

Abstract
Helicases are a class of nucleic acid motors that catalyze NTP-dependent unwinding of nucleic acid duplexes into single-strands, a reaction essential to all areas of nucleic acid metabolism. In the last decade, single-molecule technology has proven to be highly useful in revealing mechanistic insight into helicase activity that is not always detectable via ensemble assays. A combination of methods based on fluorescence, optical and magnetic tweezers, and flow-induced DNA stretching have enabled the study of helicase conformational dynamics, force-generation, step-size, pausing, reversal, and repetitive behaviors during translocation and unwinding by helicases working alone and as part of multi-protein complexes. The contributions of these single-molecule investigations to our understanding of helicase mechanism and function will be discussed.
1. Introduction
Helicases are enzymes that catalyze the NTP-dependent unwinding of duplex nucleic acids (NA) into single strands (Fig. 1). This activity is ubiquitous in all pathways of nucleic acid metabolism ranging from DNA replication, repair, and recombination to transcription, splicing, and translation. DNA helicases and certain viral RNA helicases possess three intrinsic activities: NA-dependent NTP hydrolysis, NTPase-powered unidirectional translocation along single-stranded NA, and NTPase-dependent unwinding of duplex NA. These helicases can be thought of as nucleic acid motors that translocate along ssNA and upon reaching a ss-dsNA junction, continue to move through and separate the duplex. However, RNA helicases in the DEAD-box classification do not catalyze NTPase-dependent translocation, but rather perform local strand separation induced upon NTP-dependent binding of helicase to RNA with limited NTP hydrolysis required for enzyme recycling (Chen et al., 2008; Hilbert, Karow & Klostermeier, 2009; Liu, Putnam & Jankowsky, 2008). Helicases have been classified into six superfamiles (SFI-VI) based on conserved sequence motifs characteristic of proteins that catalyze directional translocation on NA. SFI and SFII helicases generally operate as monomers or dimers on a diverse range of DNA and RNA substrates, while SFIII-SFVI helicases adopt hexameric structures and function in replication. All helicases utilize one of two NTP-binding catalytic domains: the RecA fold or the AAA+ fold. Helicase structure and activity has been the subject of several recent reviews (Berger, 2008; Enemark & Joshua-Tor, 2008; Lohman, Tomko & Wu, 2008; Patel & Donmez, 2006; Pyle, 2008; Singleton, Dillingham & Wigley, 2007).
Fig. 1. Helicase Activity involving translocation along ssNA.
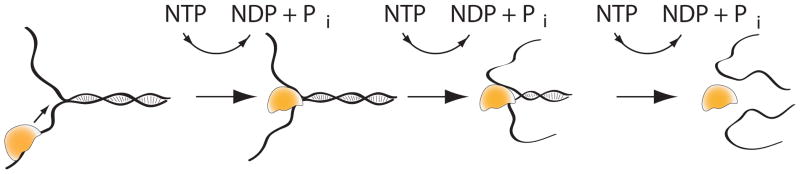
A helicase (yellow) will typically bind a single-stranded region (for example the tail in the fork substrate shown) and use its intrinsic NTPase activity to translocate unidirectionally (either 3′–5′ or 5′–3′) along one of the single stands towards the ssNA-dsNA junction. Upon reaching the junction, the helicase will continue translocating and concurrently unwind the duplex into separate single strands, again powered by NTP hydrolysis.
It is important to distinguish bona fide helicases that actually catalyze duplex NA unwinding from other helicase-like proteins that possess the ability to translocate along NA but cannot actually separate the strands. Some examples of the latter class of DNA motors include restriction enzyme, EcoR1241, Swi2/SWI chromatin remodelers, and branch migration complex RuvAB. While single-molecule techniques have been applied to study the latter class of DNA motors (Hopfner & Michaelis, 2007; Lionnet et al., 2006), we will limit the scope of this review to bona-fide helicases.
Nucleic acid melting/annealing in the absence of a protein catalyst is controlled by energy and entropic parameters. Melting is favored by conditions that result in higher entropy such as high temperature and the increased strand flexibility generated by unwinding, whereas annealing is promoted by energetically favorable conditions that stabilize the duplex such as high ionic strength and GC content. As the cell cannot change its temperature or ionic environment, it relies on helicases to overcome the energetic barrier associated with base-pair melting. Under physiological conditions (8 mM ATP, 8 mM ADP, 0.4 mM Pi), the energy of NTP hydrolysis is 12.1 kcal/mole or ~ 20 kBT which translates into 86 pN-nm at 310 K (Phillips, Kondev & Theriot, 2008). For a maximally efficient helicase, then, unwinding 3–10 bp (i.e. moving ~1–3.4 nm) translates into enzyme force generation of 25–86 pN. These estimates of helicase-induced force are in a range similar to experimental measurements of 10–20 pN force required to unzip nucleic acid in the absence of enzymes (Essevaz-Roulet, Bockelmann & Heslot, 1997). Another factor to consider regarding the physical work done by helicases is the role of NTP hydrolysis and its efficiency of utiliziation. If, for example, a helicase hydrolyzed one ATP per bp unwound, the ~ 20 kBT of energy released would likely be amply sufficient to unwind a single bp (~3 kBT energy barrier per GC bp unwound) and also in many cases to power translocation along NA - all of which would require energy-dependent enzyme conformational changes. Such estimates help provide a theoretical framework for the physical parameters under which helicases operate, and thus guide experimental designs to probe helicase activity.
To date, a large body of knowledge on helicase structure, substrate specificity, kinetics, and mechanisms of unwinding has been obtained through structural and ensemble-level studies (Lohman et al., 2008; Pyle, 2008; Singleton et al., 2007). More recently, single-molecule (sm) approaches have been applied to probe mechanistic details and heterogeneity of helicase action that are undetectable by ensemble-averaged measurements (Hopfner & Michaelis, 2007; Lionnet et al., 2006; Rasnik, Myong & Ha, 2006). Optical/magnetic trapping/tweezer-based methods have yielded information about differences in force generation by helicases, step size of helicase movement along DNA, and detected pausing and reversal during helicase unwinding. Single-molecule FRET-based methods have allowed observations of conformational dynamics in protein-NA and protein-protein interactions during translocation and unwinding, yielded data on helicase step size, and led to the discovery of repetitive behavior in helicase translocation and unwinding. Lastly, flow-stretch, tweezer-based, and sm fluorescence methods have helped elucidate how helicases function in multi-protein complexes. The significant increase in our level of understanding and exploration of helicase mechanisms achieved through single-molecule technology is the focus of the current review.
2. Single-Molecule Techniques used to Probe Helicase Function
During the last several decades, ensemble studies have yielded considerable information on helicase function and mechanism. Among the most widely used techniques for measuring unwinding in bulk are gel electrophoresis-based monitoring of complete unwinding of radioactively-labeled nucleic acid substrates as well as fluorimeter-based measurement of unwinding of fluorescently-labeled substrates. These assays have helped elucidate helicase functional properties regarding substrate specificity and steady-state kinetic parameters. Adaptation of these assays to stopped-flow technology has allowed for measurement of unwinding under single-turnover conditions providing more accurate measurement of translocation step size and unwinding kinetics in a synchronized ensemble population (see Section 3.3.1 for more details). These bulk techniques, however, are not able to detect unwinding reaction intermediates nor any heterogeneity in activity within the population. In reality, the unsynchronized motion of enzymes and macromolecules calls for the use of sm techniques to observe, in real time, reaction dynamics and heterogeneous and novel behaviors as well as to evaluate force-dependence in this class of enzymes.
The last decade has yielded a vast variety of sm techniques that allow researchers to study physical biology problems on individual molecules. On the one hand, sm force spectroscopy with AFM, optical and magnetic tweezers allows for the manipulation and observation of force-induced mechanical changes, yielding important information about the energy landscape of nucleic acid (NA) and protein-NA interactions. These studies perturb the free energy landscape of single molecules with force such that certain reactions are sped up or slowed down to mimic intracellular forces or to match the experimentally observed time scale. On the other hand, sm fluorescence and smFRET (Förster resonance energy transfer) are tools to watch relatively unperturbed molecular dynamics on the length scale of 2- 8 nm (Roy, Hohng & Ha, 2008). Currently, smFRET is typically used to study protein-NA interactions, protein folding, and enzyme kinetics (Roy et al., 2008). These investigations observe NTP-dependent conformational changes, equilibrium fluctuations or the relaxation of individual molecules after a perturbation of their equilibrium. In this section we will present an overview of which sm techniques are applied in current studies of helicases (Fig. 2 and Table 1). Our goal is to provide an overview of how and why these techniques are applied in various studies that will be discussed further in Section 3.
Fig. 2. Single-molecule techniques used to probe helicase function.

(a) TIR smFRET: Single-molecule Förster resonance energy transfer (smFRET) is used in surface tethered assays such that dsNAs are specifically attached to a passivated surface via biotin-streptavidin interaction. Time-traces of individual helicases converting dsNA (with donor and acceptor initially in close proximity) to ssNA are obtained over tens of seconds via EMCCD cameras and total internal reflection illumination. (b) Confocal smFRET: Alternatively, single helicase molecules can be dual-labeled with donor and acceptor, and cofactor-induced conformational changes can be monitored by confocal microscopy of individual diffusing molecules. (c) Flow-Stretch Method: In flow-stretch experiments dsDNA is stretched via laminar flow while attached to the surface. The extension of the stretched DNA is monitored using light microscopy to follow a microsphere attached to the other end of the surface-tethered dsDNA. The molecular elasticity of dsDNA is significantly different from that of ssDNA and thus the conversion of dsDNA to ssDNA results in a shortening or lengthening of the extension. (d) Flow-Stretch with Optical Tweezers: A derivative flow-stretch experiment follows the release of a dsDNA intercalator (illustrated as *) upon conversion of dsDNA to ssDNA. In this approach, the dsDNA is specifically attached to a microsphere held in solution by optical tweezers and stretched via laminar flow. (e) Magnetic Tweezers (nicked linear substrate): Here, DNA is specifically bound to the surface as well as a paramagnetic microsphere. Applying a magnetic field can stretch the DNA at a specific force. Again the conversion of dsDNA to ssDNA will lead to a change in the height of the microsphere above the surface at a constant force. This change can be monitored with light microscopy and converted to a length change from dsDNA to ssDNA. (f) Magnetic Tweezers (hairpin substrate): Alternatively magnetic tweezers can directly apply a force on a NA hairpin substrate and thus assist the helicase in opening of the duplex. dsNA opening is measured using light-microscopy to follow microsphere height above the surface. (g) Tethered Particle Assay: A force-free approach to monitor helicase activity in which the helicase is specifically bound to a microsphere and unwinding is monitored via light microscopy of the microsphere height above the surface. (h) Optical Tweezers: Optical tweezers offer the highest resolution and are most commonly used with a hairpin substrate to probe helicase activity. The dsNA hairpin is tethered between a microsphere and a surface/microsphere. Once the helicase opens the pre-tensioned dsNA hairpin, the additional length results in a force decrease that can be compensated by a feedback system. In this configuration the external force assists the helicase in unwinding the dsNA. (i) Optical Tweezers (helicase surface-tethered): A different approach for optical tweezers requires the specific attachment of the helicase to a passivated surface. The dsNA molecule is attached to a microsphere held by optical tweezers. This assay allows the study of translocation and unwinding properties of helicases with force acting along the translocation direction. See text and Table 1 for further details.
