


Abstract
Methylation of the CpG-rich region (CpG island) overlapping a gene’s promoter is a generally accepted mechanism for silencing expression. While recent technological advances have enabled measurement of DNA methylation and expression changes genome-wide, only modest correlations between differential methylation at gene promoters and expression have been found. We hypothesize that stronger associations are not observed because existing analysis methods oversimplify their representation of the data and do not capture the diversity of existing methylation patterns. Recently, other patterns such as CpG island shore methylation and long partially hypomethylated domains have also been linked with gene silencing. Here, we detail a new approach for discovering differential methylation patterns associated with expression change using genome-wide high-resolution methylation data: we represent differential methylation as an interpolated curve, or signature, and then identify groups of genes with similarly shaped signatures and corresponding expression changes. Our technique uncovers a diverse set of patterns that are conserved across embryonic stem cell and cancer data sets. Overall, we find strong associations between these methylation patterns and expression. We further show that an extension of our method also outperforms other approaches by generating a longer list of genes with higher quality associations between differential methylation and expression.
INTRODUCTION
DNA methylation is an important factor in transcriptional regulation, playing a role in genomic imprinting, X-inactivation, retrotransposon silencing and the control of tissue-specific genes during differentiation (1). DNA methylation patterns are frequently altered in tumors (2), and there is great interest in understanding how changes to these patterns contribute to human disease (3). Even so, how alterations to DNA methylation affect gene transcription remains poorly characterized. Over 60% of genes have a CpG-rich region, termed a CpG island, overlapping their promoter (4). Classically, it is thought that hypermethylation of promoter-associated CpG islands silences transcription. However, it was recently shown that cancer- and tissue-specific methylation variation in adjacent regions, termed CpG island shores, is also associated with gene expression change (5). Additionally, genes are more likely to be repressed when they are located in partially methylated domains (6) or long hypomethylated domains (7,8) in cancer.
Techniques such as whole-genome bisulfite sequencing (WGBS) (9) and Methyl-MAPS (10) have recently been developed to map methylation at single-base resolution genome-wide. Methods to interpret this data, however, are lacking. Current computational techniques are mostly concerned with the visualization of genome-level correlations between DNA methylation and other epigenetic marks, or with the identification of regions that are differentially marked between samples [recently reviewed in (11)]. These tools have elucidated the genomic organization of these marks, but they do not sufficiently address how changes at individual loci associate with and potentially affect function.
The most common approach for characterizing methylation changes between two samples uses a sliding window to identify differentially methylated regions (DMRs) (7,9). A gene with a hypermethylated DMR near its promoter is assumed to exhibit a decrease in expression, while a gene near a hypomethylated DMR should exhibit an increase in expression. In practice, the Pearson correlation coefficient between the methylation level of the DMR and the expression of its associated gene is around −0.3 (11,12). It has been assumed that better anticorrelation is precluded owing to noise from experimental error, mixed cellular populations, copy number variations, chromatin modifiers or other regulation events. Another explanation, however, is that contemporary analysis methods are not sophisticated enough to recognize relationships involving more complex methylation patterns. Existing approaches summarize their representation of methylation change in the promoter region to simplify analysis, but this sacrifices potentially important spatial information contained in the locations of the constituent sites.
To discover DNA methylation changes that associate with gene expression changes, we propose a new method that uses the entire differential methylation profile in the vicinity of a gene’s promoter (Figure 1). We represent the differential methylation for a fixed area around each gene's transcription start site (TSS) as a continuous curve, or signature, capturing the shape of the methylation changes. We then apply a curve similarity metric, the discrete Fréchet distance, to compare differential methylation signatures for all genes. Using an unsupervised clustering technique, we arrange the signatures according to their shapes and identify which clusters of genes exhibit statistically significant changes in expression. Generalized patterns of differential methylation can be extrapolated from the resulting clusters. Because the approach is unsupervised, no assumptions need to be made about the direction of a correlation between methylation and expression. Although designed for pattern discovery, the method is easily extended to identify a list of genes potentially regulated by methylation. These gene lists are of markedly greater length and have higher quality associations between differential methylation and expression than those generated by existing methods. A current implementation of the approach can be found on the project website at http://epigenomics.wustl.edu/WIMSi/.
Figure 1.

Method overview and example methylation signatures. (A) Overview of the approach for associating spatially similar DNA methylation changes with corresponding changes in transcription. (B, C) Example methylation signatures from HMEC-HCC1954 WGBS data for the tumor suppressor gene CDH4 and the LPCAT3 gene. Conversion of methylation data to signatures allows direct comparison of the data between genes with different distributions of CpG sites relative to their TSSs. Top panels show methylation (blue is unmethylated and red is methylated) and RNA-Seq expression data on the UCSC genome browser. Bottom panels show interpolated and smoothed methylation signatures (black curve) that are used to calculate the discrete Fréchet distance. Blue tick marks show locations of all CpG sites. Black dots mark experimentally measured differences in methylation between the two samples.
MATERIALS AND METHODS
Methylation signatures
Using WGBS or Methyl-MAPS data (single-base resolution) to compare methylation patterns between different genes’ promoter regions is complicated by the variability in the locations of their CpGs. To enable comparisons between genes, we standardize the representation of differential methylation data for each gene by creating a methylation signature across a fixed-width region centered at the TSS (Figure 1). In a single sample, the methylation level at each probed CpG is represented as a continuous value between 0 and 1, denoting fully unmethylated and methylated, respectively. To compare methylation between two samples, we subtract these values at each site to produce a differential methylation score that ranges from −1 to 1, denoting complete hypomethylation and hypermethylation, respectively.
We create a gene's methylation signature on a fixed-width target region by interpolating the differential methylation scores using piecewise cubic Hermite interpolation (Figure 1B and C) across all CpG sites within the region and between one and five CpG sites flanking the region on either side. Because methylation is highly correlated over short distances (Supplementary Figure S1) (13), interpolation provides a suitable estimate for differential methylation in areas with missing data. Such missing data points can occur owing to insufficient coverage or experimental limitations (e.g. data collected only at specific restriction sites). Interpolated values always lie between −1 and 1. Regions with fewer than five CpG sites with sufficient coverage and regions with fewer than one flanking site per side are discarded. We also discard regions containing no CpG sites with absolute differential methylation >0.2. Lastly, we apply Gaussian smoothing on the curves (σ = 50 bp) to help moderate noise due to experimental artifacts such as missing or inaccurate measurements. One advantage of using a combination of interpolation and smoothing is that it improves performance of the method for low coverage data. Previously it was shown that statistical smoothing was an effective method to analyze low coverage WGBS data (14). The resulting methylation signatures for each gene are bounded between −1 and 1 on a fixed region relative to the TSS.
