenesis; rad14 rev1, lacking Rev1; rad14 rev3, lacking Polζ.
bRatio between AAF-induced mutation frequency and spontaneous mutation frequency.
DISCUSSION
In yeast cells, Polζ is clearly required for error-prone translesion synthesis across from AAF-dG DNA adducts (12,13). However, the purified yeast Polζ is rather inefficient in performing translesion synthesis across from a template AAF-dG adduct (22). Thus, effective bypass of AAF-dG adducts in cells may require accessory factor(s). Supporting this notion, much more efficient bypass of the AAF-dG adduct was achieved by combining purified yeast Polζ and Rev1. Rev1 alone is unable to catalyze either nucleotide insertion opposite the AAF-dG adduct or extension synthesis from opposite this lesion. Furthermore, the Rev1 C-terminal truncation of 205 aa abolished its stimulatory activity but retained its dCMP transferase activity. Therefore, Rev1 most likely plays a non-catalytic function by stimulating the Polζ-catalyzed translesion synthesis across from the lesion. Indeed, both the Polζ-catalyzed nucleotide insertion step and the extension synthesis step were stimulated by Rev1. Further supporting its non-catalytic role, Rev1 did not alter the Polζ specificity of nucleotide incorporation at both the nucleotide insertion and extension synthesis steps. If the dCMP transferase activity of Rev1 were involved in synergistic bypass by the Rev1–Polζ combination, C incorporation during nucleotide insertion opposite the AAF-dG adduct and extension from opposite the lesion would have been significantly elevated. This was not the case. However, whether the dCMP transferase active site is required for the stimulatory function of Rev1 on Polζ remains to be determined.
If the stimulatory function of Rev1 is important in vivo, mutagenesis induced by AAF-dG DNA adducts in yeast cells would depend on both the Rev1 and Polζ proteins. This was shown to be the case. Without Polζ or Rev1, AAF-induced mutagenesis was reduced by ∼4-fold in yeast cells. Hence, Rev1 is equally important as Polζ for error-prone translesion synthesis across from AAF-dG DNA adducts in vivo. Missing either Rev1 or Polζ will inactivate the major mechanism of error-prone translesion synthesis, thus, underscoring the importance of the functional interaction between these two polymerases in response to AAF-DNA adducts. Using a plasmid containing a site-specific AAF-dG adduct, Baynton et al. (13) also observed that Rev1 is required for non-slipped translesion synthesis at the two sequence contexts, 5′-GGGAAF-3′ and 5′-GGCGAAFCC-3′, in yeast cells.
Recently, it has been observed that multiple DNA polymerases are involved in translesion synthesis of TT (6–4) photoproducts, AAF-dG adducts and benzo[a]pyrene diol epoxide in cells (14,28,29). However, it is not clear how such a multiple-polymerase mode of translesion synthesis takes place in cells. Are these polymerases recruited to the lesion site simultaneously, cooperatively or independently? Does the in vivo lesion bypass reflect a two-polymerase two-step mechanism (8,22,30,31)? Are there functional interactions among Polζ and the Y family DNA polymerases? Our studies suggest that there exists a functional interaction between two DNA polymerases for translesion synthesis. Thus, functional interactions between two polymerases may represent an important feature for translesion synthesis during cellular response to certain types of DNA lesions.
The precise mechanism of the functional interaction between Rev1 and Polζ is unknown at the present time. One possibility is that Rev1 may somehow facilitate the binding of Polζ to its DNA substrate and/or decrease the dissociation of Polζ from the DNA template. Consistent with such a mechanism, Rev1 also strongly stimulated the deficient synthesis by Polζ at the very end of undamaged DNA templates. Since human REV1 could not substitute for the yeast Rev1 for its stimulatory activity on the yeast Polζ, the Rev1 effect could not have simply resulted from the Rev1–DNA interaction. It is not surprising that human Rev1 could not stimulate yeast Polζ, because even the four most conserved regions of the two proteins share only 21–35% sequence identity and 43–59% sequence similarity (7). More likely, the functional interaction between Rev1 and Polζ during synergistic bypass of the AAF-dG DNA adducts may be mediated by physical interactions between them. Physical interaction between human REV1 and human REV7, the non-catalytic subunit of Polζ, has indeed been observed (32). Whether the yeast Rev1 similarly interacts with the yeast Rev7, however, remains to be determined.