Table 1.
Parameters for Single-Molecule Methods Used to Probe Helicase Activity
| Technique | Temporal resolution | Spatial resolution | Through-put | Force | Comments | Example Reference(s) |
|---|---|---|---|---|---|---|
| TIR smFRET | 30 ms(8 ms) | <3 bp | 200–400 | 0 | Limited spatial range 2–8nm | (Myong et al., 2007) |
| Confocal smFRET | 1 ms | <3 bp | >100 | 0 | No time trajectories of individual molecules | (Theissen et al., 2008) |
| Flow-Stretch Method | 500 ms | 100 bp | >100 | 2–5 pN | Reconstitution of whole replisomes; Study of processivity | (Hamdan et al., 2007) |
| Flow-Stretch+ Optical Tweezers | 300 ms | 800 bp | 1 | 0.2 pN | Reconstitution of whole replisomes; Study of processivity | (Bianco et al., 2001) |
| Magnetic tweezers | 50 ms | 10 bp | 10–20 | 5–40 pN | Force method with moderate throughput | (Dessinges et al., 2004) (Lionnet et al., 2007) (Sun et al., 2008) |
| Tethered Particle Assay (TPA) | 200–500 ms | 100 bp | >100 | 0 | Protein engineering allows specific targeting of protein subunits | (Dohoney and Gelles, 2001) |
| Optical tweezers | 20 ms | Up to 1 bp | 1 | 1–40 pN | Highest spatial resolution, but only 1 molecule at a time | (Cheng et al., 2007) (Johnson et al., 2007) (Perkins et al., 2004) |
| AFM imaging | N.A. | 1 nm | >50 | N.A. | No trajectories; snapshots of trapped complexes | (Henn et al., 2001) |
| AFM force spectroscopy | 1 ms | >5 bp (force resolution limited) | 1 | 15–100 pN | Low throughput; difficult cantilever functionalization | (Marsden et al., 2006) |
2.1 TIR and Confocal smFRET
Fig. 2a illustrates a typical design of a surface-tethered smFRET helicase study. For this assay it is crucial to ensure that the protein interacts as little as possible with the surface. Therefore, over the past years, several protocols for surface passivation were developed. In the example shown, a glass surface is covered with polyethylenglycol (PEG) to reduce non-specific protein surface interactions. The specificity is then provided by biotinylated-PEG to ensure NA tethering at a desired surface density via a highly specific biotin-avidin interaction. Typically the donor (green) and acceptor (red) are attached at the junction of the illustrated fork structure. Once a helicase (yellow) starts to open the double stranded NA (dsNA), the donor gradually separates from the acceptor, with a corresponding reduction in FRET efficiency. The FRET efficiency is a measure of how much energy is transferred from the excited donor to the acceptor. Since FRET is due to a dipole-dipole interaction, it is strongly distance-dependent. The FRET efficiency is given by , with R as the distance between the donor and acceptor and R0 as the intrinsic characteristic distance for the energy transfer. R0 depends on the choice of the fluorophores and their environment and is typically ~50 Å. Thus, FRET is an excellent tool to track distance changes from 20 – 80 Å with a typical time resolution of 30 ms. Information regarding ssNA-dsNA junction binding and opening, mechanisms of unwinding and step size, for example, can be addressed with smFRET achieving a relatively high throughput (200–400 molecules in a typical imaging area) if smFRET data is taken with CCD cameras. Extremely fast processes can barely be captured with currently available sm CCD cameras, but can be recorded, for instance, molecule-by-molecule using confocal microscopy with single photon detectors (Roy et al., 2008). While local processivity of a few tens of base pairs can be studied well with sm FRET, more processive helicase unwinding on templates of several hundred bp has only been studied in one case to date (Wickersham et al., 2010)
An alternative approach for smFRET is a non-tethered assay using confocal microscopy as illustrated in Fig. 2b. Typically, the helicase is dual-labeled and thus conformational dynamics are probed. In this assay, the reaction buffer contains varying concentrations of cofactors and substrates, and the FRET efficiency of individual molecules diffusing through the confocal volume is measured. The observation time is strongly limited by the diffusion of the molecule through the confocal volume (~ 0.1 ms). Hence, this smFRET technique does not monitor the trajectory of an individual molecule over time, but rather samples an ensemble average on a single-molecule basis. Advantages of this technique over a more traditional ensemble assay are that singly-labeled molecules can be excluded from the analysis, that it allows for probing dimerization/multimerization effects, and that subpopulations of individual molecules can be detected.
2.2 Flow-induced DNA stretching
Flow-induced DNA stretching exploits the effect of laminar flow acting on a μm-sized bead to create controlled low stretching forces (< 5 pN) on the attached DNA. Fig. 2c shows a sketch of this tethered-particle method. In this setup, typically a 15–50 kbp long dsDNA substrate is used. The ends of this substrate are modified such that it specifically binds to the surface via a biotin modification at the lagging strand creating a replication fork–like structure. The other end of the dsDNA is modified for specific binding to the microsphere. In order to prevent nonspecific interaction between the microsphere and the glass surface, a paramagnetic microsphere is used combined with a permanent magnet placed above the experimental flow cell. The data collection uses the elasticity/force-extension relationship of ssDNA and dsDNA. At forces below ~5 pN the extension of ssDNA is shorter than that of dsDNA. The helicase unwinding reaction – that creates a longer ssDNA portion – is monitored by the microsphere position during the flow experiment. The microsphere will come closer to the tether point as more dsDNA is unwound. Although the spatial resolution of this assay is only on the order of 100 bp with 200 – 500 ms temporal resolution (50–100 molecules in parallel), it enables the studies of helicases with high processivity and their interactions partners. The reconstitution of multiple interaction partners within the replication machinery (> 10 proteins, > 1 MDa) is also possible (Lee et al., 2006).
Another stretching flow setup is illustrated in Fig. 2d. In this setup a long dsNA is specifically bound to a microsphere held by an optical trap. The dsDNA is stretched via linear flow forces on the order of 0.2 pN. At this force, dsDNAs are mostly extended, which can be verified by fluorescence microscopy with a dsDNA intercalator (i.e. YOYO). Unlike the previous flow experiment, dsDNA length is tracked by fluorescence decrease since the dsDNA intercalator binds with a higher affinity to dsDNA than ssDNA. Thus tracking fluorescence decrease provides a measurement of helicase unwinding with a temporal resolution of ~300 ms and a spatial resolution of ~800 bp. Again, this allows the measurement of high processivity and pauses of helicase longer than 300 ms, however, only one molecule at a time can be assayed (Bianco et al., 2001).
2.3 Magnetic Tweezers
A high-resolution method for studying single helicases in action is provided by magnetic tweezers. Fig. 2e illustrates such an experimental scheme where a dsDNA is tethered between the surface and a paramagnetic microsphere. By applying a constant stretching force to the DNA, the transition between dsDNA and ssDNA can be followed by imaging the magnetic microsphere under bright field microscopy. In contrast to flow experiments, the exerted force on the microsphere is generally kept above 5 pN resulting in ssDNA lengthening instead of coiling. Thus a helicase converting the dsDNA to ssDNA will result in a microsphere moving towards the magnets that can be studied with approximately 10 bp spatial and 50 ms temporal resolution. In this assay, external force does not directly help dsDNA unwinding if it is kept lower than the B–S transition force of dsDNA (Dessinges et al., 2004).
An alternative approach for monitoring helicase unwinding activity with magnetic tweezers is shown in Fig. 2f. Here, a hairpin is tethered to the surface and a paramagnetic microsphere with specific biotin-avidin and digoxigenin-anti-digoxigenin interactions. The microsphere allows tracking of the helicase unwinding activity via monitoring its diffraction pattern, because as in the previous case the percentage of ssDNA is changed over time. By changing the strength of the magnetic field, one can change the force acting on the hairpin and therefore change the amount of “helping force” for the helicase. A spatial resolution of ~10 bp and as well as a temporal resolution of ~50 ms together with a throughput of approximately 10–20 molecules per experiment enables a detailed, efficient study of helicase unwinding (Lionnet et al., 2007; Ramanathan et al., 2009).
2.4 Tethered Particle Assay
Another approach to study single-helicase NA unwinding is termed the tethered particle assay (TPA), as illustrated in Fig. 2g. The helicase of interest is specifically tethered to a microsphere via a biotin-avidin interaction, while the substrate is bound to the glass surface using digoxigenin-antidigoxigenin interaction. The microsphere position over time can be monitored by differential interference contrast video microscopy. Thus, the time trajectory of the average microsphere height above the surface can be recorded with a time resolution of 100 – 500 ms. Furthermore, this technique enables a large throughput since in the field of view at least 100 individual molecules can be studied at the same time. By specifically engineering biotinylation to individual subunits of helicase complexes, this technique can be used probe the composition of functional unwinding complexes (Dohoney & Gelles, 2001).
2.5 Optical Tweezers
The last tethered-particle method to introduce is the optical tweezers setup. Optical tweezers thus far has been used in two ways to study single helicase unwinding. In the first case sketched in Fig. 2h, a NA hairpin is tethered to two microspheres. Typically one microsphere is held in a focused laser beam. This microsphere is used to monitor length and force changes on the NA hairpin. By moving the other microsphere that is held, in this case with a micropipette, closer or farther apart, the force acting on the hairpin can be varied. Instead of using a micropipette and a second microsphere, one could also tether the second end of the hairpin to a microscope slide. Both, micropipette and microscope slide are in direct contact with the experimental setup and the resulting drift and vibration therefore limit the spatial resolution to a few bp (Cheng et al., 2007; Johnson et al., 2007). Recently, dual-trap optical tweezers with both microspheres held by a focused laser beam have completely decoupled the system of interest from the experimental setup. These studies achieved a single bp resolution at 20 ms temporal resolution for the movements of RNA polymerase and viral DNA packaging motor (Abbondanzieri et al., 2005; Moffitt et al., 2008).
In Fig. 2i we show an alternative approach to use optical tweezers to study single helicase unwinding. In this case the helicase is not free in solution for binding to the forked substrate, but is specifically bound to the microscope slide surface, for example, via a biotin-avidin bond. The forked substrate, on the other hand, is specifically bound to the microsphere that is held in the focused laser beam. Once a helicase starts unwinding, the force acting on the microsphere rises but can also be kept constant during the experiment. In using this approach, it is important to ensure that surface tethering of the helicase activity does not affect its activity. Since the force can be directly applied to the helicase translocation domains, a combined study of unwinding and translocation is the most likely outcome in this case. Any additional force applied in this case will most likely not assist in opening the dsNA. The resolutions are on the order of a few bp and 20 ms (Perkins et al., 2004).