Determining clusters of methylation signatures with significant expression changes
We compare methylation signatures between genes using the discrete Fréchet distance, also known as the coupling distance (15,16). The Fréchet distance is informally known as the dog–man distance because it represents the minimum length of leash necessary for a person traversing one curve to walk a dog along another, assuming neither party is allowed to walk backwards. The Fréchet metric is advantageous because it is efficiently computable, while still taking into account the entire course of the curves. In particular, it appeals to an intuitive notion of similarity between methylation signatures in that two curves with similar shape will have a low distance, even if one curve is shifted slightly from the other relative to the TSS (Supplementary Figure S2). Because Fréchet distance is calculated in Euclidean space, a scaling parameter must specify the relationship between the x-axes of the curves, in bp, and the y-axes, in differential methylation level. Intuitively, this parameter controls how far peaks in the y-direction are allowed to slide across the x-axis and still be identified as similar between two genes.
Formally, a methylation signature, or curve, can be described as a continuous mapping 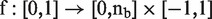 where
where 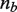 is the fixed length of the region in base pairs. For two curves A and B, the Fréchet distance is defined as follows:
is the fixed length of the region in base pairs. For two curves A and B, the Fréchet distance is defined as follows:
where 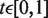 ,
, 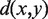 is the Euclidean distance between x and y, and
is the Euclidean distance between x and y, and 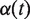 and
and 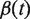 are continuous, monotonically increasing functions from [0,1] to [0,1] such that
are continuous, monotonically increasing functions from [0,1] to [0,1] such that 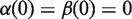 and
and 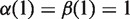 . For any two differential methylation signatures, we calculate similarity using the coupling distance, which can be computed on polygonal curves in quadratic time using a dynamic programming algorithm (16). Each continuous interpolated curve is converted into a polygonal curve by averaging every 10 bp, yielding
. For any two differential methylation signatures, we calculate similarity using the coupling distance, which can be computed on polygonal curves in quadratic time using a dynamic programming algorithm (16). Each continuous interpolated curve is converted into a polygonal curve by averaging every 10 bp, yielding 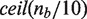 vertices for the nb bases in the target region. For sparsely sampled areas, this fixed resolution greatly limits the discretization error between our discrete Fréchet distance and the continuous version because this error is bounded by the maximum edge length for each pair of curves (17). Averaging every 10 bp allows the algorithm to run faster than sampling every 2 bp. Because the averaging interval is considerably smaller than the width of the Gaussian smoothing kernel, it has little effect on the resulting distances. We compared the results of clustering for several experiments using 10 bp averaging and dinucleotide sampling, and found no differences in the type and number of patterns found for the HMEC-HCC1954 data set. By default, scaling between the x- and y-axes was set such that 2500 bp along the x-direction was equivalent to one unit of differential methylation in the y-direction. We tested a large number of values for this ratio. Manual inspection of resulting clusterings showed that the final set of patterns discovered was not substantially altered despite moderate changes to the scaling ratio.
vertices for the nb bases in the target region. For sparsely sampled areas, this fixed resolution greatly limits the discretization error between our discrete Fréchet distance and the continuous version because this error is bounded by the maximum edge length for each pair of curves (17). Averaging every 10 bp allows the algorithm to run faster than sampling every 2 bp. Because the averaging interval is considerably smaller than the width of the Gaussian smoothing kernel, it has little effect on the resulting distances. We compared the results of clustering for several experiments using 10 bp averaging and dinucleotide sampling, and found no differences in the type and number of patterns found for the HMEC-HCC1954 data set. By default, scaling between the x- and y-axes was set such that 2500 bp along the x-direction was equivalent to one unit of differential methylation in the y-direction. We tested a large number of values for this ratio. Manual inspection of resulting clusterings showed that the final set of patterns discovered was not substantially altered despite moderate changes to the scaling ratio.
Methylation signatures are arranged using unsupervised complete-linkage agglomerative hierarchical clustering based solely on the discrete Fréchet distance between curve pairs. After clustering, we introduce the differential expression data to identify clusters of genes with similar expression differences. Expression data can be obtained through any method, including RNA-Seq and expression array analysis. To focus on genes for which differential methylation and expression could be related, we consider only genes with greater than 2-fold expression change.
We identify clusters from the dendrogram where methylation is significantly associated with expression as follows. For each cluster, we evaluate the likelihood that the observed group of expression values is atypical compared with the distribution of expression values over the entire training set [a dendrogram on ns signatures contains exactly (ns-1) clusters]. We determine significance using a two-sample Kolmogorov–Smirnov test between the set of differential expression values in each cluster and the entire population of expression values. The false discovery rate is controlled at 0.05 using the Benjamini–Hochberg procedure. This estimate of significance is conservative because one sample is a subset of the other.
After all statistically significant clusters are identified, we select a set of nonoverlapping clusters using an iterative algorithm to define the trade-off between grouping similar patterns together versus breaking them into distinct clusters. We seek a balance between the selection of larger clusters that embody more general patterns and the conformity between differential expression values, called purity. The purity of a set of genes is defined as the fraction of genes that have expression change in the same direction as the majority. The full process is described in the Supplementary Methods. We used a minimum purity of 0.85 unless otherwise stated.
Generating a gene list
To produce a list of genes with associated differential methylation and expression, we ran the discovery method on a set of overlapping 5 kb regions centered at a fixed set of locations around the TSS (Figure 6A). The scaling factor between the x- and y-dimensions, minimum cluster purity and all interpolation parameters were the same as previously described. We used 22 regions, spanning an area from [−25 kb, 25 kb] relative to the TSS. The area [−10 kb, 10 kb] relative to the TSS was covered more densely because this was where the majority of relevant differential methylation features were observed by the pattern discovery tool. The leftmost boundaries (in kb, relative to the TSS) were: −25, −20, −15, −10, −9, −8, −7, −6, −5, −4, −3, −2, −1, 0, 1, 2, 3, 4, 5, 10, 15 and 20. For each region, we recorded the set of genes identified as positives (i.e. changing in the same direction as the majority of their cluster). Genes that are identified for at least m regions are added to the final list. Unless otherwise stated, an m of two regions was used.
