

Abstract
DNA sequencing is increasingly being used to assist in species identification in order to overcome taxonomic impediment. However, few studies attempt to compare the results of these molecular studies with a more traditional species delineation approach based on morphological characters. Mitochondrial DNA Cytochrome oxidase subunit 1 (CO1) gene was sequenced, measuring 636 base pairs, from 47 ants of the genus Pheidole (Formicidae: Myrmicinae) collected in the Brazilian Atlantic Forest to test whether the morphology-based assignment of individuals into species is supported by DNA-based species delimitation. Twenty morphospecies were identified, whereas the barcoding analysis identified 19 Molecular Operational Taxonomic Units (MOTUs). Fifteen out of the 19 DNA-based clusters allocated, using sequence divergence thresholds of 2% and 3%, matched with morphospecies. Both thresholds yielded the same number of MOTUs. Only one MOTU was successfully identified to species level using the CO1 sequences of Pheidole species already in the Genbank. The average pairwise sequence divergence for all 47 sequences was 19%, ranging between 0–25%. In some cases, however, morphology and molecular based methods differed in their assignment of individuals to morphospecies or MOTUs. The occurrence of distinct mitochondrial lineages within morphological species highlights groups for further detailed genetic and morphological studies, and therefore a pluralistic approach using several methods to understand the taxonomy of difficult lineages is advocated.
Keywords : CO1, DNA-barcoding; morphospecies; MOTU; taxonomy
Introduction
Identifying species can be difficult, often requiring specialized knowledge and thereby representing a limiting factor in biodiversity inventories (Monaghan et al. 2005). Therefore, based on the growing concern over the threats to biodiversity, recent publications have emphasized the need to accelerate the analysis of biodiversity (Brooks et al. 2004; Smith et al. 2005) either by using morphospecies (Hammond 1994; Oliver and Beattie 1996; Barratt et al. 2003; Krell 2004) or DNA-based methods (Floyd et al. 2002; Hebert et al. 2003a; Tautz et al. 2003; Blaxter 2004). Both morphological and molecular approaches have faced criticism (Pires and Marinoni 2010) due to the deficiencies encountered when using only a single approach for species identification (Knowlton 1993; Jarman and Elliot 2000; Rubinoff 2006; Rubinoff et al. 2006). The comparison of results obtained by various approaches can aid in overcoming methodological issues in species identification (Mengual et al. 2006; Smith et al. 2008). A further advantage of integrating molecular and morphological approaches (Dayrat 2005; Cardoso et al. 2009) is that it promotes taxonomic stability (Padial et al. 2010).
In this paper, a single gene, Cytochrome oxidase subunit 1 (CO1) (as proposed by Hebert et al. 2003a, b), was used for barcoding on morphologically pre-defined species (morphosecies) of the hyperdiverse ant genus Pheidole (Formicidae: Myrmicinae). The aim was to evaluate how DNA barcoding enables the definition of Molecular Taxonomic Units (MOTUs); Floyd et al. 2002; Blaxter et al. 2005), and then link the delineated MOTUs to the morphospecies in order to assess congruence success. This study focused on Pheidole samples from a region in the Brazilian Atlantic Forest because the region is considered as one of the “hottest hotspots” of biodiversity (Myers et al. 2000).
Materials and Methods
Research area
The study was carried out in the Rio Cachoeira Nature Reserve (25° 18′ 51″ S, 48° 41′ 45″ W) located near the city of Antonina, in the coastal region of the Brazilian state of Paranà. Specimens were obtained between June and September 2003 from leaf litter ants sampled across 12 study sites, representing four stages of secondary forest succession. There were three replicates (sites) for each succession stage, and the replicate sites of a particular succession stage were separated by a mean distance of 4 km (range = 1–6 km). At each study site, two 50 m transects (parallel, separated by 20 m) were established, and leaf litter samples were collected (1 m2) at 5 m intervals along these transects (10 sampling points for each transect). For more details on sampling methodology, see Bihn et al. (2008, 2010).
The landscape varies from littoral plains with isolated hills to the uplands of the Serra do Mar mountain range. Lowland and submontane forests originally covered this area, but these dense ombrophilous forests have been intensely exploited. Old growth forests remain only in the hillside regions. The resulting landscape mosaic consists of old growth forests and secondary forests in various stages of succession and pastures (Bihn et al. 2008, 2010).