2.6 Atomic force microscopy
Atomic force microscopy (AFM) has been used in two different configurations to study helicase activity at the sm level. The first approach has been to use the high resolution imaging capability of AFM (~1 nm/1 Å lateral/vertical resolution) to provide direct visualization of unwinding activity by helicases. Bulk unwinding reactions are applied to the surface and dried, followed by imaging in air with ssNA easily distinguished from dsNA by the imaging height, while proteins attached to NA usually appear as ‘blobs.’ This method has been used to confirm ATP-dependent RNA unwinding by DEAD-box helicase DbpA (Henn et al., 2001), and to trap unwinding intermediates on plasmid DNA formed by PcrA in the presence of its initiator protein, RepD, SSB, and ATP (Zhang et al., 2007). Direct imaging in air has also been applied to determine the UvrD dimer Kd via a quantitative volume AFM analysis method (Ratcliff & Erie, 2001). AFM has also been used to measure helicase activity under force (pN resolution) using substrates tethered to the surface at one end and to the cantilever at the other end. This technique was applied to determine the force exerted by Ded1 and eIF4A/B - RNA helicases involved in eukaryotic translocation initiation - to unwind RNA stem loop structures (Marsden et al., 2006). Results showed that both helicases reduced the force required to unwind a 25 bp stem loop by ~35 pN, however three-fold less Ded1 was required to achieve this effect than eIF4A/B, consistent with the former’s stronger ATPase activity. Despite these efforts, AFM only allows a coarse probe of helicase activity due to limitations in throughput and force resolution plus the inability to measure real-time kinetics and track enzyme motion (Table 1).
3. Helicase Questions Addressed through Single-Molecule Investigations
3.1 Scope of Review
As stated earlier, to limit the scope of this review, we will focus solely on bona-fide helicases that catalyze nucleic acid (NA) duplex separation and exclude motors that translocate along NA but do not unwind it. The approach of this review will be to focus on the contributions of sm studies to the elucidation of major questions regarding helicase function and mechanism. We will focus on five major topics – 1) Active versus passive unwinding, 2) Step size of helicase translocation and unwinding, 3) Pausing, reversal, and repetition during helicase activity, 4) Conformational dynamics during helicase activity, and 5) Helicases functioning in complexes with other proteins. For each topic, we will present case studies that illustrate how sm techniques have been utilized to reveal insights into these questions.
3.2 Active versus passive unwinding
3.2.1 Defining active and passive modes of unwinding
The mechanistic coupling of ssNA translocation to NA unwinding has been described in terms of two extreme cases – passive and active unwinding (Delagoutte & von Hippel, 2001; Lohman & Bjornson, 1996; Singleton & Wigley, 2002). A purely passive helicase will not enter the duplex region until thermal fluctuations cause the nucleic acid (NA) at the junction to open. The passive helicase can then move forward through the duplex binding to the newly available single-stranded (ss) ssNA, preventing it from reannealing. Thus, a purely passive helicase should translocate on ssNA faster than it can unwind the duplex. In contrast, a purely ‘active’ helicase is able to directly destabilize duplex NA at the junction by lowering its free energy such that the unwinding rate for a maximally active helicase would be expected to match its translocation rate on ssNA. A mathematical description of helicase activity in terms of active and passive unwinding has been developed (Betterton & Julicher, 2003; Betterton & Julicher, 2005) in which the degree of activeness ideally follows a scale of free energy reductions, ΔGd, (d refers to destabilization) induced by the helicase per bp unwound from 0 kBT for a purely passive helicase to 3.4 kBT (i.e. G–C base-pairing energy) for an optimally active helicase.
To date, tweezer-based assays on three different helicases (Cheng et al., 2007; Johnson et al., 2007; Lionnet et al., 2007) have yielded information on the range of activeness in helicase-promoted unwinding by analyzing the dependence of unwinding kinetics on NA sequence and external force. As the larger bond energy of G–C versus A–T/U base pairs can influence both thermal fraying of ends as well as duplex destabilization, sequence-dependence of unwinding velocity is expected for both active and passive cases. Increasing force is predicted to increase the unwinding rate to a larger extent for an initially passive helicase. The experimentally measured effects of these variables on unwinding kinetics were interpreted in terms of the aforementioned theoretical model (Betterton & Julicher, 2005). Results show that helicase unwinding mechanisms range from the highly active case of hepatitis NS3 helicase (Cheng et al., 2007) to the partially active phage T7 gp4 helicase (Johnson et al., 2007) to the mostly passive phage T4 gp41 helicase (Lionnet et al., 2007).
3.2.2 Hepatitis C Virus NS3 helicase
Bustamante and co-workers (Cheng et al., 2007) used optical tweezers to investigate sequence- and force-dependence of unwinding of an RNA hairpin substrate by NS3 helicase. The hairpin contained a 60 bp duplex region that was either 30 GC followed by 30 AU or vice-versa, and was tethered at both ends at constant tension, such that an increase in length of the DNA/RNA handles was measured upon helicase-promoted unwinding (setup similar to Fig. 2h). Consistent with their previous study (Dumont et al., 2006), the unwinding velocity was independent of force, however a strong sequence-dependence of unwinding rate was observed at multiple forces (7 and 17 pN) with pausing upon encountering a sequence barrier (i.e. GC stretch). When the measured NS3 stepping velocity was compared to that expected for a passive helicase it was 14000-fold faster for unwinding on a GC template (assuming 4 bp unwinding per step (Dumont et al., 2006)) which translates into ΔGd = 2.5 kBT. These observations indicate that NS3 unwinds actively, and does not wait passively for thermal fraying to occur before stepping. These results are summarized in an active duplex destabilization model which invokes barrier sensing by NS3 followed by strand separation occurring in substeps via an inchworm type mechanism. An smFRET study on NS3 (Myong et al., 2007) suggested that DNA translocation in single nt steps leads to accumulation of tension in the protein such that near-simultaneous unwinding of several bp occurs rapidly once enough elastic energy has built up. This study might helped explain the observed force-independence of unwinding for NS3 (Dumont et al., 2006).
3.2.3 Phage T7 replicative helicase
Wang and colleagues ((Johnson et al., 2007) reviewed in (Ha, 2007)) used optical trapping to investigate the nature of unwinding by phage T7 replicative hexameric ring-shaped helicase in a setup similar to Fig. 2h. Discrete measurements of ssDNA translocation as well as duplex DNA unwinding were made under constant tension, with translocation occurring faster than unwinding at low forces. As with the NS3 study (Cheng et al., 2007), a periodic sequence dependence of unwinding rate was observed corresponding to the positioning of pseudorepeat sequences in the template, indicating that high GC content presents a barrier to unwinding. However, in contrast to NS3, the T7 helicase unwinding rate increased 10-fold, approaching the ssDNA translocation rate, as force was increased from 5 to 11 pN. This was paralleled by a corresponding increase in total unwinding distance (i.e. processivity). While these results appear to suggest that T7 helicase is intrinsically passive and external force allows it to become more active, the unwinding rate force-dependence data are best fit by a hybrid active-passive model with ΔGd = 1–2 kBT per bp that assumes a translocation step size of 2–4 nts and takes into account the measured ssDNA translocation rate, ssDNA elasticity, and mechanical stability of the dsDNA. The increase in unwinding rate with force parallels previous findings in which the presence of DNA polymerase also speeds up unwinding (Lee & Yang, 2006; Stano et al., 2005) suggesting that one mechanism by which polymerases improve helicase processivity is by mechanically preventing negative regulation of the helicase by the unwound 3′ strand at the fork (Ha, 2007).
3.2.4 Phage T4 gp41 helicase
Another study on a replicative hexameric helicase (Lionnet et al (2007) uses a magnetic tweezers set-up similar to that shown in Fig. 2f to measure unwinding of a DNA hairpin tethered between a glass surface and magnetic bead by phage T4 gp41. The measured unwinding rate was 10-fold slower than the ssDNA translocation rate (i.e. rezipping rate in this system) at 3 pN, but it increased ten-fold as force was increased to 11 pN. When the measured data was fit to the global model for helicase activity put forth by (Betterton & Julicher, 2005), the ΔGd = 0.05 kBT, consistent with passive unwinding. These data support gp41 following a strand exclusion model for unwinding by ring-shaped hexameric replicative helicases (Donmez & Patel, 2006) and are also consistent with the low processivity of gp41 without the gp43 polymerase (Dong, Weitzel & von Hippel, 1996; Schrock & Alberts, 1996). The higher ΔGd observed in the similar T7 system (Johnson et al., 2007) was postulated to be due to differences in DNA sequence and elasticity between the substrates used in the two studies (Lionnet et al., 2007). A direct comparison of the unwinding velocity force-dependence for the two phage replicative helicases (Fig. 3) suggests that below 7 pN, the unwinding by T4 helicase is slightly more force-dependent (i.e. shows a greater slope in the unwinding rate verus force curve) relative to T7 helicase; whereas, above 7 pN, T7 helicase actually displays greater force dependence (and thus passivity) relative to T4 gp41. One possibility for the opposite interpretation presented by the aforementioned research is that this may be due to differences in the theoretical modeling of the data; thus, careful attention must be paid to this aspect when interpreting such studies.
Fig. 3. Degree of Activeness in helicase activity probed by force techniques.

Overlay of rescaled data of the ring-shaped helicases of T4 and T7 phages. Unwinding rate vs. force for T7 helicase (Johnson et al., 2007) was rescaled to match the force-free unwinding rate of T4 helicase (28.4 bp/s) (Lionnet et al., 2007). The plot indicates that the two helicases have similar force-dependence below 7 pN while above this level, T7 helicase unwinding is more sensitive to external force. See text for further details.
3.2.5 Implications for helicase mechanisms
A general implication from these studies is that how actively or processively a helicase can unwind is regulated by factors such as the DNA sequence and the presence of accessory proteins like DNA polymerase. It may also provide a mechanistic distinction between structurally different helicase superfamilies. The non-ring shaped SFI helicases UvrD, Rep, and PcrA have been shown to translocate along ssDNA faster than they unwind duplex regions, indicating that energy of base pairing is an unwinding barrier (Bertram, Hayes & Soultanas, 2002; Brendza et al., 2005; Cheng et al., 2002; Dillingham, Wigley & Webb, 2000; Fischer, Maluf & Lohman, 2004; Maluf, Fischer & Lohman, 2003; Myong et al., 2005) similar to the aforementioned studies on phage T7 and T4 SFIV hexameric ring-shaped replicative helicases (Johnson et al., 2007; Lionnet et al., 2007). The force-dependence of the unwinding rate, however, varies between superfamilies with the T7 and T4 helicases being strongly force-dependent (Johnson et al., 2007; Lionnet et al., 2007), the non-ring shaped SFII hepatitis NS3 helicase reported as force-independent (Dumont et al., 2006), and UvrD reported both as force-independent (Dessinges et al., 2004) as well as impeded by force (Sun et al., 2008). Force-independence of unwinding indicates that, in some cases, strand separation alone is not the rate-limiting step. These differences might indicate a mechanistic distinction in how and why these helicases slow down upon encountering the junction. One possibility is that the non-ring shaped helicases may undergo a rate-limiting dimerization step in order to unwind (Brendza et al., 2005; Fischer et al., 2004; Ha et al., 2002; Maluf et al., 2003; Myong et al., 2005).
