

Abstract
Concentrations of acetyl–coenzyme A and nicotinamide adenine dinucleotide (NAD+) affect histone acetylation and thereby couple cellular metabolic status and transcriptional regulation. We report that the ketone body d-β-hydroxybutyrate (βOHB) is an endogenous and specific inhibitor of class I histone deacetylases (HDACs). Administration of exogenous βOHB, or fasting or calorie restriction, two conditions associated with increased βOHB abundance, all increased global histone acetylation in mouse tissues. Inhibition of HDAC by βOHB was correlated with global changes in transcription, including that of the genes encoding oxidative stress resistance factors FOXO3A and MT2. Treatment of cells with βOHB increased histone acetylation at the Foxo3a and Mt2 promoters, and both genes were activated by selective depletion of HDAC1 and HDAC2. Consistent with increased FOXO3A and MT2 activity, treatment of mice with βOHB conferred substantial protection against oxidative stress.
Cellular metabolites such as acetyl–coenzyme A (acetyl-CoA) and nicotinamide adenine dinucleotide (NAD+) influence gene expression by serving as cofactors for epigenetic modifiers that mediate posttranslational modification of histones (1). The activity of histone acetyltransferases (HATs) is dependent on nuclear acetyl-CoA concentrations (2, 3) and the deacetylase activity of class III HDACs, also called sirtuins, is dependent on NAD+ concentrations (4). Class I (HDAC1, 2, 3, 8), class II (HDAC4, 5, 6, 7, 9, 10), and class IV (HDAC11) HDACs are zincdependent enzymes, but endogenous regulators are not known.
Small-molecule inhibitors of class I and class II HDACs include butyrate, a product of bacterial anaerobic fermentation (5). Butyrate is closely related to β-hydroxybutyrate (βOHB) (Fig. 1A), the major source of energy for mammals during prolonged exercise or starvation (6). Accumulation of βOHB in blood increases to 1 to 2 mM during fasting when the liver switches to fatty acid oxidation (7, 8), and to even higher concentrations during prolonged fasting (6 to 8 mM) (6) or in diabetic ketoacidosis (>25 mM) (9).
Fig. 1.
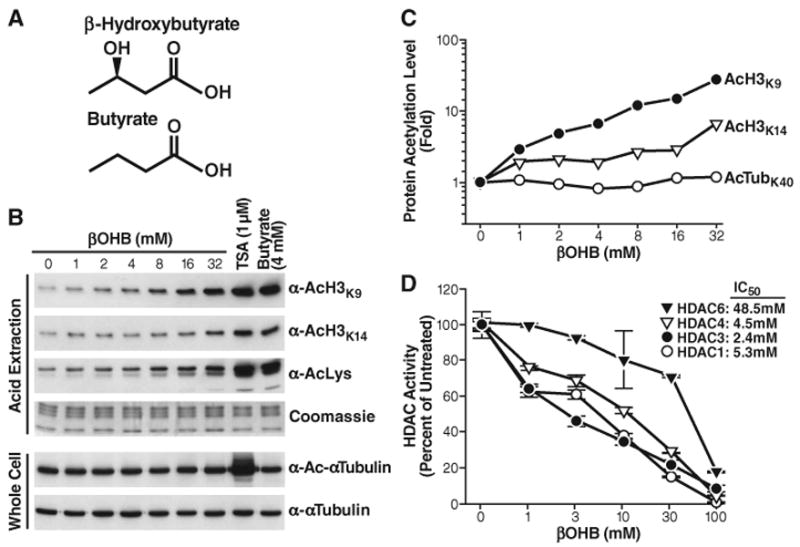
Inhibition of HDACs by βOHB in vitro and in vivo. (A) Structures of β-hydroxybutyrate and butyrate. (B) Effect of βOHB, TSA, or butyrate on acetylation of histone H3 and tubulin. HEK293 cells were treated with the indicated concentrations of drugs for 8 hours. Histones were acid-extracted, and their acetylation was assessed by protein immunoblotting with anti-AcH3K9, anti-AcH3K14, or anti-acetyllysine (AcLys). Proteins from whole-cell extracts were analyzed by immunoblotting with antibodies to α-tubulin or Ac-α-Tubulin. (C) Quantification of acetylation levels from blots in (B), shown relative to untreated cells (βOHB 0 mM). (D) Inhibition of immunopurified HDACs by βOHB in vitro. Flag-tagged HDACs were expressed in HEK293 cells, immunoprecipitated, and incubated in vitro with a 3H-labeled acetylated histone H4 peptide and the indicated concentrations of βOHB. HDAC activity is relative to the activity of each enzyme without βOHB. The IC50 values of βOHB are shown.
