

Abstract
Background
Trypanosoma cruzi is a protist parasite that causes Chagas disease. Several proteins that are essential for parasite virulence and involved in host immune responses are anchored to the membrane through glycosylphosphatidylinositol (GPI) molecules. In addition, T. cruzi GPI anchors have immunostimulatory activities, including the ability to stimulate the synthesis of cytokines by innate immune cells. Therefore, T. cruzi genes related to GPI anchor biosynthesis constitute potential new targets for the development of better therapies against Chagas disease.
Methodology/Principal Findings
In silico analysis of the T. cruzi genome resulted in the identification of 18 genes encoding proteins of the GPI biosynthetic pathway as well as the inositolphosphorylceramide (IPC) synthase gene. Expression of GFP fusions of some of these proteins in T. cruzi epimastigotes showed that they localize in the endoplasmic reticulum (ER). Expression analyses of two genes indicated that they are constitutively expressed in all stages of the parasite life cycle. T. cruzi genes TcDPM1, TcGPI10 and TcGPI12 complement conditional yeast mutants in GPI biosynthesis. Attempts to generate T. cruzi knockouts for three genes were unsuccessful, suggesting that GPI may be an essential component of the parasite. Regarding TcGPI8, which encodes the catalytic subunit of the transamidase complex, although we were able to generate single allele knockout mutants, attempts to disrupt both alleles failed, resulting instead in parasites that have undergone genomic recombination and maintained at least one active copy of the gene.
Conclusions/Significance
Analyses of T. cruzi sequences encoding components of the GPI biosynthetic pathway indicated that they are essential genes involved in key aspects of host-parasite interactions. Complementation assays of yeast mutants with these T. cruzi genes resulted in yeast cell lines that can now be employed in high throughput screenings of drugs against this parasite.
Author Summary
Chagas disease, considered one of the most neglected tropical diseases, is caused by the blood-borne parasite Trypanosoma cruzi and currently affects about 8 million people in Latin America. T. cruzi can be transmitted by insect vectors, blood transfusion, organ transplantation and mother-to-baby as well as through ingestion of contaminated food. Although T. cruzi causes life-long infections that can result in serious damage to the heart, the two drugs currently available to treat Chagas disease, benznidazole and nifurtimox, which have been used for more than 40 years, have proven efficacy only during the acute phase of the disease. Thus, there is an urgent need to develop new drugs that are more targeted, less toxic, and more effective against this parasite. Here we described the characterization of T. cruzi genes involved in the biosynthesis of GPI anchors, a molecule responsible for holding different types of glycoproteins on the parasite membrane. Since GPI anchored proteins are essential molecules T. cruzi uses during infection, besides helping understand how this parasite interacts with its host, this work may contribute to the development of better therapies against Chagas disease.
Introduction
Glycosylphosphatidylinositol (GPI) is an abundant component of the plasma membrane of protist parasites. In most eukaryotic cells, GPIs are found as free molecules or as lipid anchor for proteins that are bound to the cell surface [1]. They are complex molecules that are synthesized in the ER by sequential addition of sugar residues and other substituents, e.g. ethanolamine-phosphate, to the phosphatidylinositol (PI) precursor and transported to the cell surface, as a free GPI also known as GIPL (glycoinositolphospholipid) or linked to the C-terminus of a protein that contains a GPI signal sequence [2]. Numerous studies with different parasites clearly show that GIPLs and GPI-anchored proteins play important roles in different processes related to host-parasite interaction. Also, it has been suggested that, because of the existence of differences in the structure of GPI from several parasite species as well as between GPIs of the parasite and their host cells [2], [3], [4], these molecules constitute promising targets for studies towards the development of new anti-microbial drugs [5].
Trypanosoma cruzi is a parasitic protist that causes Chagas disease, an illness not only prevalent in Latin America, where an estimated 8 million people are infected, but a worldwide health issue for which there is an urgent need for the development of new chemotherapeutic agents and more effective prophylactic methods (www.who.int/topics/chagas_disease/en/). The surface of T. cruzi is covered by a large amount of GPI-anchored proteins whose structure and chemical composition have been extensively studied [6] and are expressed in all developmental stages of the parasite life cycle [3], [7]. Analysis of the T. cruzi genome indicated that 12% of the parasite genes encode proteins anchored by GPI, a percentage that is much higher when compared with other organisms [8]. Many of these proteins play important roles in the invasion process and, since they show varying sequences, they could also participate in the processes responsible for evasion of the host immune response [9], [10]. Two main components of the T. cruzi surface, the trans-sialidases and mucins, which act, respectively, as enzymes responsible for the transfer and acceptors for sialic acid molecules, are GPI-anchored glycoproteins [11]. It has also been demonstrated that T. cruzi GPI-anchored mucins as well as free GPI anchors act as potent pro-inflammatory agents that are recognized by Toll like receptors [12] and, because of their role in activating the innate immune response, they have been used as adjuvants in immunization protocols [13].
In Saccharomyces cerevisiae, biosynthesis of GPI is essential for cell growth and occurs in eleven steps beginning with the transfer of a molecule of N-acetyl-glucosamine (GlcNAc) from UDP-GlcNAc to PI [14], [15]. After the addition of mannose molecules using dolichol-P-mannose as a donor, followed by the transfer of ethanolamine-phosphate (EtNP) to the third mannose residue, GPI is transferred to proteins that have a predicted GPI addition signal at their C-terminal end, in a reaction catalyzed by the GPI-transamidase complex [16]. Genes encoding enzymes involved in GPI pathway from various organisms, including protist parasites such as Trypanosoma brucei, Leishmania mexicana and Plasmodium falciparum have been cloned and their products characterized by functional complementation in mammalian cells and in yeast mutants [17], [18], [19], [20]. Although the main structure of GPI is conserved in all organisms, several studies have shown differences in the biosynthetic pathway and additional modifications to GPI structures present in mammalian and parasite cells [2], [3], [4]. Substrate analogues of enzymes of the GPI biosynthetic pathway showing trypanocidal activity have been described [21]. Since enzymes involved in the basic steps common to the biosynthesis of GPI in the different organisms have different sensitivities to various inhibitors [22], [23], [24], [25], [26], [27], we sought to characterize the genes involved in biosynthesis of GPI anchors in T. cruzi. Orthologous sequences of all genes involved in biosynthesis of T. cruzi GPI anchors were identified and, for three of them, we were able to show that they complement yeast conditional mutants of genes of this pathway. Unsuccessful attempts to generate T. cruzi knockouts for three of these genes suggest that GPI is an essential component of the parasite. Since specific inhibition of GPI biosynthesis may affect the expression of a large number of T. cruzi proteins that are essential for host-parasite interactions, targeting this pathway can be considered a promising strategy for the development of new chemotherapy against Chagas disease. The availability of yeast mutants expressing T. cruzi enzymes constitutes the first step in that direction.
Methods
Parasite cultures
Epimastigotes of the CL Brener clone of T. cruzi were maintained in logarithmic growth phase at 28°C in liver infusion tryptose (LIT) medium supplemented with 10% fetal bovine serum as described by Camargo [28]. Metacyclic trypomastigotes were obtained after metacyclogenesis in LIT medium, observed after 15–20 days of culture [28] and were used to infect Vero cells. Intracellular amastigotes and tissue culture derived trypomastigotes were obtained from Vero cells grown in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 5% fetal bovine serum, at 37°C and 5% CO2 as previously described [29].
In silico analysis of T. cruzi genes
Sequence analyses were conducted using the T. cruzi genome database (www.tritrypdb.org) to identify all orthologous genes involved in the parasite GPI biosynthesis. Sequences from different organisms, such as T. brucei, P. falciparum and S. cerevisiae [16], [20], were used as queries in Blastp analyses (www.ncbi.nlm.nih.gov/blast/Blast.cgi) and ClustalW (www.clustal.org/) for multiple alignments between the predicted T. cruzi protein sequences and homologous sequences present in other organisms.
DNA and RNA extraction, northern blot and RT-PCR assays
Total DNA was purified from 109 T. cruzi epimastigotes that were harvested from exponentially growing cultures, according to previously described protocols [29]. Total RNA was isolated from epimastigotes, tissue culture derived trypomastigotes and amastigotes using the RNeasy kit (Qiagen). For northern blot analyses, 10 µg of total RNA/lane was separated in 1.2% agarose/MOPS/formaldehyde gel. The RNA was transferred to Hybond-N membrane (GE-Healthcare) and hybridized with GPI8, GPI10 and 24Sα rRNA probes previously labeled with [α-32P]-dCTP using the Amersham Ready-to-Go DNA Labeling Beads (GE-Healthcare), according to the suppliers protocol. The hybridization was carried out as previously described [30] in 50% formamide buffer at 42°C. After washing twice with 2X SSC/0.2% SDS at 60°C for 20 min, the membranes were exposed to a phosphor screen of the STORM 820 phosphor image (GE-Healthcare). Reverse-transcription amplifications (RT-PCR) were carried out with total RNA isolated from transfected yeast mutants and T. cruzi epimastigotes according to published protocols [30]. After first strand cDNA synthesis using oligo (dT)18 or gene-specific primers (see primer sequences in supplementary material, Table S1) and the SuperScript II Reverse Transcriptase (Life Technologies), the cDNAs were amplified using Taq Polymerase (Promega) and primers specific for each gene and analyzed in 1% agarose gels stained with ethidium bromide.
Yeast strains and culture media
The S. cerevisiae strain used in this work were: YPH499 (Mat a, ura3-52, lys2-801amber, ade2-101ochre, trp1-63, his3-200, leu2-1) (Stratagene), used as a control, and conditional lethal yeast mutants for GPI biosynthesis (YPH499-HIS-GAL-DPM1, YPH499-HIS-GAL-GPI3, YPH499-HIS-GAL-GPI8, YPH499-HIS-GAL-GPI10, YPH499-HIS-GAL-GPI12, YPH499-HIS-GAL-GPI14, YPH499-HIS-GAL-GAA1, and YPH499-HIS-GAL-AUR1), which were generated by replacement of the endogenous yeast promoter by a galactose regulated promoter, as described [31]. S. cerevisiae strains were grown in YPGR medium (1% w/v yeast extract, 2% w/v bacto-peptone, 2% w/v galactose, 1% w/v raffinose), or in SD medium (0.17% yeast nitrogen base, 0.5% ammonium sulfate, 2% glucose, containing the nutritional supplements necessary to complement the auxotrophic samples or to allow selection of transformants). Before complementation, yeast clones were cultivated in SGR medium (4% galactose, 2% raffinose, 0.17% yeast nitrogen base, 0.5% ammonium sulfate) in which glucose is replaced by galactose/raffinose as a carbon source.
Transformation of conditional lethal S. cerevisiae mutants
Sequences encompassing the full-length coding regions of TcDPM1, TcGPI3, TcGPI8, TcGPI10, TcGPI12, TcGPI14, TcGAA-1, and TcIPCS were PCR amplified from total DNA of T. cruzi epimastigotes prepared as described above, using primers specific for each gene (Table S1). The amplicons were inserted into the S. cerevisiae expression vector pRS426Met [32]. Full-length coding sequences corresponding to orthologous S. cerevisiae genes were also PCR amplified with specific primers (Table S1) and cloned into the same vector. Transformation of yeast mutants were carried out using the standard lithium acetate procedure [33]. Conditional lethal mutants were transformed with pRS426Met plasmids carrying either the S. cerevisiae (Sc) or the T. cruzi (Tc) genes and transformed cells were plated on minimal medium lacking histidine and uracil containing either galactose (SGR) or glucose (SD) and incubated at 30°C.