Definition of morphospecies
Pheidole specimens were identified to species with the key for neo-tropical species given by Wilson (2003). In cases where identification was not possible with this identification key (e.g., when major workers were not collected) or led to ambiguous results, ants were sorted into morphospecies using characters described by Wilson (2003). In addition, morphometric measurements were made to aid in the assignment of specimens into morphospecies (for details on the set of measurements taken and their definition, see Longino 2008). The morphometric characters used included: head width, distance between eyes, head length, anterior head length, scape length, mandible length, eye length, eye width, mesosoma length, promesonotal groove depth, propodeal spine length, femur length, tibia length, propodeal spiracle width, petiole width, and postpetiole width. The morphometric data were first standardize to mean = 0 and var = 1, and a hierarchical clustering of the morphospecies occurring in the Rio Cachoeira Nature Reserve was effected using the average linkage method (Figure 1).
Figure 1.
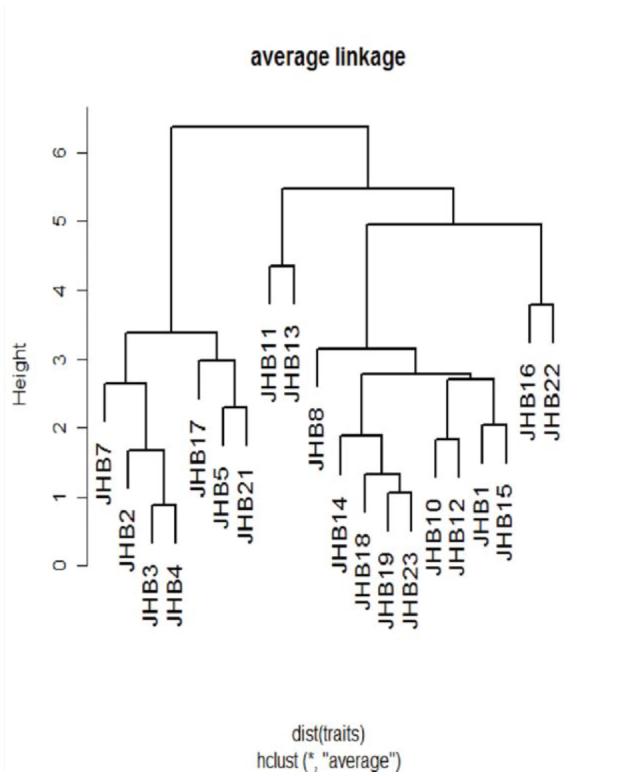
Hierarchical clustering using average linkage method on morphometric characters of the genus Pheidole from Rio Cachoeira Nature Reserve in the Brazilian Atlantic Forest, which defines 20 morphospecies. High quality figures are available online.
DNA extraction, amplification, and sequencing
Field collections were preserved in 95% ethanol until the time for DNA extraction. Specimens already examined and identified to be Pheidole morphospecies using morphological taxonomy were used for DNA extraction. Mitochondrial DNA was isolated for at least two minor workers from each morphospecies using the Qiagen DNeasy tissue extraction kit (Qiagen, www.qiagen.com) following the manufacturer's protocols. In cases where rare species were involved, DNA was extracted from a single individual, in which case either two legs or the whole individual was used.
