

Abstract
Platelets are chief effector cells in hemostasis. In addition, they are multifaceted inflammatory cells with functions that span the continuum from innate immune responses to adaptive immunity. Activated platelets have key “thromboinflammatory” activities in a variety of vascular disorders and vasculopathies. Recently-identified inflammatory and immune activities provide insights into the biology of these versatile blood cells that are directly relevant to human vascular diseases.
Keywords: Platelets, Inflammation, Immune Responses, Vasculopathy, Vessel Diseases
“For blood clot turns into pus and excites inflammation, and prevents agglutination of the wound.”
– Cornelius Celsus 1
Introduction
Platelets are highly specialized for hemostasis, and are first responders in vascular injury and endothelial disruption. This is traditionally the most well-known function of platelets, and platelet-dependent hemostasis is intensely studied 2. Nevertheless, platelets are also inflammatory effector cells, and have activities across the spectrum from acute inflammation to adaptive immunity 3-6. There is evidence that the inflammatory specializations of platelets – like their specializations for hemostasis – are evolutionarily-driven adaptations that facilitate host defense, tissue repair, and protection against pathogens5, 6. Furthermore, platelets provide key, central links between the inflammatory and hemostatic limbs of the host defense system, contributing to integration of vascular and wound repair and maintenance of tissue integrity 5, 7, 8. The inflammatory activities of platelets also contribute to many organ-localized and systemic diseases with inflammatory components, however, based on existing evidence and reasonable inference where observations are incomplete 6. It is intriguing that the ancient author Celsus, writing in Rome in the first century A.D., described an apparent link between blood clots and pathologic inflammation (see introductory quote)1, although platelets had not yet been discovered in his time 9 and delineation of their activities in vascular diseases and inflammatory syndromes is much more recent.
Human platelets respond with multiple innate immune effector activities when signaled by endogenous agonists generated or released in hemostasis and inflammation, by microbial toxins, or by many microbial pathogens. Key innate immune activities of platelets include release of chemokines, cytokines, and other mediators and interactions with endothelial cells, monocytes, and neutrophils, which are also critical responders in innate immunity and acute inflammation3-6, 10-12 (Figure 1, Online Table I). (In this review we use “neutrophil” interchangeably with “polymorphonuclear leukocyte” and “PMN”, although eosinophils and basophils are sometimes classified as polymorphonuclear leukocytes as well. In addition, however, activated human platelets operate across the immune continuum, and have a substantial armamentarium of adaptive immune activities3-6. These include synthesis and/or release of pleiotropic immune mediators including interleukin-1β, CD40 ligand (CD40L), and RANTES, and interaction with lymphocyte subclasses and dendritic cells (reviewed in 5, 6).
Figure 1.

The repertoire of activated platelets includes diverse mechanisms and molecular pathways that mediate inflammation and immune modulation in addition to hemostasis. Most functional responses shown contribute to both thrombosis and inflammation and to “thromboinflammatory” activities in vascular diseases. Each is discussed in the text.
Newly-recognized inflammatory activities of platelets, including toll-like receptor-mediated responses and the ability to trigger neutrophil extracellular trap (NET) formation (Figure 2), continue to emerge. Here we emphasize some of the recently-discovered and rapidly-evolving aspects of the biology of platelets in the immune continuum and the spectrum of inflammation, and provide an overview of inflammatory activities of platelets in vascular diseases and vasculopathies. Because of space limitations we telegraphically profile many topics that have been previously reviewed in comprehensive fashion. We also include additional online supplemental information to expand some of these issues.
Figure 2.

Human neutrophils form NETs in response to signals from activated platelets in addition to other stimuli (A). NETs have antimicrobial functions, but also mediate inflammatory vascular and tissue injury. Human β-defensin1 immunocaptured from platelets (A) and recombinant hBD-1 (B) trigger NET formation, detected by extracellular DNA staining. Reproduced from 99 with permission of the publisher.
The Inflammatory Repertoire of Platelets
Thrombosis and Inflammation: Linked Outcomes of Platelet Activation
Many functions of activated platelets that are induced by classic hemostatic agonists such as thrombin and ADP and that, for many years, were thought to exclusively mediate hemostasis and thrombosis are now known to also contribute to inflammatory and immune responses (reviewed in 5, 13) (Figure 1; Online Table I). “Traditional” prothrombotic activities that are also proinflammatory include adhesion and aggregation 14, display of surface molecules that favor generation of thrombin15, binding of fibrinogen 16, 17, and interaction with the fibrin mesh of clots17 (Campbell R, Weyrich AS, manuscript in preparation). Thrombin and fibrinogen are key proinflammatory factors in addition to being essential components of the coagulation cascade18, 19. Activated platelets can also synthesize tissue factor (reviewed in 5), which has complex roles in inflammation in addition to its critical procoagulant activities20.
Platelets likely evolved from multifunctional defensive cells with the capacity to immobilize and kill pathogens, seal wounds, and repair tissues3-5, 21. “Modern” procoagulant and proinflammatory specializations of platelets, and the ability to link inflammation and hemostasis and sense pathogens5, 21, 22, may thus have ancient origins in host defense. Nevertheless, “thromboinflammatory” activities of platelets contribute to a large number of vascular diseases.
Platelet Receptors, Activation Pathways, and Integrins
Platelets have a diverse array of surface receptors that are linked to intracellular signaling cascades and that trigger cellular activation and key functional responses23, 24. Many induce and/or regulate hemostatic functions of activated platelets 23-27 and are targets for anti-platelet therapies in thrombotic diseases 26, 28, 29. Many also link activation of platelets to proinflammatory responses5, 6, in addition to triggering prothrombotic signaling cascades 23-29. G protein-coupled receptors (GPCR) and their intracellular pathways24, 26 are chief examples (Online Table I), but a variety of receptor-mediated pathways of platelets induce both inflammation and thrombosis 5, 23. Inside-out signaling, also termed activation, of integrin αIIbβ3, a central downstream effector response that is essential for normal hemostasis16, mediates pathologic thrombosis25, 28, 29 and can amplify inflammation5, 6. Activation of integrin αIIbβ3 is induced by a signaling cascade terminating in RAP1, Kindlin 3, and Talin24, 26, 30, 31. Several other integrins also mediate adhesive activities of activated platelets17, 23, 27 and may participate in inflammatory interactions5, 31. Integrin signaling is bidirectional, and components of the inside-out cascade that mediates activation of integrin αIIbβ3 16, 30, 32, including Kindlin 333 and RAP134, participate in outside-in signaling. Outside-in signaling of αIIbβ3 triggers or amplifies functional responses of platelets that have both hemostatic16, 24, 32 and inflammatory5, 6 consequences.
Platelet Immunoreceptors and Toll-Like Receptors
Some platelet surface receptors have been termed immunoreceptors because of their ligand recognition specificities and/or features of their molecular structures35. Platelet Fc receptors (FcγRIIA, FcεRI, FcαRI), glycoprotein VI (GPVI), and C-type lectin-like receptor 2 (CLEC-2) are among these5, 6, 23, 35. Platelet Fc receptors bind immunoglobulins and immune complexes, providing direct mechanisms for immune recognition and signaling 23, 35, 36. GPVI, which is uniquely expressed by platelets, is a major receptor for collagen with signaling activities in hemostasis and inflammation 6, 37 (Online Table I). CLEC-2, like GPVI, has emerging activities in platelet-dependent hemostasis, vascular integrity, and inflammation, including regulation of lymphangiogenesis in mouse models6, 37.
Human platelets, and platelets and thrombocytes from other mammals and vertebrates, express TLRs (reviewed in 4-6, 23, 38, 39). Intracellular and cell surface TLRs detect microbes and microbial products and transmit signals to inflammatory and immune transduction pathways. TLRs also recognize endogenous, host-derived “danger-associated molecular patterns” (DAMPs), in addition to microbial pathogen-associated molecular pattern (PAMP) motifs 40-42. The presence of TLRs on platelets is persuasive evidence that they evolved as immune effector cells and provides a molecular basis for their activities as pathogen sensors 4, 5, 21, 38, 39. Together with other classes of receptors with immune signaling activities5, 23, 35, platelet TLRs provide a molecular basis for intravascular immune surveillance43 and for pathologic inflammatory and immune activities4-6, 44. As with “prothrombotic” receptors24-26, platelet TLRs may be engaged in parallel or in sequence with other receptors and may act cooperatively5, 6, 36. Megakaryocytic cell lines and megakaryocytes also express TLRs38, indicating that the TLR signaling may regulate thrombopoiesis and other megakaryocyte functions in the marrow4 and/or lungs8.
Human and mouse platelets express multiple TLRs at the messenger RNA (mRNA) and/or protein levels4-6, 38, 39. TLR4 is the most extensively studied 6, 21, 23. Early reports indicated that platelets from several species bind or respond to lipopolysaccharide (LPS), suggesting the presence of one or more receptors6. In vitro experiments with isolated human platelets indicated that LPS amplifies responses triggered via Fc receptors36 and that the endotoxic backbone of LPS, Lipid A, can directly induce aggregation and serotonin secretion45. After molecular characterization of TLR4 – the major immune receptor for LPS 42, 46 – its expression was reported on human and murine platelets47-50, although it was not detected in one early study51. In vitro experiments also demonstrated binding of LPS to platelet TLR450, 52, 53. Binding of LPS to platelets may increase their consumption and phagocytosis in hemolytic uremic syndrome and autoimmune thrombocytopenic purpura 52, 54. Variable changes in surface expression of TLR4 on freshly-isolated platelets in response to thrombin have been reported39, 50, 52. One study indicated that binding of LPS to freshly-isolated, washed human platelets increases with thrombin stimulation and that TLR4 and P-selectin on the surfaces of activated human platelets cooperatively bind LPS52. Surface display of TLR4 on human platelets in stored concentrates was reported to decrease after activation48.
An early report found no evidence for direct activation of human platelets by TLR4 or for modulation of GPCR-triggered platelet activation responses by LPS, and concluded that TLRs may be vestigial molecular remnants bequeathed to platelets by megakaryocytes49. Subsequent studies of platelet responses to LPS in vitro describe contradictory results that are both perplexing and intriguing (Supplementary Table 2). In some experiments LPS did not induce typical activation responses of human or murine platelets, including shape change, aggregation, or surface translocation of P-selectin and did not enhance activation triggered by classic agonists 47, 53, 55, 56 – similar to results in the previous survey49. In others, however, LPS potentiated activation responses triggered by subthreshold concentrations of thrombin or collagen, and directly induced actin cytoskeleton rearrangement, aggregation, binding of PAC-1 antibody (which recognizes activated integrin αIIbβ3), fibrinogen binding, degranulation of alpha and dense granule markers, display of CD40 ligand (CD40L), adhesion of platelets to cultured endothelial monolayers, platelet-neutrophil interaction and neutrophil extracellular trap (NET) formation, and splicing of pre-mRNAs coding for IL-1β and tissue factor and synthesis of these protein products48, 52, 53, 56-59. (Most of these functional activities will be discussed in later sections of this review.)There is as yet no clear explanation for these discrepancies, but some of the factors may include: variable potencies and structural features of LPS types from different bacterial sources 45, 52, 58; delayed and more protracted responses of platelets to TLR4 engagement compared to activation via classic “prothrombotic” receptors (Online Table I)57; use of isolated, washed platelets versus platelet-rich plasma (PRP) or platelets supplemented with plasma, serum, or recombinant or purified CD14 and LPS binding protein (LBP) in key experiments52, 57-59. CD14 and LBP are blood proteins that facilitate recognition of LPS by TLR4 42, 46, and are likely critical in the efficiency of binding of LPS to platelet TLR4 as with other cells that express this TLR family member. As examples, platelets in plasma but not washed platelets adhered to LPS-treated cultured endothelium52, the concentration of LPS required to potentiate collagen-induced platelet aggregation was 5-10 fold lower in PRP than in washed platelets58, and rCD14 and LBP facilitated pre-mRNA splicing by LPS-treated platelets 57, 59.