SDS-PAGE of [2-3H]myo-inositol labeled yeast proteins
Control YPH499 cells, mutant yeasts (YPH499-HIS-GAL) and mutant yeasts carrying pRS426Met containing yeast or T. cruzi genes were grown in SGR to saturation and used to inoculate SD (2% glucose), in which they were grown for about 16 h. Cells (1×108) were washed twice in SD without inositol medium (2% glucose), resuspended in 1 ml of SD without inositol (2% glucose) and depleted of inositol for 20 min before the addition of 30 µCi of [2-3H]myo-inositol (American Radiolabeled Chemicals, St. Louis, USA). Cells were labeled for 1 hour. Protein extraction was done according to Damasceno et al. [34] with the following modifications: radiolabeled cells were harvested, washed twice in phosphate-buffered saline (PBS 1X) at pH 7.4, and resuspended in 100 µl of Yeast Breaking Buffer [50 mM sodium phosphate, pH 7.4; 1 mM phenylmethylsulfonyl fluoride (PMSF); 1X protease inhibitor cocktail (Amresco, Solon, USA); 1 mM EDTA, and 5% (v/v) glycerol]. Yeast cells were lysed by the addition of acid-washed glass beads (425–600 µm) vortexing for 1 min with 1 min intervals on ice, repeated twenty times. The lysate was centrifuged at 2,000×g for 5 min at 4°C and the supernatant was collected. The remaining pellet containing cell debris and glass beads was resuspended in 75 µl of Yeast Breaking Buffer containing 2% (w/v) sodium dodecyl sulfate (SDS) by vortexing for 1 min with 1 min intervals on ice, repeated five times. After removing cellular debris by centrifugation, the lysates were combined and the proteins were then separated by 10% SDS-polyacrylamide gel electrophoresis. Protein bands containing labeled inositol were detected by fluorography.
Dol-P-Man synthase assays
Wild type and yeast mutant cell lysates were prepared as previously described [35]. Briefly, exponential-phase yeast cultures corresponding to 1.5×107 cells/ml of cells grown in glucose-containing medium (nonpermissive) or in galactose-containing medium (permissive medium) were lysed after incubation in 1.0 ml of 1 M sorbitol/1 mM EDTA containing Zymolyase at 37°C and glass beads for 30 min, harvested by centrifugation (1800×g, 10 min, 4°C) and resuspended in 200 µl of TM buffer (50 mM Tris/HCl, pH 7.5, containing 5 mM MgCl2 and 0.2% 2-mercaptoethanol). Ninety µl for lysates (corresponding to 3×108 cells for each assay) were assayed directly for Dol-P-Man synthase activity as described [36]. Briefly, incubation mixtures contained 5 µl of GDP-[3H]Man (1 µCi/ml), 1 µl of Dol-P (5 mg/ml dispersed in 1.0% Triton X-100 by sonication) and water to give a final volume of 10 µl. Amphomycin and tunicamycin (final concentrations 1 mg/ml) were added to some samples. After the addition of 90 µl of cell lysates and incubation at 30°C for 30 min, the reactions were terminated by the addition of 1.5 ml of ice-cold chloroform/methanol (2∶1, v/v). The reactions were centrifuged (1500×g, 5 min, 4°C) and the pellet extracted twice with 500 µl of chloroform/methanol. Equivalent amounts of radiolabeled, chloroform/methanol extractable reaction products were analyzed by TLC on Silica 60 plates (Merck) with chloroform/methanol/acetic acid/water (25∶15∶4∶2, by vol.) as solvent and Dol-P-Man as a reference. Plates were screened for radioactivity with a Berthold LB 2842 Automatic TLC-Linear Analyzer.
Parasite transfections and cellular localization of GFP fusion proteins
Full-length TcDPM1, TcGPI3, and TcGPI12 coding sequences were PCR amplified from genomic DNA purified from cultures of the T. cruzi epimastigotes, using forward and reverse primers carrying XbaI and EcoRI restriction sites, respectively (Table S1). The amplicons were inserted into the XbaI-EcoRI sites of the T. cruzi expression vector pTREXnGFP [37], generating pTREX-TcDPM1-GFP, pTREX-TcGPI3-GFP, and pTREX-TcGPI12-GFP that contain TcDPM1, TcGPI3 and TcGPI12 genes fused to the N-terminus of the green fluorescent protein (GFP). A total of 100 µg of each plasmid construction was used to transfect T. cruzi epimastigotes as previously described [37]. Twenty four hours post-transfection, parasites were fixed with 4% paraformaldehyde for 30 min at 4°C, permeabilized with 0.1% Triton X-100 for 5 min at room temperature and blocked with 5% fetal bovine serum in PBS (blocking solution) for 20 min at 4°C. Staining of the parasite ER was done with rabbit anti-T. brucei BiP antibody ([38]; kindly provided by Renato Mortara, Universidade Federal de São Paulo), at a 1∶1000 dilution, and secondary goat anti-rabbit IgG antibody conjugated to Alexa Fluor 555 (1∶1000 dilution) (Molecular Probes/Life Technologies). After nuclei staining with 1 µg/ml of 4′,6-diamidino-2-phenylindole (DAPI, Molecular Probes/Life Technologies), cover slides were mounted with 90% glycerol, 10% 1 M Tris HCl pH 9.0, and 2.3% DABCO (Sigma). Images were obtained with a fluorescence microscope (Nikon Eclipse Ti) or with the 5 LIVE confocal microscope (Zeiss), both at the Center of Electron Microscopy (CEMEL), at the Instituto de Ciências Biológicas, UFMG. Transfections of HT1080 human fibrosarcoma cells were done with 1 µg of pcDNA3.1/NT-GFP-TOPO (Life Technologies) containing the different T. cruzi genes inserted in fusion with GFP (for primer sequences, see Table S1) and the FuGENE transfection reagent (Roche), following the manufacturer's instructions. All plasmids were co-transfected with pGAG-DsRed-ER, a mammalian expression vector that encodes the Discosoma sp. red fluorescent protein (DsRed) in fusion with ER targeting sequences and the ER retention sequence, KDEL (Clontech).
Disruption of T. cruzi genes
DNA constructs designed to delete both TcGPI8 alleles in the T. cruzi CL Brener genome by homologous recombination were prepared after PCR amplification of the 5′ and 3′ regions of the TcGPI8 gene (for primer sequences, see Table S1). The generated PCR products (with 487 bp and 647 bp, respectively) were cloned sequentially into the SacI/SpeI and XhoI/XbaI sites of pCR2.1 TOPO vector (Invitrogen), flanking the neomycin phosphotransferase (NeoR) or hygromycin phosphotransferase (HygR) resistance markers that were cloned into this vector. To improve mRNA expression in the parasite, the 3′ UTR plus downstream intergenic sequences of the T. cruzi gliceraldehyde-3-phosphate dehydrogenase (gapdh) gene was inserted downstream from the HygR marker. Similar constructs using 5′ and 3′ flanking sequences derived from TcGPI3 and TcGPI10 genes were generated. Epimastigote transfections were performed by electroporation with 50 µg DNA as described previously [37]. Twenty-four hours after transfection, 200 µg/ml of hygromycin B or G418 was added to the cultures and selected populations were obtained approximately 30 days after transfection. Cloned cell lines were obtained by plating on semisolid blood agar plates, after another 30 days of incubation at 28°C.
Electron microscopy analyses of T. cruzi
Epimastigotes were fixed in 5% glutaraldehyde in 0.1 M cacodylate buffer pH 7.2 and processed following standard protocols, including post-fixation in osmium tetroxide followed by block counterstained with uranyl acetate and embedding in Epon resin. Ultrathin sections were counterstaining with lead citrate and analyzed in the Transmission Electron Microscope Tecnai G2-12 - SpiritBiotwin FEI - 120 kV located at the Center of Microscopy at the Universidade Federal de Minas Gerais, Belo Horizonte, Brazil.
Cell membrane preparation, immunoblot and flow cytometry analyses
Approximately 109 epimastigotes were lysed in 20 mM Hepes, 10 mM KCl, 1.5 mM MgCl2, 250 mM sucrose, 1 mM DTT, 0.1 mM PMSF, with five cycles of freezing in liquid nitrogen and thawing at 37°C. Total cell lysate was centrifuged at a low speed (2,000×g) for 10 min and the supernatant was subjected to ultracentrifugation (100,000×g) for one hour. The resulting supernatant was analyzed as soluble, cytoplasmic fraction (C) whereas the pellet, corresponding to the membrane fraction (M) was resuspended in lysis buffer. Volumes corresponding to 20 µg of proteins from total parasite cell lysate (T), cytoplasmic (C) and membrane (M) fractions were loaded onto a 12.5% SDS-PAGE gel, transferred to nitrocellulose membranes, blocked with 5.0% non-fat dry milk and incubated with the anti-mucin antibody 2B10 (gently provided by Nobuko Yoshida, Universidade Federal de São Paulo), at 1∶200 dilution followed by incubation with peroxidase conjugated anti-mouse IgG and the ECL Plus reagent (GE-Healthcare). For flow cytometric analysis, epimastigotes were stained with anti-mucin 2B10 (dilution 1∶450) and Alexa Fluor 488 conjugated secondary antibodies. Data were acquired on a FACScan flow cytometer (Becton Dickinson).
Results
In silico identification of T. cruzi genes involved in the GPI biosynthetic pathway
Eighteen T. cruzi genes involved in 8 steps of the GPI biosynthetic pathway were identified based on their similarities to the yeast, mammals, Trypanosoma brucei and Plasmodium falciparum sequences [15], [16], [17], [20], (Table 1). For the majority of these genes, annotated as putative T. cruzi orthologs in the TriTrypDB (www.tritrypdb.org), both alleles, belonging to the two CL Brener haplotypes, were identified. Since CL Brener is a hybrid strain, as described by El-Sayed et al. [39], the two haplotypes corresponding to the two ancestral genomes that originated the CL Brener genome, named Esmeraldo-like and non-Esmeraldo-like, were separated during the T. cruzi genome assembly. In Table 1, the genes corresponding to the non-Esmeraldo haplotype were indicated by their identification numbers in the TriTrypDB database. For all listed genes, the amino acid identities between the two alleles were greater than 94%. Based on these sequences and the known structure of the GPI anchor in this parasite (Figure 1A) [3], we proposed that the T. cruzi GPI biosynthetic pathway occurs in the ER according to the diagram shown in Figure 1B.