3.3 Step Size for Helicase Unwinding and Translocation
3.3.1 Step size definitions and measuring techniques
One of the key mechanistic questions regarding helicases is elucidation of step size or how many bp/nts are unwound/translocated per NTP hydrolyzed, also referred to as chemical step size or NTP coupling ratio. Other definitions of step size include: 1) physical step size or average distance moved by the motor each time it moves on the NA lattice, and 2) kinetic step size - the average number of bp unwound (or nts translocated) between two successive rate-limiting steps in the unwinding or translocation cycle (assuming it consists of n sequential steps of identical rates). Kinetic step size is not necessarily equivalent to chemical step size or the physical distance translocated. For example, a kinetic step size of 3 bp/nt might reflect a series of fast translocation steps in 1 nt increments followed by a slow rate-limiting step such as a conformational change that occurs every 3 bp. Physical translocation could also occur in larger steps comprised of multiple steps with different step sizes (Fischer & Lohman, 2004; Lohman et al., 2008).
Four methods have been used to determine or predict helicase step size:
Crystallography is used primarily to predict NTP coupling ratios by structural comparisons of helicase-DNA co-crystals in the nucleotide-bound form versus apo-form.
Ensemble multiple-turnover kinetics can be used to obtain NTP coupling ratios by comparing steady-state rates of unwinding or translocation to rates of NTP hydrolysis.
Ensemble pre-steady-state single-turnover kinetics of unwinding/translocation and NTP hydrolysis (described below) allow for more accurate determination of kinetic and physical step sizes and NTP coupling ratios under conditions where the helicase binds once and does not rebind.
Single-molecule methods allow for determination of kinetic and physical step sizes.
In ensemble pre-steady-state single turnover assays for unwinding and translocation, (reviewed in (Fischer & Lohman, 2004; Lucius et al., 2003)) the helicase and DNA substrate are allowed to form an initial complex and then ATP is added using a stopped flow apparatus and translocation/unwinding is measured as a function of time. Single-round kinetics is usually ensured by the addition of a protein trap such as heparin or excess DNA substrate to prevent any dissociated enzyme from rebinding the substrate. Unwinding assays measure release of radioactively-labeled single-strand from the duplex or decrease in FRET between a donor and acceptor fluorophore pair situated in close proximity on opposing strands of duplex substrates of varying lengths (Lucius et al., 2003). Direct translocation assays monitor the arrival of a helicase at the end of a ssDNA by measuring protein-induced changes in the intensity of a fluorophore positioned at the end of ssDNAs of varying lengths (Dillingham, Wigley & Webb, 2002). In addition, a single-turnover fluorescent-based ATPase assay performed as a function of DNA cofactor length (Dillingham et al., 2000; Tomko et al., 2007) allows for calculation of the ATP coupling ratio.
Bulk kinetic determination of helicase step size can vary due to differences, for instance, in helicase oligomerization state, efficiency of NTP hydrolysis usage, enzyme pausing, and solution conditions. In some cases, translocation step size is not equivalent to unwinding step size, and large steps comprised of smaller sub-steps have been measured for a given enzyme. Single-molecule studies have helped resolve controversy in the literature regarding step sizes predicted by crystallographic and bulk kinetic studies. To explain this, we will focus on two examples: SFIA helicases PcrA/UvrD and SFII replicative RNA helicases such as HCV NS3.
3.3.2 SFI Helicases UvrD and PcrA
Crystallographic studies suggest that binding/hydrolysis of 1 ATP causes inchworm-like translocation along ssDNA in 1 nt increments for B. stearothermophilus PcrA (Velankar et al., 1999), and unwinding and translocation in 1 bp/nt increments for E. coli UvrD (Lee & Yang, 2006). In these examples, as crystal structures were obtained with monomeric species, the conclusion is that the small 1 bp stepping is achieved by helicase monomers.
In contrast, ensemble single-turnover kinetic assays performed on these helicases resulted in larger step sizes for unwinding and translocation. Pre-steady state assays led to determination of translocation kinetic step sizes of ~4 nts for UvrD (Fischer et al., 2004; Tomko et al., 2007) and PcrA (Niedziela-Majka et al., 2007) and of unwinding kinetic step sizes of 4–6 bp for UvrD (Ali & Lohman, 1997) and 4 bp for PcrA (Yang et al., 2008). Although the step size for translocation and unwinding correlates for each helicase, the protein/DNA ratios and length of ssDNA tail in substrates used in these studies along with numerous bulk kinetic studies suggest that translocation is performed by a monomeric species whereas unwinding requires dimerization (Ali, Maluf & Lohman, 1999; Fischer et al., 2004; Maluf et al., 2003; Yang et al., 2008).
Further support for a larger kinetic step size was obtained from a sm magnetic tweezer study of UvrD unwinding ((Dessinges et al., 2004) setup similar to Fig. 2e) in which statistical analysis of helicase-induced noise in unwinding burst kinetics yielded a kinetic step size for unwinding of ~6 bp. However, due to the large force (35 pN) applied to the ends of the nicked substrate in this work, it is possible that this measurement actually represented a translocation kinetic step size (Fischer et al., 2004). This was also suggested in a subsequent magnetic tweezer investigation that monitored UvrD unwinding of a hairpin substrate ((Sun et al., 2008) setup similar to Fig. 2f), in which the unwinding rate was 4-fold lower than that reported by (Dessinges et al., 2004). In this second sm study on UvrD (Sun et al., 2008), a similar statistical fluctuation analysis to that of (Dessinges et al., 2004) was applied to calculate an unwinding kinetic step size of 4.2 bp. Xi and colleagues also suggest, based on the nonexponential distribution of off-times between unwinding bursts, that a UvrD dimer catalyzes unwinding in their assay (Sun et al., 2008).
The ATP coupling ratios for translocation calculated from single-turnover ATPase time courses in the presence of varying length oligonucleotides were ~ 1 nt/ATP hydrolyzed for PcrA (Dillingham et al., 2000) and also for UvrD (Tomko et al., 2007). Recently, an ATP coupling ratio for unwinding by PcrA in the presence of initiator protein RepD was determined to be 1 bp per ATP hydrolyzed via similar assays with linear partial duplex and plasmid substrates (Slatter, Thomas & Webb, 2009). The differences between the small ATP coupling ratio predicted by crystallographic studies and single-turnover ATPase kinetics and the larger kinetic step size calculated from ensemble single-round pre-steady state kinetics and sm studies suggest that these helicases move 1 nt/bp per ATP hydrolyzed with a rate-limiting step in translocation or unwinding occurring every 4 ATPase cycles.
3.3.3 Replicative RNA Helicases
In our second example of replicative RNA helicases, an ATP coupling ratio of 1 bp unwound per ATP hydrolyzed has also been predicted based on comparison of ATP-bound and unbound crystal structures of four SFII RNA helicases including HCV NS3 helicase (reviewed in (Hopfner & Michaelis, 2007). However, similar to the SFIA helicase family, ensemble kinetic and sm studies have shown larger step sizes and unwinding bursts. Single-round pre-steady state kinetic measurements of unwinding of dsRNA of different lengths by Vaccinia virus NPH-II revealed a 6 bp RNA unwinding kinetic step size (Jankowsky et al., 2000). A combination of ensemble steady-state and pre-steady-state kinetic measurements of NS3 unwinding of RNA of different lengths revealed both a physical and kinetic step size for RNA unwinding of 18 bp (Serebrov & Pyle, 2004). A complementary sm approach using optical tweezers to monitor NS3 unwinding of RNA hairpins (Dumont et al., 2006) found extension occurred with a physical step size for unwinding of ~11 bp that was independent of ATP concentration and force. However, the optical tweezer experiment also detected three smaller substeps of 3.6 bp within each physical step, suggesting that NS3 is not unwinding in a single motion but in discrete smaller motions coordinated and triggered by the ATPase cycle, consistent with ATP dependence of the stepping velocity and duration of pauses between steps (Dumont et al., 2006) (Fig. 4a).
Fig. 4. Single Molecule Studies on Helicase Step Size.

(a) From (Dumont et al., 2006). (left) Optical tweezer setup to monitor NS3 helicase unwinding of an RNA hairpin (see Fig. 2h); (center) Pairwise distance distribution for unwinding extension time traces reveals an 11 ± 3 bp periodicity in pausing during extension. (right) Unwinding substeps of 3.6 ± 1.3 bp that appear within the 11 bp step observed at limiting ATP concentrations. (b) From (Myong et al., 2007). (far left) Experimental smFRET setup to measure NS3 unwinding of 18 bp partial duplex DNA; (mid-left) FRET unwinding time trace fit to step-finding algorithm quantitates FRET values and dwell times for six steps; (mid-right) Total Density Plot representing two-dimensional histograms for pairs of FRET values entering and exiting each transition reveals six peaks; (far-right) Gamma distribution fitting to collected dwell times at each plateau pause reveals three irreversible steps within the 3 bp step indicating 1 nt elemental step size within each 3 bp transition. See text for further details.
A separate smFRET-based assay of DNA unwinding by NS3 helicase (Myong et al., 2007) revealed unwinding of a partial duplex 18 bp DNA in 3 bp steps. Dwell time analysis of pause durations at each step was consistent with three irreversible steps that need to occur before 3 bp unwinding, indicating that the elemental step size within each 3 bp is 1 nt. This led to development of a model that translocation occurs in 1 nt increments, building up tension in ‘spring-loaded’ NS3 such that every 3 nts, unwinding occurs in 3 bp bursts (Myong et al., 2007). These sm studies thus provide a way to understand 1 nt translocation coupling ratio predicted by crystal structures with respect to larger 3.6 bp unwinding steps observed (Fig. 4b). However, what about the larger kinetic steps sizes of 11 and 18 bp previously observed (Dumont et al., 2006; Serebrov & Pyle, 2004)? In another series of pre-steady state stopped flow experiments using RNA of varying lengths and sequence (Serebrov, Beran & Pyle, 2009), the step size reduced from 18 to 11 bp upon lowering ionic strength. The NS3 oligomeric state was correlated with the ionic strength used in the previous studies, with it operating as a monomer at low ionic strength (Dumont et al., 2006) but as a dimer at higher ionic strength (Serebrov & Pyle, 2004). As the smFRET study (Myong et al., 2007) also showed that NS3 remained associated with unwound strand, Pyle and co-workers (Serebrov et al., 2009) then proposed that the 11 bp or 18 bp steps corresponded to 5′ unwound tail product release in pulses after RNA unwinding has reached a certain length.
3.3.4 Implications for helicase mechanisms
The combined work on NS3 provides evidence that helicases can move in 1 nt/bp steps which are linked to large conformational physical motions that occur during unwinding such as product release that are apparent as much bigger ‘steps.’ In general for all the helicases described, the smaller kinetic steps sizes of 3–6 nt/bp that are observed through sm and pre-steady-state single-turnover kinetics represent ATP-dependent rate-limiting events such as unwinding bursts which occur after moving a discrete number of bp in single-nt increments. Nonlinear global fitting of the single-cycle kinetic ensemble data is limited by two factors. First is the assumption that helicase movement occurs without slippage or pausing, which would lead to an overestimation of step size (Fischer et al., 2004). Second is the uncertainty in step size estimation caused by analyzing multiple data sets as a group (each with a limited amount of data) as opposed to a single data set with less variation (Lucius et al., 2002). Both of these problems can be addressed by sm techniques which in fact have detected pausing by many helicases (Lionnet et al., 2006) as well as derived kinetic information from data sets derived from single-molecule(s) (Myong et al., 2007; Myong et al., 2005).