The signaling pathways that mediate responses of platelets to LPS have not been comprehensively defined, although key components that are required for TLR4 signaling in other cells are present. In addition to TLR4, MD2 – a lipoprotein binding protein family member that associates with TLR4 near its extracellular N-terminal domain23 – is expressed on human platelets58, 60. CD14 has also been detected on the plasma membranes of resting human platelets in some, but not all studies, and may be primarily soluble CD14 derived from plasma52, 58, 59. Isolated human platelets or platelets in PRP did not bind anti-CD14 antibody in the resting state or when stimulated with thrombin, but binding of the antibody to platelets in PRP increased in response to pretreatment with LPS52. At the intracellular face, MyD88 – which is essential for downstream signaling by most TLRs 42 – is present in human platelets58, 60, and small molecule inhibitors of MyD88 interrupt LPS signaling59. Further, MyD88, in addition to TLR4, is required for LPS signaling of platelets based on studies of mice with mutations in TLR4 that make them hyporesponsive or unresponsive to LPS, TLR4−/− mice, or MyD88−/− mice47, 50, 52, 58, 59. None of these experiments examined platelet-specific genetic modifications of TLR4 or MyD88. Distal components of the canonical MyD88 pathway42, including TIRAP, IRAK1 and 4, and TRAF6, are also present in human platelets based on experimental inhibition of their activities and other approaches 22, 59, 60. Downstream from the MyD88 cascade there is evidence for rapid phosphorylation of JNK and AKT (within minutes) that is associated with more delayed functional responses59. A TLR4/MyD88-dependent activating pathway involving cyclic GMP, protein kinase G, and nitric oxide synthase has also been described, and it was suggested that this mechanism can inhibit or activate platelets depending on the concentrations of LPS 58. This might explain inhibition of platelet activities by LPS in some earlier reports61, 62. Taken together, the studies reported to date suggest that there are gaps in our understanding of signaling cascades triggered by platelet TLR4, and that novel pathways may be involved. It is unknown if recently-identified platelet signaling cascades such as those involving mammalian target of rapamcyin (mTOR) 63-65, ADP-ribosylation factor 6 (Arf6-GTP)66-68, or others24 mediate LPS-triggered responses.
In vivo experiments also indicate that LPS activates platelets, although complex signaling interactions involving parallel activation of myeloid leukocytes and endothelial cells occur as well. Challenge of mice or rabbits with LPS causes thrombocytopenia and time-dependent accumulation of platelets in the lungs and liver with the magnitude influenced by the microbial origin of the LPS 47, 50, 53, 69-73. Platelet accumulation in microvessels of mice injected with LPS has been demonstrated using spinning-disc confocal microscopy and transgenic mice expressing endogenously-labeled integrin subunit αIIb or in vivo labeling of platelets with anti-α2 integrin subunit (CD49) antibody73. In other models, LPS induced accumulation of platelets in mesenteric vessels of rats 74 and cremasteric veins of mice 55, 73, and promoted platelet-dependent thrombus formation in ferric chloride-injured carotid arteries of mice58. These responses were abrogated in mice with disabling mutations of TLR4 or in animals genetically deficient in TLR4 or MyD88 47, 50, 55, 58. Maximal tumor necrosis factor α (TNFα) generation is dependent on platelet TLR4 in mice challenged with LPS50. Thrombocytopenia induced by LPS in mice may involve effects on megakaryocytes in addition to circulating platelets 75. Administration of LPS to human subjects under controlled conditions causes thrombocytopenia, alterations in CD40L on platelets, and formation of platelet-monocyte aggregates 76-78, consistent with some of the studies with surrogate models and establishing translational relevance for them.
TLR2 on platelets has not been examined as extensively as TLR4, but it is clearly an important inflammatory receptor. TLR2, which heterodimerizes with TLR1 and TLR6, may be the most promiscuous of TLRs in terms of ligand recognition, and binds lipopeptides, lipoproteins, peptidoglycans, and lipoteichoic acids from gram positive bacteria, some LPS species, and some fungal and mycobacterial products38, 58. Human and mouse platelets express TLR2, and human platelets express TLR1 and TLR648-50. Surface display of TLR2 may increase with cellular activation39. Most, but not all49, reports indicate that treatment of human platelets with ligands for TLR2 triggers thromboinflammatory activities. These responses appear to be differentially regulated by intracellular pathways distinct from those activated by classic prothrombotic receptors (Online Table I), and include inside-out signaling of integrin αIIbβ3, aggregation, adhesion to collagen, P-selectin translocation, generation of reactive oxygen intermediates, and splicing of IL-1β pre-mRNA 57, 79-81. In addition, the synthetic TLR2/TLR1 agonist PAM3CSK4 induces formation of platelet-neutrophil aggregates in whole human blood80. PAM3CYSK4 and a bacterium that is recognized by TLR2 triggered platelet activation in wild type mice that was reduced in TLR2−/− animals80. Engagement of TLR2 on murine megakaryocytes and a megakaryocyte cell line altered mechanisms that regulate megakaryocyte maturation; in addition, treatment of mice with Pam3CSK4 increased megakaryocyte maturation and platelet numbers in wild type animals but not those deficient in TLR2, indicating the TLR2 on megakaryocytes may regulate megaryopoiesis and thrombopoiesis in inflammation 38, 82.
TLR9 is the most intriguing member of the platelet TLR repertoire. TLR9 is basally expressed on the plasma membranes and in the cytoplasm of resting human platelets, and its surface display is increased when platelets are activated by thrombin5, 48, 50, 83. In other cell types, TLR9 is exlusively intracellular and is restricted to endosomal domains40, 42. Recent studies of mouse and human platelets and megakaryocytes demonstrate that TLR9 is distributed to a newly-identified compartment, termed the T granule, during proplatelet production and that Type IV collagen increases TLR9 surface expression. Certain oligodeoxynucleotides (ODN), which are known synthetic ligands for TLR9, induce P-selectin translocation and also increase TLR9 surface display 83. TLR9 recognizes unmethylated CpG islands in viral and bacterial DNA5, 83, suggesting a previously-unrecognized pathogen sensing system in platelets. In addition, platelet TLR9 was recently reported to bind a carboxyalkylpyrrole protein adduct that may act as a DAMP under conditions of oxidant stress, and to transmit signals triggering aggregation and degranulation via the MyD88 pathway84.
Noncanonical Activities of Immune Transcriptional Regulators
In nucleated cells, signaling via the TLR/MyD88 pathway is a major mechanism of expression of cytokines and chemokines. Synthesis of many of these proteins is controlled by the nuclear factor kappa B (NF-κB) transcriptional regulatory system41, 42. Platelets are anucleate cells, however, and there is no evidence for transcriptional activity aside from mitochondrial transcription. Nevertheless, several NF-KB proteins are expressed by human platelets60, 85-89 and one, BcL-3, is induced in human and mouse platelets as a result of signal-dependent translation of constitutive BcL-3 transcripts63, 64, 85 (Online Table III). Interestingly, levels of transcripts for some NF-κB pathway members and TLR2 are altered in platelets from subjects with cardiovascular risk factors and obesity90. Together, these observations suggest that NF-κB family members have physiologic activities in anucleate platelets independent of their traditional roles in transcription85, 86, although transcriptional regulation clearly occurs in megakaryocytes38, 88. Consistent with this prediction, Bcl-3 was found to be an intracellular regulator of clot retraction by human and mouse platelets63. More recently, a complex of NF-κB, IKB, and protein kinase A was reported to exert negative feedback activities that modulate cytoskeletal reorganization and aggregation in platelets stimulated by thrombin or collagen87-89. It is not yet known if pathways regulating – or regulated by – NF-κB are altered by upstream signaling from TLRs 2, 4, or 9 and MyD88 in platelets. Additional nuclear factors with noncanonical activities are also expressed by these cells 89.
Degranulation, Secretion, and Translocation: Inflammatory Signaling and Effector Mechanisms of Activated Platelets
Activated platelets have diverse mechanisms for inflammatory signaling and transcellular transfer of biologically-active factors and molecular information (reviewed in 3, 5, 6). This is in part engendered by diverse mechanisms for release of signaling factors or their display on the platelet surface (Online Table III).
Rapid degranulation and secretion of preformed soluble factors are a classic activation responses of platelets 4, 5, 44, 91. Over 300 proteins may be released, depending on the agonist. Alpha and dense granules are the most rigorously characterized intracellular storage organelles of platelets91. Distinct subpopulations of alpha granules have been reported in murine platelets4, 92, but differential release of α granule cargo may predominantly be a kinetic variable when human platelets are stimulated93. Factors released from alpha and dense granules contribute to hemostasis, thrombosis, angiogenesis, endothelial barrier modulation, and inflammatory signaling 3-6, 8, 44, 91 (Online Table III). Examples of recently reported activities of secreted platelet factors include increased vascular permeability in experimental arthritis mediated by serotonin94, modification of inflammation-associated hemorrhage by TREM-like transcript 195, and modulation of angiogenesis 96.
Some factors that are degranulated by activated platelets are not immediately released into solution but are retained on the plasma membrane after surface translocation. P-selectin, which mediates platelet-leukocyte interactions and intercellular signaling is a key example (Online Table III)5, 44. Studies in mice and humans indicate that fibrinogen, another alpha granule constituent, contributes to maintenance of intracellular and cell surface of P-selectin97, 98. Additional constitutive or newly-synthesized factors can also mediate spatially-regulated juxtacrine intercellular signaling at the platelet surface. Platelet activating factor (PAF) – a phospholipid signaling molecule – CD40L, and IL-1β are examples5, 6.
New platelet mediators, compartments, and intercellular signaling mechanisms continue to be identified. Human β-defensin 1 (hBD-1), an antimicrobial peptide not previously-known to be present in platelets, is released in response to staphylococcal α-toxin, induces formation of NETs (Figure 2), and kills S. aureus in vitro99. In resting human platelets, hBD-1 is found in an intracellular compartment distinct from classic granules99. As noted previously, TLR9 is also localized in a unique intracellular compartment83.
Synthetic Pathways, the Platelet Transcriptome, and Signal-Dependent Alterations in the Platelet Proteome
The platelet is a synthetic cell when it is activated. Traditional synthetic pathways include those that regulate production of TXA2 13 PAF100-102, and reactive oxygen species 13, 80, 103. Most investigators discarded the possibility that platelets synthesize new proteins, however, because they are anucleate 104 – even though platelets were known to have messenger RNAs (mRNAs) in their intracellular arsenal 64, 105. More recently, we and others found that many of these mRNA transcripts can be translated in a signal-dependent fashion by activated platelets and that this alters the platelet proteome in functionally significant ways 63, 64, 85, 106. This led to the unexpected discovery that platelets also have pre-mRNAs and a spliceosome-dependent mechanism for their processing to mature transcripts and subsequent translation107-109 (Online Table IV). The platelet transcriptome is now being comprehensively examined in the basal state and in disease using new approaches including genome-wide next generation RNA sequencing (RNAseq)110. Diversity in the constitutive platelet transcriptome continues to emerge, and it is now known that it includes microRNAs in addition to processed mRNA and pre-mRNA transcripts108, 110, 111. There is evidence that megakaryocytes differentially sort transcripts to platelets during thrombopoiesis and that this is a mechanism for regulating protein synthesis by the circulating cells112. In parallel, diversity in the post-transcriptional pathways for transcript processing and regulated translation has been identified in the platelet repertoire64, 113. There is evidence that these newly-discovered mechanisms influence the platelet proteome and functions in thrombosis, inflammation, and immune responses(reviewed in 5, 6, 64, 113). There is also evidence that the platelet transcriptome is altered in infection and disease5, 6, 56, 90, 110 (Rondina M, et al, manuscript in preparation).
Protein products with functional relevance in inflammation and hemostasis are synthesized in a signal-dependent fashion by activated human platelets (Online Table III) 56, 57, 59, 63, 64, 85, 106-109, 112-116. Synthesis of IL-1β provides a physiologically-relevant example of intricate post-transcriptional pathways that are involved (reviewed in 5, 6, 64). Quiescent human platelets have the IL-1β pre-mRNA but little or no processed mature IL-1β transcript or Il-1β protein108. When they are activated by thrombin or other classic agonists in the presence of fibrinogen they synthesize pro-IL-1β protein, some of which is processed to the mature IL-1β cytokine107, 108. Human platelets have key components of the inflammasome6, which mediates conversion of pro-IL-1β to mature Il-1β117. Dissection of the molecular steps upstream from IL-1β protein synthesis and processing yielded a regulatory mechanism unexpected in mammalian biology118. Key studies demonstrated evidence that megakaryocytes deliver the IL-1β pre-mRNA and a functional spliceosome to platelets during proplatelet formation and that spliceosome activity is triggered in a signal-dependent fashion in activated platelets108. The signal-dependent splicing pathway is controlled by Cdc-like kinase 1 (CLK1), a kinase not previously known to be present in platelets57, 59, 109. TLRs and immunoreceptors also activate this mechanism: LPS in the presence of CD14 and LBP (see previous section)57, 59, engagement of TLR257, and engagement of FcαRI114 – an immunoreceptor for IgA on platelets – trigger splicing of IL-1β pre-mRNA and synthesis of IL-1β protein. IL-1β is a pleiotropic cytokine that has been called the “gatekeeper of inflammation”119. Thus, this regulated synthetic pathway provides platelets with a powerful mechanism to influence inflammatory and immune events 5, 6, 107, 108. In vitro and in vivo studies indicate that activated human platelets signal endothelial cells, leukocytes, and vascular smooth muscle cells by releasing Il-1β in solution or its transport in microvesicles (see below), or by juxtacrine signaling (reviewed in 6). Recent in vivo observations also indicate that release of IL-1β in platelet microvesicles is one mechanism by which platelets communicate with and activate extravascular cells120, 121.