Table 1. T. cruzi genes encoding enzymes of the GPI biosynthetic pathway.
| Step | T. cruzi gene | Gene ID (TriTrypDB*) | Number of amino acids | % Identity at protein level (**) | |
| Yeast | Human | ||||
| Dolicholphosphate-mannose synthase | DPM1 | Tc00.1047053506581.10 | 260 aa | 50% (DPM1) | 31% (DPM1) |
| N-acetyl-glucosamine transferase (GlcNAc-PI) (Step 1) | GPI1 | Tc00.1047053510329.200 | 827 aa | 15% (GPI1) | 16% (PIG-Q) |
| GPI2 | Tc00.1047053503781.20 | 336 aa | 14% (GPI2) | 32% (PIG-C) | |
| GPI3 | Tc00.1047053509215.16 | 455 aa | 41% (GPI3) | 49% (PIG-A) | |
| GPI15 | Tc00.1047053511655.10 | 307 aa | 12% (GPI15) | 18% (PIG-H) | |
| GPI19 | Tc00.1047053508307.100 | 142 aa | 10% (GPI19) | 27% (PIG-P) | |
| DPM2 | Tc00.1047053510043.29 | 100 aa | – | 17% (DPM2) | |
| GlcNAc-PI de-N-acetylase (Step 2) | GPI12 | Tc00.1047053511481.40 | 252 aa | 31% (GPI12) | 32% (PIG-L) |
| α-1,4-Mannosyltransferase I (Step 3) | GPI14 | Tc00.1047053511507.50 | 472 aa | 30% (GPI14) | 30% (PIG-M) |
| α-1,6-Mannosyltransferase II (Step 4) | GPI18 | Tc00.1047053503521.89 | 529 aa | 17% (GPI18) | 20% (PIG-V) |
| α-1,2-Mannosyltransferase III (Step 5) | GPI10 | Tc00.1047053510299.50 | 584 aa | 21% (GPI10) | 24% (PIG-B) |
| α-1,2-Mannosyltransferase IV (?) (Step 6) | ni | ni | – (SMP3) | – (PIG-Z) | |
| Ethanolamine phosphotransferase (Step 7) | GPI13 | Tc00.1047053503979.10 | 691 aa | 15% (GPI13) | 21% (PIG-O) |
| GPI transamidase (Step 8) | GAA1 | Tc00.1047053504069.60 | 462 aa | 10% (GAA1) | 13% (GAA1) |
| GPI8 | Tc00.1047053511277.450 | 325 aa | 31% (GPI8) | 29% (PIG-K) | |
| GPI16 | Tc00.1047053510877.180 | 684 aa | 8% (GPI16) | 9% (PIG-T) | |
| TTA1 | Tc00.1047053510435.40 | 387 aa | – | – | |
| TTA2 | Tc00.1047053508661.60 | 414 aa | – | – | |
| GPI-inositol deacylase | GPIdeAc2 | Tc00.1047053508153.1040 | 804 aa | 9% (BST1) | 12% (PGAP1) |
| Inositol phosphorylceramide synthase | IPCS | Tc00.1047053510729.290 | 335 aa | 10% (AUR1) | – |
(*) Gene ID numbers refer to the non-Esmeraldo-like haplotype, except for TcGPI16 and TcGPI19, for which only the Esmeraldo-like alleles were identified.
(**) Names for the yeast and human orthologs are shown in parentheses.
ni: not identified.
Figure 1. Structure and the biosynthesis of T. cruzi GPI anchors.

(A) Structure of a T. cruzi GPI anchor, according to Previato et al. [3]. (B) Proposed biosynthetic pathway of GPI anchor in the endoplasmic reticulum of T. cruzi. N-acetylglucosamine (GlcNAc) is added to phosphatidylinositol (PI) in step 1 and, during the following steps, deacetylation and addition of four mannose residues occur. The addition of ethanolamine-phosphate on the third mannose (step 7) enables the transferring of the completed GPI anchor to the C-terminal of a protein (step 8). Dolichol-P-mannose acts as a mannose donor for all mannosylation reactions that are part of the GPI biosynthesis. This pathway was based on the structure of the T. cruzi GPI and sequence homology of T. cruzi genes with genes known to encode components of this pathway in Saccharomyces cerevisiae, Homo sapiens, Trypanosoma brucei and Plasmodium falciparum. Not shown in the figure, free glycoinositolphospholipids (GIPLs), also present in the T. cruzi membrane, are likely to be by-products of the same GPI biosynthetic pathway.
Dolichol-phosphate mannose synthase (DPM1), also named dolichol-phosphate-β-D-mannosyltransferase, catalyses the transfer of a mannose residue from GDP-mannose to dolichol-phosphate (Dol-P) generating Dol-P-mannose, used as a donor for all mannosylation reactions that are part of the GPI biosynthetic pathway [40], [41]. Comparisons among DPM1 of various organisms [42], [43], [44] showed that, together with S. cerevisiae, T. brucei, and Leishmania mexicana [45] and in contrast to P. falciparum DPM1, T. cruzi DPM1 belongs to a group that includes monomeric enzymes that have a C-terminal hydrophobic tail. The glycosyltransferase complex that is responsible for transferring N-acetylglucosamine (GlcNAc) from UDP-GlcNAc to phosphatidylinositol (PI) to generate N-acetylglucosaminyl-PI (GlcNAc-PI) has six and seven proteins, respectively, in yeast and mammalian cells [16]. TcGPI3 was identified as the gene encoding the catalytic subunit of the T. cruzi glycosyltransferase complex since it shares 41% and 49% of sequence identity with the yeast GPI3 and mammalian PIG-A, respectively. Among other components of the glycosyltransferase complex present in yeast, we identified the T. cruzi orthologs of GPI1, GPI2, GPI15, and GPI19. In mammalian cells, DPM2, a non-catalytic subunit of dolichol-P-mannose synthase, is physically associated with PIG-A, PIG-C and PIG-Q and enhances GlcNAc-PI transferase activity [46]. A T. cruzi gene encoding a protein with 17% identity to human DPM2 and containing a DPM2 domain, which probably acts as a regulatory component of the N-acetyl-glucosamine transferase complex, was also identified. Only one component of this complex, named ERI1 in yeast [47], and PIG-Y in mammals [48], was not identified either in T. cruzi, P. falciparum or T. brucei. The T. cruzi ortholog of yeast GPI12 (named PIG-L in mammals) [49], encoding the enzyme responsible for the de-N-acetylation of GlcNAc-PI, which has been well characterized in T. brucei [50], [51], was also identified. Since differences in substrate recognition among the mammal and T. brucei enzyme have been described [52], this enzyme has been considered as a suitable target for drug development.
As depicted in Figure 1B, the first two reactions of the GPI biosynthetic pathway occur on the cytoplasmic face of the ER, whereas mannosylation reactions occur in the ER lumen. After deacetylation, the GPI precursor is transported across the ER membrane to the ER lumen, a step that requires distinct flippases [53]. In yeast and mammalian cells, the addition of mannose residues to GlcN-PI after flipping this precursor into the ER lumen requires acylation of the inositol ring and, after mannosylation and the attachment of GPIs to proteins, this group is removed [54]. In contrast, in T. brucei, inositol acylation occurs after the addition of the first mannose residue [55] since both acylated and non-acylated GPI intermediates exist during transfer of the Man2 and Man3 to GPI intermediates [56]. Although analyses of GPI precursors synthesized in T. cruzi cell-free systems indicated that this organism also has the ability to acylate the inositol ring [57], sequences encoding an enzyme responsible for acylation of the inositol ring, named PIG-W in mammals and GWT1 in yeast [54], [58] were not identified either in T. cruzi or in T. brucei [2]. In spite of that, the two alleles encoding the ortholog of the enzyme responsible for inositol deacylation, named GPIdeAc2 in T. brucei [56], were found in the T. cruzi genome (Tc00.1047053508153.1040 and Tc00.1047053506691.22).
All three genes encoding mannosyltransferases, responsible for the addition of the first, second and third mannose residues to GlcN-PI, named TcGPI14 (α-1,4-mannosyltransferase), TcGPI18 (α-1,6-mannosyltransferase) and TcGPI10 (α-1,2-mannosyltransferase), were identified in the T. cruzi genome. Since the predicted T. cruzi proteins exhibit sequence identities with yeast and human proteins ranging from 17% to 30%, for some of these genes, functional assays are necessary to confirm these predictions. It is noteworthy that no T. cruzi ortholog encoding the enzyme responsible for the addition of the fourth residue of mannose (step 6), named SMP3 in yeast and PIG-Z in human, was identified. Similarly, no ortholog of the SMP3 gene was found in P. falciparum, even though the presence of a fourth mannose residue has been shown by structural studies of the GPI anchor from both organisms [3], [20], [59]. Furthermore, genes encoding an essential component of the mannosyltransferase I complex named PBN1 in yeast and PIG-X in mammals, have not been identified either in T. cruzi or in T. brucei [60], [61].
In mammals and yeasts there are three enzymes that add ethanolamine-phosphate (EtNP) to different mannose residues: PIG-N/MCD4 (EtNP addition to Man1), PIG-G/GPI7 (Man2), and PIG-O/GPI13 (Man3) [2], resulting in the structure to which the protein will be linked. In T. cruzi, T. brucei and P. falciparum, EtNP addition occurs only at the third mannose [2], [20] and, as expected, only a T. cruzi GPI13 ortholog was identified. However, it has also been shown in different T. cruzi strains, that GPI-linked proteins as well as free GIPLs have 2-aminoethylphosphonate (AEP) replacing EtNP at the third mannose residue and that an additional AEP is linked to GlcN in T. cruzi GPI anchors (for recent reviews, see [62], [63]).
After being assembled, the transfer of the GPI anchor to the C-terminal end of a protein is mediated by a transamidase complex that cleaves the GPI-attachment signal peptide of the nascent protein. In human and yeast, this complex consists of five ER membrane proteins, PIG-K/GPI8, PIG-T/GPI16, PIG-S/GPI17, PIG-U/GAB1 and GAA1 [64] in which GPI8 is considered the catalytic subunit [16], [65]. As shown in Table 1, we identified T. cruzi GPI8, GAA1 and GPI16 orthologs. Although orthologs of GPI17 and GAB1 were not identified in other trypanosomatids, genes encoding two other components of the transamidase complex, known as trypanosomatid transamidase 1 (TTA1) and TTA2, were also found in T. cruzi [66].
Besides differences in the glycan core, in T. cruzi GPI anchors, the phosphatidylinositol (PI) is replaced by inositolphosphorylceramide (IPC), a molecule also present in plants, fungi but not present in mammals [4]. This change in the lipid portion of the anchor occurs during the differentiation of epimastigotes into metacyclic trypomastigotes [67] and is observed in members of the large family of trans-sialidases [68]. Although it may not be considered part of the GPI biosynthetic pathway, the T. cruzi IPC synthase (TcIPCS) is thought to be a highly attractive drug target [69]. Based on that, Denny and collaborators [70] identified the ortholog of AUR1, that encodes the yeast IPC synthase [71], in Leishmania major and two closely related T. cruzi sequences encoding proteins sharing 52–53% identity with the Leishmania IPC synthase [70]. Our analysis confirmed that the two sequences described by Denny and collaborators [70] correspond to the two alleles of the T. cruzi IPC synthase (TcIPCS) gene present in the CL Brener genome, which are synthenic with the L. major and T. brucei orthologs.
mRNA expression and subcellular localization analyses of T. cruzi enzymes
To verify whether the genes identified through the in silico analyses described above are expressed in T. cruzi, sequences encoding two enzymes of the GPI biosynthetic pathway were used as probes in northern blot hybridizations performed with total RNA purified from epimastigote, trypomastigote and amastigote forms of the parasite. As shown in Figure 2, transcripts with 1,300 nt and 2,100 nt, approximately, corresponding to TcGPI8 and TcGPI10 mRNAs were detected in all three stages of the parasite life cycle. As expected, increased levels of both transcripts were found in the two proliferative stages, epimastigotes and amastigotes, compared to the infective, nonproliferative trypomastigote stage.