Although Rev1 acts as a DNA polymerase on repeating template G sequences and has therefore been suggested as a specialized DNA polymerase (8,9), its dCMP transferase activity is remarkable. With respect to its catalytic function, Rev1 recognizes various damaged and undamaged template bases with very different affinity, but it preferentially inserts dCMP without an exception (6,8,9,33). Hence, the rules of geometry constraints and hydrogen bonding that governs nucleotide selection during normal DNA synthesis simply do not apply to Rev1. The structural basis for the unique C insertion opposite the various undamaged and damaged template bases is a mystery. Clues about the structural basis of the Rev1 dCMP transferase activity would have to wait for the crystal structural studies of the protein. Like its human counterpart, yeast Rev1 also efficiently inserts a C opposite a template 1,N6-ethenoadenine adduct. The essentially unresponsiveness of human REV1 to a template AAF-dG adduct was also observed with the yeast Rev1. Thus, although the protein sequences of yeast Rev1 and human REV1 are not overwhelmingly conserved, their biochemical properties appear to be highly conserved. So far, direct participation of the Rev1 dCMP transferase activity at the nucleotide insertion step has been demonstrated in only one case in cells: translesion synthesis of AP sites (10). It is of great interest to determine in the future whether such an involvement of the Rev1 dCMP transferase is only limited to cellular response to AP sites or constitutes a more general mechanism in the Polζ mutagenesis pathway. Regardless of the extent to which the catalytic dCMP transferase function is employed during in vivo translesion synthesis of various types of DNA lesions, a non-catalytic role appears to be an important and may well be the predominant function of Rev1 in the Polζ mutagenesis pathway. Our studies suggest one such non-catalytic function for Rev1 in the translesion synthesis of AAF-dG DNA adducts. Extended studies into other types of DNA lesions could bring in more insights into our understanding of this fascinating member of the Y family DNA polymerases in translesion synthesis.
Acknowledgments
ACKNOWLEDGEMENT
This work was supported by NIH grant CA92528.
REFERENCES
- 1.Zhang Y., Wu,X., Guo,D., Rechkoblit,O., Taylor,J.S., Geacintov,N.E. and Wang,Z. (2002) Lesion bypass activities of human DNA polymerase µ. J. Biol. Chem., 277, 44582–44587. [DOI] [PubMed] [Google Scholar]
- 2.Havener J.M., McElhinny,S.A., Bassett,E., Gauger,M., Ramsden,D.A. and Chaney,S.G. (2003) Translesion synthesis past platinum DNA adducts by human DNA polymerase µ. Biochemistry, 42, 1777–1788. [DOI] [PubMed] [Google Scholar]
- 3.Ohmori H., Friedberg,E.C., Fuchs,R.P. P., Goodman,M.F., Hanaoka,F., Hinkle,D., Kunkel,T.A., Lawrence,C.W., Livneh,Z., Nohmi,T. et al. (2001) The Y-family of DNA polymerases. Mol. Cell, 8, 7–8. [DOI] [PubMed] [Google Scholar]
- 4.Lawrence C. (1994) The RAD6 DNA repair pathway in Saccharomyces cerevisiae: what does it do and how does it do it? Bioessays, 16, 253–258. [DOI] [PubMed] [Google Scholar]
- 5.Wang Z. (2001) Translesion synthesis by the UmuC family of DNA polymerases. Mutat. Res., 486, 59–70. [DOI] [PubMed] [Google Scholar]