To determine whether βOHB might have HDAC inhibitor activity, we treated human embryonic kidney 293 (HEK293) cells with different amounts of βOHB for 8 hours, and measured histone acetylation levels by Western blot with antibodies to acetylated histone H3 lysine 9 (AcH3K9) and to acetylated histone H3 lysine 14 (AcH3K14) (Fig. 1, B and C, and figs. S1 and S2). βOHB increased histone acetylation in a dose-dependent manner, even at 1 to 2 mM, which can occur in humans after a 2- to 3-day fast or strenuous exercise (6, 8, 10). Like butyrate, βOHB did not increase acetylation of α-tubulin, indicating that it inhibits class I HDACs but not the class IIb tubulin deacetylase, HDAC6.
To test the HDAC inhibitor activity of βOHB and its possible selectivity, we purified recombinant human HDACs after transient transfection of expression vectors for human epitope-tagged (FLAG) HDAC1, HDAC3, HDAC4, and HDAC6 in HEK293T cells. We purified the HDACs, incubated them with 3H-labeled acetylated histone H4 peptides, and measured their deacetylase activity (Fig. 1D) (11). βOHB inhibited HDAC1, HDAC3, and HDAC4 in a dose-dependent manner with a median inhibitory concentration (IC50) of 5.3, 2.4, and 4.5 mM, respectively. The HDAC6 IC50 was much higher (48.5 mM) (Fig. 1E), and βOHB did not inhibit HDAC6 activity on its natural substrate tubulin (fig. S3). To examine the possibility that histone acetylation was enhanced by increased concentration of acetyl-CoA (because βOHB is catabolized into acetyl-CoA in target tissues), we directly measured abundance of acetyl-CoA in βOHB-treated HEK293 cells, but no change was observed (fig. S4). We also tested a possible activating effect of βOHB on histone acetyltransferase activity of p300 and PCAF (P300/CBP-associated factor) but did not detect any change induced by βOHB (fig. S5). Thus, millimolar concentrations of βOHB appear to increase histone acetylation directly through HDAC inhibition. High concentrations of acetoacetate (AcAc) also inhibit class I and class IIa HDACs in vitro (fig. S6A) and in HEK293 cells (fig. S6B). However, the concentration of AcAc in blood is one-third or less than that of βOHB during fasting and therefore less likely to reach concentrations that would inhibit HDACs under physiological conditions (12). To test the relative contribution of βOHB, acetoacetate, and acetyl-CoA to histone acetylation in response to βOHB treatment, we depleted cells of βOHB dehydrogenases (BDH1, 2) with small interfering RNA. Both enzymes catalyze the transformation of βOHB into aceteto-acetate, and their suppression had no effect on histone acetylation in response to βOHB up to 3mM. At higher concentrations of βOHB (10 and 30 mM), however, further increase in his-tone acetylation was suppressed by depletion of βOHB dehydrogenases (fig. S7, A and B), indicating that either AcAc or acetyl-CoA might also contribute to histone acetylation in cells exposed to concentrations of βOHB above the IC50 for HDACs.