Figure 2. mRNA expression of T. cruzi genes encoding enzymes of the GPI biosynthetic pathway.
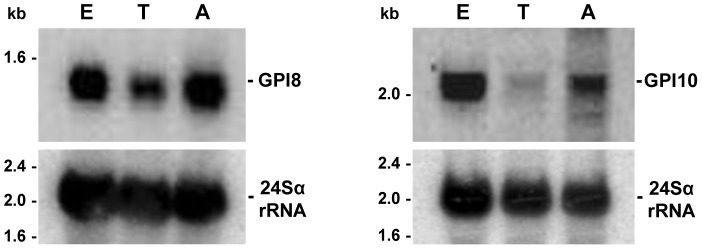
Total RNA extracted from epimastigotes (E), trypomastigotes (T) and amastigotes (A) were separated in agarose gels, transferred to nylon membranes and hybridized with [α-32P]-labeled probes specific for TcGPI8 and TcGPI10 genes. The bottom panel shows hybridization with a probe for 24Sα rRNA, used as loading control. The size of ribosomal RNA bands are indicated on the left.
To provide further evidence for the role of the proteins encoded by the T. cruzi genes identified through in silico analyses as components of the GPI biosynthetic pathway, we determined the subcellular localization of three of these proteins expressed as GFP fusion in T. cruzi epimastigotes. The coding regions of TcDPM1, TcGPI3 and TcGPI12 genes were cloned in the T. cruzi expression vector pTREXnGFP and, after transfection into epimastigotes, the cells were examined by fluorescence microscopy. Figure 3 shows that all three fusion proteins in transfected parasites that were stained with anti-BiP antibodies [38] co-localize with BiP, a known ER marker. Similar results were obtained with confocal microscopy analyses (not shown), thus confirming that these enzymes are part of the GPI biosynthetic pathway. In addition, transfection of T. cruzi genes TcDPM1, TcGPI3, TcGPI8 and TcGPI12 in fusion with GFP in the HT1080 human fibrosarcoma cells also resulted in the expression of the GFP fusion T. cruzi proteins with a cellular localization compatible with the ER (Figure S1).
Figure 3. Cellular localization of T. cruzi enzymes of the GPI biosynthetic pathway.

Epimastigotes were transiently transfected with the plasmids pTREX-TcDPM1-GFP (A), pTREX-TcGPI3-GFP (B), pTREX-TcGPI12-GFP (C) or pTREXnGFP as a control plasmid (D) and (E). Transfected parasites were fixed with 4% paraformaldehyde, incubated with the ER marker anti-BiP (1∶1000) and the secondary antibody conjugated to Alexa 555 (1∶1000). Cells were also stained with DAPI showing the nuclear and kinetoplast DNA. In panel E, parasites that were not incubated with the primary, anti-BiP antibody are shown as negative controls. Images were captured with the Nikon Eclipse Ti fluorescence microscope. Scale bars: 5 µm.
Functional analyses of T. cruzi genes expressed in yeast
One of the main goals of this work is to develop a strategy for high-throughput screening of drugs against T. cruzi enzymes involved in the GPI biosynthetic pathway. S. cerevisiae has been largely used as surrogate system to express heterologous proteins from diverse parasites including Leishmania spp and T. brucei. Therefore, not only to assay for the functions of the T. cruzi genes but also to create yeast cells expressing T. cruzi target enzymes for future drug studies, conditional lethal yeast mutants were transformed with an expression vector containing the coding sequences for the T. cruzi genes TcDPM1, TcGPI3, TcGPI12, TcGPI14, TcGPI10, TcGAA1, TcGPI8 as well as with the TcIPCS. These mutants were constructed by replacing the endogenous promoter of each one of the GPI genes by the GAL 1 promoter, resulting in yeast cell lines that could only grow in the presence of galactose [31]. By inhibiting the expression of the endogenous GPI genes in medium containing glucose, the complementation of yeast cells with the T. cruzi genes can be easily accessed by comparing the growth of transformed colonies in glucose and galactose-containing medium. As shown in Figure 4A and Table 2, we tested eight T. cruzi genes for which yeast mutants were available. Three of them, TcDPM1, TcGPI10 and TcGPI12, once transformed into yeast, allowed the yeast mutants to grow on plates containing glucose as well as galactose. For all tested yeast mutants, we verified that transformation with plasmids containing the orthologous yeast gene allows them to grow on glucose-containing medium. Figure 4A also shows that, when the mutants were plated on glucose-containing medium supplemented with uracil, none of them were able to grow. As expected, wild type yeast, which has histidine deficiency, does not grow in minimum media lacking histidine. As an additional control, we verified, by RT-PCR analyses, the expression of two T. cruzi genes transformed into yeast mutants, for which we did not observed the complementation, i.e., that did not grow in nonpermissive media. Transcripts derived from the T. cruzi TcGPI8 or TcIPCS genes, as well as from the orthologous yeast genes, were detected in the corresponding yeast mutants growing in galactose-containing media (Figure S2), indicating that the inability of these mutants to grow in the presence of glucose is not due to the lack of expression of the T. cruzi genes in the transfected yeasts.
Figure 4. Yeast complementation with T. cruzi genes encoding enzymes of the GPI biosynthetic pathway.

(A) DPM1, GPI10 and GPI12 yeast conditional lethal mutants (YPH499-HIS-GAL-DPM1, YPH499-HIS-GAL-GPI10 and YPH499-HIS-GAL-GPI12, respectively) were transformed with pRS426Met plasmids carrying either T. cruzi or S. cerevisiae genes encoding DPM1, GPI10 and GPI12 (TcDPM1 or ScDPM1, TcGPI10 or ScGPI10, and TcGPI12 or ScGPI12, respectively). Wild-type (WT), non-transformed mutants and transformed yeast mutants were streaked onto plates with nonpermissive, glucose-containing SD medium lacking histidine, with or without uracil or in galactose-containing medium (with uracil) and incubated at 30°C for 3 days. In the bottom panel, yeast mutants (YPH499-HIS-GAL-GPI14) transformed with pRS426Met plasmid carrying T. cruzi gene (TcGPI14), which could not restore cell growth of GPI14 deficient yeast are shown. (B) GPI-anchored proteins synthesized by the conditional lethal yeast mutants expressing T. cruzi genes were separated by SDS-PAGE and analyzed after fluorography. Wild-type (WT), non-transformed yeast mutants and yeast mutants that were transformed with plasmids containing the corresponding yeast genes (ScDPM1 or ScGPI12) or with the T. cruzi genes (TcDPM1 or TcGPI12), were cultivated in medium glucose-containing in the presence of [2-3H]myo-inositol for 1 hour. Total protein extract corresponding to 1×108 cells were loaded on each lane of a 10% SDS-PAGE and the labeled proteins were visualized by fluorography (top panels). As a loading control, Coomassie Blue stained gels prepared with equivalents amounts of total proteins are shown in the bottom panels. Untransfected DPM1 and GPI12 mutants were grown in the presence of galactose for 2 days and then switched to glucose-containing medium for 16 hours before addition of [2-3H]myo-inositol. Molecular weight markers (M) are shown on the left.
Table 2. Functional complementation of yeast mutants by T. cruzi genes.
| Yeast mutants | pRS Tc |
| YPH499 DPM1 | + |
| YPH499 GPI3 | − |
| YPH499 GPI12 | + |
| YPH499 GPI14 | − |
| YPH499 GPI10 | + |
| YPH499 GAA1 | − |
| YPH499 GPI8 | − |
| YPH499 AUR1 | − |
The (+) signs indicate the ability of transformed mutants to grow in nonpermissive glucose-containing media.
To evaluate whether the expression of T. cruzi enzymes in yeast results in the correct synthesis of GPI anchor precursors by the complemented mutants, SDS-PAGE and fluorography analyses of yeast proteins containing [2-3H]myo-inositol were performed. As shown in Figure 4B, after 1 hour growing in medium containing glucose and [2-3H]myo-inositol, a complex pattern of proteins is visualized by fluorography in wild type cells as well as in yeast mutants expressing the T. cruzi genes. The protein patterns in yeast mutants expressing TcDPM1 and TcGPI12 genes growing in glucose-containing medium were indeed indistinguishable from the pattern observed with molecules synthesized by wild type yeasts or by mutants transformed with the orthologous yeast genes. On the other hand, a much weaker signal was detected in non-transformed yeast mutants, indicating that the expression of T. cruzi orthologs encoding enzymes of the GPI biosynthetic pathway restores the mutants' ability to synthesize GPI molecules.
Corroborating the functional complementation of yeast mutants with the TcDPM1 gene, thin layer chromatography (TLC) of yeast mutants expressing the T. cruzi gene or the yeast ScDPM1gene, as a positive control, showed the presence of dolichol-P-mannose. Yeast cell extracts were preincubated with dolichol-phosphate and labeled in vitro with GDP-[2-3H]mannose. Labeled dolichol-P-mannose was detected in wild type yeast cells as well as in DPM1 mutants that were transfected with the TcDPM1 or with the yeast ScDPM1 gene, confirming that the expression of the T. cruzi enzyme rescues the mutant ability to synthesize dolichol-P-mannose (Figure S3).
T. cruzi GPI8 mutants have altered cell surface
Knockout parasites of GPI8, GPI16 and GPI10 were generated in T. brucei whereas a GPI8 knockout was described in L. mexicana [18], [19], [72], [73]. To further investigate the role of GPI anchors in T. cruzi, we tried to generate parasite cell lines in which both alleles of TcGPI3, TcGPI8 and TcGPI10 genes were deleted by homologous recombination. Although we were able to generate heterozygote epimastigotes carrying a drug resistance marker inserted in each one of the TcGPI8 alleles (Figure 5A–B), several attempts to generate double-resistant, null mutant epimastigotes with both TcGPI8 alleles deleted were unsuccessful. Also unexpectedly, transfection with plasmid constructs containing TcGPI3 and TcGPI10 sequences flanking the neomycin resistance gene did not result in G418 resistant parasites, indicating that disruption of even one allele of a gene involved in the initial steps of the GPI biosynthesis pathway results in non-viable parasites (not shown). Thus, our results suggest that, in contrast to T. brucei and L. mexicana, the GPI biosynthesis may be an essential pathway in epimastigotes of T. cruzi. In agreement with PCR analyses that showed the disruption of single alleles of TcGPI8 (Figure 5B), northern blot assays (Figure 5C) showed that both heterozygous TcGPI8 mutants have the expression of TcGPI8 mRNA reduced by about 40%. Although a few double-resistant epimastigote clones were generated and PCR analyses indicated that the neomycin and hygromycin resistance genes were inserted into both TcGPI8 alleles, PCR amplifications also indicated that additional sequences corresponding to the TcGPI8 gene were present in a different genomic location in the double resistant parasites (Figure 6A–B). It should be noted that it was possible to generate the double resistant parasites only after we prepared different plasmid constructs in which the resistance genes were linked to trans-splicing and polyadenylation signals from the glyceraldehyde-3-phosphate dehydrogenase (gapdh) and the ribosomal protein TcP2β (HX1) genes and performing drug selection by gradually increasing drug concentrations. Northern blot analyses (Figure 6C) indicate that the recombination events that resulted in viable, double resistant parasites allowed the expression of an aberrant TcGPI8 mRNA population. Among this TcGPI8 mRNA population transcribed in the double resistant mutants, mature, trans-spliced mRNAs were detected by RT-PCR using primers specific for TcGPI8 sequences and the T. cruzi spliced leader (Figure 6D), thus indicating that this gene is still active in these mutants. Although no significant changes in either growing or overall morphology of the TcGPI8 mutants were observed, transmission electron microscopy showed striking alterations in the dense glycocalyx that covers the parasite surface. As shown in Figure 7, cell membranes of epimastigotes from TcGPI8 heterozygous mutants (+/−N) present a thinner layer of the surface glycocalyx compared to wild type (WT) epimastigotes. In contrast, cell membranes from both clones of double resistant parasites (N/H), which may have suffered recombination events involving TcGPI8 sequences, present an increased thickness of their glycocalyx compared to the heterozygous mutants (Figure 7). Although no significant differences in the levels of mucins were detected in the heterozygous mutants, western blot analyses of membrane proteins of WT and double resistant TcGPI8 mutants using the anti-mucin monoclonal antibody 2B10 [74] showed increased amounts of the 35–50 kDa glycoproteins (also known as Gp35/50 mucins) expressed on the surface of epimastigotes of the double resistant clones (Figure 8). Flow cytometry of epimastigotes stained with 2B10 antibodies also showed increased amounts of surface mucins in the double resistant parasites (Figure S4).