- 6.Nelson J.R., Lawrence,C.W. and Hinkle,D.C. (1996) Deoxycytidyl transferase activity of yeast REV1 protein. Nature, 382, 729–731. [DOI] [PubMed] [Google Scholar]
- 7.Lin W., Xin,H., Zhang,Y., Wu,X., Yuan,F. and Wang,Z. (1999) The human REV1 gene codes for a DNA template-dependent dCMP transferase. Nucleic Acids Res., 27, 4468–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y., Wu,X., Rechkoblit,O., Geacintov,N.E., Taylor,J.-S. and Wang,Z. (2002) Response of human REV1 to different DNA damage: preferential dCMP insertion opposite the lesion. Nucleic Acids Res., 30, 1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haracska L., Prakash,S. and Prakash,L. (2002) Yeast Rev1 protein is a G template-specific DNA polymerase. J. Biol. Chem., 277, 15546–15551. [DOI] [PubMed] [Google Scholar]
- 10.Nelson J.R., Gibbs,P.E., Nowicka,A.M., Hinkle,D.C. and Lawrence,C.W. (2000) Evidence for a second function for Saccharomyces cerevisiae Rev1p. Mol. Microbiol., 37, 549–554. [DOI] [PubMed] [Google Scholar]
- 11.Morrison A., Christensen,R.B., Alley,J., Beck,A.K., Bernstine,E.G., Lemontt,J.F. and Lawrence,C.W. (1989) REV3, a Saccharomyces cerevisiae gene whose function is required for induced mutagenesis, is predicted to encode a nonessential DNA polymerase. J. Bacteriol., 171, 5659–5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baynton K., Bresson-Roy,A. and Fuchs,R.P. (1998) Analysis of damage tolerance pathways in Saccharomyces cerevisiae: a requirement for Rev3 DNA polymerase in translesion synthesis. Mol. Cell. Biol., 18, 960–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baynton K., Bresson-Roy,A. and Fuchs,R.P. (1999) Distinct roles for Rev1p and Rev7p during translesion synthesis in Saccharomyces cerevisiae. Mol. Microbiol., 34, 124–133. [DOI] [PubMed] [Google Scholar]
- 14.Xie Z., Braithwaite,E., Guo,D., Bo,Z., Geacintov,N.E. and Wang,Z. (2003) Mutagenesis of benzo[a]pyrene diol epoxide in yeast: requirement for DNA polymerase ζ and involvement of DNA polymerase η. Biochemistry, 42, 11253–11262. [DOI] [PubMed] [Google Scholar]
- 15.Gibbs P.E., McGregor,W.G., Maher,V.M., Nisson,P. and Lawrence,C.W. (1998) A human homolog of the Saccharomyces cerevisiae REV3 gene, which encodes the catalytic subunit of DNA polymerase ζ. Proc. Natl Acad. Sci. USA, 95, 6876–6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin W., Wu,X. and Wang,Z. (1999) A full-length cDNA of hREV3 is predicted to encode DNA polymerase ζ for damage-induced mutagenesis in humans. Mutat. Res., 433, 89–98. [DOI] [PubMed] [Google Scholar]
- 17.Gibbs P.E., Wang,X.D., Li,Z., McManus,T.P., McGregor,W.G., Lawrence,C.W. and Maher,V.M. (2000) The function of the human homolog of Saccharomyces cerevisiae REV1 is required for mutagenesis induced by UV light. Proc. Natl Acad. Sci. USA, 97, 4186–4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Z., Zhang,H., McManus,T.P., McCormick,J.J., Lawrence,C.W. and Maher,V.M. (2002) hREV3 is essential for error-prone translesion synthesis past UV or benzo[a]pyrene diol epoxide-induced DNA lesions in human fibroblasts. Mutat. Res., 510, 71–80. [DOI] [PubMed] [Google Scholar]
- 19.Larimer F.W., Perry,J.R. and Hardigree,A.A. (1989) The REV1 gene of Saccharomyces cerevisiae: isolation, sequence and functional analysis. J. Bacteriol., 171, 230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rajpal D.K., Wu,X. and Wang,Z. (2000) Alteration of ultraviolet-induced mutagenesis in yeast though molecular modulation of the REV3 and REV7 gene expression. Mutat. Res., 461, 133–143. [DOI] [PubMed] [Google Scholar]