Polymerase chain reaction (PCR) was conducted under the following reaction volumes: 2–4 µl DNA template, 2 µl in 10× PCR buffer, 1.6 µl of dNTPs in 10 mM concentra- tion, 1 µl of each primer in 10 mM concentration, 0.2 µl of Taq DNA polymerase, and distilled water for a total reaction volume of 20 µl. Reactions conditions included: initial denaturation at 95° C for 5 min; 33 cycles at 95° C (30 sec), 45–52° C at 40–48 sec (annealing time and temperature depended on primer used), and 72° C at 1 min; a final elongations at 72° C for 10 min reactions were done using an Eppendorf Thermal Cycler. Full-length sequences were amplified using primer pair LCO1490— GGTCAACAAATCAAAAGATATTGG and HCO2198— TAAACTTTCAGGGTGACCAAAAAATCA (Folmer et al. 1994). Primer pair LF1— ATTCAACCAATCATAAAGATATTGG and LR1— ATTTGAAGACCTACAGGTTTTTTAGT (Herbert et al. 2004a) was also used on specimens that were difficult to amplify using primers HCO/LCO. The two primer pairs gave the same length of base pairs. Products were visualized on a 2% agarose gel, and samples containing clean, single bands were purified using QIAquick PCR purification kit (Qiagen). The purified samples were sent for sequencing (AGOWA genomics, www.agowa.de), whereby the primers used in each case for amplification served as sequencing primers. All samples were sequenced in both directions, and the obtained sequences aligned using BioEdit version 7.0.9.0 (Hall 1999). The resultant fragments were approximately 658 base pairs (bp), and were identified as CO1 fragments for the ant genus Pheidole, with BLAST procedure search in GenBank (Altschul et al. 1997) done between 2008 and 2009. After trimming, the aligned sequences were 636 bp long and free from gaps. A translation with the invertebrate mitochondrial code returned uninterrupted amino acid sequences. These observations support the conclusion that the sequences analyzed were mitochondrial DNA and not nuclear pseudogenes (Bensasson et al. 2001). All sequences were deposited in the GenBank under accession numbers JF825012–JF825054 and JF914928–JF914931.
Phylogenetic analysis
Sequence divergences were calculated using the Kimura two parameter distance model (Kimura 1980), and the relationships between sequences were visualized by a Neighbor-Joining tree (Saitou and Nei 1987) using MEGA software version 4 (Tamura et al. 2007). A bootstrap test of phylogeny was effected by 100,000 replications and a similar random seed. To further infer relationships among the supposed morphospecies of Pheidole, phylogenetic analyses were performed using MrBayes version 3.1.1 (Huelsenbeck and Ronquist 2001), using the default value of four Markov chains and the General Time Reversible model. The Markov chain Monte Carlo length was 2,000,000 generations, sampled every 100 generations (burn-in = 4,500). Convergence of the chains was confirmed in the two runs by the examination of the average standard deviation of split frequencies, which in the present study had approached 0.007. Bayesian posterior probabilities were estimated as the proportion of the trees sampled after the burn-in that contained each of the observed bipartitions (Rannala and Yang 1996; Larget and Simon 1999). The phylogenetic tree was rooted using two species of the tribe Pheidolini, Aphaenogaster texana and Messor julianus.
The MOTU delineation from the CO1 sequences relied on two aspects. First, individuals were considered to be the same MOTU if sequences from the same morphospecies clustered together in the phylogenetic tree. A MOTU was thus defined as the least inclusive terminal groups (i.e. closest to the tips). Second, sequence clusters with a mean divergence value less than or equal to a threshold of 2% and 3%, as proposed by Herbert et al. (2004b), were considered as MOTUs. In this case, if sequences from two different morphospecies formed the same cluster, they only qualified to be a single MOTU if their mean sequence divergence was below or equal to thresholds 2% and 3%.
Match success of the 47 sequences was further examined in relation to the CO1 sequence of species in the genus Pheidole already present in the CO1 Genbank library (NCBI, GenBank, http://www.ncbi.nlm.nih.gov/) (searches done between 2008 and 2009). In cases where the match success was above 95%, the species name for that MOTU was allocated. To establish the distribution of genetic divergence and positioning of MOTUs in relation to Pheidole species from other regions, all CO1 sequences (genus Pheidole) that contained 640 or more bp (sequences retrieved on 2 March 2011) were extracted from the Genbank. A total of 141 sequences were obtained and combined with 47 sequences from this study for further alignment. The final set of 188 sequences was trimmed to 636 bp, and a histogram and a Neighbor-Joining tree were constructed (tree in Figure 4) using Kimura two parameter distance (Kimura 1980). This was implemented using data application Package ape (Paradis et al. 2004) available in R (R Development Core Team 2009).
Figure 4.

A combined phylogeny of CO1 sequences all from the genus Pheidole from the Genbank, and 47 sequences (taxa in blue) from Rio Cachoeira Nature Reserve. The 47 sequences formed distinct clusters in relation to those from the Genbank. The tree is rooted using the taxa in red. High quality figures are available online.