An important feature of signal-dependent translation of proteins by platelets is that it can continue for many hours after they are activated 56, 85, 107-109, 112. In addition, some proteins can be basally synthesized continuously over many days by platelets stored ex vivo122. These and other aspects of regulated post-transcriptional protein expression are features of the “new biology” of platelets64, 121 and also have clinical relevance to thromboinflammatory diseases6 and transfusion medicine123. Prolonged synthesis of proteins by platelets also raises interesting questions regarding their contributions to the biochemical milieu of clots and thromboinflammatory lesions in vivo. While the lifespan of platelets in the circulation and in vitro is known124 their longevity in thombi and wounds has not been precisely determined, and they may persist and synthesize proteins and other factors for extended periods of time and in variable temporal patterns. In vitro studies indicate that thrombi may be reservoirs for IL-1β synthesized by platelets6, 107 – suggesting a mechanism for inflammatory properties of clots proposed by Celsus1.
Platelet-Leukocyte Interactions
Activated platelets interact with circulating myeloid leukocytes and with lymphocytes of several classes 5, 14. Binding of activated platelets to leukocytes enhances their adhesion to endothelium and transmigration under some experimental conditions, and is an important mechanism that facilitates accumulation of neutrophils and monocytes in inflamed vessels and infected or injured tissues 5, 6. As recent examples, platelets enhanced adhesion of CD14highCD16+ monocytes to endothelial cells in vitro125, facilitated human and murine neutrophil transmigration across IL-1β-activated endothelial monolayers and in an inflamed cornea model126, and selectively enhanced monocyte recruitment to endothelium in mouse and human models of vascular inflammation127. Alternatively, platelets were recruited to sinusoidal microvessels in the livers of mice injected with LPS primarily by interacting with neutrophils that were already adherent to the endothelial surface73. Platelet depletion reduced monocyte emigration and parasite killing in a mouse model of Leishmania infection128. These observations illustrate diverse defensive and pathologic consequences that may result from platelet-leukocyte interactions5, 6. In addition, platelet binding to exposed subendothelium is a mechanism to recruit myeloid leukocytes to injured vessels 6, and may be one that allows neutrophil accumulation at shear stresses that retard or prevent their adhesion to endothelium under flow129.
Activated platelets also bind to leukocytes in circulating blood, forming multicellular “heterotypic” aggregates5, 6, 44. In each of these interactions, binding of P-selectin on the plasma membrane of the activated platelet to P-selectin glycoprotein ligand 1 (PSGL-1) on the leukocyte is a dominant molecular event (Figure 3), although other adhesive mechanisms have also been described depending on the specific leukocyte subtype, species of origin, and experimental condition5, 6. Binding of P-selectin on activated platelets to PSGL-1 on leuckoytes not only tethers the cells together but also delivers signals that induce integrin activation, trigger gene expression pathways, and induce other functional responses by the leukocytes 5, 6, 44 (see below). Studies using whole human blood indicated that reciprocal platelet activation also occurs5, 6. While such interactions of activated platelets with leukocytes have physiologic inflammatory consequences, there is also evidence that they contribute to microvascular occlusion, thrombosis, and propagation of inflammatory damage in disease6, 44, 130, 131.
Figure 3.

Human platelets adhere to monocytes via P-selectin engagement of PSGL-1 and deliver signals that induce expression of pro-inflammatory gene products. From 5 with permission.
Interactions of platelets with monocytes and neutrophils have been studied the most. Platelet-monocyte aggregates are more stable than platelet-neutrophil complexes in part because of differential regulation of PSGL-1132, 133, and formation of platelet-monocyte aggregates may be the most sensitive and durable index of in vivo platelet activation in experimental and clinical conditions134. Platelet-monocyte aggregates have been detected in the blood of humans with a variety of diseases 6 (Online Table V).
Early studies demonstrated that P-selectin mediates binding of activated platelets to monocytes135, 136. Both intercellular adhesion and signaling are consequences of this molecular interaction (Figure 3)5, 44. Signals delivered to the monocyte by ligated PSGL-1 can be amplified by, and integrated with, signaling by PAF, RANTES, or other factors from the activated platelet or autocrine signaling by the monocyte136-141. New expression of key proinflammatory cytokines and metalloproteinases, synthesis of tissue factor, and expression of cyclooxygenase 2 (COX-2) with generation of COX-2-dependent eicosanoids occur in response to signaling of human monocytes by platelets (Figure 3) 130, 137, 140-147. Differential signaling and monocyte synthetic responses can be detected in model conditions mimicking ruptured atherosclerotic plaques 138. Molecular interactions between activated platelets and monocytes provide specific targets for therapeutic interruption of thromboinflammatory activities in vascular diseases 28, 29, 125, 138, 140, 145. As with monocytes, binding of activated platelets to neutrophils, eosinophils, and basophils via P-selectin/PSGL-1 induces signaling as well as intercellular adhesion5. Platelet-neutrophil aggregates have been detected in blood samples from patients with inflammatory syndromes6, although as noted above they may be more transient than platelet-monocyte aggregates. PSGL-1 engagement on human neutrophils triggers kinase cascades and chemokine synthetic pathways 5, 44, 148, and in murine models induces activation of β2 integrins on neutrophils44, 149, 150. Activation of leukocyte αMβ2 integrin in this fashion may stabilize aggregation of activated platelets and leukocytes by allowing αMβ2 to recognize GPIb on the platelet or fibrinogen bound to platelet integrin αIIbβ3 44 . Activation of platelets in whole blood induces platelet-leukocyte aggregate formation and binding of tissue factor, factor Xa, and fibrinogen to the surfaces of neutrophils and monocytes; aggregates may thus be vehicles for local and systemic delivery of coagulation factors151, 152. In vitro modeling indicates that human neutrophils enhance fibrin deposition under flow in a fashion that is partly platelet-dependent153. Observations in humans and mouse models indicate that platelet-neutrophil aggregates contribute to increased pulmonary vascular permeability and a chronic inflammatory state in sickle cell disease154. Platelet-leukocyte interactions of this nature contribute to pathologic vascular inflammation and thrombosis in a variety of experimental and clinical syndromes 6 (Figures 4-6; Online Table V).
Figure 4.

Each of the effector responses of activated platelets (Figure 1) may be pivotal in the acute inflammatory vasculopathy of experimental and clinical sepsis. Reproduced from 184 with permission.
Figure 6.

Platelets are effectors of inflammatory and immune injury in the evolution of atherosclerosis, and in acute thromboinflammatory complications triggered by plaque rupture. Platelets also contribute to pathologic consequences of vascular stenting and angioplasty. From 182 with permission.
NETs are extracellular lattices of chromatin, histones, and granule enzymes extruded by neutrophils via a unique cell death pathway sometimes termed “NETosis”155, 156. NETs capture and kill a variety of microbes, but there is also evidence that they mediate infectious and sterile vascular injury in sepsis, immune vasculitis, transfusion-related acute lung injury (TRALI), venous thrombosis, and other syndromes6, 10, 155, 156. LPS-stimulated human platelets trigger NET formation under flow conditions and platelets are required for in vivo formation of NETs in mice challenged with LPS53, 157. Platelets and leukocytes accumulate and NETs are formed in lung microvessels in a murine model of TRALI, and platelet depletion, aspirin treatment, or blockade of integrin αIIbβ3 decreases NET formation and lung damage158, 159 (Online Table V). The signals from activated platelets that induce NETosis are not completely defined. NET formation by human neutrophils is induced by hBD-1 from platelets activated by Staphylococcol α-toxin (Figure 2)99, but this may not occur in mice because of species differences in defensin expression160
Interactions of activated platelets with lymphocytes, dendritic cells, and macrophages are key events with the potential to influence adaptive immune responses and contribute to chronic inflammatory and immune syndromes3, 5. Adaptive immune facets of the platelet repertoire complement the innate immune armamentarium of these cells. This has been progressively appreciated in recent evolution of the field3-5. Discovery that platelets express and release CD40L – a pivotal immune signaling molecule that is critical in T-cell-dependent isotype switching and generation of antibody subclasses by B cells, and for dendritic cell activation – has been central in recognition of platelets as adaptive immune effector cells3, 5, 13, 39, 161. Additional activities of platelets, such as synthesis of IL-1β, also have the potential to initiate, amplify, or modify adaptive immune pathways (reviewed in 5).
Interactions of Platelets and Endothelium
Endothelial cells (EC) have potent mechanisms for retarding platelet activation at the uninjured vascular luminal surface, including enzymatic generation of prostacyclin and nitric oxide and degradation of ADP and ATP 13, 162. There is also evidence that platelets signal uninjured endothelium and are critical for maintenance of basal endothelial barrier function in pulmonary and systemic vessels6, 8. Basal endothelial barrier properties regulate transvascular flux of water and solutes, contributing to the prevention of edema, and also prevent red blood cells extravasation and bleeding. In several experimental models thrombocytopenia leads to “leakiness” of vessels8. Increased endothelial permeability secondary to disrupted barrier properties is a cardinal feature of inflammation mediated by LPS, cytokines, and other factors. This often occurs in the absence of profound thrombocytopenia, but in some cases inflammation and thrombocytopenia negatively influence barrier function in concert6, 95, 163. In mice, dramatic reduction in platelet numbers ( to <2-3% of control) sufficient to prolong tail clip bleeding times did not cause spontaneous bleeding; in contrast, spontaneous hemorrhage into inflamed tissues occurred when thrombocytopenic animals were subjected to the cutaneous arthus reaction, LPS-induced lung inflammation, or cerebral ischemia163. A relatively small increase in platelet numbers (~10%) afforded protection against spontaneous bleeding, and this was independent of classic mechanisms of platelet-dependent hemostasis mediated by integrin αIIbβ3, GPIb, GPVI, and Von Willebrand Factor163. This protective effect may result from local delivery of barrier stabilizing factors and/or inhibition of neutrophil-mediated endothelial injury by platelets8, 164. Changes in these components of the platelet repertoire in inflammatory syndromes have not been examined8. More recently, GPV1 and CLEC2 were reported to be required for prevention of inflammatory hemorrhage by murine platelets165, at variance with some of the earlier observations163, 164.
Paradoxically, platelets also contribute to disruption of endothelial barrier integrity in inflammation. Many of these observations come from models of acute lung injury and the acute respiratory distress syndrome8, 166 (Online Table V). In one study, formation of platelet-neutrophil aggregates and their interaction with endothelium mediated increased pulmonary vascular permeability and alveolar inflammation in murine models of acid aspiration and sepsis167. In other lung or systemic models, platelets or platelet lysates trigger inflammation and edema or are required for maximal vascular barrier disruption6, 8, 94, 158, 159, 168, 169. Serotonin delivered by platelets was recently reported to increase synovial vascular permeability in murine experimental arthritis94. Serotonin also had endothelial barrier destabilizing properties or – in contrast – promoted barrier stabilization in several earlier studies8. As outlined previously, activated platelets synthesize and release IL-1β, a potent mediator of endothelial barrier destabilization and increased endothelial permeability5, 6. Thus, platelets have multiple mechanisms by which they can influence endothelial barrier integrity in inflammatory diseases6, 8.