Figure 5. Generation of TcGPI8 heterozygous mutants.

(A) DNA constructs generated to delete both TcGPI8 alleles by homologous recombination are shown with the NeoR or HygR genes flanked by 5′ and 3′ sequences of the TcGPI8 gene and the SacI/SpeI and XhoI/XbaI cloning sites from the pCR2.1TOPO vector. After transfecting epimastigotes with the purified DNA fragments, parasites were selected in LIT medium containing 200 µg/ml of G418 or hygromycin. Total DNA, isolated from G418 or hygromycin resistant parasites was analyzed by PCR amplifications, using the primers indicated by arrows. Below the schemes of DNA constructs, the sizes of the NeoR or HygR genes and the 5′ and 3′ sequences of the TcGPI8 gene are shown. (B) PCR amplification products analyzed on 1% agarose gel electrophoresis were obtained from DNA isolated from epimastigotes transfected with the GPI8-Neo (top panel) or GPI8-Hyg construct (bottom panel) and using pairs of primers showed in A. Amplicons derived from PCR using the primer pair 1F/7R that amplify a T. cruzi GPI8 allele which was not deleted are shown. On lanes indicated by (-), loaded samples were from PCR in which no template DNA was added. (C) Expression levels of TcGPI8 mRNA in WT and TcGPI8 single knockout of each allele interrupted by NeoR or HygR genes (+/− N or +/− H, respectively). Total RNAs purified from epimastigotes were hybridized to [α-32P]-labeled probes specific for the TcGPI8 gene (top panel) or for the 24Sα rRNA (bottom panel) used as loading control. The size of ribosomal RNA bands are indicated on the left and a graph with the quantification of the signals from the TcGPI8 probe after normalization using the 24Sα rRNA probe is shown below.
Figure 6. Translocation of the TcGPI8 gene in T. cruzi mutants.

(A) DNA constructs generated to delete both TcGPI8 alleles are shown with the NeoR or HygR genes flanked by 5′ and 3′ sequences of the TcGPI8 and spliced leader (SL) addition and polyadenylation signals from TcP2β (HX1) and gapdh genes. (B) DNA isolated from two cloned epimastigote cell lines that have been sequentially transfected with NeoR and HygR constructs and selected with G418 and hygromycin (double resistant mutants, N/H) were PCR amplified with primers shown in (A). Amplicons generated using primers 2F/2R indicate the integration of the NeoR in one of the TcGPI8 alleles whereas amplicons generated with primers 1F/4R and 5F/2R indicate the integration of the HygR sequences in the second TcGPI8 allele. PCR amplification using primers 1F/8R shows that double resistant parasite cell lines still maintained at least one intact copy of the TcGPI8 gene. As positive controls, DNA from NeoR (for primers 2F/2R), HygR (for primers 1F/4R and 5F/2R) single knockout and wild-type parasites (for primers 1F/8R) were used. (C) Expression levels of TcGPI8 mRNA in WT, TcGPI8 single knockout NeoR (+/− N1) and two double resistant clones (N/H1 and N/H2). RNA purified from epimastigotes were hybridized to [α-32P]-labeled TcGPI8 (top panel) or 24Sα rRNA (middle panel) probes. (D) RT-PCR amplification of TcGPI8 sequences. Reverse transcribed TcGPI8 mRNA obtained from WT, single knockout NeoR (+/− N1) and two double resistant clones (N/H1 and N/H2) were PCR amplified with primers annealing with TcGPI8 sequences and the T. cruzi SL sequence. First strand cDNA synthesis reactions were done with primers complementary to TcGPI8 in the presence (+) or absence (−) of reverse transcriptase. PCR products, separated on 1% agarose gels were stained with ethidium bromide. Molecular weight DNA markers are shown on the left.
Figure 7. Cell membrane morphology of T. cruzi GPI8 mutants.

Transmission electron microscopy showing cellular membranes of wild type T. cruzi epimastigotes (WT), TcGPI8 single allele knockout, neomycin resistant (+/− N1) and double resistant TcGPI8 epimastigote mutants (N/H1). Although displaying similar morphologies, representative images show that single allele TcGPI8 mutants present a thinner layer of parasite glycocalyx, when compared to wild type cells, whereas cell membranes of double resistant parasites present a glycocalyx layer that is slightly thicker than the glycocalyx of wild type parasite membranes (indicated by the arrows).
Figure 8. Cell membrane mucins in T. cruzi GPI8 mutants.
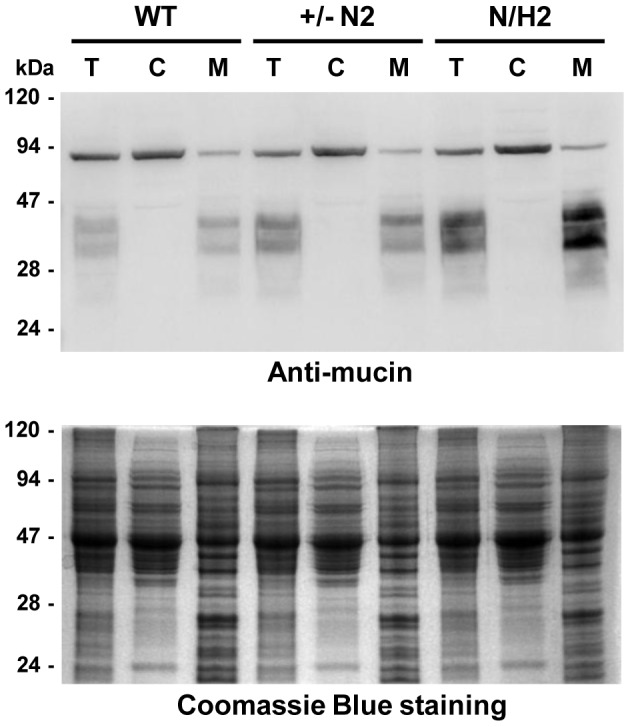
Immunoblot of total (T), cytoplasmic (C) and membrane (M) fractions of WT epimastigotes, TcGPI8 single allele knockout, neomycin resistant (+/−N2) and double resistant TcGPI8 (N/H2) mutant cell lines. Equivalent amounts of protein from each fraction, as showed by the Coomassie blue stained bands (bottom panel), were transferred to nitrocellulose membranes and incubated with anti-mucin antibodies and revealed with horseradish peroxidase conjugated secondary antibodies.
Discussion
Several T. cruzi surface proteins known to be involved in parasite infectivity or escape from the host immune response are anchored to the parasite membrane by covalent linkage to glycosylphosphatidylinositol (GPI). T. cruzi GPI anchors are also strong proinflammatory molecules, being critical in the modulation of the host immune response against this parasite. T. cruzi belongs to a group of early branching eukaryotic protists that are responsible for several neglected human tropical diseases, for which there is a strong need for new drug treatments. The elucidation of the structures of molecules that play a direct role in host-parasite interactions and the understanding of the biosynthetic pathways that generate these specific parasite molecules, such as the T. cruzi GPI biosynthetic pathway, represent a significant contribution towards this goal. Using a combination of sequence similarity analyses based on yeast, mammal, T. brucei and P. falciparum previously characterized genes, cellular localization and functional expression in yeast mutants we identified 18 orthologous genes encoding components of the GPI biosynthetic pathway present in the published T. cruzi CL Brener genome database. In addition, the gene encoding the IPC synthase, an enzyme responsible for a key modification that occurs in the lipid anchor during in the infective, trypomastigote stage of the parasite, was also identified. Although most sequences were correctly annotated in the TriTrypDB genome database, TcGPI15, TcGPI19, TcDPM2 and TcGPI16 were not correctly identified. It should be noted however that, because the assembly of the CL Brener genome is not complete, our in silico analyses might still contain a few missing genes whose presence or absence can only be unambiguously determined by a total sequencing read based analysis or through degenerate PCR experiments. Efforts towards identifying additional genes, such as the ones encoding a component of the mannosyltransferase I complex, α-1,2-mannosyltransferase IV or the acyltransferase, responsible for acylation of the inositol ring, are underway.
By expressing these genes in yeast mutants, we generated yeast cell lines that can now be used in high throughput screening assays for drugs that are specifically targeted to T. cruzi enzymes. Using yeast as a tool for drug screening against parasites is a strategy that has been successfully employed [5], [21], [75]. This system allows the identification of drugs acting specifically on the parasite enzyme since their effect on transfected yeast mutants growing in permissive and nonpermissive media can be compared (for a recent review, see [76]). Alternatively, specific inhibitors can be discovered using cell-free system assays, as it was shown for T. brucei and L. major enzymes involved with GlcNAc-PI de-N-acetylation, mannosylation and inositol acylation [23], [24], [25]. It is noteworthy that, among all tested genes, we observed functional complementation in yeast only for those whose products are not part of a protein complex. Among the T. cruzi genes that we were able to show complementation is the DPM1 gene. Since all four mannose residues are likely to be transferred from dolichol-P-mannose, DPM1, a gene encoding the dolichol-P-mannose synthase is considered an excellent candidate gene to be targeted for drug test studies. In contrast to DPM1, for which the T. cruzi homologous protein has high levels of amino acid identity with the yeast enzyme, TcGPI10 was also able to complement the yeast mutation even though it has only 21% identity with the yeast enzyme. On the other hand, the T. cruzi IPC synthase, which presents 10% identity with the yeast enzyme and is also a promising target for chemotherapy against trypanosomiases, is not functional in yeast. This is an unexpected result, since it has been shown that the Leishmania major IPC synthase gene (also known as AUR1 gene) restored the growth of yeast AUR1 mutants in nonpermissive, glucose-containing media [70].
We further confirmed the role of these genes by analyzing the cellular localization and mRNA expression of their gene products. Sequences corresponding to TcDPM1, TcGPI3 and TcGPI12 genes, expressed as GFP-fusion proteins in epimastigotes, showed a cellular localization compatible with ER. Interestingly, the T. cruzi sequences containing ER localization signals can be recognized by the mammalian protein trafficking machinery, since we were also able to show similar localization of GFP fusions of TcDPM1, TcGPI3, TcGPI8 and TcGPI12 in the ER of transfected HT1080 human fibrosarcoma cells. As expected, analyses of mRNA levels of TcGPI8 and TcGPI10 indicated that the components of the GPI biosynthetic pathway are more actively produced in the two proliferative stages of the parasite life cycle, epimastigotes and amastigotes.