Figure 6.

Gene lists generated by our method are of markedly greater length and quality than those generated by alternative methods. (A) Schematic of our process for generating gene lists. (B–D) Comparison of gene lists generated using our approach with those from optimized DMR and promoter-based methods for (B) HMEC-HCC1954 WGBS, (C) IMR90-H1 WGBS and (D) MCF7-T47D Methyl-MAPS data. The plot shows the trade-off between the number of genes identified as being associated with differential expression based on their methylation (x-axis) and the quality of the associations (y-axis), which is the fraction of identified genes for which the direction of expression change matches the expected direction based on methylation. Points up and to the right indicate better performance; 50% quality is equivalent to random guessing. Only optimal parameter choices with an inverse correlation between methylation and expression are shown for DMR- and promoter-based approaches (see Supplementary Figure S23 for further information about this optimization). Promoter-based approaches were optimized across both all CpG sites (All Sites) and all significant CpG sites (All Sig. Sites). The minimum number of required windows m was varied in our method from 2 to 5 to show the effects of increased stringency.
RESULTS
Pattern discovery in high-resolution methylation data
We evaluated our technique using three primary and nine additional comparisons (17 total data sets) with high-resolution methylation and RNA-Seq expression data (Supplementary Table S1). The first primary data set was WGBS of nontumorigenic human mammary epithelial cells (HMEC) and breast cancer cells (HCC1954) (6) containing methylation data for 84.7% of genomic CpGs with a coverage level of at least 10 in each sample (Supplementary Table S1, GEO: GSE29127). We focused many of the analyses in this article on this HMEC-HCC1954 data set because it has high coverage and contains examples of all the methylation patterns we discovered (Supplementary Data 1). To examine how the method performed on a lower coverage data set, we examined WGBS data for H1 embryonic stem (ES) cells and IMR90 fetal lung fibroblasts (9) (GEO: GSE16256). While the genomic coverage level of this data was high, the data was sparsely sampled at promoters: <40% of CpGs had coverage of at least 10 (Supplementary Figure S3). By including all CpGs with coverage as low as a single read, the data covered 93.5% of genomic CpGs. However, methylation scores are expected to be less accurate in regions with lower sampling. WGBS data was processed and methylation scores computed as in (6). Analysis was limited to only CpG methylation (complete results are in Supplementary Data 2). Lastly, to validate our findings using data from an alternative experimental method, we generated Methyl-MAPS data from MCF7 and T47D breast cancer cells. Methyl-MAPS uses methylation-sensitive and -dependent restriction enzyme digests followed by high-throughput sequencing to identify methylation levels at individual CpGs (10). Libraries were constructed, sequenced and analyzed as in (10) (see Supplementary Methods for further details). We limited our analysis to sites interrogated by both digests, which included 24.9% of genomic CpGs with coverage of at least five to ensure an adequate number of CpGs with data around each promoter. Expression data for each sample came from poly(A) selected RNA-Seq experiments (see Supplementary Methods for further details). All Methyl-MAPS and RNA-Seq data are available from GEO, accession GSE45337. Complete results are in Supplementary Data 3.
In addition to these three primary comparisons, we applied our method to WGBS and RNA-Seq data comparing H9 ES cells (18,19) to IMR90 cells, H1 cells to four H1-derived differentiated cell types, female adipose-derived stem cells (ADS) to ADS-derived adipocytes and ADS-derived induced pluripotent stem cells (ADS-iPSCs) (19) and primary mouse ES cells to isolated sperm and oocytes (20).
We selected an initial region for the discovery of differential methylation patterns that associate with expression changes based on two criteria. First, average CpG density increases roughly 2 kb upstream and downstream of the TSS, suggesting that sites in this region may have regulatory importance (21). Second, increased variability of differential methylation in CpG island shores, defined as the regions up to 2 kb away from a CpG island, has been linked to differential expression (5). To search for patterns across this entire area, we chose a conservative initial region of 10 kb centered on the TSS. Using a cluster purity threshold of 0.85, we identified 27 clusters that were significantly correlated with differential expression, containing 519 genes, in the HMEC-HCC1954 data set (Figure 2; complete clustering results are in Supplementary Data 1). A cartoon depiction of each of the patterns observed is shown in Figure 3. Applying our method to the H1-IMR90 and MCF7-T47D Methyl-MAPS data sets showed that our method could still identify clusters corresponding to each of the patterns discovered in the higher quality HMEC-HCC1954 data set even with low promoter coverage or substantially reduced sampling (Supplementary Data 2 and 3). It is likely that interpolation and Gaussian smoothing are helpful for analyzing low coverage data. Limiting HMEC-HCC1954 data to only sites probed by Methyl-MAPS showed the data reduction had no impact on the ability to detect each of the identified patterns. As a negative control, we randomly scrambled the expression values for all genes in each data set; any cluster identified as significant was a false positive. For 1000 random permutations, our technique identified a false-positive cluster in 1.7–2.3% of the experiments (Supplementary Table S2).
Figure 2.