To determine whether changes in βOHB concentrations in vivo might affect histone acetylation, we measured βOHB concentration in mouse serum after a 24-hour fast or in mice on calorie restriction (CR). We also used implanted osmotic pumps to administer exogenous βOHB or phosphate-buffered saline (PBS). βOHB concentrations increased to 1.5 ± 0.1 mM after a 24-hour fast, 0.6 ± 0.1 mM in mice on CR and 1.2 ± 0.1 mM with administration of βOHB via an intraperitoneal pump (Fig. 2A). We collected tissues from fed or 24-hour–fasted mice and measured histone acetylation by immunoblotting. Acetylation of H3K9 and H3K14 reflect the competing activities of HATs and HDACs and influence gene expression in several species, including humans (13). Acetylation of H3K9 and H3K14 increased significantly in several organs in fasted mice, particularly kidney (Fig. 2B and figs. S8 and S9). In kidney, histone acetylation (H3K9 and H3K14) also increased two- to fivefold in mice under CR. Kidney histone acetylation and serum βOHB concentrations were strongly correlated for both histone H3K9 (R2 = 0.772) and histone H3K14 (R2 = 0.863) (Fig. 2C). We first focused on kidney, the organ with the largest changes in histone acetylation, to investigate the effects of βOHB on gene expression and cellular phenotype.
Fig. 2.

Serum βOHB and global histone acetylation in kidney of mice deprived of food. (A) Serum concentration of βOHB was determined in age-matched groups of three C57Bl6 mice fed or fasted (16 weeks old, fasted for 24 hours), implanted with a pump delivering either PBS or βOHB (16 weeks old, 24 hours of pump treatment), fed ad libitum (AL) or on calorie restriction to 60% of AL (CR) (8 months old, 6 months on CR). Mean ± SE, *P < 0.05 by t test between paired conditions. (B) Histones were purified from the kidneys of the same mice as in (A). Acetylation was assessed with anti-AcH3K9, anti-AcH3K14, or anti-Ac-α-tubulin (fig. S8). Acetylation is normalized to total histone H3 and shown relative to the control condition in each pair (e.g., fed versus fasted). Mean ± SE, *P < 0.05 by t test between paired conditions. (C) Serum βOHB concentrations [from (A)] plotted against acetylation of histone H3K9 and H3K14 [from (B)]; correlation coefficient R2 = 0.772 for H3K9 and 0.863 for H3K14.
Histone acetylation induced by HDAC inhibitors is associated with transcriptional activation and repression of a subset of cellular genes (14). To identify genes whose expression changed in response to βOHB, we extracted mRNA for microarray analysis from mouse kidneys treated with βOHB or PBS for 24 hours. As βOHB is more abundant in fasting conditions, gene expression changes induced by βOHB may be a subset of those induced by fasting. Of 35,556 genes tested, 284 increased transcription in response to fasting (false discovery rate <0.2, table S1). Four of the five genes with the largest changes in expression in response to βOHB were also activated in response to fasting as determined both by microarray and quantitative real-time polymerase chain reaction (QPCR) (P < 0.001 for such overlap via binomial distribution, tables S2 and S3). Ingenuity pathway analysis identified two of the five βOHB-induced genes (Mt2 and Lcn2) as regulated by FOXO3A. Overall, we found five genes in the FOXO3A network (Foxo3a, Mt2, Lcn2, Lemd3, and Hbp1) that had increased transcription in response to βOHB via QPCR (fig. S10). Foxo3a, a transcription factor, induces cell-cycle arrest and resistance to oxidative stress (15). Metallothionein 2 (Mt2), the gene with the greatest transcriptional response to βOHB treatment as determined by QPCR, also protects against oxidative stress (16). Foxo3a and Mt2 mRNA expression is also modestly increased during CR as determined by QPCR (Fig. 3, A and B). Chromatin immunoprecipitation (ChIP) analysis of the Foxo3a and Mt2 promoters with two distinct primer pairs for each promoter revealed increased histone H3K9 acetylation at both promoters after treatment of HEK293 cells with a high dose of βOHB (10 mM) (Fig. 3C).
Fig. 3.