Results
This study produced a final aligned 636 bp fragment, characterized with no gaps for all the 47 sequences. Sequences were heavily AT biased (especially in the third codon position), as is expected in insect mitochondrial DNA (Crozier and Crozier 1993; Table 1). The average pairwise sequence divergence of all 47 sequences was 19%, ranging from 0–25% (Figure 2a). The distribution of Kimura two parameter distances for 47 sequences showed one peak near zero and another between 18 and 24%, while the 141 CO1 sequences from Genbank had a peak between 16 and 24% of sequence divergence (Figure 2b).
Table 1.
Sequence statistics for the 47 specimens used in the analysis of ant genus Pheidole.
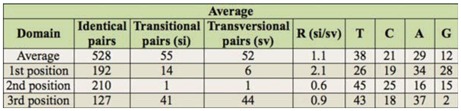
Figure 2.
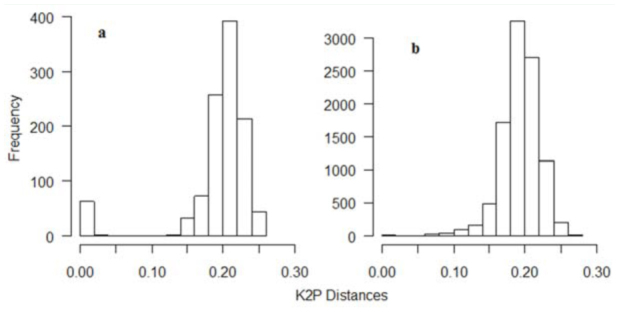
Distribution of pairwise distances of ant genus Pheidole calculated using Kimura two-parameter model (Kimura 1980) among (a) 47 Cytochrome oxidase 1 (CO 1) sequences from Rio Cachoeira Nature Reserve in Brazil and (b) 141 CO1 sequences from the Genbank. The 47 sequences have a peak near zero and another between 18% and 24%, while the 141 sequences have a major peak between 16% and 24% of sequence divergence. High quality figures are available online.
Through morphology-based taxonomy, 20 morphospecies were identified (Figure 1), three of which were allocated species names (see Figure 3). DNA analysis identified 19 MOTUs, 15 of which matched with the morphospecies—about 79% match success. Fortysix sequences showed a matching of below 95% with CO1 sequences from the Genbank, which ranged from 83 to 87%. Only the sequence for morphospecies JHB14 showed a 96% match with the Genbank sequence of Pheidole laticornis. A phylogeny containing a combination of 141 CO1 sequences of Pheidole species from the Genbank and our 47 sequences showed distinct clusters for the MOTUs (Figure 4, taxa in blue). Specimens with only one sequence were regarded as a MOTU using the 2% or 3% criterion. In four MOTUs, morphological taxonomy did not match the results of the DNA-based approach.
Figure 3.

Linearized Bayesian tree of the ant genus Pheidole from Rio Cachoeira Nature Reserve in the Brazilian Atlantic Forest, which defines 19 MOTUs. The clustering of individual sequences in the tree indicates the membership of each MOTU. MOTUs were inferred from a tree dependent clustering process, coupled with thresholds of 2%. Each colored bar represent a MOTU. The five green bars represent cases where only one individual was sequenced, two red bars indicate possible cryptic taxa, and the other two dark blue bars indicate MOTUs with shared taxa. The three names in front of the bars represent Pheidole species whose names were assigned based on morphological taxonomy, and the numbers preceded by JHB represent the different morphospecies. Posterior probability values for Bayesian tree and bootstrap support values for Neighbor-joining tree (in bold) above 50% are indicated on the nodes. A dash (-) indicates bootstrap values below 50%. High quality figures are available online.
The clustering of the 47 CO1 sequences in NJ and Bayesian trees showed congruence with most morphospecies groups, with most nodes immediately below (i.e., defining) clusters showing a bootstrap support and a posterior probability of 100 (Figure 3). Divergences between sequences making up different CO1 clusters (MOTUs) were far higher than divergences within a cluster of MOTUs (11-fold higher), with average Kimura two parameter divergences within and between clusters being 1.8% and 20% respectively (Figure 2). Exceptions occurred where deep sequence divergences were apparent between individuals identified as the same morphospecies (the two red bars in Figure 3, JHB03285G01 and JHB01425G01). These have a mean sequence divergence of 12.6%, a divergence 6–4 fold higher than the 2% and 3% thresholds respectively, which were used to allocate individuals into their respective clusters.