Studies of cultured human EC, cell lines, and animal models indicate that activated platelets can signal endothelium, inducing the expression of adhesion molecules, chemokines, and other proinflammatory factors, and that platelets interact directly or indirectly with the endothelium and with the subendothelium of inflamed or injured vessels (Figures 4-6). Functional consequences, inflammatory responses, and molecular mechanisms involved in these interactions have been reviewed in detail6, 14, 131, 170. Relevant reports are also cited here52, 53, 57, 58, 72-74, 107, 125-127, 129, 158, 159 (Online Table V). A recent study indicates that platelets may deliver RNA to endothelium and leukocytes171, suggesting a potential mechanism for complex intercellular information transfer.
Platelet Microparticles: Intravascular and Extravascular Signaling
Microparticles shed by megakaryocytes and platelets are subcellular vehicles for delivery of biologically-active factors to vessels and to extravascular cells in hemostasis, thrombosis, and inflammation 3-5, 113, 172. A variety of examples illustrate the potential for platelet-derived microparticles to drive inflammatory and immune events. Platelet microparticles mediate intercellular arachidonic acid transfer to other platelets and to EC, potentially amplifying activation responses173 and modifing endothelial-monocyte interactions174. Platelet microparticles can deliver IL-1β 59, 107 and RANTES175 to endothelium and transport newly-synthesized TF 109. CD40L carried by platelet microparticles is a mechanism for lymphoid germinal center formation and antigen-specific IgG production161, 176. Microparticles from activated platelets bearing IL-1β and IL-1α signal synoviocytes and induce chemokine synthesis in vitro, and appear to fuel synovial inflammation in vivo120, 121. A variety of other experimental observations and clinical correlates indicate that microparticles are key components of the platelet repertoire in thromboinflammatory signaling and intravascular and extravascular communication121, 172.
Platelets as Mediators of Inflammation in Vascular Diseases
There are many examples of inflammatory vasculopathies, which are syndromes that involve intravascular, intimal, and/or vessel wall inflammation. They range in frequency from extremely common to rare and arcane, in presentation from acute to chronic, and in topography from localized to systemic. Inflammatory vasculopathies are profiled in Online Table V.
Sepsis is a common, lethal, and informative example of acute systemic vasculopathy. Intravascular fibrin deposition and thrombosis, disrupted endothelial barrier function, and intravascular inflammation are key pathogenetic events6, 177, 178. Although pathogens, LPS, and other microbial toxins can directly induce the pathologic features of septic vasculopathy, clinical observations and animal studies support the central concept that molecular and cellular responses of the host are key to the pathogenesis of sepsis 6, 177. There is substantial evidence that platelets are pivotal cellular effectors (reviewed in 4-6, 10, 170, 179) (Figure 6).
Each of the activities in the repertoire of activated platelets outlined in this review (Figure 1) have been implicated in the vasculopathy of sepsis6 (Online Table V). Activated platelets and platelet-leukocyte aggregates are detected in clinical samples and in animal models (which, however, do not absolutely recapitulate clinical sepsis 6, 177), and there is evidence that platelet sequestration and deposition of platelet-fibrin thrombi in microvessels are central mechanisms 6, 53, 69-73. Sequestration of activated platelets bound to fibrin and leukocytes likely contributes to thrombocytopenia in sepsis, which is common and multi-factorial. TLR-dependent signaling of platelets by LPS and other bacterial factors provides one mechanism for platelet activation in sepsis 45-60, 69-81 (Online Table II). Activation of platelets by host factors generated in sepsis, including thrombin and PAF 6 (Online Table I), provides a second mechanism. Pathologic platelet responses including degranulation, secretion, and synthesis (Online Tables III, IV) may contribute to altered endothelial barrier function that is central to sepsis and to involvement of multiple organs, including the lungs, in septic syndromes 6, 8, 95, 163, 166. Platelet synthetic responses (TxA2, PAF, IL-1β) have the potential to drive pathologic interactions with endothelium and leukocytes and microvascular thrombosis (Figure 4)6. Platelet-neutrophil interactions and platelet-triggered NET formation (Figure 2) are mechanisms of endothelial barrier destabilization and injury based on animal models and in vitro experiments 10, 53, 58, 73, 80, 99, 167. Clinical samples and human cell experiments indicate that platelet-monocyte interactions occur in sepsis6, and animal models suggest that platelets regulate cytokine generation in this condition50. The platelet transcriptome is altered in sepsis 56 (Rondina M, et al, manuscript in preparation), potentially imparting pathologically-relevant changes in the platelet proteome in addition to diagnostic signatures. Platelet-derived microparticles may amplify systemic inflammation and thrombosis in sepsis 4. Experimental and clinical observations establish a rationale for assessment of anti-platetlet therapies in human sepsis 179. Nevertheless, studies of this nature will be challenging because sepsis is a group of syndromes with poorly-characterized subsets, a feature that often confounds clinical trials.
A number of other acute and subacute infectious vasculopathies (Online Table V) provide important but incomplete insights into inflammatory activities of platelets. Severe malaria involving the brain and lungs has features of acute vasculopathy similar to those of sepsis, including intravascular accumulation of platelets, leukocytes, and fibrin and endothelial barrier disruption. There is substantial evidence that platelets are key immune effectors in severe malarial vasculopathy 6, 180 (Online Table V). Intravascular interactions of platelets and monocytes may be central in this syndrome6 (Figure 5). Dengue provides an example of an acute viral-induced vasculopathy in which platelets may be critical (Online Table V). The roles of platelets in many syndromes of infection-induced vasculitis and thrombosis 181 have not been comprehensively examined, however.
Figure 5.
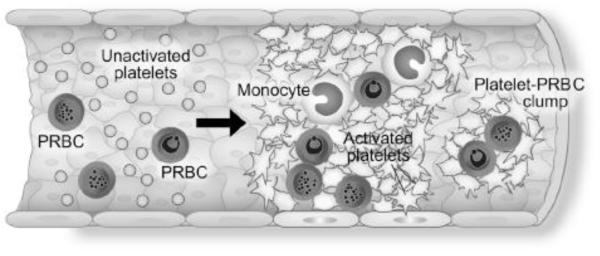
Platelets, parasitized red blood cells (PRBC), and monocytes accumulate in cerebral and lung vessels in the vasculopathy of severe malaria. Platelet signaling of leukocytes and endothelium may be a key pathophysiologic mechanism. Reproduced from 6 with permission.
Atherosclerosis, which is chronic and affects vessels in a segmental fashion, is at the other extreme of the spectrum of inflammatory and immune vasculopathies (Figure 6). Inflammation of the vessel wall is central to atherogenesis. As in sepsis, most of the activities in the platelet inflammatory and immune repertoire (Figure 1) have been implicated in initiation and progression of atherosclerotic plaques and in thromboinflammatory consequences of plaque rupture, which frequently punctuates the natural history of atherosclerosis (reviewed in 13, 170, 182) (Online Table V). Both innate and adaptive immune activities of platelets 3, 5 may contribute to the evolution of atherosclerotic lesions 182, although this is incompletely studied.
Evidence for contributions of activated platelets to initiation and progression of atherosclerotic lesions includes experimental and clinical observations that platelets commonly adhere to fatty streaks and established plaques and that platelets are activated by biochemical mediators thought to be important in atherogenesis, including oxidized low density lipoproteins and oxidatively-modified lipids (reviewed in 182). Discovery that TLRs are in the platelet repertoire and that endogenous molecules and DAMPs with potential importance in atherogenesis may activate platelets via TLRs84 – as with other cells41, 42 – extends the potential initiating mechanisms. In Apoe−/− mice, a commonly-used model of atherosclerosis, platelets were found to adhere to the intimas of arteries before lesions were detectable, genetic deletion of the αIIb subunit of integrin αIIbβ3 reduced lesional development, and circulating activated platelets secreted pro-inflammatory chemokines and formed platelet-monocyte aggregates in a P-selectin-dependent fashion, with leukocyte deposition on endothelial surfaces. These and other observations in murine models support key contributions of platelets to initiation and evolution of atherosclerotic plaques (reviewed in 131, 170, 182). Clinical studies, including immunohistochemical analysis of atherosclerotic vascular tissue and assay of plasma and urine samples for markers of platelet release, suggest persistent platelet activation in atherosclerosis and in conditions that increase risk or accelerate the syndrome and that this drives atherosclerotic vascular inflammation13, 182. In parallel, many clinical and experimental observations indicate that platelet activation, cell-cell interactions, and inflammatory signaling are central in acute coronary syndromes and other thromboinflammatory complications of plaque rupture, and in responses to vascular intervention, including angioplasty and stent placement13, 25-29, 134, 152, 182 (Online Table V). Thus, activated platelets may be ubiquitous effector cells in all phases of vascular inflammation in atherosclerosis. Acute ischemic stroke resulting from cerebral artery stenosis, thromboembolic vascular occlusion, and cerebral infarction is now recognized as a thromboinflammatory syndrome with platelets contributing to inflammation in addition to vascular occlusion (reviewed in 183), as in acute coronary syndromes (Online Table V).
Many features of the inflammatory and hemostatic repertoire of activated platelets (Figure 1, Online Table I) seem to be common ininflammatory vasculopathies, yet the syndromes are unique (Figures 4-6; Online Table V). This in part clearly reflects the activities of other key cells 10-12. It likely also reflects unique patterns of platelet responses to agonists that trigger and drive vascular inflammation – bacteria, fungi, and their products together with host factors in sepsis, parasitized erythrocytes and plasmodial toxins in malaria, oxidatively-modified lipids, metabolic DAMPs, and plaque components in atherosclerosis. It likely also reflects the fact that the platelet repertoire is not yet completely catalogued, and that new activities and mechanism will undoubtedly be identified.
Supplementary Material
Online Table I. Activation and Signaling Pathways of Human Platelets Mediate Both Hemostasis and Inflammation1-17
Online Table II. Functional Responses of Human and Mouse Platelets Induced by LPS In Vitro18-28
Online Table III. Activated Human Platelets Have Diverse Mechanisms for Release or Surface Display of Inflammatory Factors1-3, 16, 29-32
Online Table IV. Examples of Signal Dependent Translation by Human Platelets that Yields Proteins with Relevance to Thrombosis and Inflammation1, 2, 12, 27, 33-44
Online Table V. Platelet Activities in Selected Inflammatory Vasculopathies
Acknowledgements
The authors thank Alexandra Greer for skillful preparation of the manuscript and Diana Lim for artistic expertise and creative contributions in drafting the figures. We thank our colleagues, collaborators, research team, and the junior members of our group for critical discussions and scientific interactions.
Sources of funding Work discussed here was supported by the National Institutes of Health and the National Center for Research Resources (R37HL044525, R01HL092746, R01HL066277, R01HL100121, R01HL091754, K23HL092161, and ULI-RR025764).