Increased expression of oxidative stress resistance genes in cells exposed to βOHB. (A) Expression of Foxo3a under various conditions (see Fig. 2 for details) measured by QPCR. Foxo3 expression is normalized to abundance of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Mean ± SE, *P < 0.05 by t test between paired conditions. (B) Expression of Mt2, measured as in (A). (C) Promoters of Mt2 and Foxo3a are enriched for acetylated his-tone H3K9 after βOHB treatment. HEK293 cells were treated with 10 mM βOHB or PBS for 24 hours. Chromatin was immunoprecipitated with anti-H3 or anti-AcH3K9, and the purified DNA was analyzed with primer pairs specific for the Foxo3a or Mt2 promoters. Results are the ratios of AcH3K9 to total histone H3. Mean ± SE, *P < 0.05 by t test between βOHB and PBS conditions. (D) HDAC depletion increases Foxo3a and Mt2 mRNAs abundance. HEK293 cells were transfected with shRNAs specific for each class I or class II HDAC, and mRNA abundance was measured by QPCR 72 hours after transfection. Mean ± SE, *P < 0.05 by t test versus control shRNA. (E) HDAC1, but not HDAC6, is enriched at the promoters of Mt2 and Foxo3a. ChIP analysis of the Foxo3a and Mt2 promoters (two primer pairs per promoter) and Gapdh (one primer pair) from HEK293 cells with control immunoglobulin G (IgG), anti-HDAC1, or anti-HDAC6. Relative promoter binding of each HDAC is normalized to input Gapdh. Mean ± SE, *P < 0.05 by t test versus IgG control.
Next, we depleted cells of each class I and II HDAC with selective short hairpin–mediated RNAs (shRNAs). The shRNAs selective for each HDAC suppressed expression of their cognate HDAC by at least 60% (fig. S11). Depletion of HDAC1 or HDAC2 caused up-regulation of Foxo3a and Mt2 mRNA by 1.8-fold and 1.5-fold, respectively (Fig. 3D). Depletion of both HDAC1 and 2 further increased accumulation of Foxo3a and Mt2 mRNA (Fig. 3D). ChIP analysis revealed that HDAC1, but not HDAC6, was recruited to the Foxo3a and Mt2 promoters and not recruited to the Gapdh promoter (Fig. 3E). Binding of HDAC1 to the Foxo3a promoter was unchanged by βOHB treatment; thus, HDAC catalytic activity appears not to be necessary for promoter binding of HDAC1 (fig. S12). βOHB appears to induce local histone acetylation at the promoter of oxidative stress resistance genes, Foxo3a and Mt2, by inhibiting activity of HDACs 1 and 2.
Mitochondrial superoxide dismutase (Mn-SOD) and catalase are two other well-defined FOXO3A targets that contribute to its protective activity against oxidative stress (15, 17). Protein immunoblotting of kidney tissue isolated from PBS- or βOHB-treated mice showed increased expression of FOXO3A, Mn-SOD, and catalase (Fig. 4A and figs. S13 and S14).
Fig. 4.