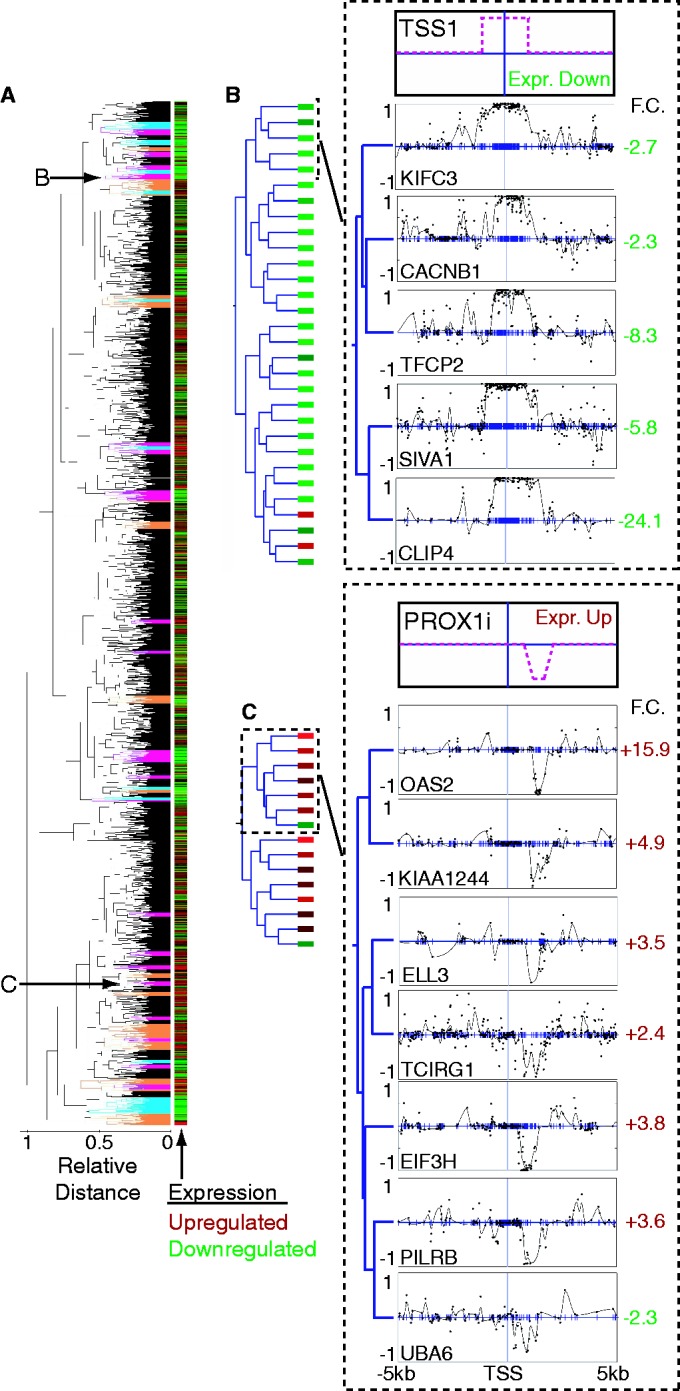
Example of clustering methylation signatures from HMEC-HCC1954 WGBS data. (A) Complete dendrogram for clustering 3566 methylation signatures. Clusters highlighted in orange, magenta and cyan indicate significant clusters with purity of at least 0.75, 0.85 and 0.95, respectively. Subclusters featured in (B, C) are indicated with arrows. A heat map of expression data is plotted in bars alongside the dendrogram. F.C. denotes expression fold change; green indicates downregulation, red indicates upregulation. (B, C) Left side shows the complete cluster with boxes to indicate expression. On the right side, the top panels are cartoons depicting the relevant pattern; the bottom panels show example signatures from subclusters of a significant HMEC-HCC1954 cluster. Clustering was performed on 10 kb regions. (B) Subcluster showing a pattern of methylation increase across the TSS, similar to the classic methylation of a CpG island across the TSS. Patterns from the entire cluster are in Supplementary Figure S4. (C) Subcluster showing a pattern defined by decrease in methylation 3′ of the TSS, similar to a methylation change at a CpG island shore. The entire cluster is in Supplementary Figure S11.
Figure 3.
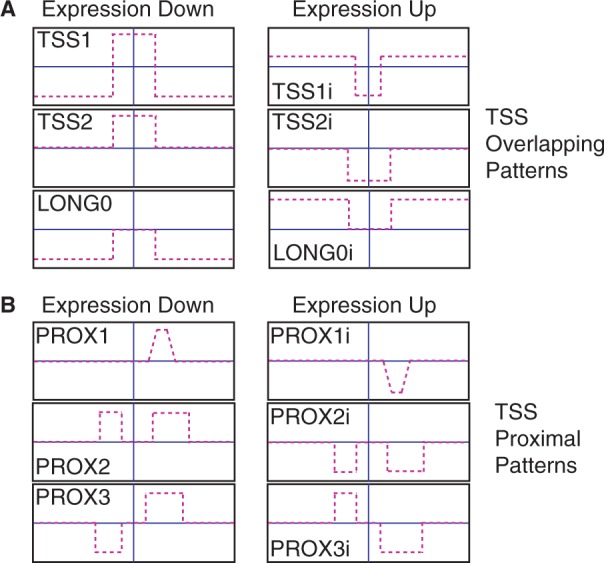
Summary of the diverse patterns found in the data sets analyzed. One version of each pattern is shown to the left with its corresponding inverted pattern to the right. (A) Patterns that overlap the TSS (TSS1, TSS2 and LONG0), and their respective inverted versions (TSS1i, TSS2i, and LONG0i). (B) Patterns proximal to the TSS all include a region of change downstream of the TSS and are separated by their upstream differences. Shown are PROX1, PROX2, PROX3 and their respective inverted versions: PROX1i, PROX2i and PROX3i.
Patterns overlapping the TSS
From the resulting sets of significant clusters, we sought to characterize the common features of the methylation signatures that may be responsible for the observed relationships with expression change. As expected, many clusters contained patterns with a region of strong hyper- or hypomethylation spanning the TSS that negatively correlated with expression change (Figure 2B). Further inspection of HMEC-HCC1954 data revealed several distinct patterns overlapping the TSS. After rerunning our method using methylation signatures based on a 30 kb region centered at the TSS, three distinct patterns emerged (Figure 3A): a hypermethylated region at the TSS surrounded by long hypomethylated domains (TSS1; Figure 4A), a hypermethylated region at the TSS set in a region with invariant methylation levels (TSS2; Figure 2B) and a pattern of long hypomethylated domains, but with no change in methylation at the TSS (LONG0; Figure 4F). The rarest pattern, LONG0, was also observed in the H9-IMR90 (Supplementary Figure S7) and H1-IMR90 (Supplementary Data 2) comparisons. Hypomethylated domains associated with these patterns (TSS1, LONG0) extend up to several Mb in both directions (Figure 4C). In HMEC-HCC1954 data, we found a hypomethylated region at the TSS set in a hypermethylated region (TSS1i; Figure 4B). While not all patterns were observed in every comparison we analyzed, the overall set of patterns was common across all data sets. For instance, MCF7-T47D and H1-IMR90 data sets show an inverted TSS2 pattern (TSS2i; Supplementary Data 2 and 3). IMR90 fibroblasts exhibit a TSS1 pattern relative to H1 stem cells (Supplementary Data 2), although the hypomethylated domains in fibroblasts are much smaller (Figure 4E). It has been suggested that the observed positive correlation between gene-body methylation and expression may be due to similar domains (8). Inspection of individual genes with the LONG0 pattern revealed that promoters were hypomethylated in both samples, leading us to speculate that long hypomethylated domains could contribute to gene silencing, possibly through the recruitment of factors that lead to the formation of repressive chromatin (6). Clusters representing the inverse patterns—a hypomethylated region at the TSS set in either long hypermethylated or invariant regions—were also observed (TSS1i, TSS2i; Figure 4B).