To gain further insights into the role of GPI molecules as well as GPI-anchored proteins, we tried to generate T. cruzi null mutants for some of these genes. Because a large number of T. cruzi proteins involved in host-parasite interactions such as members of the large trans-sialidase, mucin and MASP families are GPI anchored, the availability of T. cruzi cell lines with disrupted genes of the GPI biosynthetic pathway would allow us to perform a number of studies regarding the effect of the absence of these proteins on the parasite surface during infection. Given that it encodes the catalytic subunit of the GPI:protein transamidase complex, responsible for transferring GPI anchor to the proteins, we sought to disrupt the TcGPI8 gene, which would have resulted in parasites containing only surface GIPLs, but no GPI-anchored proteins. Not surprisingly, the deletion of a single TcGPI8 allele could be easily achieved by homologous recombination between sequences from each allele flanking the neomycin or hygromycin resistance genes. Accordingly, mRNA expression analyses showed that both TcGPI8 heterozygous mutants have decreased mRNA levels. On the other hand, several attempts to delete the second TcGPI8 allele did not result in viable parasites. When the plasmid constructs were modified and drug selection protocol was conducted in such a way that drug concentrations were increased gradually, rare double resistant cell lines were obtained. However, these parasites seem to have undergone large gene rearrangement involving GPI8 sequences. Although frequently described in Leishmania spp, where gene amplification and overexpression of sequences have been observed after disruption of essential genes [45], [77], this phenomenon has been rarely reported for T. cruzi [78]. Together with the results of northern blot and RT-PCR analyses, preliminary data on pulse field gel electrophoresis and southern blot hybridizations (not shown) suggested that the amplification of TcGPI8 sequences involved the production of episomal DNA molecules. Thus, the anomalous expression of TcGPI8 mRNA sequences from distinct genomic locations, indicated by a large smear of high molecular weight RNA bands in northern blots and the amplification of spliced leader containing TcGPI8 mRNA allowed the growth of mutants in which both TcGPI8 alleles were disrupted by drug resistance markers. Surprisingly, although no major morphological alterations were evident, electron microscopy analyses of cell membrane structures of epimastigotes showed that TcGPI8 mutants have changes in their glycocalyx layer. Even though the small reduction in the glycocalyx layer observed in the heterozygous mutants could not be correlated with changes in the levels of mucins, western blot with membrane fractions, confirmed by flow cytometry using anti-mucin antibodies indicated that double-resistant parasites present a small increase in the amount of surface glycoproteins, most likely due to an increased expression of the translocated copies of TcGPI8 gene. Mucins play a critical role during infection, since they are the acceptors of sialic acid that allows trypomastigotes to build a negatively charged coat that protects them from killing by host anti-α-galactopyranosyl antibodies [79]. Whether the genomic rearrangements that resulted in the expression of TcGPI8 from different genomic locations have affected the expression of other T. cruzi genes, it remains to be determined. It will be also important to determine which are the mechanisms employed by the parasite that resulted in the genomic rearrangement observed with the double resistant clones.
Interestingly, despite being viable in culture, T. brucei mutants lacking TbGPI8 resulted in the absence of GPI-anchored surface proteins, accumulation of non-protein-linked GPI and incapacity of procyclic forms to establish infections in the tsetse midgut [80]. In contrast, GPI8 RNAi knock-down in bloodstream forms resulted in accumulation of unanchored variant surface glycoprotein (VSG) and cell death with a phenotype indicative of blocking cytokinesis [72]. On the other hand, L. mexicana GPI8 knockouts, although deficient of GPI-anchored proteins, display normal growth in culture, are capable of differentiating into amastigotes, and are able to infect mice [19]. In addition to GPI8, procyclic T. brucei lacking the TbGPI12 and TbGPI10 were also obtained. Although unable to synthesize GPI structures beyond GlcNAc-PI, TbGPI12−/− parasites are viable in culture, but are not able to colonize the tsetse midgut [51]. Deletion of TbGPI10 also interferes with the ability of procyclic mutants to infect tsetse flies [18]. These reports are in contrast with our results indicating that disruption of only one allele of a gene involved in the initial steps of the GPI pathway such as TcGPI3 or TcGPI10 resulted in non-viable T. cruzi epimastigotes. On the other hand, similarly to the genomic alterations we observed in the T. cruzi double resistant TcGPI8 mutants, an attempt to create a L. mexicana knockout by targeted deletion of the gene encoding the dolichol-phosphate-mannose synthase resulted in amplification of this chromosomal locus [45]. Thus, our contrasting results attempting to generate T. cruzi null mutants of genes involved with GPI biosynthesis, compared to similar studies described in T. brucei and L. mexicana, suggest that, although considered closely related organisms, the different members of the trypanosomatid family have significant peculiarities that deserve detailed analyses of major biochemical pathways in each parasite species.
Supporting Information
Cellular localization of T. cruzi proteins expressed in mammalian cells. The T. cruzi genes TcDPM1, TcGPI3, TcGPI12, and TcGPI8 were cloned in fusion with GFP in the vector pcDNA3.1/NT-GFP-TOPO and transfected into HT1080 human fibrosarcoma cells. Forty eight hours after transfections with pcDNA-GFP-TcDPM1 (A), pcDNA-GFP-TcGPI3 (B), pcDNA-GFP-TcGPI12 (C), pcDNA-GFP-TcGPI8 (D) or after mock transfections (E), cells were stained with DAPI and visualized under fluorescence microscopy. All plasmids were cotransfected with the pGAG-DsRed-ER plasmid to visualize cellular ER compartments. Scale bars: 20 µm.
(TIF)
RT-PCR mRNA analysis of yeast mutants transformed with T. cruzi genes. Reverse-transcription and PCR amplifications (RT-PCR) of total RNA isolated from non-transformed yeast mutants or mutants transformed with T. cruzi genes were analyzed by agarose gel electrophoresis. Total RNA was isolated from GPI8 yeast mutants (top panel) or AUR1 mutants (bottom panel). mRNA expression was analyzed in non-transformed mutants (GPI8 mutants or AUR1 mutants) or mutants transformed with pRS426Met plasmids carrying either the T. cruzi (TcGPI8 or TcIPCS) that were grown in galactose-containing media. For each RNA sample, pair of primers used for cDNA amplifications, which are specific for the TcGPI8, TcIPCS, the endogenous ScGPI8 or ScAUR1, as well as for the yeast 26S rRNA genes, are indicated above each lane of the gel and are listed in Table S1. It is also indicated above each lane, whether the amplicons were generated in presence (+) or in the absence (−) of reverse transcriptase (RT). Molecular weight DNA markers are shown on the left.
(TIF)
Synthesis of dolichol-P-mannose in yeast mutants expressing the TcDMP1 gene. Thin Layer Chromatography (TLC) of dolichol-phosphate-mannose in vitro labeled with GDP-[2-3H]mannose was performed using membrane fractions from: wild type yeast expressing the DPM1 endogenous gene (A), grown in the complete medium and preincubated with dolichol-phosphate; (B) DPM1 mutant grown in SD medium supplemented with uracil (nonpermissive conditions); (C) wild type yeast, expressing the DPM1 endogenous gene, grown in the YPGR medium and preincubated with amphomycin and dolichol-phosphate; (D) DPM1 mutant transformed with the recombinant plasmid pRS426Met containing the ScDPM1 grown in nonpermissive medium; (E) WT yeast, containing the DPM1 endogenous gene, grown in complete but not preincubated with amphomycin and dolichol-phosphate; (F) DPM1 mutant transformed with the recombinant plasmid pRS426Met containing the TcDPM1 grown in nonpermissive medium. The position of the dolichol-P-mannose (Dol-P-Man) in the TLC is indicated by an arrow.
(TIF)
Flow cytometry analyses of T. cruzi mutants. Wild type epimastigotes (WT), two TcGPI8 single knockouts NeoR (+/− N1 and +/− N2) and double resistant clones (N/H1 and N/H2) were stained with the anti-mucin monoclonal antibody 2B10 (dilution 1∶450) and analyzed by flow cytometry. The values of mean fluorescence intensity (MFI) for each parasite cell line are shown below.
(TIF)
Sequences of oligonucleotides used for PCR amplications and to generate plasmid constructs.
(PDF)
Acknowledgments
We are indebted to Marco Antonio S. Campos for helpful discussions. R.T. Schwartz is a visiting Professor at University of Lille.