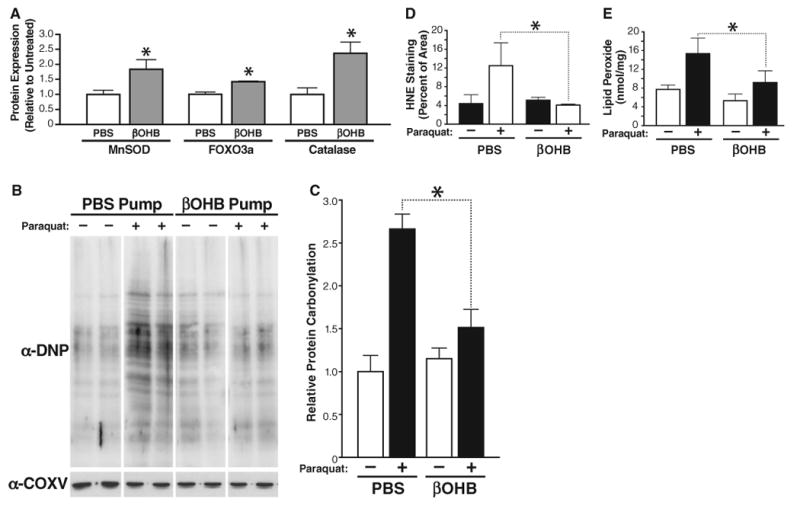
Protective effect of βOHB treatment against oxidative stress. (A) Amounts of catalase, MnSOD, or FOXO3A measured by protein immunoblotting in kidney tissue from 16-week-old mice implanted with an osmotic pump delivering PBS or βOHB (as in Fig. 2; n = 3); mean ± SE, *P < 0.05 by t test between PBS and βOHB conditions. (B) Protein carbonylation in kidney samples from mice implanted with an osmotic pump delivering PBS or βOHB (as in Fig. 2; n = 3) and treated with paraquat (50 mg/kg) or vehicle for 2 hours. Car-bonylation was measured by immunoblotting with anti-DNP. All samples were run on a single gel; after imaging, lanes were rearranged for presentation. (C) Quantification of protein carbonylation in (B). Mean ± SE, *P < 0.05 by t test between PBS and βOHB conditions. (D) Sections of kidney obtained from the same mice as in (B) were stained with anti-4-HNE and quantified (see fig. S16 for primary picture). Mean ± SE, *P < 0.05 by t test between PBS and βOHB conditions. (E). Lipid peroxides were quantified in mice kidneys (LPO assay kit, Cayman, Ann Arbor, MI). Mean ± SE, *P < 0.05 by t test between PBS and βOHB conditions.
The effect on expression of MT2, FOXO3A, MnSOD, and catalase indicated that βOHB might have protective activity against oxidative stress. Carbonyl derivatives are formed by a direct metal-catalyzed oxidative attack on the amino acid side chains of proline, arginine, lysine, and threonine. Carbonylation is irreversible and unrepairable and accumulates as organisms age (18). To test the possible protective role of βOHB against oxidative stress, we implanted mice with a subcutaneous pump delivering either βOHB or PBS for 24 hours. They then received an intravenous injection of paraquat, which induces accumulation of reactive oxygen species. Kidney tissue was isolated after 2 hours, and protein carbonylation was assayed by protein immunoblotting with an antibody to dinitrophenyl (DNP) after derivatization of protein with dinitrophenylhydrazine (DNPH) (Fig. 4B). Paraquat treatment of control mice receiving a PBS infusion led to a twofold increase in carbonylated proteins. This increase in protein carbonylation was significantly suppressed (54 ± 9% decrease) in mice receiving βOHB (Fig. 4C).
We also examined another marker of oxidative stress: lipid peroxidation. 4-Hydroxynonenal (4-HNE) is a degradation product of polyunsat-urated lipid and accumulates in response to oxidative stress (19). Kidney tissue sections from PBS- or paraquat-treated mice were stained with an antibody to 4-HNE, and the amount of 4-HNE staining was quantified with imaging software (fig. S15). Paraquat treatment increased 4-HNE staining threefold in control mice (PBS) (Fig. 4D). This increase was completely suppressed in mice treated with βOHB. Lipid peroxides were also directly quantified in an enzyme-linked immunosorbent assay that measures conversion of ferrous ions to ferric ions. A twofold increase in lipid peroxide in response to paraquat was suppressed significantly by βOHB treatment (Fig. 4E). Thus, βOHB protects against paraquat-induced oxidative stress in mouse kidney.
Our observation that βOHB is an endogenous HDAC inhibitor present in organisms at millimolar concentrations during prolonged fasting and CR reveals an example of integration between metabolic status and epigenetic changes. We show that changes in histone acetylation and gene expression caused by βOHB promote stress resistance in the kidney. Future studies should investigate the specific gene expression and physiological effects of βOHB in other tissues. For example, low-carbohydrate diets that induce substantial ketogenesis are broadly neuroprotective and enhance resistance of neurons to oxidative damage (20). In addition, reduction in HDAC activity by either genetic manipulation or chemical inhibition extends life span in Drosophila (21, 22). Inhibition of HDACs by βOHB might contribute to the beneficial effect of ketogenic diets and may be one mechanism by which calorie restriction confers health benefits.