Figure 4.

Analysis of patterns overlapping the TSS. (A, B) Top panel is a cartoon depicting the relevant pattern. Bottom panel shows an example subcluster from an HMEC-HCC1954 cluster. Clustering was performed on 30 kb regions. F.C. denotes expression fold change; green indicates downregulation, red indicates upregulation. (A) Subcluster characterized by hypermethylation at the TSS set in a long hypomethylated domain (TSS1). The entire cluster is in Supplementary Figure S6. (B) Subcluster characterized by hypomethylation at the TSS set in a long hypermethylated domain (TSS1i). The entire cluster is in Supplementary Figure S8. (C) Average differential methylation and CpG density for genes from clusters identified as exhibiting three TSS patterns (TSS1, TSS1i and TSS2). Example signatures for each of the three patterns are shown in parts (A), (B) and Figure 2B. (D) Alu elements and RefSeq genes are depleted in the regions around genes with the TSS1 pattern. No enrichment or depletion of other repeats was found (Supplementary Figure S9). (E) The TSS1 pattern is also found in the H1-IMR90 comparison (Supplementary Data 2). (F) Example subcluster showing the LONG0 pattern from the HMEC-HCC1954 comparison. The entire cluster is in Supplementary Figure S6.
Patterns proximal to the TSS
In addition to patterns with differential methylation across the TSS, we identified multiple clusters in all data sets characterized by a change in methylation downstream of the TSS (Figure 2C, Figure 5A and B). Analysis of all genes in clusters associated with this pattern indicated that it predominantly occurs within 3 kb of the TSS (Figure 5C). This 3′ pattern was observed in several distinct clusters due to variations in other parts of the differential methylation curves (see examples in Figure 5A and B). The TSS-proximal patterns found by our discovery method are in agreement with the variation in methylation found at CpG island shores. Interestingly though, we find no significant association between the proximal methylation patterns and whether promoters are classified as CpG-rich or -poor (Supplementary Figure S10). This may suggest that these proximal methylation changes are not confined to island shores, or it may be due to the fact that the patterns we discover are anchored to the TSS.
Figure 5.

Patterns containing 3′ methylation changes are inversely correlated with expression, independent of changes 5′ of the TSS. (A, B) Top panel is a cartoon depicting the relevant pattern. Bottom panel shows an example subcluster from the default HMEC-HCC1954 clustering on a 10 kb region. Entire clusters for both parts are displayed in Supplementary Figure S11. (A) Subcluster exhibiting genes with a decrease in methylation on the 3′ side of the TSS and an increase in methylation 5′ of the TSS and with a significant increase in expression (PROX3i). (B) Subcluster exhibiting genes with an increase in methylation on both 3′ and 5′ sides of the TSS and with a significant decrease in expression (PROX2). (C) Meta-gene analysis of genes from significant clusters whose dominant pattern was identified as including a 3′-proximal component shows that the 3′ pattern is typically confined to within 3 kb of the TSS. From top to bottom, the three panels depict averages for HMEC-HCC1954, MCF7-T47D and H1-IMR90 data sets. (D) The number of new genes identified increases downstream of the TSS, but not upstream. We started by identifying genes in significant clusters for a 5 kb region centered at the TSS. We iteratively moved the 5 kb-wide region upstream of the TSS and identified all genes in significant clusters not found previously. This process was repeated for each of the region positions used by the gene list tool. The entire process was then repeated for downstream of the TSS.
Although some clusters with 3′ patterns also displayed methylation change upstream of the TSS, no independent relationship was observed between a 5′ pattern and differential expression (Figure 5A and B). Differential methylation downstream of the TSS was consistently linked with expression change, regardless of upstream hyper- or hypomethylation. To further probe whether distinct correlative patterns occur upstream of the TSS, we ran our method using signatures defined on the region from −5 kb to the TSS. The majority of identified clusters were characterized by changes in methylation at the TSS. Genes in clusters with methylation changes 5′ of the TSS often had correlative 3′ changes as well. We found no clusters in any of the data sets that supported a convincing association between 5′ changes and expression. Examining 5 kb regions shifted downstream of the TSS discovered more patterns than those upstream of the TSS, consistent with the existence of the 3′ pattern (Figure 5D) and absence of a 5′ pattern. From the cumulative evidence, we speculate that CpG island shore-like regions upstream of the TSS may not be independently associated with expression changes. Furthermore, while we observed that differential methylation patterns across the TSS were generally associated with gene silencing, 3′ methylation patterns correlated more often with downregulation than complete silencing (Supplementary Figure S12). 3′ hypermethylation events have previously been shown to affect expression (22), which supports the idea that the 3′ changes we observe could be potentially functional.
Genomic features associated with particular DNA methylation patterns
Gene ontology and expression signature analysis (23) of the genes found to have correlated differential methylation and expression showed that there was enrichment for cancer-related genes in HMEC-HCC1954 data, while there was enrichment for genes associated with differentiation in H1-IMR90 data. Long hypomethylated and partially methylated domains in cancer cells were previously observed to have distinct sequence contexts (6,7). We find that genes with the TSS1 pattern are associated with a depletion of CpG density, Alu elements and gene density relative to all gene promoters (Figure 4C and D). These depletions are observed in regions ranging from 10 kb up to 0.5 Mb from the TSS. In summary, genes exhibiting the TSS1 pattern have the same distinct sequence properties as genes found in long hypomethylated domains (7).
There has been significant interest in addressing whether specific sequences direct or inhibit methylation at particular promoters or CpG islands (5,24–26). One possibility is that different sequences may direct different methylation patterns. A preliminary search for motifs associated with each discovered pattern did not find a significant enrichment for any novel or known motifs associated with one pattern more than another. In addition, we find no significant association between each of the different observed methylation patterns and whether promoters are classified as CpG-rich or CpG-poor (Supplementary Figure S10).