3.4 Pausing, Reversal and Repetition during helicase activity
One of the advantages of sm versus bulk measurements is that it allows for detection of unsynchronized behavior of individual molecules such as enzyme and substrate conformational dynamics, chemical reaction intermediates, and behavioral heterogeneity of individual molecules that normally would be masked in ensemble-averaged measurements. Examples of such behaviors revealed in sm investigations of helicases include enzyme pausing and reversal during translocation and unwinding. These findings have been made for a broad spectrum of helicases using a variety of sm techniques. We describe below how discovery of these novel behaviors has provided insight into unwinding mechanisms and functions of several helicases.
3.4.1 Pausing and Reversal by RecBCD
E. coli RecBCD is a dual helicase/nuclease required in recombinational repair of dsDNA breaks. The helicase initially binds at the break site and proceeds to unwind ~ 30 kbp using two motors, RecD and RecB, which translocate with opposite polarity. As it unwinds DNA, the enzyme degrades both strands at differential rates. Upon encountering a χ recombination hotspot, RecBCD undergoes a conformational change resulting in decreased helicase/nuclease activity and production of a long 3′ ss tail for loading of RecA and continuation of homologous recombination. Results of several sm studies have helped elucidate the mechanism and χ-site regulation of RecBCD translocation.
Two non-force sm experimental systems have provided evidence for highly processive unidirectional movement by RecBCD along dsDNA. A tethered-particle-motion (TPA) investigation (Dohoney & Gelles, 2001) monitored changes in Brownian motion of a streptavidin-coated polystyrene bead linked to RecBCD through a biotin-tagged RecD as it unwound/degraded a 1 kb tethered DNA (in an assay similar to Fig. 2g). No enzyme pausing was detected regardless of whether the DNA contained an internal χ site, with RecBCD dissociating upon reaching the tether, thus ruling out a model where the χ site-induced conformational change involves RecD subunit ejection. Another study (Bianco et al., 2001) used optical trapping fluorescence microscopy to measure RecBCD movement through 40-fold longer λ DNA (Fig. 2d). Again, no force was applied, but tethering was achieved by placing a bead linked to one DNA end in an optical trap, while the remaining DNA was visible by intercalation of fluorescent probe YOYO. A microfluidics setup was used in which the DNA-RecBCD complex in one channel is captured in the optical trap and then moved to a second channel where ATP is flowed in. RecBCD unwinding/degradation was followed by monitoring changes in fluorescence while YOYO was released upon dsDNA shortening. Again, highly processive unwinding with no pausing was observed at saturating ATP over the length of the entire λ DNA.
This optical trapping fluorescence method was next applied to analyze RecBCD unwinding of 30 kb DNA containing χ sequences (Spies et al., 2003), and ~5 sec χ-specific pauses were observed followed by unwinding at a reduced rate rate (Fig. 5a). This phenomenon was postulated to reflect either uncoupling of RecD from the holoenzyme or a conformational change in RecBCD that inactivates RecD. The detection of χ-induced pauses in this setup as opposed to the TPA experiment was probably due to the higher spatio-temporal resolution. An extension of this experiment was performed without YOYO but instead using RecBCD tagged through its RecD subunit with a fluorescent bead (Handa et al., 2005). Again, processive unwinding was observed prior to a 6 sec pause at χ site after which the unwinding rate decreased two-fold. The fluorescent nanoparticle stayed with the RecBCD for the entire duration of the pause, thus agreeing with the conclusion made by (Dohoney & Gelles, 2001) that RecD is not dissociating at χ. Further analysis using the same assay with a RecBCD mutant with a nonfunctional RecD subunit (Spies et al., 2007) led to the finding that RecD is the lead motor prior to χ recognition whereupon RecD becomes uncoupled from the holoenzyme and RecB becomes the lead motor. The unwinding rates correlated with a model in which there is an initial requirement for fast unwinding of damaged or foreign DNA but χ recognition signals the need to slow down in order to properly assemble the RecA filament.
Fig. 5. Single-Molecule Detection of Pausing and Reversal during helicase unwinding.

(a) From (Spies et al., 2003). Optical Trap Fluorescence Microscopy (see Fig. 2d) to monitor DNA unwinding by RecBCD detects χ-site-specific pausing followed by unwinding at a slower velocity by a RecD-uncoupled holoenzyme. (b) From (Sun et al., 2008). Magnetic Tweezer study of UvrD unwinding of DNA hairpin shows multiple patterns in unwinding bursts (extension) followed by rapid rehybrization (not shown) or slower rezipping. (left) Pauses (P) during unwinding may reflect transient dimer dissocation; (center) Transitions from a slow to fast rewinding rate may reflect dimer dissociation during translocation on the opposing strand after strand switching. (right) Inverse force-dependence of unwinding rate was incorporated into a strained inchworm model for unwinding in which UvrD dimers bend the 3′ ssDNA tail to form an active unwinding species. (c) From (Yodh et al., 2009). smFRET study of unwinding of forked DNA by human BLM helicase (left) Example of partial repetitive unwinding behavior by a single helicase. (center) Histogram of the change in FRET values in repetitive unwinding events shows a peak at 0.3 ΔFRET efficiency. (right) A threshold ΔFRET of 0.2 must be achieved before BLM reverses unwinding. These data suggest BLM ‘measures’ the amount of bp it unwinds before it reverses unwinding. See text for further details.
As a follow up to the original TPA study (Dohoney & Gelles, 2001), a force-TPA assay was developed in which a stretching force was applied by continuous laminar flow to the DNA-bead complex (Fan & Li, 2009). Using this force-TPA setup with low ATP and 0.2 pN stretching force, RecBCD unwound a 4.1 kb DNA at the same rate observed previously except in this case χ-specific pausing was observed followed by a rate decrease, suggesting that the lack of pausing seen by (Dohoney & Gelles, 2001) was likely due to the more limited resolution of that experimental setup.
Block and colleagues applied a different force-based approach to investigate RecBCD activity (Perkins et al., 2004) in which a biotinylated RecBCD was tethered to a coverslip via a streptavidin linkage and a 7 kb DNA substrate lacking χ sites was attached at its distal end to a polystyrene bead held inside an optical trap over a varying force range as shown in Fig 2i. The surface-bound enzyme bound to the free DNA end and upon ATP addition, its movement along dsDNA towards the trap was monitored under constant force up to 8 pN. DNA unwinding correlated with decreasing length of the tether, and RecBCD was observed to move unidirectionally over ~1000s of bp. However in contrast to the previous zero- or low-force studies (Bianco et al., 2001; Dohoney & Gelles, 2001), χ-independent pausing was observed including transient force-dependent pauses as well as spontaneous pausing that was independent of both force and ATP. This high resolution optical trapping method revealed yet another surprising behavior – sporadic uneven RecBCD-dependent backward movement during unwinding. Releasing the force converted the reversals into forward motions indicating backsliding was caused by force. The change in tether length upon backsliding was consistent with partial ssDNA formation indicating possibly a ssDNA loop formation after unwinding but prior to degradation. In general, the force-dependent pausing and reversal during RecBCD movement were not thought to be sequence-dependent but instead to reflect kinetically distinct persistent states of the enzyme-DNA complex.
3.4.2 Force-dependent Unwinding/Rewinding Bursts by UvrD and Replicative Helicases
Magnetic tweezer studies analyzing force-dependent UvrD unwinding kinetics have revealed pausing and reversal behavior (Dessinges et al., 2004; Sun et al., 2008). Croquette and coworkers (Dessinges et al., 2004) monitored the unwinding of an 11 kb singly nicked duplex DNA that was anchored to a magnetic bead at one end of the duplex and to a glass surface at the other end (as in Fig. 2e). At UvrD < 1 nM, the extension versus time curves obtained at high force > 30 pN exhibited isolated unwinding bursts. Each burst showed a regular increase in extension corresponding to unwinding for which the slope and height of the burst was used to calculate the rate of unwinding (U) and extent of unwinding, respectively. Unwinding proceeded processively without pausing. At a certain point, the extension signal decreased back to baseline in one of two fashions – UH bursts show an instantaneous decrease corresponding to enzyme dissociation and rapid rehybridization (H); whereas, UZ bursts showed a much slower decrease reflecting a slower rezipping (Z) event. In UZ bursts, the slow rezipping rate was equivalent to the unwinding rate, and was ATP-dependent indicating that ssDNA translocation might be involved. These findings led the authors to propose a model in which UvrD is capable of reversing unwinding by switching strands and translocating 3′–5′ on the opposing single strand, thus inducing reannealing behind the helicase. The high force required to achieve unwinding at low UvrD in this study most likely helps prevent reannealing during unwinding and was proposed to be the reason unwinding is achieved at sub-nM concentrations unlike in bulk studies (Maluf et al., 2003).
A second study (Sun et al., 2008) analyzed force-dependent UvrD unwinding of a λ DNA hairpin situated between a λ dsDNA tethered to the surface and a 3′ ssDNA oligonucleotide tethered to a magnetic bead (similar to Fig. 2f). At saturating ATP, UvrD < 10 nM and forces > 4 pN, UvrD produced the UH and UZ unwinding burst patterns observed previously (Dessinges et al., 2004), however additional patterns were also observed. First, pausing was observed in 25% of unwinding events during both U and Z phases as well as between them. The pausing frequency was inversely proportional to UvrD concentration, thus relating pausing to the enzyme oligomeric state. Second, slow and fast rezipping (Z) rates were observed, indicating there may be two kinds of translocases operating. The U and slow Z rates in the hairpin study (Sun et al., 2008) were 10-fold lower than those measured previously on nicked DNA (Dessinges et al., 2004) which were actually closer to monomeric UvrD ssDNA translocation rate measured in ensemble studies (Fischer et al., 2004). The fast rezipping rate observed in the hairpin study was also closer to the ssDNA translocation rate. The distributions of off-times between unwinding bursts followed a nonexponential distribution which was inconsistent with single enzyme binding kinetics (Sun et al., 2008). In light of these findings Xi and colleagues proposed that their unwinding bursts reflected unwinding by a dimeric helicase with the pausing representing transient or permanent dimer dissociation (Fig. 5b) which is reminiscent of the unwinding initiation and reinitiation model proposed for Rep helicase based on sm analysis (Ha et al., 2002). The switch between slow and fast rezipping may reflect dimer dissociation/monomer-monomer reassociation, with both dimers and monomers being able to switch strands and translocate on ssDNA (Sun et al., 2008).
Perhaps the most intriguing result of the second magnetic tweezer study (Sun et al., 2008) was that the UvrD unwinding rate decreased with increasing force, indicating that force kinetically impeded unwinding (Fig. 5b). From this the authors proposed that UvrD uses a strained inchworm mechanism for unwinding involving a UvrD dimer that must bend the 3′ ssDNA tail in order to achieve a modeled interaction of both 2B domains required for activation of unwinding. The experimental force on the ssDNA tail in this system may hinder the bending of the DNA at the junction and thus unwinding.