- 21.Stofko-Hahn R.E., Carr,D.W. and Scott,J.D. (1992) A single step purification for recombinant proteins. Characterization of a microtubule associated protein (MAP 2) fragment which associates with the type II cAMP-dependent protein kinase. FEBS Lett., 302, 274–278. [DOI] [PubMed] [Google Scholar]
- 22.Guo D., Wu,X., Rajpal,D.K., Taylor,J.-S. and Wang,Z. (2001) Translesion synthesis by yeast DNA polymerase ζ from templates containing lesions of ultraviolet radiation and acetylaminofluorene. Nucleic Acids Res., 29, 2875–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y., Yuan,F., Wu,X., Wang,M., Rechkoblit,O., Taylor,J.-S., Geacintov,N.E. and Wang,Z. (2000) Error-free and error-prone lesion bypass by human DNA polymerase κ in vitro. Nucleic Acids Res., 28, 4138–4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xin H., Lin,W., Sumanasekera,W., Zhang,Y., Wu,X. and Wang,Z. (2000) The human RAD18 gene product interacts with HHR6A and HHR6B. Nucleic Acids Res., 28, 2847–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Z., Svejstrup,J.Q., Feaver,W.J., Wu,X., Kornberg,R.D. and Friedberg,E.C. (1994) Transcription factor b (TFIIH) is required during nucleotide-excision repair in yeast. Nature, 368, 74–76. [DOI] [PubMed] [Google Scholar]
- 26.Wang Z., Buratowski,S., Svejstrup,J.Q., Feaver,W.J., Wu,X., Kornberg,R.D., Donahue,T.F. and Friedberg,E.C. (1995) Yeast TFB1 and SSL1 genes, which encode subunits of transcription factor IIH, are required for nucleotide excision repair and RNA polymerase II transcription. Mol. Cell. Biol., 15, 2288–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Z., Wu,X. and Friedberg,E.C. (1996) A yeast whole cell extract supports nucleotide excision repair and RNA polymerase II transcription in vitro. Mutat. Res., 364, 33–41. [DOI] [PubMed] [Google Scholar]
- 28.Zhang H. and Siede,W. (2002) UV-induced T→C transition at a TT photoproduct site is dependent on Saccharomyces cerevisiae polymerase η in vivo. Nucleic Acids Res., 30, 1262–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bresson A. and Fuchs,R.P. (2002) Lesion bypass in yeast cells: Pol η participates in a multi-DNA polymerase process. EMBO J., 21, 3881–3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuan F., Zhang,Y., Rajpal,D.K., Wu,X., Guo,D., Wang,M., Taylor,J.-S. and Wang,Z. (2000) Specificity of DNA lesion bypass by the yeast DNA polymerase η. J. Biol. Chem., 275, 8233–8239. [DOI] [PubMed] [Google Scholar]
- 31.Johnson R.E., Washington,M.T., Haracska,L., Prakash,S. and Prakash,L. (2000) Eukaryotic polymerases ι and ζ act sequentially to bypass DNA lesions. Nature, 406, 1015–1019. [DOI] [PubMed] [Google Scholar]
- 32.Murakumo Y., Ogura,Y., Ishii,H., Numata,S., Ichihara,M., Croce,C.M., Fishel,R. and Takahashi,M. (2001) Interactions in the error-prone post-replication repair proteins, hREV1, hREV3 and hREV7. J. Biol. Chem., 276, 35644–35651. [DOI] [PubMed] [Google Scholar]
- 33.Masuda Y., Takahashi,M., Fukuda,S., Sumii,M. and Kamiya,K. (2001) Mechanisms of dCMP transferase reactions catalyzed by mouse Rev1 protein. J. Biol. Chem., 277, 3040–3046. [DOI] [PubMed] [Google Scholar]