Abreviations
- CD40L
CD40 ligand (CD154)
- DAMP
danger-associated molecular pattern
- EC
endothelial cell
- IL-1β
interleukin 1β
- LPS
lipopolysaccharide
- NET
neutrophil extracellular trap
- PAMP
pathogen-associated molecular pattern
- PMN
polymorphonuclear leukocyte
- PSGL-1
P-selectin glycoprotein ligand 1
- RANTES
Regulated upon Activation, Normal T cell expressed presumed Secreted
- TLR
toll-like receptor
Footnotes
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Majno G. The healing hand: Man and wound in the ancient world. Harvard University Press; Cambridge, Massachusetts: 1975. p. 367. [Google Scholar]
- 2.Platelets. 3rd edition Elsevier; London: 2013. pp. 1–1353. [Google Scholar]
- 3.Weyrich AS, Zimmerman GA. Platelets: Signaling cells in the immune continuum. Trends Immunol. 2004;25:489–495. doi: 10.1016/j.it.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 4.Semple JW, Italiano JE, Jr., Freedman J. Platelets and the immune continuum. Nat Rev Immunol. 2011;11:264–274. doi: 10.1038/nri2956. [DOI] [PubMed] [Google Scholar]
- 5.Vieira-de-Abreu A, Campbell RA, Weyrich AS, Zimmerman GA. Platelets: Versatile effector cells in hemostasis, inflammation, and the immune continuum. Seminars in immunopathology. 2012;34:5–30. doi: 10.1007/s00281-011-0286-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vieira-de-Abreu A, Rondina MT, Weyrich AS, Zimmerman GA. Platelets in disease: Inflammation. In: Michelson A, editor. Platelets. 3rd edition Elsevier; London: 2013. pp. 733–766. [Google Scholar]
- 7.Nurden AT. Platelets, inflammation and tissue regeneration. Thrombosis and haemostasis. 2011;105(Suppl 1):S13–33. doi: 10.1160/THS10-11-0720. [DOI] [PubMed] [Google Scholar]
- 8.Weyrich AS, Zimmerman GA. Platelets in lung biology. Annual review of physiology. 2013;75:569–591. doi: 10.1146/annurev-physiol-030212-183752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coller BS. Historical perspective and future directions in platelet research. Journal of thrombosis and haemostasis : JTH. 2011;9(Suppl 1):374–395. doi: 10.1111/j.1538-7836.2011.04356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med. 2011;17:1381–1390. doi: 10.1038/nm.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 12.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7:803–815. doi: 10.1038/nri2171. [DOI] [PubMed] [Google Scholar]
- 13.Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. 2007;357:2482–2494. doi: 10.1056/NEJMra071014. [DOI] [PubMed] [Google Scholar]
- 14.Bergmeier W, Wagner DD. Inflammation. In: Alan D, Michelson M, editors. Platelets. 2nd edition Elsevier; London: 2007. pp. 713–726. [Google Scholar]
- 15.Bouchard BA, J.R. S, Tracey P. Interactions between platelets and the coagulation system. In: Michelson A, editor. Platelets. 3rd edition Elsevier; London: 2013. pp. 425–451. [Google Scholar]
- 16.Coller BS, Shattil SJ. The gpiib/iiia (integrin alphaiibbeta3) odyssey: A technology-driven saga of a receptor with twists, turns, and even a bend. Blood. 2008;112:3011–3025. doi: 10.1182/blood-2008-06-077891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruggeri ZM, Jackson SP. Platelet thrombus formation in flowing blood. In: Michelson A, editor. Platelets. 3rd edition Elsevier; London: 2013. pp. 399–423. [Google Scholar]
- 18.Ma L, Dorling A. The roles of thrombin and protease-activated receptors in inflammation. Seminars in immunopathology. 2012;34:63–72. doi: 10.1007/s00281-011-0281-9. [DOI] [PubMed] [Google Scholar]
- 19.Davalos D, Akassoglou K. Fibrinogen as a key regulator of inflammation in disease. Seminars in immunopathology. 2012;34:43–62. doi: 10.1007/s00281-011-0290-8. [DOI] [PubMed] [Google Scholar]
- 20.Rothmeier AS, Ruf W. Protease-activated receptor 2 signaling in inflammation. Seminars in immunopathology. 2012;34:133–149. doi: 10.1007/s00281-011-0289-1. [DOI] [PubMed] [Google Scholar]
- 21.Yeaman MR, Bayer AS. Antimicrobial host defense. In: Michelson A, editor. Platelets. 3rd edition Elsevier; London: 2013. pp. 767–801. [Google Scholar]
- 22.Garraud O, Berthet J, Hamzeh-Cognasse H, Cognasse F. Pathogen sensing, subsequent signalling, and signalosome in human platelets. Thrombosis research. 2011;127:283–286. doi: 10.1016/j.thromres.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 23.Clemetson KJ, Clemetson JM. Platelet receptors. In: Michelson A, editor. Platelets. 3rd edition Elsevier; London: 2013. pp. 169–194. [Google Scholar]
- 24.Brass LF, Newman DK, Wannemacher KM, Zhu L, Stalker TJ. Signal transduction during platelet plug formation. In: Michelson A, editor. Platelets. 3rd edition Elsevier; London: 2013. pp. 367–398. [Google Scholar]
- 25.Freedman JE. Molecular regulation of platelet-dependent thrombosis. Circulation. 2005;112:2725–2734. doi: 10.1161/CIRCULATIONAHA.104.494468. [DOI] [PubMed] [Google Scholar]
- 26.Smyth SS, Woulfe DS, Weitz JI, Gachet C, Conley PB, Goodman SG, Roe MT, Kuliopulos A, Moliterno DJ, French PA, Steinhubl SR, Becker RC. G-protein-coupled receptors as signaling targets for antiplatelet therapy. Arteriosclerosis, thrombosis, and vascular biology. 2009;29:449–457. doi: 10.1161/ATVBAHA.108.176388. [DOI] [PubMed] [Google Scholar]
- 27.Varga-Szabo D, Pleines I, Nieswandt B. Cell adhesion mechanisms in platelets. Arteriosclerosis, thrombosis, and vascular biology. 2008;28:403–412. doi: 10.1161/ATVBAHA.107.150474. [DOI] [PubMed] [Google Scholar]
- 28.Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nature reviews. Drug discovery. 2003;2:15–28. doi: 10.1038/nrd985. [DOI] [PubMed] [Google Scholar]
- 29.Michelson AD. Antiplatelet therapies for the treatment of cardiovascular disease. Nature reviews. Drug discovery. 2010;9:154–169. doi: 10.1038/nrd2957. [DOI] [PubMed] [Google Scholar]
- 30.Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: The end game. Nature reviews. Molecular cell biology. 2010;11:288–300. doi: 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harris ES, Weyrich AS, Zimmerman GA. Lessons from rare maladies: Leukocyte adhesion deficiency syndromes. Curr Opin Hematol. 2013;20:16–25. doi: 10.1097/MOH.0b013e32835a0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takizawa H, Nishimura S, Takayama N, Oda A, Nishikii H, Morita Y, Kakinuma S, Yamazaki S, Okamura S, Tamura N, Goto S, Sawaguchi A, Manabe I, Takatsu K, Nakauchi H, Takaki S, Eto K. Lnk regulates integrin alphaiibbeta3 outside-in signaling in mouse platelets, leading to stabilization of thrombus development in vivo. J Clin Invest. 2010;120:179–190. doi: 10.1172/JCI39503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moser M, Nieswandt B, Ussar S, Pozgajova M, Fassler R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med. 2008;14:325–330. doi: 10.1038/nm1722. [DOI] [PubMed] [Google Scholar]
- 34.Zhang G, Xiang B, Ye S, Chrzanowska-Wodnicka M, Morris AJ, Gartner TK, Whiteheart SW, White GC, 2nd, Smyth SS, Li Z. Distinct roles for rap1b protein in platelet secretion and integrin alphaiibbeta3 outside-in signaling. J Biol Chem. 2011;286:39466–39477. doi: 10.1074/jbc.M111.239608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kasirer-Friede A, Kahn ML, Shattil SJ. Platelet integrins and immunoreceptors. Immunol Rev. 2007;218:247–264. doi: 10.1111/j.1600-065X.2007.00532.x. [DOI] [PubMed] [Google Scholar]
- 36.Ginsberg MH, Henson PM. Enhancement of platelet response to immune complexes and igg aggregates by lipid a-rich bacterial lipopolysaccharides. J Exp Med. 1978;147:207–217. doi: 10.1084/jem.147.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pollitt AY, Hughes CE, Watson S. Gpvi and clec-2. In: Michelson A, editor. Platelets. 3rd edition Elsevier; London: 2013. pp. 215–233. [Google Scholar]
- 38.Beaulieu LM, Freedman JE. The role of inflammation in regulating platelet production and function: Toll-like receptors in platelets and megakaryocytes. Thrombosis research. 2010;125:205–209. doi: 10.1016/j.thromres.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garraud O, Cognasse F. Platelet toll-like receptor expression: The link between “danger” ligands and inflammation. Inflammation & allergy drug targets. 2010;9:322–333. doi: 10.2174/187152810793937991. [DOI] [PubMed] [Google Scholar]
- 40.Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity. 2010;32:305–315. doi: 10.1016/j.immuni.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 41.Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327:291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 43.Hickey MJ, Kubes P. Intravascular immunity: The host-pathogen encounter in blood vessels. Nat Rev Immunol. 2009;9:364–375. doi: 10.1038/nri2532. [DOI] [PubMed] [Google Scholar]
- 44.Smyth SS, McEver RP, Weyrich AS, Morrell CN, Hoffman MR, Arepally GM, French PA, Dauerman HL, Becker RC. Platelet functions beyond hemostasis. Journal of thrombosis and haemostasis : JTH. 2009;7:1759–1766. doi: 10.1111/j.1538-7836.2009.03586.x. [DOI] [PubMed] [Google Scholar]
- 45.Grabarek J, Timmons S, Hawiger J. Modulation of human platelet protein kinase c by endotoxic lipid a. J Clin Invest. 1988;82:964–971. doi: 10.1172/JCI113705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beutler B, Rietschel ET. Innate immune sensing and its roots: The story of endotoxin. Nat Rev Immunol. 2003;3:169–176. doi: 10.1038/nri1004. [DOI] [PubMed] [Google Scholar]
- 47.Andonegui G, Kerfoot SM, McNagny K, Ebbert KV, Patel KD, Kubes P. Platelets express functional toll-like receptor-4. Blood. 2005;106:2417–2423. doi: 10.1182/blood-2005-03-0916. [DOI] [PubMed] [Google Scholar]
- 48.Cognasse F, Hamzeh H, Chavarin P, Acquart S, Genin C, Garraud O. Evidence of toll-like receptor molecules on human platelets. Immunology and cell biology. 2005;83:196–198. doi: 10.1111/j.1440-1711.2005.01314.x. [DOI] [PubMed] [Google Scholar]
- 49.Ward JR, Bingle L, Judge HM, Brown SB, Storey RF, Whyte MK, Dower SK, Buttle DJ, Sabroe I. Agonists of toll-like receptor (tlr)2 and tlr4 are unable to modulate platelet activation by adenosine diphosphate and platelet activating factor. Thrombosis and haemostasis. 2005;94:831–838. [PubMed] [Google Scholar]
- 50.Aslam R, Speck ER, Kim M, Crow AR, Bang KW, Nestel FP, Ni H, Lazarus AH, Freedman J, Semple JW. Platelet toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood. 2006;107:637–641. doi: 10.1182/blood-2005-06-2202. [DOI] [PubMed] [Google Scholar]
- 51.Montrucchio G, Bosco O, Del Sorbo L, Fascio Pecetto P, Lupia E, Goffi A, Omede P, Emanuelli G, Camussi G. Mechanisms of the priming effect of low doses of lipopoly-saccharides on leukocyte-dependent platelet aggregation in whole blood. Thrombosis and haemostasis. 2003;90:872–881. doi: 10.1160/TH03-02-0085. [DOI] [PubMed] [Google Scholar]
- 52.Stahl AL, Svensson M, Morgelin M, Svanborg C, Tarr PI, Mooney JC, Watkins SL, Johnson R, Karpman D. Lipopolysaccharide from enterohemorrhagic escherichia coli binds to platelets through tlr4 and cd62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood. 2006;108:167–176. doi: 10.1182/blood-2005-08-3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD, Keys EM, Allen-Vercoe E, Devinney R, Doig CJ, Green FH, Kubes P. Platelet tlr4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13:463–469. doi: 10.1038/nm1565. [DOI] [PubMed] [Google Scholar]
- 54.Semple JW, Aslam R, Kim M, Speck ER, Freedman J. Platelet-bound lipopolysaccharide enhances fc receptor-mediated phagocytosis of igg-opsonized platelets. Blood. 2007;109:4803–4805. doi: 10.1182/blood-2006-12-062695. [DOI] [PubMed] [Google Scholar]