Discussion
The results indicated that CO1 sequences showed promising success in allocating morphologically pre-defined individuals into distinct Pheidole MOTUs. Most sequences clustered into cohesive, well-differentiated groups, most of which showed congruence with the predefined morphospecies. The majority of nodes immediately defining the clusters showed remarkably high levels of nodal support. Furthermore, most clusters remained distinct as sample sizes increased during the progress of the work, an indication that such groups included distinct CO1 lineages rather than scattered sequence variation (Hajibabaei et al. 2006). In addition, sequence divergence in CO1 mitochondrial DNA within MOTU (clusters) was usually much lower than 2%, whereas divergence between the clusters was often greater, but remained within the range of divergences between CO1 sequences of Pheidole species from the Genbank. This result is in general agreement with empirical levels of divergence found between species in barcoding studies (Hebert et al. 2003b). These aspects strengthen the fact that most of the identified morphospecies were indeed distinct lineages (Wiens and Penkrot 2002).
A total of 20 morphospecies were recovered using morphological characters, whereas 19 MOTUs were recovered using barcodes. The diversity estimates (MOTU) using threshold values of 2% and 3% were similar to the diversity estimate based on morphological characters. In cases where the 2–3% MOTU and the morphological estimation of a species differed, either different morphospecies clustered to form the same MOTU (e.g., as was the case with the two MOTUs represented by the dark blue bars in Figure 3) or sequences from the same morphospecies showed deep sequence divergence (e.g., MOTUs represented by two red bars in Figure 3) and were thus allocated as different MOTUs.
Morphological reexamination of the two MOTUs that shared morphospecies (i.e., JHB13 and JHB17, JHB12 and JHB21; MOTUs indicated with dark blue bars in Figure 3) revealed significant differences in morphometric characters between the shared species, as evidenced by the hierarchical clustering (Figure 1). The grouping into one MOTU may be due to incomplete lineage sorting or even mitochondrial introgression (Herbert and Gregory 2005; Meyer and Paulay 2005). Incomplete lineage sorting or gene introgression could be possible in taxa with shared sequences/haplotypes because the species occurred in the same locality. This explanation is further reflected by their low mean sequence divergence (below 0.02) and high bootstrap support for their respective clusters. On the contrary the two MOTUs representing possible cryptic taxa (two red bars; JHB02 in Figure 3) had high mean sequence divergence and their clustering was not strongly supported, thereby qualifying them to possibly be different species. Further reevaluation of the four MOTUs either for introgression or cryptic diversity using mitochondrial DNA was, however, hampered by the limited samples.
The results further revealed very low success when matching MOTUs with Pheidole species already in a CO1 library. After integrating the 47 sequences with those from the Genbank, clusters of the MOTUs remained stable within the phylogeny, with only four MOTUs forming monophyletic clusters with the species from the Genbank (Figure 4). The species name for MOTU JHB05239G01 was allocated as Pheidole laticornis based on the set criteria of allocating species names in this study and others (Meier et al. 2006). Overall, it was difficult to allocate species names to MOTUs based on CO1 sequences in the library. This observation does not imply that CO1 cannot be used in species identification as barcodes, but for the MOTUs in this study, other strategies will be necessary. The low success in matching these MOTUs with Genbank sequences was likely because only a few of the more than 600 described species of Pheidole are included in Genbank. Also, there are many undescribed ant species in the neo-tropical region, and there are suggestions that many species in the Mata Atlantica may be endemic (R. Brandl, personal communication).
Congruence success between the two species identification approaches was not very high, which may be attributed to the criteria applied in delimiting MOTUs. For instance, threshold approach is vulnerable to both false positives and false negatives (Meyer and Paulay 2005). Regardless of such shortcomings in both morphological taxonomy and DNA barcoding (DeSalle et al. 2005; Pires and Marinoni 2010), the low congruence success does not compromise their effective use for species identification (Smith et al. 2005); on the contrary, either approach helps to illuminate taxonomic assignments in need of further scrutiny (Herbert and Gregory 2005; Padial et al. 2010). Such scenarios call for a more thorough morphological and CO1 diversity survey among the members of the involved taxa. Moreover, in cases of introgression, the analysis of a rapidly evolving nuclear sequence, such as the internal transcribed spacer region of the ribosomal repeat, will aid taxonomic resolution (Herbert et al. 2003a). However, this study did not manage to employ other molecular markers for species delimitation, and was only limited to mitochondrial DNA.