Supplementary Material
Acknowledgments
We thank J. Carroll for graphics, G. Howard and S. Ordway for editorial assistance, V. Fonseca for administrative assistance, and A. Brunet (Stanford University) for reagents. Supported by funds from the Gladstone Institutes.
Footnotes
Supplementary Materials: http://www.sciencemag.org/cgi/content/full/science.1227166/DC1
Materials and Methods
References (23–26)
References and Notes
- 1.Ladurner AG. Mol Cell. 2006;24:1. doi: 10.1016/j.molcel.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 2.Wellen KE, et al. Science. 2009;324:1076. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takahashi H, McCaffery JM, Irizarry RA, Boeke JD. Mol Cell. 2006;23:207. doi: 10.1016/j.molcel.2006.05.040. [DOI] [PubMed] [Google Scholar]
- 4.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Nature. 2000;403:795. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 5.Candido EP, Reeves R, Davie JR. Cell. 1978;14:105. doi: 10.1016/0092-8674(78)90305-7. [DOI] [PubMed] [Google Scholar]
- 6.Cahill GF., Jr Annu Rev Nutr. 2006;26:1. doi: 10.1146/annurev.nutr.26.061505.111258. [DOI] [PubMed] [Google Scholar]
- 7.Cahill GF, Jr, et al. J Clin Invest. 1966;45:1751. doi: 10.1172/JCI105481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robinson AM, Williamson DH. Physiol Rev. 1980;60:143. doi: 10.1152/physrev.1980.60.1.143. [DOI] [PubMed] [Google Scholar]
- 9.Laffel L. Diabetes Metab Res Rev. 1999;15:412. doi: 10.1002/(sici)1520-7560(199911/12)15:6<412::aid-dmrr72>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 10.Koeslag JH, Noakes TD, Sloan AW. J Physiol. 1980;301:79. doi: 10.1113/jphysiol.1980.sp013190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.North BJ, Schwer B, Ahuja N, Marshall B, Verdin E. Methods. 2005;36:338. doi: 10.1016/j.ymeth.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 12.Owen OE, et al. J Clin Invest. 1973;52:2606. doi: 10.1172/JCI107453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Agalioti T, et al. Cell. 2000;103:667. doi: 10.1016/s0092-8674(00)00169-0. [DOI] [PubMed] [Google Scholar]
- 14.Van Lint C, Emiliani S, Verdin E. Gene Expr. 1996;5:245. [PMC free article] [PubMed] [Google Scholar]
- 15.Kops GJ, et al. Nature. 2002;419:316. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 16.Kawai K, et al. Chem Res Toxicol. 2000;13:1275. doi: 10.1021/tx000119l. [DOI] [PubMed] [Google Scholar]
- 17.Nemoto S, Finkel T. Science. 2002;295:2450. doi: 10.1126/science.1069004. [DOI] [PubMed] [Google Scholar]
- 18.Nyström T. EMBO J. 2005;24:1311. doi: 10.1038/sj.emboj.7600599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Esterbauer H, Schaur RJ, Zollner H. Free Radic Biol Med. 1991;11:81. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 20.Kim Y, et al. J Neurochem. 2007;101:1316. doi: 10.1111/j.1471-4159.2007.04483.x. [DOI] [PubMed] [Google Scholar]
- 21.Rogina B, Helfand SL, Frankel S. Science. 2002;298:1745. doi: 10.1126/science.1078986. [DOI] [PubMed] [Google Scholar]
- 22.Kang HL, Benzer S, Min KT. Proc Natl Acad Sci USA. 2002;99:838. doi: 10.1073/pnas.022631999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.