Croquette and colleagues also observed primarily UH/UZ-type events when monitoring unwinding of a hairpin substrate by T4 hexameric gp41 helicase via magnetic tweezers as shown in Fig 2f (Lionnet et al., 2007) at saturating ATP, and forces between 6–10 pN. In UH events, the helicase unwound the hairpin fully and then dissociated causing the hairpin to reanneal instantaneously. In UZ events, the slow rezipping phase was ATP-dependent signifying helicase-dependence, however this was not caused by strand switching as proposed for UvrD (Dessinges et al., 2004) because the original substrate was a hairpin. Instead, the gp41 was proposed to continue translocating along the newly unwound DNA with the hairpin reannealing in its wake. Similar reverse motions characterized by unwinding/rewinding bursts have also been observed albeit infrequently for the T7 hexameric helicase using an optical trap-based unzipping assay at forces between 7–12 pN (Johnson et al., 2007). Finally, rare step-wise rewinding events have also been detected during NS3 unwinding of an RNA hairpin measured at 17 pN using an optical tweezer-based assay (Dumont et al., 2006).
3.4.3 Reverse motions during helicase activity detected by smFRET
Single-molecule FRET studies on helicases have also revealed reverse motions and stalling during helicase activity at zero force. Initial studies on E. coli Rep helicase (Ha et al., 2002) using an unwinding substrate - a partial duplex DNA with both donor and acceptor dyes at the ss-dsDNA junction (Fig. 2a) – revealed that Rep could efficiently unwind an 18 bp duplex but not a 40 bp duplex. Seventy percent of the time, unwinding of the 40-mer was punctuated by stalls that usually led to dissociation of Rep and reannealing of the substrate. The stalls were proposed to be due to dissociation of dimeric rep, the functional oligomeric state for unwinding. In some cases, reassociation of monomers led to unwinding re-initiation on the same substrate.
Ha and co-workers have also discovered repeating unwinding patterns in smFRET time traces for two SFII helicases – HCV NS3 (Myong et al., 2007) and the human BLM helicase (Yodh et al., 2009). Using partial duplex DNA templates with an 18 bp duplex region tethered to the surface and acceptor and donor dyes situated at the junction on adjacent strands, addition of ATP caused NS3 to unwind the DNA (with a corresponding FRET decrease), however upon reaching the surface, the DNA reannealed instantaneously. This pattern was repeated in succession on the same molecule and persisted even after washing out free protein indicating it was catalyzed by a single NS3 species. Incomplete, repetitive unwinding was also observed using duplexes longer than 18 bp indicating that a steric blockade is not necessary for unwinding reversal. The authors proposed a model that NS3 remains in contact with the displaced strand during unwinding and upon reaching a steric or length-based block, returns to the junction with concomitant strand reannealing and begins the unwinding cycle again (Myong et al., 2007).
Repetitive unwinding was also catalyzed by BLM helicase, a human RecQ homologue, on forked DNA of varying duplex lengths with the dyes situated in close proximity near the fork junction (Yodh et al., 2009) (Fig. 5c). This behavior was observed for wild type BLM and a mutant comprising the RecQ catalytic core which lacks the oligomerization domain (Janscak et al., 2003). This fact as well as protein-concentration independence of unwinding times led to conclusion that successive unwinding events on the same DNA molecule are carried out by a single BLM monomer. Each unwinding event was characterized by three phases: I) a phase during which FRET decreases by a certain amount, ΔFRET, due to unwinding, followed by II) a time period during which FRET remains at a constant low value, followed by III) a reannealing phase characterized by a rapid switch back to high FRET. The ATP-dependence of reannealing time was reminiscent of the ATP-dependence of the slow-rezipping events detected for UvrD previously (Dessinges et al., 2004). It was proposed that BLM partially dissociated after unwinding a critical length of DNA, and then switched its helicase motors to the opposing strand and translocated 3′–5′ on this strand with the strands reannealing in its wake. However, unlike UvrD and T7 which terminated unwinding stochastically after a random number of bp were unwound, BLM appeared to terminate after a short but defined length of DNA was unwound - based on a defined peak observed in ΔFRET distributions. Thus, BLM appears to be able to ‘measure’ the number of bp it has unwound and once it has unwound a critical length, it rapidly reverses unwinding via strand switching (Fig. 5c). The ability to switch strands and measure DNA unwound may facilitate BLM processing of stalled replication forks and illegitimately-formed recombination intermediates.
In these investigations on NS3 and BLM, smFRET was able to detect partially unwound intermediates in real time, leading to discovery of repetitive unwinding. Repetitive behaviors during helicase translocation by Rep and RIG-I proteins (Myong et al., 2009; Myong et al., 2005) were also discovered using sm fluorescence (described in section 3.5).
3.5 Conformational dynamics during helicase activity
TPA and force-based tweezer techniques do not offer information on conformational changes in the enzyme or enzyme-substrate complexes during helicase action. In contrast, fluorescence-based sm techniques such as smFRET can detect conformational dynamics because of the ability to place donor and acceptor probes in specific locations on the enzyme and/or substrate and extract information regarding changes in probe-probe distances as the helicase operates in real time. In this section we will describe smFRET investigations that have focused on helicase dynamics during translocation (Myong et al., 2005), substrate-enzyme binding dynamics (Rothenberg et al., 2007), and helicase inter-domain arrangements (Aregger & Klostermeier, 2009; Karow & Klostermeier, 2009; Linden, Hartmann & Klostermeier, 2008; Theissen et al., 2008).
3.5.1 Repetitive shuttling during translocation
smFRET was used to measure E. coli Rep translocation on ssDNA using a partial duplex substrate with acceptor dye positioned at the junction end of the 3′ ssDNA tail while the enzyme was donor-labeled (Myong et al., 2005). These experiments were performed at sub-nM concentrations of Rep which do not allow for dimerization and therefore unwinding. In the presence of ATP, ssDNA translocation by Rep was expected to initiate with the enzyme binding to the available 3′ end of the ssDNA tail producing a low FRET signal and then moving towards the junction with a corresponding FRET increase related to the distance translocated. However, the enzyme did not dissociate upon reaching the junction but instead appeard to snap back to the 3′ end of the ssDNA (observed as an abrupt FRET decrease), and showed repeated cycles of translocation and snap-back. The same phenomenon was observed with dual-labeled Rep and an unlabeled translocation substrate indicating that Rep undergoes a conformational change during translocation from an open to closed form, possibly exposing a secondary binding site that then rebinds the free 3′ tail, transferring Rep from the junction back to the 3′ end of the ssDNA tail whence it begins another cycle (Fig. 6a). This translocation behavior was termed ‘repetitive shuttling’ and proposed to play role in removal of proteins such as RecA filaments in order to prevent unwanted recombination. Note that Rep shuttling behavior was also observed using sm protein-induced fluorescent enhancement (PIFE) with unlabeled Rep on a Cy3-labeled DNA (Myong et al., 2009).
Fig. 6. Single-Molecule Detection of Helicase Conformational Dynamics.

(a) From (Myong et al., 2005). (left) smFRET assay demonstrates dual-labeled Rep helicase catalyzes repetitive translocation along unlabeled ssDNA, indicating Rep itself undergoes a conformational change during the shuttling cycle. (right) Model for translocation cycle in which Rep transitions form open to closed forms, exposing a secondary binding site for the free 3′ ss tail, allowing Rep to transfer from the junction to the 3′ end and reinitiate the cycle. (b) From (Theissen et al., 2008). (left) Dual fluorescent labeling of YxiN, DEAD-box helicase with donor (A488) and acceptor (A546) on different sides of interdomain cleft separating the two RecA-like helicase domains (right) FRET histograms ± ligands for fluorescently labeled YxiN* S108C/S229C. FRET in the absence of any ligands or in the presence of any individual ligand is 0.5. Cooperative binding of ADPNP and RNA promotes interdomain cleft closure increasing FRET to 0.8. See text for further details.
The PIFE technique was also applied to show that the dsRNA dependent SFII DExH ATPase, RIG-I, a cytosolic viral sensor, can translocate on dsRNA in an ATP-dependent manner (Myong et al., 2009). Similar to Rep translocation along ssDNA, a single RIG-I catalyzed translocation in successive repetitions on the same RNA molecule. Although the physical mechanism of RIG-I dsRNA translocation is still unclear, the finding that this activity could be trans-activated by 5′ triphosphate caps which are typically present on viral RNA post-infection are consistent with a role for this activity in viral sensing by RIG-I.
3.5.2 Replicative helicase-DNA tail interaction dynamics
smFRET has also been applied to investigate interaction dynamics between replicative helicases and ssDNA tails at the fork (Rothenberg et al., 2007). The archaeal MCM from S. solfataricus (SsoMCM), a homologue of eukaryotic hexameric MCM replicative helicases, was previously characterized to preferentially unwind Y-shaped substrates with accessible 3′ tails. Using acceptor-labeled SsoMCM and donor-labeled substrates, smFRET was applied (Rothenberg et al., 2007) to determine the orientation of MCM along the 3′ tail, and confirm the helicase’s preference for fork substrates. Binding of unlabeled SsoMCM to forked substrates with acceptor dye placed at the end of the 5′ tail and donor dye placed at various locations on the 3′ tail increased FRET from low to high, consistent with the idea that MCM bound the 3′ tail through its central channel while interacting with the 5′ tail through an external site on the protein surface. While MCM bound very stably without dissociating for up to an hour, temporal fluctuations in FRET were also observed with N-terminally acceptor-labeled MCM and donor-labeled 5′ tail but not with donor-labeled 3′ tail, indicating a specific dynamic interaction between the helicase and the 5′ tail. This work provided evidence that a replicative helicase may also interact with the nontracking strand during unwinding, possibly playing a role in substrate specificity, helicase stability at the fork, and unwinding (Rothenberg et al., 2007), and suggesting that ‘exclusion’ of this strand may not be the main mechanistic role.
3.5.3 DEAD-Box RNA helicase conformational switching
Cofactor-induced conformational changes in DEAD-box RNA helicases have also been probed using smFRET-based techniques (Aregger & Klostermeier, 2009; Karow & Klostermeier, 2009; Linden et al., 2008; Theissen et al., 2008). Crystal structures of members of this helicase family reveal a common core structure consisting of two RecA-like helicase domains separated by an interdomain cleft, and suggest that simultaneous binding of ssRNA and nucleotide induce a conformational change from an ‘open’ state in which the helicase domains are separated to a ‘closed’ state in which the helicase domains make multiple contacts with each other. As there is no DEAD-box RNA helicase for which both open and closed structures are available, Klostermeier and colleagues utilized a confocal smFRET method (shown in Fig. 2b) to analyze interdomain cleft movement in B. subtillus YxiN, a helicase involved in ribosome biogenesis (Theissen et al., 2008). YxiN double mutants were constructed for attachment of a donor and an acceptor in each helicase domain. In the absence of ligands, double-labeled YxiN followed a unimodal mid-FRET distribution consistent with a single open conformation. In the presence of nucleotide or RNA alone, the FRET distributions remained unaltered, however simultaneous addition of ADPNP/ATP plus RNA resulted in a switch to a high FRET species for all the YxiN double mutants. Thus RNA and ATP substrates act cooperatively to induce closure of the interdomain cleft, bringing the helicase domains in close contact (Fig. 6b). Thus, smFRET captured both open and closed conformations of YxiN and correlated them with RNA/ADP-bound and RNA/ATP bound states of the helicase, respectively (Theissen et al., 2008). In a follow-up study (Aregger & Klostermeier, 2009), it was shown that YxiN remained in the closed state in the presence of ATP analogues plus RNA, suggesting that helicase reopening post-ATP hydrolysis is most likely coupled to Pi release. Furthermore, YxiN mutants deficient in unwinding were still able to undergo interdomain cleft closure, indicating that cleft closure is important but not sufficient for unwinding (Karow & Klostermeier, 2009). This assay was also applied to investigate the substrate specificity of a related DEAD-box RNA helicase, Hera from T. thermophilus (Linden et al., 2008). Two RNA substrates that were identified by their ability to induce interdomain cleft closure were RNase P RNA and 23S rRNA hairpin 92, suggesting a role for Hera in ribosome biogenesis. These studies illustrate the success of smFRET applications to determining not only conformational changes undergone by these DEAD-box RNA helicases but also information on their cellular functions (Linden et al., 2008).