- 55.Rumbaut RE, Bellera RV, Randhawa JK, Shrimpton CN, Dasgupta SK, Dong JF, Burns AR. Endotoxin enhances microvascular thrombosis in mouse cremaster venules via a tlr4-dependent, neutrophil-independent mechanism. American journal of physiology. Heart and circulatory physiology. 2006;290:H1671–1679. doi: 10.1152/ajpheart.00305.2005. [DOI] [PubMed] [Google Scholar]
- 56.Rondina MT, Schwertz H, Harris ES, Kraemer BF, Campbell RA, Mackman N, Grissom CK, Weyrich AS, Zimmerman GA. The septic milieu triggers expression of spliced tissue factor mrna in human platelets. Journal of thrombosis and haemostasis : JTH. 2011;9:748–758. doi: 10.1111/j.1538-7836.2011.04208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shashkin PN, Brown GT, Ghosh A, Marathe GK, McIntyre TM. Lipopolysaccharide is a direct agonist for platelet rna splicing. J Immunol. 2008;181:3495–3502. doi: 10.4049/jimmunol.181.5.3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang G, Han J, Welch EJ, Ye RD, Voyno-Yasenetskaya TA, Malik AB, Du X, Li Z. Lipopolysaccharide stimulates platelet secretion and potentiates platelet aggregation via tlr4/myd88 and the cgmp-dependent protein kinase pathway. J Immunol. 2009;182:7997–8004. doi: 10.4049/jimmunol.0802884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brown GT, McIntyre TM. Lipopolysaccharide signaling without a nucleus: Kinase cascades stimulate platelet shedding of proinflammatory il-1beta-rich microparticles. J Immunol. 2011;186:5489–5496. doi: 10.4049/jimmunol.1001623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berthet J, Damien P, Hamzeh-Cognasse H, Pozzetto B, Garraud O, Cognasse F. Toll-like receptor 4 signal transduction in platelets: Novel pathways. Br J Haematol. 2010;151:89–92. doi: 10.1111/j.1365-2141.2010.08292.x. [DOI] [PubMed] [Google Scholar]
- 61.Saba HI, Saba SR, Morelli G, Hartmann RC. Endotoxin-mediated inhibition of human platelet aggregation. Thrombosis research. 1984;34:19–33. doi: 10.1016/0049-3848(84)90103-8. [DOI] [PubMed] [Google Scholar]
- 62.Sheu JR, Hung WC, Kan YC, Lee YM, Yen MH. Mechanisms involved in the antiplatelet activity of escherichia coli lipopolysaccharide in human platelets. Br J Haematol. 1998;103:29–38. doi: 10.1046/j.1365-2141.1998.00938.x. [DOI] [PubMed] [Google Scholar]
- 63.Weyrich AS, Denis MM, Schwertz H, Tolley ND, Foulks J, Spencer E, Kraiss LW, Albertine KH, McIntyre TM, Zimmerman GA. Mtor-dependent synthesis of bcl-3 controls the retraction of fibrin clots by activated human platelets. Blood. 2007;109:1975–1983. doi: 10.1182/blood-2006-08-042192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zimmerman GA, Weyrich AS. Signal-dependent protein synthesis by activated platelets: New pathways to altered phenotype and function. Arteriosclerosis, thrombosis, and vascular biology. 2008;28:s17–24. doi: 10.1161/ATVBAHA.107.160218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aslan JE, Tormoen GW, Loren CP, Pang J, McCarty OJ. S6k1 and mtor regulate rac1-driven platelet activation and aggregation. Blood. 2011;118:3129–3136. doi: 10.1182/blood-2011-02-331579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Choi W, Karim ZA, Whiteheart SW. Arf6 plays an early role in platelet activation by collagen and convulxin. Blood. 2006;107:3145–3152. doi: 10.1182/blood-2005-09-3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Karim ZA, Choi W, Whiteheart SW. Primary platelet signaling cascades and integrin-mediated signaling control adp-ribosylation factor (arf) 6-gtp levels during platelet activation and aggregation. J Biol Chem. 2008;283:11995–12003. doi: 10.1074/jbc.M800146200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kanamarlapudi V, Owens SE, Saha K, Pope RJ, Mundell SJ. Arf6-dependent regulation of p2y receptor traffic and function in human platelets. PloS one. 2012;7:e43532. doi: 10.1371/journal.pone.0043532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Endo Y, Shibazaki M, Nakamura M, Takada H. Contrasting effects of lipopolysaccharides (endotoxins) from oral black-pigmented bacteria and enterobacteriaceae on platelets, a major source of serotonin, and on histamine-forming enzyme in mice. The Journal of infectious diseases. 1997;175:1404–1412. doi: 10.1086/516473. [DOI] [PubMed] [Google Scholar]
- 70.Shibazaki M, Kawabata Y, Yokochi T, Nishida A, Takada H, Endo Y. Complement-dependent accumulation and degradation of platelets in the lung and liver induced by injection of lipopolysaccharides. Infect Immun. 1999;67:5186–5191. doi: 10.1128/iai.67.10.5186-5191.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhao L, Ohtaki Y, Yamaguchi K, Matsushita M, Fujita T, Yokochi T, Takada H, Endo Y. Lps-induced platelet response and rapid shock in mice: Contribution of o-antigen region of lps and involvement of the lectin pathway of the complement system. Blood. 2002;100:3233–3239. doi: 10.1182/blood-2002-01-0252. [DOI] [PubMed] [Google Scholar]
- 72.Kiefmann R, Heckel K, Schenkat S, Dorger M, Goetz AE. Role of p-selectin in platelet sequestration in pulmonary capillaries during endotoxemia. Journal of vascular research. 2006;43:473–481. doi: 10.1159/000095247. [DOI] [PubMed] [Google Scholar]
- 73.Jenne CN, Wong CH, Petri B, Kubes P. The use of spinning-disk confocal microscopy for the intravital analysis of platelet dynamics in response to systemic and local inflammation. PloS one. 2011;6:e25109. doi: 10.1371/journal.pone.0025109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Katayama T, Ikeda Y, Handa M, Tamatani T, Sakamoto S, Ito M, Ishimura Y, Suematsu M. Immunoneutralization of glycoprotein ibalpha attenuates endotoxin-induced interactions of platelets and leukocytes with rat venular endothelium in vivo. Circulation research. 2000;86:1031–1037. doi: 10.1161/01.res.86.10.1031. [DOI] [PubMed] [Google Scholar]
- 75.Jayachandran M, Brunn GJ, Karnicki K, Miller RS, Owen WG, Miller VM. In vivo effects of lipopolysaccharide and tlr4 on platelet production and activity: Implications for thrombotic risk. J Appl Physiol. 2007;102:429–433. doi: 10.1152/japplphysiol.01576.2005. [DOI] [PubMed] [Google Scholar]
- 76.Li N, Soop A, Sollevi A, Hjemdahl P. Multi-cellular activation in vivo by endotoxin in humans--limited protection by adenosine infusion. Thrombosis and haemostasis. 2000;84:381–387. [PubMed] [Google Scholar]
- 77.Wilson M, Blum R, Dandona P, Mousa S. Effects in humans of intravenously administered endotoxin on soluble cell-adhesion molecule and inflammatory markers: A model of human diseases. Clinical and experimental pharmacology & physiology. 2001;28:376–380. doi: 10.1046/j.1440-1681.2001.03463.x. [DOI] [PubMed] [Google Scholar]
- 78.Kalsch T, Elmas E, Nguyen XD, Suvajac N, Kluter H, Borggrefe M, Dempfle CE. Endotoxin-induced effects on platelets and monocytes in an in vivo model of inflammation. Basic research in cardiology. 2007;102:460–466. doi: 10.1007/s00395-007-0667-y. [DOI] [PubMed] [Google Scholar]
- 79.Berg M, Offermanns S, Seifert R, Schultz G. Synthetic lipopeptide pam3cysser(lys)4 is an effective activator of human platelets. The American journal of physiology. 1994;266:C1684–1691. doi: 10.1152/ajpcell.1994.266.6.C1684. [DOI] [PubMed] [Google Scholar]
- 80.Blair P, Rex S, Vitseva O, Beaulieu L, Tanriverdi K, Chakrabarti S, Hayashi C, Genco CA, Iafrati M, Freedman JE. Stimulation of toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circulation research. 2009;104:346–354. doi: 10.1161/CIRCRESAHA.108.185785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rex S, Beaulieu LM, Perlman DH, Vitseva O, Blair PS, McComb ME, Costello CE, Freedman JE. Immune versus thrombotic stimulation of platelets differentially regulates signalling pathways, intracellular protein-protein interactions, and alpha-granule release. Thrombosis and haemostasis. 2009;102:97–110. doi: 10.1160/TH08-08-0513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Beaulieu LM, Lin E, Morin KM, Tanriverdi K, Freedman JE. Regulatory effects of tlr2 on megakaryocytic cell function. Blood. 2011;117:5963–5974. doi: 10.1182/blood-2010-09-304949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Thon JN, Peters CG, Machlus KR, Aslam R, Rowley J, Macleod H, Devine MT, Fuchs TA, Weyrich AS, Semple JW, Flaumenhaft R, Italiano JE., Jr. T granules in human platelets function in tlr9 organization and signaling. J Cell Biol. 2012;198:561–574. doi: 10.1083/jcb.201111136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Panigrahi S, Ma Y, Hong L, Gao D, West XZ, Salomon RG, Byzova TV, Podrez EA. Engagement of platelet toll-like receptor 9 by novel endogenous ligands promotes platelet hyperreactivity and thrombosis. Circulation research. 2013;112:103–112. doi: 10.1161/CIRCRESAHA.112.274241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Weyrich AS, Dixon DA, Pabla R, Elstad MR, McIntyre TM, Prescott SM, Zimmerman GA. Signal-dependent translation of a regulatory protein, bcl-3, in activated human platelets. Proc Natl Acad Sci U S A. 1998;95:5556–5561. doi: 10.1073/pnas.95.10.5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu F, Morris S, Epps J, Carroll R. Demonstration of an activation regulated nf-kappab/i-kappabalpha complex in human platelets. Thrombosis research. 2002;106:199–203. doi: 10.1016/s0049-3848(02)00130-5. [DOI] [PubMed] [Google Scholar]
- 87.Gambaryan S, Kobsar A, Rukoyatkina N, Herterich S, Geiger J, Smolenski A, Lohmann SM, Walter U. Thrombin and collagen induce a feedback inhibitory signaling pathway in platelets involving dissociation of the catalytic subunit of protein kinase a from an nfkappab-ikappab complex. J Biol Chem. 2010;285:18352–18363. doi: 10.1074/jbc.M109.077602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Spinelli SL, Casey AE, Pollock SJ, Gertz JM, McMillan DH, Narasipura SD, Mody NA, King MR, Maggirwar SB, Francis CW, Taubman MB, Blumberg N, Phipps RP. Platelets and megakaryocytes contain functional nuclear factor-kappab. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:591–598. doi: 10.1161/ATVBAHA.109.197343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Spinelli SL, Maggirwar SB, Blumberg N, Phipps RP. Nuclear emancipation: A platelet tour de force. Science signaling. 2010;3:pe37. doi: 10.1126/scisignal.3144pe37. [DOI] [PubMed] [Google Scholar]
- 90.Freedman JE, Larson MG, Tanriverdi K, O’Donnell CJ, Morin K, Hakanson AS, Vasan RS, Johnson AD, Lafrati MD, Benjamin EJ. Relation of platelet and leukocyte inflammatory transcripts to body mass index in the framingham heart study. Circulation. 2010;122:119–129. doi: 10.1161/CIRCULATIONAHA.109.928192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Flaumenhaft R. Platelet secretion. In: Michelson A, editor. Platelets. 3rd edition Elsevier; London: 2013. pp. 343–366. [Google Scholar]
- 92.Italiano JE, Jr., Richardson JL, Patel-Hett S, Battinelli E, Zaslavsky A, Short S, Ryeom S, Folkman J, Klement GL. Angiogenesis is regulated by a novel mechanism: Pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood. 2008;111:1227–1233. doi: 10.1182/blood-2007-09-113837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jonnalagadda D, Izu LT, Whiteheart SW. Platelet secretion is kinetically heterogeneous in an agonist-responsive manner. Blood. 2012;120:5209–5216. doi: 10.1182/blood-2012-07-445080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cloutier N, Pare A, Farndale RW, Schumacher HR, Nigrovic PA, Lacroix S, Boilard E. Platelets can enhance vascular permeability. Blood. 2012;120:1334–1343. doi: 10.1182/blood-2012-02-413047. [DOI] [PubMed] [Google Scholar]