Five rare morphospecies were represented with only a single sequence (MOTUs represented by the green bars in Figure 3) due to the used sampling techniques and were coded by the 2% and 3% criterion as MOTUs. A previous study on DNA barcoding of ants using these thresholds (Smith et al. 2005) recommended that only by sampling multiple individuals from supposed species, or MOTUs, will inter-specific variation be properly assessed. Otherwise, it is impossible to test the hypothesis of species-level monophyly (Funk and Omland 2003) and could lead to biodiversity overestimation. This is a valid concern in an analysis of MOTUs from inventories of hyperdiverse groups such as ants, which often include many taxa known only from single individuals (Fisher 1999; Longino et al. 2002). Morphological identification of such rare species also calls for the use of multiple individuals in order to assess the conformity in taxonomic characters within individuals of a given taxa. With additional inventories in the future, many of these rare species will be represented in collections by more specimens.
The aim of this study was to investigate the efficacy of DNA barcoding in delimiting predefined species. The results provide an example of the complementarity with which DNA barcoding can be applied together with a more conventional morphological approach, without competing or replacing the latter approach (Hebert and Gregory 2005). Moreover, thresholds of 2% and 3% proved to be effective in delineating species in the genus Pheidole. Despite the shortcoming in match success, this study demonstrated that diversity estimates using CO1 MOTU together with morphological taxonomy offer a means to map the occurrence of ant species that still wait to be formally described and included into keys for identification.
Acknowledgments
We thank AG Brandl lab team for the support in lab procedures for DNA work. Funding for this study was provided by the German government. We also thank J. H. Bihn from Philipps-Universitaet Marburg, for assisting in grouping and identification of the morphospecies, not forgeting the editors and the anonymous reviewers who made this publication a success.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of Molecular Biology. 1990;215:403–403. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Barratt BIP, Derraik JGB, Rufaut CG, Goodman AJ, Dickinson KJM. Morphospecies as a substitute for Coleoptera species identification, and the value of experience in improving accuracy. Journal of the Royal Society of New Zealand. 2003;33:583–590. [Google Scholar]
- Bensasson D, Zhang DX, Hartl DL, Hewitt GM. Mitochondrial pseudogenes: evolution's misplaced witnesses. Trends in Ecology and Evolution. 2001;16:314–321. doi: 10.1016/s0169-5347(01)02151-6. [DOI] [PubMed] [Google Scholar]
- Bihn JH, Gebauer G, Brandl R. Loss of functional diversity of ant assemblages in secondary tropical forests. Ecology. 2010;91:782–792. doi: 10.1890/08-1276.1. [DOI] [PubMed] [Google Scholar]
- Bihn JH, Verhaagh M, Brändle M, Brandl R. Do secondary forests act as refuges for old-growth forest animals? Recovery of ant diversity in the Atlantic forest of Brazil. Biological Conservation. 2008;141:733–743. [Google Scholar]
- Blaxter M, Mann J, Chapman T. Defining operational taxonomic units using DNA barcode data. Philosophical Transactions of the Royal Society of London, Series B, Biological Sciences. 2005;360:1935–1943. doi: 10.1098/rstb.2005.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaxter ML. The promise of DNA taxonomy. Philosophical Transactions of the Royal Society of London, Series B, Biological Sciences. 2004;359:669–679. doi: 10.1098/rstb.2003.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks T, Da Fonseca GAB, Rodrigues ASL. Species, data, and conservation planning. Conservation Biology. 2004;18:1682–1688. [Google Scholar]
- Cardoso A, Serrano A, Vogler AP. Morphological and molecular variation in tiger beetles of the Cicindela hybrida complex: is an ‘integrative taxonomy’ possible? Molecular Ecology. 2009;18:648–664. doi: 10.1111/j.1365-294X.2008.04048.x. [DOI] [PubMed] [Google Scholar]
- Crozier RH, Crozier YC. The Mitochondrial Genome of the Honeybee Apis mellifera - Complete Sequence and Genome Organization. Genetics. 1993;133:97–117. doi: 10.1093/genetics/133.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayrat B. Towards integrative taxonomy. Biological Journal of the Linnean Society. 2005;85:407–415. [Google Scholar]