3.6 Helicases working in multiprotein complexes
The sm results described thus far have yielded important information on unwinding/translocation parameters and mechanisms of helicases by themselves. However, in vivo, helicases do not work as isolated molecules but in complexes with, for example, replication, recombination, and repair machinery. Thus, it is essential to observe helicases operating in conjunction with their required partners in these processes such as primase, polymerase, and single-stranded DNA binding proteins. This section describes recent applications of sm methods towards this goal.
3.6.1 Phage T4 primosome assembly
Single-molecule techniques have been used to probe the assembly of the bacteriophage T4 primosome, comprised of the hexameric helicase (gp41), primase (gp61), single-stranded binding protein (gp32), and accessory helicase loader protein (gp59). This complex works together with DNA polymerase (gp43) which is aided by sliding clamp (gp45) to carry out DNA synthesis as a replisome. While gp41 can bind to forked templates by itself, its binding is stimulated by gp59 which recruits and loads gp41 helicase onto gp32-coated DNA during recombinational replication. Hammes and colleagues determined the order of assembly of primosome components using a combination of ensemble and smFRET techniques (Zhang et al., 2005). In this work, all four primosome proteins were single-labeled with donor or acceptor fluorophores and analyzed for interactions between the various proteins in the presence of ssDNA (ensemble FRET) to obtain binding stoichiometries, or with a biotinylated forked DNA containing both leading and lagging strand junctions (smFRET) to prove direct interactions between proteins and the order in which they were required for assembly. The results led to a model in which a 1:1 complex of gp32 and gp59 first forms on forked DNA followed by recruitment of gp41 helicase and its hexamerization. Subsequent ATP hydrolysis by gp41 leads to dissociation of gp32 and gp59. Primase (gp61) then binds to gp41-DNA complex to complete primosome assembly. Recently, sm photobleaching experiments showed that the hexameric form of gp59 required for helicase loading (Arumugam, Lee & Benkovic, 2009). The same group has also used this assay to further probe the role of helicase loader (gp59) in initiation of lagging strand replication (Xi et al., 2005a; Xi et al., 2005b)
3.6.2 Phage T7, T4, and E. coli replisome dynamics
van Oijen and colleagues have used sm flow-stretch techniques to investigate replisome activity in the bacteriophage T7 and E. coli systems (Hamdan et al., 2007; Hamdan et al., 2009; Lee et al., 2006; Tanner et al., 2008; Tanner et al., 2009). The experimental setup is as shown in Fig 2c in which a forked λ DNA template is tethered to the surface via a ssDNA region at the 5′ end of the lagging strand template; its 3′ end is attached to a tracking bead; and the entire template subject to laminar flow at ~ 3 pN. A DNA primer is annealed to the other strand creating a primer-template junction for leading strand synthesis. Leading strand synthesis by T7 polymerase was measured in this system (Lee et al., 2006) by assembling a minimal replisome (helicase-primase (gp4) and thioredoxin-polymerase (trx-gp5)) and monitoring DNA shortening of the single-stranded lagging strand template during fork unwinding/DNA synthesis. The rate of DNA synthesis matched that measured in bulk assays, however the processivity of DNA synthesis/unwinding in this concerted system was 1–2 orders of magnitude higher relative to that measured with polymerase or helicase alone. When rNTPs were added to this system to enable primase, short pauses during leading strand synthesis trajectories were observed. These were interpreted to reflect pausing of the polymerase while primers were being formed on the lagging strand template. When excess polymerase was additionally added to this system to enable lagging strand synthesis, the pauses were followed by continued shortening of the DNA and then abrupt lengthening. This pattern was interpreted to represent pausing during primer formation followed by creation and release of a DNA loop during lagging strand synthesis (i.e. trombone model). These real-time trajectories of DNA length changes created during action of the minimal T7 replisome were incorporated into a primase-brake model to explain how lagging and leading strand synthesis are synchronized (Lee et al., 2006) (Fig. 7a).
Fig. 7. Single-Molecule Studies of Helicases within Replisome Complexes.
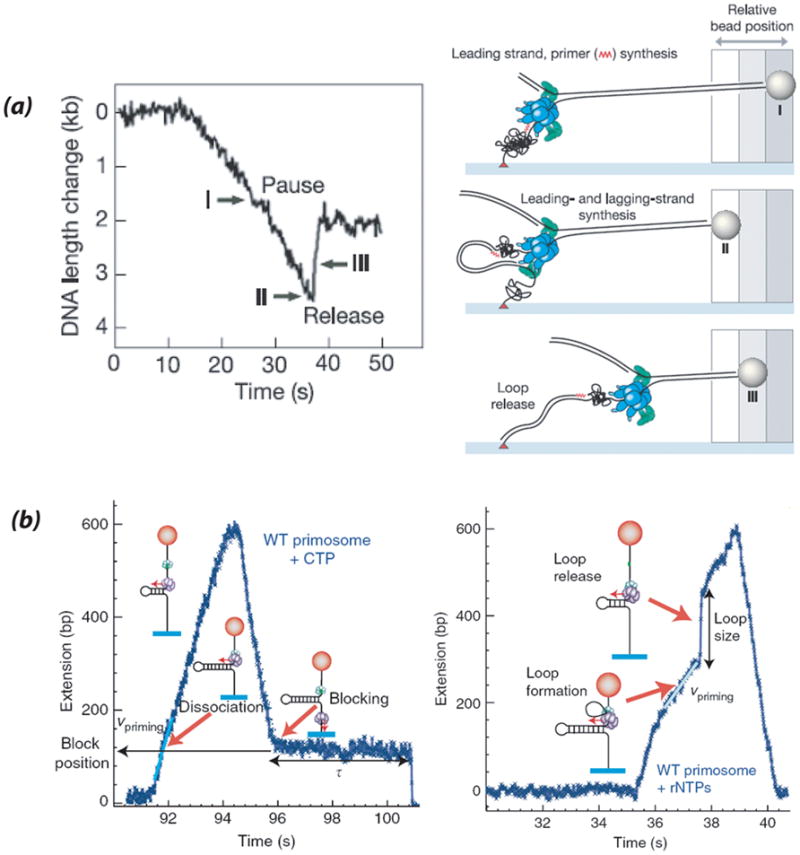
(a) From (Lee et al., 2006). Flow-Stretch assay (see Fig. 2c) of phage T7 replisome activity monitoring DNA shortening of the lagging strand template generated during leading strand synthesis. Pauses (I) were interpreted to reflect primer synthesis after which lagging strand synthesis continues with formation of a lagging strand replication loop (II). Loop release (III) is observed as lengthening of the DNA. (b) From (Manosas et al., 2009). Magnetic Tweezers Assay (see Fig. 2f) of phage T4 primosome (gp41 helicase/gp61 primase) activity using a hairpin substrate. Trajectories show an increase in DNA extension representing hairpin unwinding followed by fast or slow (shown) rehybridization (decrease in DNA extension) as helicase continues moving around the hairpin. (left) T4 primosome does not pause while priming (i.e. unwinding velocity remains constant) and rezipping traces are interrupted by primer-specific periods of constant extension interpreted to represent blocks in reannealing caused by a dissociated primase-primer complex. (right) Detection of priming loop formation in DNA extension time traces represented by periods of slower unwinding velocity followed by jumps back to original rate reflecting loop release. The priming loop is created by intact primosomes such that the unwound lagging strand template forms a loop which is released upon primer completion and transfer to the lagging strand polymerase. See text for further details.
This flow-stretch assay also confirmed that an electrostatic interaction between the C-terminal tail of the helicase and a basic region in the trx-binding region of the polymerase was crucial for T7 replisome processivity via retention of trx-gp5 in the event that it dissociates during leading strand synthesis (Hamdan et al., 2007). The kinetics of T7 trombone loop formation and dissolution were further analyzed (Hamdan et al., 2009) suggesting that the trombone loop is released in response to either new primer formation or Okazaki fragment completion. Loop size also decreased with increasing stretching force consistent with idea that the loop is held together by predicted interactions between helicase at the fork and leading and lagging strand polymerases. Imaging of a dsDNA intercalator, YOYO, confirmed that DNA shortening represents both leading and lagging strand formation.
This experimental system was also used to measure primer extension and leading strand synthesis by the E. coli PolIII holoenzyme (Tanner et al., 2008). Whereas primer extension on a ssDNA template occurred in repeated bursts of ~1 kb separated by pauses, the leading strand synthesis on dsDNA template by the replisome which also included DnaB, helicase loader DnaC and a minimal clamp loading complex, occurred in ~10 kb increments. The addition of primase (DnaG) decreased the processivity of DNA synthesis three-fold through its interaction with DnaB, but no pausing was observed suggesting an alternate mechanism for slowing down leading strand synthesis during slow primer synthesis on the lagging strand.
A magnetic tweezer setup similar to Fig. 2f was recently used to monitor phage T4 primosome (i.e. gp41-gp61 helicase-primase) activity on a DNA hairpin substrate (Manosas et al., 2009). In this system, unwinding activity is reported as an increase in DNA extension, followed by either rapid rezipping or slow reannealing of the hairpin after helicase dissociation. No pausing was observed during the unwinding phase even when primers are created. Two classes of behavior were observed during primer synthesis. In the first, primase dissocated from the helicase as suggested by periods of no extension during reannealing which were shown to be caused by transient blocking of hairpin rezipping by the primase-primer complex (Fig. 7b). In the second, unwinding traces showed periods of slower velocity unwinding followed by a switch back to the original unwinding rate. This pattern was interpreted to represent formation of a priming loop by an intact primosome in which the unwound DNA forms a loop between the helicase and primase that is released once the primer is synthesized. The slower unwinding velocity is due to the fact that only one of the two unwound strands contributes to the extension (Fig. 7b). A fused primosome (i.e. single gp41-gp61 polypeptide) worked entirely via the priming loop mechanism and addition of T4 replisome accessory proteins increased the probability of priming loop formation. This work has thus provided evidence for two types of primosome behavior during primer synthesis involving either primosome disassembly or a priming loop.