Funding Statement
This study was supported by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brazil), Fundação de Amparo a Pesquisa do Estado de Minas Gerais (FAPEMIG, Brazil), The Instituto Nacional de Ciência e Tecnologia de Vacinas (INCTV, Brazil), Deutsche Forschungsgemeinschaft (DFG), the Deutscher Akademischer Austauschdienst (DAAD, Germany) and Bundesministerium für Bildung und Forschung (BMBF, Germany). MSC, JOP, LMP, RTG and SMRT are recipients of CNPq fellowships; PS is a recipient of fellowships from Alumni Medizin Marburg, Roland und Elfriede Schauer-Stiftung and Studien-stiftung des Deutschen Volkes (Germany). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. McConville MJ, Ferguson MA (1993) The structure, biosynthesis and function of glycosylated phosphatidylinositols in the parasitic protozoa and higher eukaryotes. Biochem J 294: 305–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fujita M, Kinoshita T (2010) Structural remodeling of GPI anchors during biosynthesis and after attachment to proteins. FEBS Lett 584: 1670–1677. [DOI] [PubMed] [Google Scholar]
- 3. Previato JO, Wait R, Jones C, DosReis GA, Todeschini AR, et al. (2004) Glycoinositolphospholipid from Trypanosoma cruzi: structure, biosynthesis and immunobiology. Adv Parasitol 56: 1–41. [DOI] [PubMed] [Google Scholar]
- 4. De Lederkremer RM, Agusti R, Docampo R (2011) Inositolphosphoceramide metabolism in Trypanosoma cruzi as compared with other trypanosomatids. J Eukaryot Microbiol 58: 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ferguson MA (2000) Glycosylphosphatidylinositol biosynthesis validated as a drug target for African sleeping sickness. Proc Natl Acad Sci U S A 97: 10673–10675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Previato JO, Jones C, Xavier MT, Wait R, Travassos LR, et al. (1995) Structural characterization of the major glycosylphosphatidylinositol membrane-anchored glycoprotein from epimastigote forms of Trypanosoma cruzi Y-strain. J Biol Chem 270: 7241–7250. [DOI] [PubMed] [Google Scholar]
- 7. Giorgi ME, de Lederkremer RM (2011) Trans-sialidase and mucins of Trypanosoma cruzi: an important interplay for the parasite. Carbohydr Res 346: 1389–1393. [DOI] [PubMed] [Google Scholar]
- 8. Nakayasu ES, Yashunsky DV, Nohara LL, Torrecilhas AC, Nikolaev AV, et al. (2009) GPIomics: global analysis of glycosylphosphatidylinositol-anchored molecules of Trypanosoma cruzi. Mol Syst Biol 5: 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yoshida N (2006) Molecular basis of mammalian cell invasion by Trypanosoma cruzi. An Acad Bras Cienc 78: 87–111. [DOI] [PubMed] [Google Scholar]
- 10. Alves MJ, Colli W (2008) Role of the gp85/trans-sialidase superfamily of glycoproteins in the interaction of Trypanosoma cruzi with host structures. Subcell Biochem 47: 58–69. [DOI] [PubMed] [Google Scholar]
- 11. Frasch AC (2000) Functional diversity in the trans-sialidase and mucin families in Trypanosoma cruzi. Parasitol Today 16: 282–286. [DOI] [PubMed] [Google Scholar]
- 12. Almeida IC, Gazzinelli RT (2001) Proinflammatory activity of glycosylphosphatidylinositol anchors derived from Trypanosoma cruzi: structural and functional analyses. J Leukoc Biol 70: 467–477. [PubMed] [Google Scholar]
- 13. Junqueira C, Guerrero AT, Galvão-Filho B, Andrade WA, Salgado AP, et al. (2012) Trypanosoma cruzi adjuvants potentiate T cell-mediated immunity induced by a NY-ESO-1 based antitumor vaccine. PLoS One 7: e36245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eisenhaber B, Maurer-Stroh S, Novatchkova M, Schneider G, Eisenhaber F (2003) Enzymes and auxiliary factors for GPI lipid anchor biosynthesis and post-translational transfer to proteins. Bioessays 25: 367–385. [DOI] [PubMed] [Google Scholar]
- 15. Pittet M, Conzelmann A (2007) Biosynthesis and function of GPI proteins in the yeast Saccharomyces cerevisiae. Biochim Biophys Acta 1771: 405–420. [DOI] [PubMed] [Google Scholar]
- 16. Kinoshita T, Inoue N (2000) Dissecting and manipulating the pathway for glycosylphosphatidylinositol-anchor biosynthesis. Curr Opin Chem Biol 4: 632–638. [DOI] [PubMed] [Google Scholar]
- 17. Ferguson MA (1999) The structure, biosynthesis and functions of glycosylphosphatidylinositol anchors, and the contributions of trypanosome research. J Cell Sci 112 Pt 17: 2799–2809. [DOI] [PubMed] [Google Scholar]
- 18. Nagamune K, Nozaki T, Maeda Y, Ohishi K, Fukuma T, et al. (2000) Critical roles of glycosylphosphatidylinositol for Trypanosoma brucei. Proc Natl Acad Sci U S A 97: 10336–10341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hilley JD, Zawadzki JL, McConville MJ, Coombs GH, Mottram JC (2000) Leishmania mexicana mutants lacking glycosylphosphatidylinositol (GPI):protein transamidase provide insights into the biosynthesis and functions of GPI-anchored proteins. Mol Biol Cell 11: 1183–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Delorenzi M, Sexton A, Shams-Eldin H, Schwarz RT, Speed T, et al. (2002) Genes for glycosylphosphatidylinositol toxin biosynthesis in Plasmodium falciparum. Infect Immun 70: 4510–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. de Macedo CS, Shams-Eldin H, Smith TK, Schwarz RT, Azzouz N (2003) Inhibitors of glycosyl-phosphatidylinositol anchor biosynthesis. Biochimie 85: 465–472. [DOI] [PubMed] [Google Scholar]
- 22. Sütterlin C, Horvath A, Gerold P, Schwarz RT, Wang Y, et al. (1997) Identification of a species-specific inhibitor of glycosylphosphatidylinositol synthesis. EMBO J 16: 6374–6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Smith TK, Sharma DK, Crossman A, Dix A, Brimacombe JS, et al. (1997) Parasite and mammalian GPI biosynthetic pathways can be distinguished using synthetic substrate analogues. EMBO J 16: 6667–6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Smith TK, Sharma DK, Crossman A, Brimacombe JS, Ferguson MA (1999) Selective inhibitors of the glycosylphosphatidylinositol biosynthetic pathway of Trypanosoma brucei. EMBO J 18: 5922–5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smith TK, Crossman A, Borissow CN, Paterson MJ, Dix A, et al. (2001) Specificity of GlcNAc-PI de-N-acetylase of GPI biosynthesis and synthesis of parasite-specific suicide substrate inhibitors. EMBO J 20: 3322–3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smith TK, Crossman A, Paterson MJ, Borissow CN, Brimacombe JS, et al. (2002) Specificities of enzymes of glycosylphosphatidylinositol biosynthesis in Trypanosoma brucei and HeLa cells. J Biol Chem 277: 37147–37153. [DOI] [PubMed] [Google Scholar]
- 27. Morita YS, Paul KS, Englund PT (2000) Specialized fatty acid synthesis in African trypanosomes: myristate for GPI anchors. Science 288: 140–143. [DOI] [PubMed] [Google Scholar]
- 28. Camargo EP (1964) Growth and differentiation in Trypanosoma cruzi. I. Origin of metacyclic trypanosoma in liquid media. Rev Inst Med Trop Sao Paulo 6: 93–100. [PubMed] [Google Scholar]
- 29. Teixeira SM, Kirchhoff LV, Donelson JE (1995) Post-transcriptional elements regulating expression of mRNAs from the amastin/tuzin gene cluster of Trypanosoma cruzi. J Biol Chem 270: 22586–22594. [DOI] [PubMed] [Google Scholar]
- 30.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, et al.. (1995) Current Protocols in Molecular Biology. New York: John Wiley & Sons.
- 31. Mazhari-Tabrizi R, Blank M, Mumberg D, Funk M, Schwarz RT, et al. (1999) Chromosomal promoter replacement in Saccharomyces cerevisiae: construction of conditional lethal strains for the cloning of glycosyltransferases from various organisms. Glycoconj J 16: 673–679. [DOI] [PubMed] [Google Scholar]
- 32. Christianson TW, Sikorski RS, Dante M, Shero JH, Hieter P (1992) Multifunctional yeast high-copy-number shuttle vectors. Gene 110: 119–122. [DOI] [PubMed] [Google Scholar]
- 33. Gietz RD, Schiestl RH, Willems AR, Woods RA (1995) Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11: 355–360. [DOI] [PubMed] [Google Scholar]
- 34. Damasceno LM, Anderson KA, Ritter G, Cregg JM, Old LJ, et al. (2007) Cooverexpression of chaperones for enhanced secretion of a single-chain antibody fragment in Pichia pastoris. Appl Microbiol Biotechnol 74: 381–389. [DOI] [PubMed] [Google Scholar]
- 35. Mazhari-Tabrizi R, Eckert V, Blank M, Müller R, Mumberg D, et al. (1996) Cloning and functional expression of glycosyltransferases from parasitic protozoans by heterologous complementation in yeast: the dolichol phosphate mannose synthase from Trypanosoma brucei brucei. Biochem J 316 Pt 3: 853–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sharma CB, Babczinski P, Lehle L, Tanner W (1974) The role of dolicholmonophosphate in glycoprotein biosynthesis in Saccharomyces cerevisiae. Eur J Biochem 46: 35–41. [DOI] [PubMed] [Google Scholar]
- 37. DaRocha WD, Silva RA, Bartholomeu DC, Pires SF, Freitas JM, et al. (2004) Expression of exogenous genes in Trypanosoma cruzi: improving vectors and electroporation protocols. Parasitol Res 92: 113–120. [DOI] [PubMed] [Google Scholar]
- 38. Bangs JD, Uyetake L, Brickman MJ, Balber AE, Boothroyd JC (1993) Molecular cloning and cellular localization of a BiP homologue in Trypanosoma brucei. Divergent ER retention signals in a lower eukaryote. J Cell Sci 105: 1101–1113. [DOI] [PubMed] [Google Scholar]
- 39. El-Sayed NM, Myler PJ, Bartholomeu DC, Nilsson D, Aggarwal G, et al. (2005) The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science 309: 409–415. [DOI] [PubMed] [Google Scholar]
- 40. Orlean P (1990) Dolichol phosphate mannose synthase is required in vivo for glycosyl phosphatidylinositol membrane anchoring, O mannosylation, and N glycosylation of protein in Saccharomyces cerevisiae. Mol Cell Biol 10: 5796–5805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maeda Y, Kinoshita T (2008) Dolichol-phosphate mannose synthase: structure, function and regulation. Biochim Biophys Acta 1780: 861–868. [DOI] [PubMed] [Google Scholar]
- 42. Tomita S, Inoue N, Maeda Y, Ohishi K, Takeda J, et al. (1998) A homologue of Saccharomyces cerevisiae Dpm1p is not sufficient for synthesis of dolichol-phosphate-mannose in mammalian cells. J Biol Chem 273: 9249–9254. [DOI] [PubMed] [Google Scholar]
- 43. Colussi PA, Taron CH, Mack JC, Orlean P (1997) Human and Saccharomyces cerevisiae dolichol phosphate mannose synthases represent two classes of the enzyme, but both function in Schizosaccharomyces pombe. Proc Natl Acad Sci U S A 94: 7873–7878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shams-Eldin H, de Macedo CS, Niehus S, Dorn C, Kimmel J, et al. (2008) Plasmodium falciparum dolichol phosphate mannose synthase represents a novel clade. Biochem Biophys Res Commun 370: 388–393. [DOI] [PubMed] [Google Scholar]
- 45. Ilgoutz SC, Zawadzki JL, Ralton JE, McConville MJ (1999) Evidence that free GPI glycolipids are essential for growth of Leishmania mexicana. EMBO J 18: 2746–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Watanabe R, Murakami Y, Marmor MD, Inoue N, Maeda Y, et al. (2000) Initial enzyme for glycosylphosphatidylinositol biosynthesis requires PIG-P and is regulated by DPM2. EMBO J 19: 4402–4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sobering AK, Watanabe R, Romeo MJ, Yan BC, Specht CA, et al. (2004) Yeast Ras regulates the complex that catalyzes the first step in GPI-anchor biosynthesis at the ER. Cell 117: 637–648. [DOI] [PubMed] [Google Scholar]