- 95.Washington AV, Gibot S, Acevedo I, Gattis J, Quigley L, Feltz R, De La Mota A, Schubert RL, Gomez-Rodriguez J, Cheng J, Dutra A, Pak E, Chertov O, Rivera L, Morales J, Lubkowski J, Hunter R, Schwartzberg PL, McVicar DW. Trem-like transcript-1 protects against inflammation-associated hemorrhage by facilitating platelet aggregation in mice and humans. J Clin Invest. 2009;119:1489–1501. doi: 10.1172/JCI36175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Battinelli EM, Markens BA, Italiano JE., Jr. Release of angiogenesis regulatory proteins from platelet alpha granules: Modulation of physiologic and pathologic angiogenesis. Blood. 2011;118:1359–1369. doi: 10.1182/blood-2011-02-334524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yang H, Lang S, Zhai Z, Li L, Kahr WH, Chen P, Brkic J, Spring CM, Flick MJ, Degen JL, Freedman J, Ni H. Fibrinogen is required for maintenance of platelet intracellular and cell-surface p-selectin expression. Blood. 2009;114:425–436. doi: 10.1182/blood-2008-03-145821. [DOI] [PubMed] [Google Scholar]
- 98.Schwertz H, Zimmerman GA, Weyrich AS. Fibrinogen selects selectins. Blood. 2009;114:234. doi: 10.1182/blood-2009-04-216135. [DOI] [PubMed] [Google Scholar]
- 99.Kraemer BF, Campbell RA, Schwertz H, Cody MJ, Franks Z, Tolley ND, Kahr WH, Lindemann S, Seizer P, Yost CC, Zimmerman GA, Weyrich AS. Novel anti-bacterial activities of beta-defensin 1 in human platelets: Suppression of pathogen growth and signaling of neutrophil extracellular trap formation. PLoS pathogens. 2011;7:e1002355. doi: 10.1371/journal.ppat.1002355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Touqui L, Hatmi M, Vargaftig BB. Human platelets stimulated by thrombin produce platelet-activating factor (1-o-alkyl-2-acetyl-sn-glycero-3-phosphocholine) when the degrading enzyme acetyl hydrolase is blocked. Biochem J. 1985;229:811–816. doi: 10.1042/bj2290811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ostrovsky L, King AJ, Bond S, Mitchell D, Lorant DE, Zimmerman GA, Larsen R, Niu XF, Kubes P. A juxtacrine mechanism for neutrophil adhesion on platelets involves platelet-activating factor and a selectin-dependent activation process. Blood. 1998;91:3028–3036. [PubMed] [Google Scholar]
- 102.Prescott SM, Zimmerman GA, Stafforini DM, McIntyre TM. Platelet-activating factor and related lipid mediators. Annu Rev Biochem. 2000;69:419–445. doi: 10.1146/annurev.biochem.69.1.419. [DOI] [PubMed] [Google Scholar]
- 103.Chakrabarti S, Varghese S, Vitseva O, Tanriverdi K, Freedman JE. Cd40 ligand influences platelet release of reactive oxygen intermediates. Arteriosclerosis, thrombosis, and vascular biology. 2005;25:2428–2434. doi: 10.1161/01.ATV.0000184765.59207.f3. [DOI] [PubMed] [Google Scholar]
- 104.Libby P, Simon DI. Inflammation and thrombosis: The clot thickens. Circulation. 2001;103:1718–1720. doi: 10.1161/01.cir.103.13.1718. [DOI] [PubMed] [Google Scholar]
- 105.Weyrich AS, Schwertz H, Kraiss LW, Zimmerman GA. Protein synthesis by platelets: Historical and new perspectives. Journal of thrombosis and haemostasis : JTH. 2009;7:241–246. doi: 10.1111/j.1538-7836.2008.03211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pabla R, Weyrich AS, Dixon DA, Bray PF, McIntyre TM, Prescott SM, Zimmerman GA. Integrin-dependent control of translation: Engagement of integrin alphaiibbeta3 regulates synthesis of proteins in activated human platelets. J Cell Biol. 1999;144:175–184. doi: 10.1083/jcb.144.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lindemann S, Tolley ND, Dixon DA, McIntyre TM, Prescott SM, Zimmerman GA, Weyrich AS. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J Cell Biol. 2001;154:485–490. doi: 10.1083/jcb.200105058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Denis MM, Tolley ND, Bunting M, Schwertz H, Jiang H, Lindemann S, Yost CC, Rubner FJ, Albertine KH, Swoboda KJ, Fratto CM, Tolley E, Kraiss LW, McIntyre TM, Zimmerman GA, Weyrich AS. Escaping the nuclear confines: Signal-dependent pre-mrna splicing in anucleate platelets. Cell. 2005;122:379–391. doi: 10.1016/j.cell.2005.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schwertz H, Tolley ND, Foulks JM, Denis MM, Risenmay BW, Buerke M, Tilley RE, Rondina MT, Harris EM, Kraiss LW, Mackman N, Zimmerman GA, Weyrich AS. Signal-dependent splicing of tissue factor pre-mrna modulates the thrombogenicity of human platelets. J Exp Med. 2006;203:2433–2440. doi: 10.1084/jem.20061302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rowley JW, Schwertz H, Weyrich AS. Platelet mrna: The meaning behind the message. Curr Opin Hematol. 2012;19:385–391. doi: 10.1097/MOH.0b013e328357010e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Provost P. Platelet microrna. In: Michelson A, editor. Platelets. 3rd edition Elsevier; London: 2013. pp. 91–101. [Google Scholar]
- 112.Cecchetti L, Tolley ND, Michetti N, Bury L, Weyrich AS, Gresele P. Megakaryocytes differentially sort mrnas for matrix metalloproteinases and their inhibitors into platelets: A mechanism for regulating synthetic events. Blood. 2011;118:1903–1911. doi: 10.1182/blood-2010-12-324517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Smith MC, Schwertz H, Zimmerman GA, Weyrich AS. The platelet proteome. In: Michelson A, editor. Platelets. 3rd edition Elsevier; London: 2013. pp. 103–116. [Google Scholar]
- 114.Qian K, Xie F, Gibson AW, Edberg JC, Kimberly RP, Wu J. Functional expression of iga receptor fcalphari on human platelets. J Leukoc Biol. 2008;84:1492–1500. doi: 10.1189/jlb.0508327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Brogren H, Karlsson L, Andersson M, Wang L, Erlinge D, Jern S. Platelets synthesize large amounts of active plasminogen activator inhibitor 1. Blood. 2004;104:3943–3948. doi: 10.1182/blood-2004-04-1439. [DOI] [PubMed] [Google Scholar]
- 116.Gerrits AJ, Koekman CA, van Haeften TW, Akkerman JW. Platelet tissue factor synthesis in type 2 diabetic patients is resistant to inhibition by insulin. Diabetes. 2010;59:1487–1495. doi: 10.2337/db09-1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 118.Meshorer E, Misteli T. Splicing misplaced. Cell. 2005;122:317–318. doi: 10.1016/j.cell.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 119.Dinarello CA. A clinical perspective of il-1beta as the gatekeeper of inflammation. European journal of immunology. 2011;41:1203–1217. doi: 10.1002/eji.201141550. [DOI] [PubMed] [Google Scholar]
- 120.Boilard E, Nigrovic PA, Larabee K, Watts GF, Coblyn JS, Weinblatt ME, Massarotti EM, Remold-O’Donnell E, Farndale RW, Ware J, Lee DM. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. 2010;327:580–583. doi: 10.1126/science.1181928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zimmerman GA, Weyrich AS. Immunology. Arsonists in rheumatoid arthritis. Science. 2010;327:528–529. doi: 10.1126/science.1185869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Thon JN, Devine DV. Translation of glycoprotein iiia in stored blood platelets. Transfusion. 2007;47:2260–2270. doi: 10.1111/j.1537-2995.2007.01455.x. [DOI] [PubMed] [Google Scholar]
- 123.Schubert P, Devine DV. De novo protein synthesis in mature platelets: A consideration for transfusion medicine. Vox Sang. 2010;99:112–122. doi: 10.1111/j.1423-0410.2010.01333.x. [DOI] [PubMed] [Google Scholar]
- 124.Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, Kelly PN, Ekert PG, Metcalf D, Roberts AW, Huang DC, Kile BT. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128:1173–1186. doi: 10.1016/j.cell.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 125.Passacquale G, Vamadevan P, Pereira L, Hamid C, Corrigall V, Ferro A. Monocyte-platelet interaction induces a pro-inflammatory phenotype in circulating monocytes. PloS one. 2011;6:e25595. doi: 10.1371/journal.pone.0025595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lam FW, Burns AR, Smith CW, Rumbaut RE. Platelets enhance neutrophil transendothelial migration via p-selectin glycoprotein ligand-1. American journal of physiology. Heart and circulatory physiology. 2011;300:H468–475. doi: 10.1152/ajpheart.00491.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kuckleburg CJ, Yates CM, Kalia N, Zhao Y, Nash GB, Watson SP, Rainger GE. Endothelial cell-borne platelet bridges selectively recruit monocytes in human and mouse models of vascular inflammation. Cardiovascular research. 2011;91:134–141. doi: 10.1093/cvr/cvr040. [DOI] [PubMed] [Google Scholar]
- 128.Goncalves R, Zhang X, Cohen H, Debrabant A, Mosser DM. Platelet activation attracts a subpopulation of effector monocytes to sites of leishmania major infection. J Exp Med. 2011;208:1253–1265. doi: 10.1084/jem.20101751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kuijper PH, Gallardo Torres HI, van der Linden JA, Lammers JW, Sixma JJ, Koenderman L, Zwaginga JJ. Platelet-dependent primary hemostasis promotes selectin- and integrin-mediated neutrophil adhesion to damaged endothelium under flow conditions. Blood. 1996;87:3271–3281. [PubMed] [Google Scholar]
- 130.Freedman JE, Loscalzo J. Platelet-monocyte aggregates: Bridging thrombosis and inflammation. Circulation. 2002;105:2130–2132. doi: 10.1161/01.cir.0000017140.26466.f5. [DOI] [PubMed] [Google Scholar]
- 131.van Gils JM, Zwaginga JJ, Hordijk PL. Molecular and functional interactions among monocytes, platelets, and endothelial cells and their relevance for cardiovascular diseases. J Leukoc Biol. 2009;85:195–204. doi: 10.1189/jlb.0708400. [DOI] [PubMed] [Google Scholar]
- 132.Rinder HM, Tracey JL, Rinder CS, Leitenberg D, Smith BR. Neutrophil but not monocyte activation inhibits p-selectin-mediated platelet adhesion. Thrombosis and haemostasis. 1994;72:750–756. [PubMed] [Google Scholar]
- 133.Lorant DE, McEver RP, McIntyre TM, Moore KL, Prescott SM, Zimmerman GA. Activation of polymorphonuclear leukocytes reduces their adhesion to p-selectin and causes redistribution of ligands for p-selectin on their surfaces. J Clin Invest. 1995;96:171–182. doi: 10.1172/JCI118018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Michelson AD, Barnard MR, Krueger LA, Valeri CR, Furman MI. Circulating monocyte-platelet aggregates are a more sensitive marker of in vivo platelet activation than platelet surface p-selectin: Studies in baboons, human coronary intervention, and human acute myocardial infarction. Circulation. 2001;104:1533–1537. doi: 10.1161/hc3801.095588. [DOI] [PubMed] [Google Scholar]
- 135.Larsen E, Celi A, Gilbert GE, Furie BC, Erban JK, Bonfanti R, Wagner DD, Furie B. Padgem protein: A receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell. 1989;59:305–312. doi: 10.1016/0092-8674(89)90292-4. [DOI] [PubMed] [Google Scholar]
- 136.Weyrich AS, McIntyre TM, McEver RP, Prescott SM, Zimmerman GA. Monocyte tethering by p-selectin regulates monocyte chemotactic protein-1 and tumor necrosis factor-alpha secretion. Signal integration and nf-kappa b translocation. J Clin Invest. 1995;95:2297–2303. doi: 10.1172/JCI117921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Weyrich AS, Elstad MR, McEver RP, McIntyre TM, Moore KL, Morrissey JH, Prescott SM, Zimmerman GA. Activated platelets signal chemokine synthesis by human monocytes. J Clin Invest. 1996;97:1525–1534. doi: 10.1172/JCI118575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Galt SW, Lindemann S, Medd D, Allen LL, Kraiss LW, Harris ES, Prescott SM, McIntyre TM, Weyrich AS, Zimmerman GA. Differential regulation of matrix metalloproteinase-9 by monocytes adherent to collagen and platelets. Circulation research. 2001;89:509–516. doi: 10.1161/hh1801.096339. [DOI] [PubMed] [Google Scholar]
- 139.Mahoney TS, Weyrich AS, Dixon DA, McIntyre T, Prescott SM, Zimmerman GA. Cell adhesion regulates gene expression at translational checkpoints in human myeloid leukocytes. Proc Natl Acad Sci U S A. 2001;98:10284–10289. doi: 10.1073/pnas.181201398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Weyrich AS, Denis MM, Kuhlmann-Eyre JR, Spencer ED, Dixon DA, Marathe GK, McIntyre TM, Zimmerman GA, Prescott SM. Dipyridamole selectively inhibits inflammatory gene expression in platelet-monocyte aggregates. Circulation. 2005;111:633–642. doi: 10.1161/01.CIR.0000154607.90506.45. [DOI] [PubMed] [Google Scholar]