- Desalle R, Egan MG, Siddall M. The unholy trinity: taxonomy, species delimitation and DNA barcoding. Philosophical Transactions of the Royal Society of London, Series B, Biological Sciences. 2005;360:1905–1916. doi: 10.1098/rstb.2005.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher BL. Improving inventory efficiency: A case study of leaf-litter ant diversity in Madagascar. Ecological Applications. 1999;9:714–731. [Google Scholar]
- Floyd R, Abebe E, Papert A, Blaxter M. Molecular barcodes for soil nematode identification. Molecular Ecology. 2002;11:839–850. doi: 10.1046/j.1365-294x.2002.01485.x. [DOI] [PubMed] [Google Scholar]
- Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular Marine Biology andBiotechnology. 1994;3:294–299. [PubMed] [Google Scholar]
- Funk DJ, Omland KE. Species-level paraphyly and polyphyly: Frequency, causes, and consequences, with insights from animal mitochondrial DNA. Annual Review of Ecology Evolution and Systematics. 2003;34:397–423. [Google Scholar]
- Hajibabaei M, Janzen DH, Burns JM, Hallwachs W, Hebert PDN. DNA barcodes distinguish species of tropical Lepidoptera. Proceedings of the National Academy of Sciences USA. 2006;103:968–971. doi: 10.1073/pnas.0510466103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series. 1999;41:95–98. [Google Scholar]
- Hammond PM. Practical approaches to the estimation of the extent of biodiversity in speciose groups. Philosophical Transactions of the Royal Society of London, Series B, Biological Sciences. 1994;345:119–136. [Google Scholar]
- Hebert PDN, Ratnasingham S, deWaard JR. Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proceedings of the Royal Society of London, Series B, Biological Sciences. 2003a;270:S96–S99. doi: 10.1098/rsbl.2003.0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert PDN, Cywinska A, Ball SL, Dewaard JR. Biological identifications through DNA barcodes. Proceedings of the Royal Society London, Series B, Biological Sciences. 2003b;270:313–322. doi: 10.1098/rspb.2002.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert PDN, Stoeckle MY, Zemlak TS, Francis CM. Identification of birds through DNA barcodes. Public Library of Science, Biology. 2004a;2:1657–1663. doi: 10.1371/journal.pbio.0020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert PDN, Stoeckle MY, Zemlak TS, Francis CM. Identification of birds through DNA barcodes. Public Library of Science, Biology. 2004b;2:e312. doi: 10.1371/journal.pbio.0020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert PDN, Gregory TR. The promise of DNA barcoding for taxonomy. Systematic Biology. 2005;54:852–859. doi: 10.1080/10635150500354886. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- Janzen DH, Hajibabaei M, Burns JM, Hallwachs W, Remigio E, Hebert PDN. Wedding biodiversity inventory of a large and complex Lepidoptera fauna with DNA barcoding. Philosophical Transactions of the Royal Society of London, Series B, Biological Sciences. 2005;360:1835–1845. doi: 10.1098/rstb.2005.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarman SN, Elliott NG. DNA evidence for morphological and cryptic Cenozoic speciations in the Anaspididae, ‘living fossils’ from the Triassic. Journal of Evolutionary Biology. 2000;13:624–633. [Google Scholar]
- Kimura MA. Simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide-sequences. Journal of Molecular Evolution. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- Knowlton N. Sibling species in the sea. Annual Review of Ecology and Systematics. 1993;24:189–216. [Google Scholar]
- Krell F-T. Parataxonomy vs. taxonomy in biodiversity studies — pitfalls and applicability of ‘morphospecies’ sorting. Biodiversity and Conservation. 2004;13:795–812. [Google Scholar]
- Larget B, Simon DL. Markov chain Monte Carlo algorithms for the Bayesian analysis of phylogenetic trees. Molecular Biology and Evolution. 1999;16:750–759. [Google Scholar]
- Longino JT, Coddington J, Colwell RK. The ant fauna of a tropical rain forest: Estimating species richness three different ways. Ecology. 2002;83:689–702. [Google Scholar]
- Longino JT. The ants of Costa Rica. The Evergreen State College; 2008. Available online: http://academic.evergreen.edu/projects/ants/AntsofCostaRica.html. [Google Scholar]
- Meier R, Shiyang K, Vaidya G, Ng PKL. DNA barcoding and taxonomy in Diptera: A tale of high intraspecific variability and low identification success. Systematic Biology. 2006;55:715–728. doi: 10.1080/10635150600969864. [DOI] [PubMed] [Google Scholar]
- Mengual X, Stahls G, Vujic A, Marcos-Garcia MA. Integrative taxonomy of Iberian Merodon species (Diptera, Syrphidae). Zootaxa. 2006. pp. 1–26.