Quantifying the sensitivity of pattern discovery
Because the true patterns of differential methylation that correlate with expression change are unknown, an obvious question is whether we have detected all correlative methylation patterns in the data set. To address this issue and quantify the method’s ability to recover genes from known correlative patterns, we created a model to simulate differential methylation signatures and expression values. A complete description of the methods for simulated data is in the Supplemental Methods.
Simulated genes with a predefined correlation between methylation and expression were added into two kinds of background data sets: randomly generated simulated genes with no correlation with expression (Supplementary Figure S13–S15) and real data. Using wholly simulated data we explored the types of patterns our method could discover. We successfully detected a variety of introduced patterns including peaks of differential methylation with fixed widths and locations relative to the TSS (Supplementary Figure S16), peaks with varying location (Supplementary Figure S17) and peaks with different variances in height (Supplementary Figure S18). More complex patterns were more easily detected, presumably because highly constrained curves are more likely to have similar Fréchet distances.
We also explored the method’s ability to discover new patterns in a background of real methylation data. We introduced three simulated patterns (Supplementary Figure S19) into HMEC-HCC1954 WGBS data. We varied the number of genes added, and measured both the ability to discover the patterns and the number of genes correctly identified (Supplementary Figure S20). We found that the patterns were easily discoverable when at least 50–100 simulated genes were introduced, depending on the particular simulated pattern. Performance was impacted when one of the simulated patterns was too similar to the existing patterns (Supplementary Figure S21). Furthermore, we found that the fraction of simulated genes required to reliably detect a pattern decreased as the number of genes in the data set increased (Supplementary Figure S22). This suggests that one could detect rare patterns by simply concatenating multiple data sets.
Enumerating genes with correlative methylation signatures
Generating a list of genes for which expression and methylation changes are potentially linked is a primary interest of any genome-wide methylation profiling experiment. The method described above is tuned to discover patterns, not to create a gene list. Individual genes within good clusters will sometimes be false positives, and other genes will be excluded because their clusters do not meet the high purity threshold. To produce a better gene list, we executed our method on a set of overlapping 5 kb regions centered at a fixed set of locations around the TSS (Figure 6A). Comparison of gene lists from H1-IMR90 replicate WGBS data shows good concordance between replicates. To determine the extent to which genes are incorrectly included owing to the creation of errant clusters, we randomly scrambled the expression values in the HMEC-HCC1954 dendrogram. For 1000 such experiments using default clustering parameters, only seven experiments returned any false-positive genes: six reported a single false positive and a seventh returned two. We also tested the ability of the gene list method to recover introduced simulated genes (see ‘Quantifying the sensitivity of pattern discovery’ section above). For several simulated patterns, we found that the resultant gene lists included nearly all simulated genes when 50–100 were present (Supplementary Figure S20).
Comparison to other approaches
We compared the quality of the gene lists produced by our approach to lists constructed by two commonly used methods. For the DMR approach, regions of differential methylation are defined between two samples using a sliding window. DMRs are coupled to a particular gene using a cutoff for the distance between the DMR and the gene’s TSS (7,9). For the promoter-based approach, a fixed window around each gene’s TSS defines the gene’s promoter. If methylation changes substantially within this window, the gene is labeled as differentially methylated (10,27). We optimized DMR- and promoter-based approaches for each data set using 69 360 and 6174 parameter choices, respectively (Supplementary Figure S23 and Supplementary Table S3), while using a single common set of parameters for our approach across all data sets.
Judging the quality of the gene lists is difficult because there is no experimental gold standard data set for which the relationship between methylation at specific CpG sites and expression is well known for all genes. A correlation coefficient has often been used to quantify the association between methylation and expression. When applied to a list of genes for which methylation is predicted to associate with expression change, however, the correlation coefficient only judges one part of the method’s performance. For example, by varying its parameters, the DMR method can produce short gene lists with strong correlations or long lists with weak correlations. To compare methods, we directly examine the trade-off between the total number of differentially expressed genes identified as potentially correlated versus the fraction of identified genes that are actually correlated in the predicted direction (Figure 6). This trade-off is somewhat analogous to comparing the rate of total positives to the rate of true positives, with the true-negative and false-negative rates being unknown. On the basis of these criteria, our approach clearly outperforms DMR- and promoter-based methods. For example, consider the HMEC-HCC1954 data (Figure 6B). When the DMR method is examined at a level where it correctly associates the direction of expression change 92% of the time, it returns only 26 genes (0.7% of all differentially expressed genes). Our approach produces a list of 461 genes (13%) at a correct association rate of 95%. With less restrictive criteria, the DMR method gives a list of 717 genes (20%) at a correct association rate of 71%. Our technique gives a list of roughly the same size (761 genes) at a correct association rate of 93%.
Table 1 presents results from each of the different data sets we analyzed and includes data from primary cells and cell lines. All analyses were performed using the default parameters. These results imply that prior approaches have underestimated the strength of the relationship between differential methylation and expression. With a better model of the underlying patterns of methylation change, it is clear that methylation and expression data are highly associated.
Table 1.
Summary of the numbers of genes identified by the gene list tool for each comparison
| Sample Comparison | Differentially expressed genes | CpGs with methylation difference > 0.3 | Genes with correct association | Genes with incorrect association | Fraction of genes with correct association |
|---|---|---|---|---|---|
| HMEC - HCC1954 | 3584 | 6 979 726 | 1150 | 118 | 91% |
| H1 - IMR90 | 2571 | 7 210 721 | 608 | 68 | 90% |
| MCF7 - T47D | 3740 | 1 973 564 | 664 | 57 | 92% |
| ADS - ADS-Adipose | 2937 | 332 489 | 138 | 15 | 90% |
| ADS - ADS-iPSC | 3803 | 5 917 387 | 1124 | 93 | 92% |
| H1 - H1-Mesenchymal | 3714 | 2 170 686 | 559 | 40 | 93% |
| H1 - H1-Neural Progenitor | 2546 | 829 297 | 79 | 3 | 96% |
| H1 - H1-BMP4 | 4103 | 737 003 | 65 | 3 | 96% |
| H1 - H1-Mesendoderm | 2353 | 765 051 | 59 | 5 | 92% |
| H9-IMR90 | 5875 | 7 669 782 | 605 | 58 | 91% |
| oocyte - ES cell (mouse) | 4727 | 1 204 883 | 334 | 25 | 93% |
| sperm - ES cell (mouse) | 4580 | 4 364 748 | 1027 | 104 | 91% |
Gene lists for low coverage data sets
We next sought to examine how our approach performed with suboptimal data. Our technique returned similar results for the low coverage IMR90-H1 comparison, which has no minimum coverage cutoff, while DMR- and promoter-based methods struggled (Figure 6C). We also ran experiments on the HMEC-HCC1954 sample pair to test the method’s performance on downsampled data. First, we removed raw sequence reads at random, finding that WGBS data obtained with an average coverage as low as seven resulted in little loss in our method’s ability to identify genes (Supplementary Figure S24A). We also removed methylation scores at random from the mapped data, and found that 94.5% of genes were still detected when 50% of the CpGs were removed (Supplementary Figure S24B). Additionally, our method outperforms the optimized DMR- and promoter-based methods for the MCF7–T47D sample pair collected using Methyl-MAPS, which contained 37.4% of the CpG sites from the 10 kb region centered at the TSS. These results suggest that our method is robust in the context of missing or low coverage data.