- 141.Dixon DA, Tolley ND, Bemis-Standoli K, Martinez ML, Weyrich AS, Morrow JD, Prescott SM, Zimmerman GA. Expression of cox-2 in platelet-monocyte interactions occurs via combinatorial regulation involving adhesion and cytokine signaling. J Clin Invest. 2006;116:2727–2738. doi: 10.1172/JCI27209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Celi A, Pellegrini G, Lorenzet R, De Blasi A, Ready N, Furie BC, Furie B. P-selectin induces the expression of tissue factor on monocytes. Proc Natl Acad Sci U S A. 1994;91:8767–8771. doi: 10.1073/pnas.91.19.8767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Neumann FJ, Marx N, Gawaz M, Brand K, Ott I, Rokitta C, Sticherling C, Meinl C, May A, Schomig A. Induction of cytokine expression in leukocytes by binding of thrombin-stimulated platelets. Circulation. 1997;95:2387–2394. doi: 10.1161/01.cir.95.10.2387. [DOI] [PubMed] [Google Scholar]
- 144.Lindmark E, Tenno T, Siegbahn A. Role of platelet p-selectin and cd40 ligand in the induction of monocytic tissue factor expression. Arteriosclerosis, thrombosis, and vascular biology. 2000;20:2322–2328. doi: 10.1161/01.atv.20.10.2322. [DOI] [PubMed] [Google Scholar]
- 145.Steiner S, Seidinger D, Huber K, Kaun C, Minar E, Kopp CW. Effect of glycoprotein iib/iiia antagonist abciximab on monocyte-platelet aggregates and tissue factor expression. Arteriosclerosis, thrombosis, and vascular biology. 2003;23:1697–1702. doi: 10.1161/01.ATV.0000087035.46547.89. [DOI] [PubMed] [Google Scholar]
- 146.Eligini S, Barbieri SS, Arenaz I, Tremoli E, Colli S. Paracrine up-regulation of monocyte cyclooxygenase-2 by platelets: Role of transforming growth factor-beta1. Cardiovascular research. 2007;74:270–278. doi: 10.1016/j.cardiores.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 147.Christersson C, Johnell M, Siegbahn A. Tissue factor and il8 production by p-selectin-dependent platelet-monocyte aggregates in whole blood involves phosphorylation of lyn and is inhibited by il10. Journal of thrombosis and haemostasis : JTH. 2008;6:986–994. doi: 10.1111/j.1538-7836.2008.02956.x. [DOI] [PubMed] [Google Scholar]
- 148.Lindemann SW, Yost CC, Denis MM, McIntyre TM, Weyrich AS, Zimmerman GA. Neutrophils alter the inflammatory milieu by signal-dependent translation of constitutive messenger rnas. Proc Natl Acad Sci U S A. 2004;101:7076–7081. doi: 10.1073/pnas.0401901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Evangelista V, Pamuklar Z, Piccoli A, Manarini S, Dell’elba G, Pecce R, Martelli N, Federico L, Rojas M, Berton G, Lowell CA, Totani L, Smyth SS. Src family kinases mediate neutrophil adhesion to adherent platelets. Blood. 2007;109:2461–2469. doi: 10.1182/blood-2006-06-029082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zarbock A, Muller H, Kuwano Y, Ley K. Psgl-1-dependent myeloid leukocyte activation. J Leukoc Biol. 2009;86:1119–1124. doi: 10.1189/jlb.0209117. [DOI] [PubMed] [Google Scholar]
- 151.Barnard MR, Linden MD, Frelinger AL, 3rd, Li Y, Fox ML, Furman MI, Michelson AD. Effects of platelet binding on whole blood flow cytometry assays of monocyte and neutrophil procoagulant activity. Journal of thrombosis and haemostasis : JTH. 2005;3:2563–2570. doi: 10.1111/j.1538-7836.2005.01603.x. [DOI] [PubMed] [Google Scholar]
- 152.Brambilla M, Camera M, Colnago D, Marenzi G, De Metrio M, Giesen PL, Balduini A, Veglia F, Gertow K, Biglioli P, Tremoli E. Tissue factor in patients with acute coronary syndromes: Expression in platelets, leukocytes, and platelet-leukocyte aggregates. Arteriosclerosis, thrombosis, and vascular biology. 2008;28:947–953. doi: 10.1161/ATVBAHA.107.161471. [DOI] [PubMed] [Google Scholar]
- 153.Goel MS, Diamond SL. Neutrophil enhancement of fibrin deposition under flow through platelet-dependent and -independent mechanisms. Arteriosclerosis, thrombosis, and vascular biology. 2001;21:2093–2098. doi: 10.1161/hq1201.100255. [DOI] [PubMed] [Google Scholar]
- 154.Polanowska-Grabowska R, Wallace K, Field JJ, Chen L, Marshall MA, Figler R, Gear AR, Linden J. P-selectin-mediated platelet-neutrophil aggregate formation activates neutrophils in mouse and human sickle cell disease. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:2392–2399. doi: 10.1161/ATVBAHA.110.211615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Brinkmann V, Zychlinsky A. Neutrophil extracellular traps: Is immunity the second function of chromatin? J Cell Biol. 2012;198:773–783. doi: 10.1083/jcb.201203170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Saffarzadeh M, Preissner KT. Fighting against the dark side of neutrophil extracellular traps in disease: Manoeuvres for host protection. Curr Opin Hematol. 2013;20:3–9. doi: 10.1097/MOH.0b013e32835a0025. [DOI] [PubMed] [Google Scholar]
- 157.McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell host & microbe. 2012;12:324–333. doi: 10.1016/j.chom.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 158.Looney MR, Nguyen JX, Hu Y, Van Ziffle JA, Lowell CA, Matthay MA. Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. J Clin Invest. 2009;119:3450–3461. doi: 10.1172/JCI38432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, Toy P, Werb Z, Looney MR. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest. 2012;122:2661–2671. doi: 10.1172/JCI61303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ganz T. Defensins: Antimicrobial peptides of innate immunity. Nat Rev Immunol. 2003;3:710–720. doi: 10.1038/nri1180. [DOI] [PubMed] [Google Scholar]
- 161.Elzey BD, Ratliff TL, Sowa JM, Crist SA. Platelet cd40l at the interface of adaptive immunity. Thrombosis research. 2011;127:180–183. doi: 10.1016/j.thromres.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Beaulieu LM, Freedman JE. Inhibition of platelet function by endothelium. In: Michelson A, editor. Platelets. 3rd edition Elsevier; London: 2013. pp. 313–342. [Google Scholar]
- 163.Goerge T, Ho-Tin-Noe B, Carbo C, Benarafa C, Remold-O’Donnell E, Zhao BQ, Cifuni SM, Wagner DD. Inflammation induces hemorrhage in thrombocytopenia. Blood. 2008;111:4958–4964. doi: 10.1182/blood-2007-11-123620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ho-Tin-Noe B, Demers M, Wagner DD. How platelets safeguard vascular integrity. Journal of thrombosis and haemostasis : JTH. 2011;9(Suppl 1):56–65. doi: 10.1111/j.1538-7836.2011.04317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Boulaftali Y, Hess PR, Getz TM, Cholka A, Stolla M, Mackman N, Owens AP, 3rd, Ware J, Kahn ML, Bergmeier W. Platelet itam signaling is critical for vascular integrity in inflammation. J Clin Invest. 2013 doi: 10.1172/JCI65154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. 2012;122:2731–2740. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zarbock A, Singbartl K, Ley K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J Clin Invest. 2006;116:3211–3219. doi: 10.1172/JCI29499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Braunstein PW, Cuenoud HF, Joris I, Majno G. Platelets, fibroblasts, and inflammation: Tissue reactions to platelets injected subcutaneously. Am J Pathol. 1980;99:53–66. [PMC free article] [PubMed] [Google Scholar]
- 169.Kim MH, Curry FR, Simon SI. Dynamics of neutrophil extravasation and vascular permeability are uncoupled during aseptic cutaneous wounding. American journal of physiology. Cell physiology. 2009;296:C848–856. doi: 10.1152/ajpcell.00520.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Evangelista V, Smyth SS. Interactions between platelets, leukocytes, and endothelium. In: Michelson A, editor. Platelets. 3rd edition Elsevier; London: 2013. pp. 295–312. [Google Scholar]
- 171.Risitano A, Beaulieu LM, Vitseva O, Freedman JE. Platelets and platelet-like particles mediate intercellular rna transfer. Blood. 2012;119:6288–6295. doi: 10.1182/blood-2011-12-396440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Italiano JE, Jr., Mairuhu AT, Flaumenhaft R. Clinical relevance of microparticles from platelets and megakaryocytes. Curr Opin Hematol. 2010;17:578–584. doi: 10.1097/MOH.0b013e32833e77ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Barry OP, Pratico D, Lawson JA, FitzGerald GA. Transcellular activation of platelets and endothelial cells by bioactive lipids in platelet microparticles. J Clin Invest. 1997;99:2118–2127. doi: 10.1172/JCI119385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Barry OP, Pratico D, Savani RC, FitzGerald GA. Modulation of monocyte-endothelial cell interactions by platelet microparticles. J Clin Invest. 1998;102:136–144. doi: 10.1172/JCI2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Mause SF, von Hundelshausen P, Zernecke A, Koenen RR, Weber C. Platelet microparticles: A transcellular delivery system for rantes promoting monocyte recruitment on endothelium. Arteriosclerosis, thrombosis, and vascular biology. 2005;25:1512–1518. doi: 10.1161/01.ATV.0000170133.43608.37. [DOI] [PubMed] [Google Scholar]
- 176.Sprague DL, Elzey BD, Crist SA, Waldschmidt TJ, Jensen RJ, Ratliff TL. Platelet-mediated modulation of adaptive immunity: Unique delivery of cd154 signal by platelet-derived membrane vesicles. Blood. 2008;111:5028–5036. doi: 10.1182/blood-2007-06-097410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8:776–787. doi: 10.1038/nri2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Goldenberg NM, Steinberg BE, Slutsky AS, Lee WL. Broken barriers: A new take on sepsis pathogenesis. Science translational medicine. 2011;3:88ps25. doi: 10.1126/scitranslmed.3002011. [DOI] [PubMed] [Google Scholar]
- 179.Li Z, Yang F, Dunn S, Gross AK, Smyth SS. Platelets as immune mediators: Their role in host defense responses and sepsis. Thrombosis research. 2011;127:184–188. doi: 10.1016/j.thromres.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Shi G, Morrell CN. Platelets as initiators and mediators of inflammation at the vessel wall. Thrombosis research. 2011;127:387–390. doi: 10.1016/j.thromres.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Pagnoux C, Cohen P, Guillevin L. Vasculitides secondary to infections. Clinical and experimental rheumatology. 2006;24:S71–81. [PubMed] [Google Scholar]
- 182.Schwertz H, Weyrich AS, Zimmerman GA. Platelets, athersclerosis, and immunity. Encyclopedia of medical immunology. 2013 in press. [Google Scholar]
- 183.Nieswandt B, Pleines I, Bender M. Platelet adhesion and activation mechanisms in arterial thrombosis and ischaemic stroke. Journal of thrombosis and haemostasis : JTH. 2011;9(Suppl 1):92–104. doi: 10.1111/j.1538-7836.2011.04361.x. [DOI] [PubMed] [Google Scholar]
- 184.Harris ES, Rondina MT, Schwertz H, Weyrich AS, Zimmerman GA. Pathogenesis of sepsis and sepsis-induced acute lung injury. In: Choi AMK, editor. Acute respiratory distress syndrome. 2nd edition Informa Healthcare; New York, London: 2010. pp. 369–419. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Online Table I. Activation and Signaling Pathways of Human Platelets Mediate Both Hemostasis and Inflammation1-17
Online Table II. Functional Responses of Human and Mouse Platelets Induced by LPS In Vitro18-28
Online Table III. Activated Human Platelets Have Diverse Mechanisms for Release or Surface Display of Inflammatory Factors1-3, 16, 29-32
Online Table IV. Examples of Signal Dependent Translation by Human Platelets that Yields Proteins with Relevance to Thrombosis and Inflammation1, 2, 12, 27, 33-44
Online Table V. Platelet Activities in Selected Inflammatory Vasculopathies