- Meyer CP, Paulay G. DNA barcoding: Error rates based on comprehensive sampling. Public Library of Science, Biology. 2005;3:2229–2238. doi: 10.1371/journal.pbio.0030422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan MT, Balke M, Gregory TR, Vogler AP. DNA-based species delineation in tropical beetles using mitochondrial and nuclear markers. Philosophical Transactions of the Royal Society of London, Series B, Biological Sciences. 2005;360:1925–1933. doi: 10.1098/rstb.2005.1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers N, Mittermeier RA, Mittermeier CG, Da Fonseca GAB, Kent J. Biodiversity hotspots for conservation priorities. Nature. 2000;403:853–858. doi: 10.1038/35002501. [DOI] [PubMed] [Google Scholar]
- Oliver I, Beattie AJ. Invertebrate morphospecies as surrogates for species: a case study. Conservation Biology. 1996;10:99–109. [Google Scholar]
- Padial JM, Miralles A, De La Riva I, Vences M. The integrative future of taxonomy. Frontiers in Zoology. 2010;7:16. doi: 10.1186/1742-9994-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- Pires AC, Marinoni L. DNA barcoding and traditional taxonomy unified through Integrative Taxonomy: a view that challenges the debate questioning both methodologies. Biota Neotropica. 2010;10(2) [Google Scholar]
- R Development Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing; 2009. Available online: http://www.Rproject.org. [Google Scholar]
- Rannala B, Yang Z. Probability distribution of molecular evolutionary trees: a new method of phylogenetic inference. Journal of Molecular Evolution. 1996;43:304–311. doi: 10.1007/BF02338839. [DOI] [PubMed] [Google Scholar]
- Rubinoff D, Cameron S, Will K. A genomic perspective on the shortcomings of mitochondrial DNA for “Barcoding” identification. Journal of Heredity. 2006;97:581–594. doi: 10.1093/jhered/esl036. [DOI] [PubMed] [Google Scholar]
- Rubinoff D. Utility of Mitochondrial DNA Barcodes in Species Conservation. Conservation Biology. 2006;20:1026–1033. doi: 10.1111/j.1523-1739.2006.00372.x. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Molecular Biology and Evolution. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Smith MA, Fisher BL, Hebert PDN. DNA barcoding for effective biodiversity assessment of a hyperdiverse arthropod group: the ants of Madagascar. Philosophical Transactions of the Royal Society of London, Series B, Biological Sciences. 2005;360:1825–1834. doi: 10.1098/rstb.2005.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Rodriguez JJ, Whitfield JB, Deans AR, Janzen DH, Hallwachs W, Hebert PDN. Extreme diversity of tropical parasitoid wasps exposed by iterative integration of natural history, DNA barcoding, morphology, and collections. Proceedings of the National Academy of Sciences USA. 2008;105:12359–12364. doi: 10.1073/pnas.0805319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Molecular Biology and Evolution. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Tautz D, Arctander P, Minelli A, Thomas RH, Vogler AP. A plea for DNA taxonomy. Trends in Ecology and Evolution. 2003;18:70–74. [Google Scholar]
- Wiens JJ, Penkrot TA. Delimiting species using DNA and morphological variation and discordant species limits in spiny lizards (Sceloporus). Systematic Biology. 2002;51:69–91. doi: 10.1080/106351502753475880. [DOI] [PubMed] [Google Scholar]
- Wilson EO. Pheidole in the New World. A dominant, hyperdiverse ant genus; Harvard University Press: 2003. [Google Scholar]