DISCUSSION
A summary of the patterns discovered in the data sets analyzed is presented in Figure 3. One primary advance of our approach is in its use of spatial information. Several of the patterns we found demonstrate the need for using spatial information about specific CpG sites when trying to connect differential methylation to expression change. For instance, the LONG0 pattern is positively correlated with expression, while the 3′-hypomethylation pattern, commonly seen in conjunction with a 5′-hypomethylation event (PROX2i), is negatively correlated. Given methylation marks from one of these two cases, it is clear that the spatial information about specific CpG locations is necessary to successfully determine the direction of expression change. This may help explain an observation made during an analysis of 82 methylation data sets from human tissues and cell lines using reduced representation bisulfite sequencing (12). The authors observed that methylation changes in CpG island sites greater than 2 kb downstream were sometimes positively correlated with expression and sometimes negatively correlated. Based on our findings, one explanation for their observation is that they are observing a mixture of genes with LONG0 and 3′ patterns.
We also find that despite variation throughout the vicinity of a gene’s promoter, expression change is often well correlated with only the methylation changes in a confined area relative to the TSS. This point is well supported by the many examples of the 3′-proximal pattern, which is seen with a variety of methylation activity upstream including hypermethylation, hypomethylation or little activity (Figure 3B). DMR-based methods are generally tuned to find wider regions than what we observe in our 3′-proximal clusters, to better identify genes with classical differential methylation at a CpG island promoter (e.g. TSS2 and TSS2i). While these methods can be tuned to locate narrower DMRs, this would result in more cases where contradictory DMRs exist near the TSS (e.g. one hypermethylated DMR and one hypomethylated DMR, such as PROX3 in Figure 3B). In these cases, there is no obvious way to decide which DMR may be influencing transcription without introducing pre-learned spatial information. As another example, the DMR approach has difficulty discriminating between the LONG0 pattern that is associated with a decrease in expression and the PROX1i and PROX2i patterns that are associated with an increase in expression. When set to find long regions downstream of the TSS, DMRs can be identified with positive correlation between expression and methylation. When set to find short regions, DMRs can be identified with negative correlations. However, no single set of DMR parameters can easily simultaneously capture each differential methylation pattern and its correlation with expression. These examples also underscore a fundamental limitation of the DMR method: it cannot be used to discover new patterns, but can only search for a limited set of relationships that are already known to exist.
Spatial information has proven useful to the analysis of other epigenetic data sets as well. Recently, a model considering the spatial locations of chromatin marks was used to train a support vector machine to predict chromatin signals at transcription factor binding sites (28). The authors divided each fixed region into 50 bp bins, each of which was used as a feature in a vector for classification. The success of this approach demonstrates the potential of spatial models to better capture the nature of the epigenetic data than is possible with a simple window-based approach. However, for analyzing DNA methylation, it is important to account for the relationship between such bins rather than treating them as independent features. For instance, in Figure 2C, the topmost and bottommost example curves have a similar—but slightly shifted—3′-proximal decrease in methylation and are both upregulated. If we used predefined nonoverlapping bins here, they would either be so narrow that the top peak and bottom peak lie in different bins, or so wide that the salient feature of the curve is diminished. Because our shape-similarity approach tolerates some movement of peaks in the x-direction, features such as these are naturally associated with one another.
Finally, our findings have implications for how DNA methylation should be assayed to preserve all potentially useful information. Using downsampled WGBS data and Methyl-MAPS data we demonstrated that we can discover the full set of patterns from Figure 3 without needing values at all CpG sites, as long as they are removed in a relatively unbiased manner (Supplementary Figure S24, Supplementary Data 3). However, information from some sites may be more informative than others. Many experimental methods attempt to assess a gene's methylation state by restricting analysis to only CpG-rich regions, or to only a handful of CpG sites in and around the promoter. It is unclear whether such techniques measure methylation at the correct sites to discriminate the full spectrum of methylation patterns that correlate with transcriptional changes. As additional single-base resolution genome-wide DNA methylation data sets become available, we can explore which subsets of individual CpGs most inform promoter methylation patterns and thus need to be experimentally measured to accurately assess changes in a gene’s methylation state.
CONCLUSIONS
Our findings suggest that characterizing gene promoters simply as ‘methylated’ or ‘unmethylated’ is insufficient. By considering the entire set of methylation changes near the promoter, we found and described a variety of methylation patterns that correlate with expression change. The power of our method is its ability to discover and separate distinct patterns without any prior knowledge about existing relationships, which cannot be accomplished with contemporary approaches. This allows us to use the full potential of unbiased genome-wide profiling of DNA methylation to reveal previously unknown information about methylation’s functional role. Although we applied our method on regions of various widths around the TSS, all correlative patterns except those associated with the long hypomethylated domains were found within 5 kb of the TSS (Figure 5D). Interestingly, all patterns found in the cancer cell data sets were also found in the ES and iPSC cell data sets. While 5′ upstream patterns are observed, these appear to occur due to correlations with the 3′ downstream patterns. By appropriately capturing the diverse set of methylation patterns that exist, we observe a high level of association between changes in a gene’s methylation state and changes in its expression.