- 48. Murakami Y, Siripanyaphinyo U, Hong Y, Tashima Y, Maeda Y, et al. (2005) The initial enzyme for glycosylphosphatidylinositol biosynthesis requires PIG-Y, a seventh component. Mol Biol Cell 16: 5236–5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Watanabe R, Ohishi K, Maeda Y, Nakamura N, Kinoshita T (1999) Mammalian PIG-L and its yeast homologue Gpi12p are N-acetylglucosaminylphosphatidylinositol de-N-acetylases essential in glycosylphosphatidylinositol biosynthesis. Biochem J 339 Pt 1: 185–192. [PMC free article] [PubMed] [Google Scholar]
- 50. Chang T, Milne KG, Güther ML, Smith TK, Ferguson MA (2002) Cloning of Trypanosoma brucei and Leishmania major genes encoding the GlcNAc-phosphatidylinositol de-N-acetylase of glycosylphosphatidylinositol biosynthesis that is essential to the African sleeping sickness parasite. J Biol Chem 277: 50176–50182. [DOI] [PubMed] [Google Scholar]
- 51. Güther ML, Lee S, Tetley L, Acosta-Serrano A, Ferguson MA (2006) GPI-anchored proteins and free GPI glycolipids of procyclic form Trypanosoma brucei are nonessential for growth, are required for colonization of the tsetse fly, and are not the only components of the surface coat. Mol Biol Cell 17: 5265–5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sharma DK, Smith TK, Weller CT, Crossman A, Brimacombe JS, et al. (1999) Differences between the trypanosomal and human GlcNAc-PI de-N-acetylases of glycosylphosphatidylinositol membrane anchor biosynthesis. Glycobiology 9: 415–422. [DOI] [PubMed] [Google Scholar]
- 53. Sanyal S, Frank CG, Menon AK (2008) Distinct flippases translocate glycerophospholipids and oligosaccharide diphosphate dolichols across the endoplasmic reticulum. Biochemistry 47: 7937–7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Murakami Y, Siripanyapinyo U, Hong Y, Kang JY, Ishihara S, et al. (2003) PIG-W is critical for inositol acylation but not for flipping of glycosylphosphatidylinositol-anchor. Mol Biol Cell 14: 4285–4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Güther ML, Ferguson MA (1995) The role of inositol acylation and inositol deacylation in GPI biosynthesis in Trypanosoma brucei. EMBO J 14: 3080–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hong Y, Nagamune K, Morita YS, Nakatani F, Ashida H, et al. (2006) Removal or maintenance of inositol-linked acyl chain in glycosylphosphatidylinositol is critical in trypanosome life cycle. J Biol Chem 281: 11595–11602. [DOI] [PubMed] [Google Scholar]
- 57. Heise N, Raper J, Buxbaum LU, Peranovich TM, de Almeida ML (1996) Identification of complete precursors for the glycosylphosphatidylinositol protein anchors of Trypanosoma cruzi. J Biol Chem 271: 16877–16887. [DOI] [PubMed] [Google Scholar]
- 58. Umemura M, Okamoto M, Nakayama K, Sagane K, Tsukahara K, et al. (2003) GWT1 gene is required for inositol acylation of glycosylphosphatidylinositol anchors in yeast. J Biol Chem 278: 23639–23647. [DOI] [PubMed] [Google Scholar]
- 59. Güther ML, de Almeida ML, Yoshida N, Ferguson MA (1992) Structural studies on the glycosylphosphatidylinositol membrane anchor of Trypanosoma cruzi 1G7-antigen. The structure of the glycan core. J Biol Chem 267: 6820–6828. [PubMed] [Google Scholar]
- 60. Ashida H, Hong Y, Murakami Y, Shishioh N, Sugimoto N, et al. (2005) Mammalian PIG-X and yeast Pbn1p are the essential components of glycosylphosphatidylinositol-mannosyltransferase I. Mol Biol Cell 16: 1439–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kim YU, Ashida H, Mori K, Maeda Y, Hong Y, et al. (2007) Both mammalian PIG-M and PIG-X are required for growth of GPI14-disrupted yeast. J Biochem 142: 123–129. [DOI] [PubMed] [Google Scholar]
- 62. Buscaglia CA, Campo VA, Di Noia JM, Torrecilhas AC, De Marchi CR, et al. (2004) The surface coat of the mammal-dwelling infective trypomastigote stage of Trypanosoma cruzi is formed by highly diverse immunogenic mucins. J Biol Chem 279: 15860–15869. [DOI] [PubMed] [Google Scholar]
- 63. de Lederkremer RM, Agusti R (2009) Glycobiology of Trypanosoma cruzi. Adv Carbohydr Chem Biochem 62: 311–366. [DOI] [PubMed] [Google Scholar]
- 64. Orlean P, Menon AK (2007) Thematic review series: lipid posttranslational modifications. GPI anchoring of protein in yeast and mammalian cells, or: how we learned to stop worrying and love glycophospholipids. J Lipid Res 48: 993–1011. [DOI] [PubMed] [Google Scholar]
- 65. Benghezal M, Benachour A, Rusconi S, Aebi M, Conzelmann A (1996) Yeast Gpi8p is essential for GPI anchor attachment onto proteins. EMBO J 15: 6575–6583. [PMC free article] [PubMed] [Google Scholar]
- 66. Nagamune K, Ohishi K, Ashida H, Hong Y, Hino J, et al. (2003) GPI transamidase of Trypanosoma brucei has two previously uncharacterized (trypanosomatid transamidase 1 and 2) and three common subunits. Proc Natl Acad Sci U S A 100: 10682–10687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Serrano AA, Schenkman S, Yoshida N, Mehlert A, Richardson JM, et al. (1995) The lipid structure of the glycosylphosphatidylinositol-anchored mucin-like sialic acid acceptors of Trypanosoma cruzi changes during parasite differentiation from epimastigotes to infective metacyclic trypomastigote forms. J Biol Chem 270: 27244–27253. [DOI] [PubMed] [Google Scholar]
- 68. Agusti R, Couto AS, Campetella OE, Frasch AC, de Lederkremer RM (1997) The trans-sialidase of Trypanosoma cruzi is anchored by two different lipids. Glycobiology 7: 731–735. [DOI] [PubMed] [Google Scholar]
- 69. Figueiredo JM, Dias WB, Mendonça-Previato L, Previato JO, Heise N (2005) Characterization of the inositol phosphorylceramide synthase activity from Trypanosoma cruzi. Biochem J 387: 519–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Denny PW, Shams-Eldin H, Price HP, Smith DF, Schwarz RT (2006) The protozoan inositol phosphorylceramide synthase: a novel drug target that defines a new class of sphingolipid synthase. J Biol Chem 281: 28200–28209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Heidler SA, Radding JA (1995) The AUR1 gene in Saccharomyces cerevisiae encodes dominant resistance to the antifungal agent aureobasidin A (LY295337). Antimicrob Agents Chemother 39: 2765–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lillico S, Field MC, Blundell P, Coombs GH, Mottram JC (2003) Essential roles for GPI-anchored proteins in African trypanosomes revealed using mutants deficient in GPI8. Mol Biol Cell 14: 1182–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hong Y, Nagamune K, Ohishi K, Morita YS, Ashida H, et al. (2006) TbGPI16 is an essential component of GPI transamidase in Trypanosoma brucei. FEBS Lett 580: 603–606. [DOI] [PubMed] [Google Scholar]
- 74. Mortara RA, da Silva S, Araguth MF, Blanco SA, Yoshida N (1992) Polymorphism of the 35- and 50-kilodalton surface glycoconjugates of Trypanosoma cruzi metacyclic trypomastigotes. Infect Immun 60: 4673–4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bilsland E, Pir P, Gutteridge A, Johns A, King RD, et al. (2011) Functional expression of parasite drug targets and their human orthologs in yeast. PLoS Negl Trop Dis 5: e1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Norcliffe JL, Alvarez-Ruiz E, Martin-Plaza JJ, Steel PG, Denny PW (2013) The utility of yeast as a tool for cell-based, target-directed high-throughput screening. Parasitology 1–9. [DOI] [PubMed] [Google Scholar]
- 77. Boitz JM, Ullman B (2010) Amplification of adenine phosphoribosyltransferase suppresses the conditionally lethal growth and virulence phenotype of Leishmania donovani mutants lacking both hypoxanthine-guanine and xanthine phosphoribosyltransferases. J Biol Chem 285: 18555–18564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Li ZH, Alvarez VE, De Gaudenzi JG, Sant'Anna C, Frasch AC, et al. (2011) Hyperosmotic stress induces aquaporin-dependent cell shrinkage, polyphosphate synthesis, amino acid accumulation, and global gene expression changes in Trypanosoma cruzi. J Biol Chem 286: 43959–43971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Pereira-Chioccola VL, Acosta-Serrano A, Correia de Almeida I, Ferguson MA, Souto-Padron T, et al. (2000) Mucin-like molecules form a negatively charged coat that protects Trypanosoma cruzi trypomastigotes from killing by human anti-alpha-galactosyl antibodies. J Cell Sci 113 Pt 7: 1299–1307. [DOI] [PubMed] [Google Scholar]
- 80. Nagamune K, Acosta-Serrano A, Uemura H, Brun R, Kunz-Renggli C, et al. (2004) Surface sialic acids taken from the host allow trypanosome survival in tsetse fly vectors. J Exp Med 199: 1445–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cellular localization of T. cruzi proteins expressed in mammalian cells. The T. cruzi genes TcDPM1, TcGPI3, TcGPI12, and TcGPI8 were cloned in fusion with GFP in the vector pcDNA3.1/NT-GFP-TOPO and transfected into HT1080 human fibrosarcoma cells. Forty eight hours after transfections with pcDNA-GFP-TcDPM1 (A), pcDNA-GFP-TcGPI3 (B), pcDNA-GFP-TcGPI12 (C), pcDNA-GFP-TcGPI8 (D) or after mock transfections (E), cells were stained with DAPI and visualized under fluorescence microscopy. All plasmids were cotransfected with the pGAG-DsRed-ER plasmid to visualize cellular ER compartments. Scale bars: 20 µm.
(TIF)
RT-PCR mRNA analysis of yeast mutants transformed with T. cruzi genes. Reverse-transcription and PCR amplifications (RT-PCR) of total RNA isolated from non-transformed yeast mutants or mutants transformed with T. cruzi genes were analyzed by agarose gel electrophoresis. Total RNA was isolated from GPI8 yeast mutants (top panel) or AUR1 mutants (bottom panel). mRNA expression was analyzed in non-transformed mutants (GPI8 mutants or AUR1 mutants) or mutants transformed with pRS426Met plasmids carrying either the T. cruzi (TcGPI8 or TcIPCS) that were grown in galactose-containing media. For each RNA sample, pair of primers used for cDNA amplifications, which are specific for the TcGPI8, TcIPCS, the endogenous ScGPI8 or ScAUR1, as well as for the yeast 26S rRNA genes, are indicated above each lane of the gel and are listed in Table S1. It is also indicated above each lane, whether the amplicons were generated in presence (+) or in the absence (−) of reverse transcriptase (RT). Molecular weight DNA markers are shown on the left.
(TIF)
Synthesis of dolichol-P-mannose in yeast mutants expressing the TcDMP1 gene. Thin Layer Chromatography (TLC) of dolichol-phosphate-mannose in vitro labeled with GDP-[2-3H]mannose was performed using membrane fractions from: wild type yeast expressing the DPM1 endogenous gene (A), grown in the complete medium and preincubated with dolichol-phosphate; (B) DPM1 mutant grown in SD medium supplemented with uracil (nonpermissive conditions); (C) wild type yeast, expressing the DPM1 endogenous gene, grown in the YPGR medium and preincubated with amphomycin and dolichol-phosphate; (D) DPM1 mutant transformed with the recombinant plasmid pRS426Met containing the ScDPM1 grown in nonpermissive medium; (E) WT yeast, containing the DPM1 endogenous gene, grown in complete but not preincubated with amphomycin and dolichol-phosphate; (F) DPM1 mutant transformed with the recombinant plasmid pRS426Met containing the TcDPM1 grown in nonpermissive medium. The position of the dolichol-P-mannose (Dol-P-Man) in the TLC is indicated by an arrow.
(TIF)
Flow cytometry analyses of T. cruzi mutants. Wild type epimastigotes (WT), two TcGPI8 single knockouts NeoR (+/− N1 and +/− N2) and double resistant clones (N/H1 and N/H2) were stained with the anti-mucin monoclonal antibody 2B10 (dilution 1∶450) and analyzed by flow cytometry. The values of mean fluorescence intensity (MFI) for each parasite cell line are shown below.
(TIF)
Sequences of oligonucleotides used for PCR amplications and to generate plasmid constructs.
(PDF)