A strength of the technique described here is its potential for expansion to examine more general epigenetic modifications. We have confined our analysis to data from genome-wide single-base resolution methods such as WGBS or Methyl-MAPS. However, similar approaches could be used to analyze the relationships between expression and other epigenetic patterns, such as 5-hydroxymethylcytosine, non-CpG methylation, and histone modifications. All expression data used in this study came from single-end short read RNA-Seq experiments. If long-read paired-end RNA-Seq data were available, it could easily be used with our method to understand alternate-promoter and isoform-specific methylation patterns. Considering the explosion of experiments aiming to profile epigenetic landscapes, this method represents a valuable tool for exploring the relationships between changes in epigenetic patterns and transcription both in normal cellular function and in human disease.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–4, Supplementary Figures 1–24, Supplementary Methods, Supplementary Data 1–3 and Supplementary References [29–32].
FUNDING
National Institutes of Health [NIH R00 CA127360, NIH R21 LM011199 to J.R.E.]; the Department of Defense [W81XWH-11-1-0401 to J.R.E.]; a Siteman Cancer Center Breast Cancer Program career development award [to J.R.E.]. Funding for open access charge: The Internal Fund from the Department of Medicine, Washington University in St. Louis.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Yu-Tseh Chi and Tao Ju for helpful discussions about curve comparison algorithms. The authors also thank Christopher Schlosberg for helpful discussions and reading of this manuscript.
REFERENCES
- 1.Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J, et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320–331. doi: 10.1101/gr.101907.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene. 2002;21:5400–5413. doi: 10.1038/sj.onc.1205651. [DOI] [PubMed] [Google Scholar]
- 3.Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 2011;12:529–541. doi: 10.1038/nrg3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl Acad. Sci. USA. 2006;103:1412–1417. doi: 10.1073/pnas.0510310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet. 2009;41:1350–1353. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hon GC, Hawkins RD, Caballero OL, Lo C, Lister R, Pelizzola M, Valsesia A, Ye Z, Kuan S, Edsall LE, et al. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 2012;22:246–258. doi: 10.1101/gr.125872.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, Wen B, Wu H, Liu Y, Diep D, et al. Increased methylation variation in epigenetic domains across cancer types. Nat. Genet. 2011;43:768–775. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berman BP, Weisenberger DJ, Aman JF, Hinoue T, Ramjan Z, Liu Y, Noushmehr H, Lange CP, van Dijk CM, Tollenaar RA, et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat. Genet. 2012;44:40–46. doi: 10.1038/ng.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edwards JR, O'Donnell AH, Rollins RA, Peckham HE, Lee C, Milekic MH, Chanrion B, Fu Y, Su T, Hibshoosh H, et al. Chromatin and sequence features that define the fine and gross structure of genomic methylation patterns. Genome Res. 2010;20:972–980. doi: 10.1101/gr.101535.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bock C. Analysing and interpreting DNA methylation data. Nat. Rev. Genet. 2012;13:705–719. doi: 10.1038/nrg3273. [DOI] [PubMed] [Google Scholar]
- 12.Varley KE, Gertz J, Bowling KM, Parker SL, Reddy TE, Pauli-Behn F, Cross MK, Williams BA, Stamatoyannopoulos JA, Crawford GE, et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013;23:555–567. doi: 10.1101/gr.147942.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet. 2006;38:1378–1385. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansen KD, Langmead B, Irizarry RA. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 2012;13:R83. doi: 10.1186/gb-2012-13-10-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alt H, Godau M. Computing the Frechet distance between two polygonal curves. Int. J. Comput. Geom. Appl. 1995;5:75–91. [Google Scholar]
- 16.Eiter T, Mannila H. Tech. Report CS-TR-2008-0010. San Antonio, TX: University of Texas at San Antonio; 1994. Computing discrete Fréchet distance. [Google Scholar]
- 17.Aronov B, Har-Peled S, Knauer C, Wang Y, Wenk C. Proceedings of the 14th conference on Annual European Symposium - Volume 14. Switzerland: Springer-Verlag, Zurich; 2006. pp. 52–63. [Google Scholar]
- 18.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, Antosiewicz-Bourget J, O'Malley R, Castanon R, Klugman S, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kobayashi H, Sakurai T, Imai M, Takahashi N, Fukuda A, Yayoi O, Sato S, Nakabayashi K, Hata K, Sotomaru Y, et al. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 2012;8:e1002440. doi: 10.1371/journal.pgen.1002440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Majewski J, Ott J. Distribution and characterization of regulatory elements in the human genome. Genome Res. 2002;12:1827–1836. doi: 10.1101/gr.606402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Appanah R, Dickerson DR, Goyal P, Groudine M, Lorincz MC. An unmethylated 3' promoter-proximal region is required for efficient transcription initiation. PLoS Genet. 2007;3:e27. doi: 10.1371/journal.pgen.0030027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Culhane AC, Schroder MS, Sultana R, Picard SC, Martinelli EN, Kelly C, Haibe-Kains B, Kapushesky M, St Pierre AA, Flahive W, et al. GeneSigDB: a manually curated database and resource for analysis of gene expression signatures. Nucleic Acids Res. 2012;40:D1060–D1066. doi: 10.1093/nar/gkr901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Das R, Dimitrova N, Xuan Z, Rollins RA, Haghighi F, Edwards JR, Ju J, Bestor TH, Zhang MQ. Computational prediction of methylation status in human genomic sequences. Proc. Natl Acad. Sci. USA. 2006;103:10713–10716. doi: 10.1073/pnas.0602949103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bock C, Paulsen M, Tierling S, Mikeska T, Lengauer T, Walter J. CpG island methylation in human lymphocytes is highly correlated with DNA sequence, repeats, and predicted DNA structure. PLoS Genet. 2006;2:e26. doi: 10.1371/journal.pgen.0020026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feltus FA, Lee EK, Costello JF, Plass C, Vertino PM. Predicting aberrant CpG island methylation. Proc. Natl Acad. Sci. USA. 2003;100:12253–12258. doi: 10.1073/pnas.2037852100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arvey A, Agius P, Noble WS, Leslie C. Sequence and chromatin determinants of cell-type-specific transcription factor binding. Genome Res. 2012;22:1723–1734. doi: 10.1101/gr.127712.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rollins RA, Haghighi F, Edwards JR, Das R, Zhang MQ, Ju J, Bestor TH. Large-scale structure of genomic methylation patterns. Genome Res. 2006;16:157–163. doi: 10.1101/gr.4362006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.