A related study on the phage T7 system investigated coordination of leading/lagging strand synthesis via ensemble and smFRET kinetic assays (Pandey et al., 2009). Using smFRET unwinding substrates such as in Fig. 2a, and similar radiolabeled substrates for ensemble DNA elongation assays, a primase activity-dependent slowing/pausing was detected during unwinding by gp4 helicase-primase but not during unwinding/DNAsynthesis by the T7 replisome, in contrast to previous reports (Lee et al., 2006). In a separate experiment, a smFRET substrate with donor placed near the priming sequence and acceptor 40 bp away was utilized to provide evidence for primer-sequence dependent creation of a priming loop during unwinding by both gp4 and the T4 replisome. Finally, ensemble DNA elongation assays showed that lagging strand synthesis is 30% faster than leading strand synthesis by the T4 replisome. Combined, these results suggest three ways to coordinate dual strand synthesis: 1) DNA synthesis occurs concomitantly and is not interrupted by primer synthesis; 2) Primase remains engaged with the priming site during unwinding/DNA synthesis leading to loop formation in the nascent lagging strand such that the nascent primers are in close proximity for hand-off to the polymerase, and 3) the lagging strand polymerase synthesizes DNA faster allowing more time for recycling steps and to keep up with leading strand synthesis. In light of this study, prior results that relate pausing during T7 DNA synthesis to primase action (Lee et al., 2006), may actually reflect uncoupled lagging and leading strand synthesis.
3.6.3 Effect of hRPA on BLM unwinding dynamics
The human BLM helicase has been shown to interact with numerous proteins both in vitro and in vivo. One important partner is the human single-stranded binding protein, Replication factor A (hRPA), which facilitates unwinding of long duplex regions by BLM (Brosh et al., 2000). This processivity effect was postulated to be due to a direct specific interaction between BLM and hRPA. As described earlier, an smFRET study (Yodh et al., 2009) revealed a repetitive unwinding behavior by BLM on forked DNA substrates in which BLM partially unwound a critical length of DNA followed by reversal of unwinding by strand switching, and translocation on the opposing strand with concomitant reannealing of the strands in its wake. After a certain point (i.e. wait time), BLM switched back to the tracking strand and repeated the unwinding cycle. Surprisingly, the repetitive unwinding pattern persisted in the presence of hRPA with only a limited increase in unwinding processivity per cycle. The same effect was observed using the S. cerevisiae homologue, scRPA, which interacts with BLM through the same residues as hRPA but not with the structurally nonhomologous SSB from E. coli. This indicated that the hRPA effect is mediated through a direct physical interaction with BLM as opposed to solely hRPA prevention of strand reannealing. The presence of hRPA had no effect on reannealing time indicating it is most likely dissociating during this phase. And while hRPA did not change the wait time between repetitive unwinding events for wild-type BLM, it did significantly increase wait time for a mutant BLM that comprises the RecQ helicase core (Janscak et al., 2003) but lacks the high affinity hRPA binding site. The latter result indicates that hRPA facilitates BLM transfer back to the tracking strand and re-initiation of unwinding via direct physical interaction between the two proteins.
3.6.4 Translocation on hRPA-coated DNA by Archaeal XPD helicase
Archaeal Rad3 from F. acidarmanus, a homologue of human XPD helicase required in nucleotide excision repair, possesses a unique Fe-S cluster in helicase domain 1. Spies and colleagues took advantage of fluorescence quenching property of the Fe-S cluster to examine translocation by this helicase on RPA-coated DNA (Honda et al., 2009). F. acidarmanus RPA1 and RPA2 have been shown to enhance unwinding by XPD but whether this is mediated through direct interactions is unknown (Pugh et al., 2008). Using short linear ssDNA biotinylated at the 5′ end with Cy3 tethered at the 3′ end, XPD ATP-dependent ssDNA translocation was measured using sm fluorescence microscopy as distance-dependent quenching of Cy3 as a function of time. In the presence of Cy5-labeled RPA1 and RPA2, XPD continued translocation by one of two mechanisms – either with concurrent dissociation of RPA1 or while bypassing RPA2. Both of these patterns were confirmed in experiments monitoring Cy5-RPA quenching induced by translocating XPD on unlabeled DNA. These sm experiments thus provide information on general mechanisms used by DNA repair helicases when encountering substrates coated with single-stranded DNA binding proteins.
4. Conclusions and Future Directions
It is clear that sm technology is proving to be an extremely relevant tool for probing the mechanism by which helicase motors work. Force-and fluorescence based experiments have provided support for models in which helicases move in 1 bp/nt increments with rate-limiting events such as unwinding bursts occurring every 3–6 bp, and physical motions such as strand release occurring in larger steps. Measurements of unwinding force- and sequence-dependence have provided a means to determine the degree of duplex destabilization achieved by helicases across superfamilies. Optical trapping and smFRET methods and tweezer-based assays have allowed observations of sequence-specific pausing and spontaneous pausing/reversal during helicase movement as well as discovery of repetitive translocation/partial unwinding helicase activities. Fluorescence-based methods have yielded new information on conformational dynamics during helicase-NA and helicase subunit interactions. Finally, flow-stretch, tweezer-based and smFRET experiments have enabled the study of helicases as they operate within the primosome and replisome and in conjunction with relevant partners such as RPA.
Ongoing developments in single-molecule techniques such as use of multicolor FRET, fluorescence-force spectroscopy (Joo et al., 2008), and improved spatiotemporal resolution in force-based methods will no doubt further advance our understanding of helicase activity in these and other areas of investigation. Examples of questions requiring further elucidation include how helicases operate in multiprotein complexes involved in repair, recombination, and RNA metabolism; the physical basis for directional translocation; helicase action on complex substrates such as Holliday junctions, G-quadruplexes, and nucleosomes; and catalysis of alternative helicase activities such as NA single-strand annealing. However, to date, single-molecule methods have only proven useful to determine new activities and kinetic/mechanistic information about helicases in vitro. Thus, perhaps the most relevant next direction to take will be application of single-molecule imaging techniques to study helicase action in vivo.
References
- Abbondanzieri EA, Greenleaf WJ, Shaevitz JW, Landick R, Block SM. Direct observation of base-pair stepping by RNA polymerase. Nature. 2005;438:460–5. doi: 10.1038/nature04268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali JA, Lohman TM. Kinetic measurement of the step size of DNA unwinding by Escherichia coli UvrD helicase. Science. 1997;275:377–80. doi: 10.1126/science.275.5298.377. [DOI] [PubMed] [Google Scholar]
- Ali JA, Maluf NK, Lohman TM. An oligomeric form of E. coli UvrD is required for optimal helicase activity. J Mol Biol. 1999;293:815–34. doi: 10.1006/jmbi.1999.3185. [DOI] [PubMed] [Google Scholar]
- Aregger R, Klostermeier D. The DEAD box helicase YxiN maintains a closed conformation during ATP hydrolysis. Biochemistry. 2009;48:10679–81. doi: 10.1021/bi901278p. [DOI] [PubMed] [Google Scholar]
- Arumugam SR, Lee TH, Benkovic SJ. Investigation of stoichiometry of T4 bacteriophage helicase loader protein (gp59) J Biol Chem. 2009;284:29283–9. doi: 10.1074/jbc.M109.029926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger JM. SnapShot: nucleic acid helicases and translocases. Cell. 2008;134:888–888. e1. doi: 10.1016/j.cell.2008.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram RD, Hayes CJ, Soultanas P. Vinylphosphonate internucleotide linkages inhibit the activity of PcrA DNA helicase. Biochemistry. 2002;41:7725–31. doi: 10.1021/bi025755s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betterton MD, Julicher F. A motor that makes its own track: helicase unwinding of DNA. Phys Rev Lett. 2003;91:258103. doi: 10.1103/PhysRevLett.91.258103. [DOI] [PubMed] [Google Scholar]
- Betterton MD, Julicher F. Opening of nucleic-acid double strands by helicases: active versus passive opening. Phys Rev E Stat Nonlin Soft Matter Phys. 2005;71:011904. doi: 10.1103/PhysRevE.71.011904. [DOI] [PubMed] [Google Scholar]
- Bianco PR, Brewer LR, Corzett M, Balhorn R, Yeh Y, Kowalczykowski SC, Baskin RJ. Processive translocation and DNA unwinding by individual RecBCD enzyme molecules. Nature. 2001;409:374–8. doi: 10.1038/35053131. [DOI] [PubMed] [Google Scholar]
- Brendza KM, Cheng W, Fischer CJ, Chesnik MA, Niedziela-Majka A, Lohman TM. Autoinhibition of Escherichia coli Rep monomer helicase activity by its 2B subdomain. Proc Natl Acad Sci U S A. 2005;102:10076–81. doi: 10.1073/pnas.0502886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosh RM, Jr, Li JL, Kenny MK, Karow JK, Cooper MP, Kureekattil RP, Hickson ID, Bohr VA. Replication protein A physically interacts with the Bloom’s syndrome protein and stimulates its helicase activity. J Biol Chem. 2000;275:23500–8. doi: 10.1074/jbc.M001557200. [DOI] [PubMed] [Google Scholar]
- Chen Y, Potratz JP, Tijerina P, Del Campo M, Lambowitz AM, Russell R. DEAD-box proteins can completely separate an RNA duplex using a single ATP. Proc Natl Acad Sci U S A. 2008;105:20203–8. doi: 10.1073/pnas.0811075106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W, Brendza KM, Gauss GH, Korolev S, Waksman G, Lohman TM. The 2B domain of the Escherichia coli Rep protein is not required for DNA helicase activity. Proc Natl Acad Sci U S A. 2002;99:16006–11. doi: 10.1073/pnas.242479399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W, Dumont S, Tinoco I, Jr, Bustamante C. NS3 helicase actively separates RNA strands and senses sequence barriers ahead of the opening fork. Proc Natl Acad Sci U S A. 2007;104:13954–9. doi: 10.1073/pnas.0702315104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delagoutte E, von Hippel PH. Molecular mechanisms of the functional coupling of the helicase (gp41) and polymerase (gp43) of bacteriophage T4 within the DNA replication fork. Biochemistry. 2001;40:4459–77. doi: 10.1021/bi001306l. [DOI] [PubMed] [Google Scholar]
- Dessinges MN, Lionnet T, Xi XG, Bensimon D, Croquette V. Single-molecule assay reveals strand switching and enhanced processivity of UvrD. Proc Natl Acad Sci U S A. 2004;101:6439–44. doi: 10.1073/pnas.0306713101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillingham MS, Wigley DB, Webb MR. Demonstration of unidirectional single-stranded DNA translocation by PcrA helicase: measurement of step size and translocation speed. Biochemistry. 2000;39:205–12. doi: 10.1021/bi992105o. [DOI] [PubMed] [Google Scholar]
- Dillingham MS, Wigley DB, Webb MR. Direct measurement of single-stranded DNA translocation by PcrA helicase using the fluorescent base analogue 2-aminopurine. Biochemistry. 2002;41:643–51. doi: 10.1021/bi011137k. [DOI] [PubMed] [Google Scholar]
- Dohoney KM, Gelles J. Chi-sequence recognition and DNA translocation by single RecBCD helicase/nuclease molecules. Nature. 2001;409:370–4. doi: 10.1038/35053124. [DOI] [PubMed] [Google Scholar]
- Dong F, Weitzel SE, von Hippel PH. A coupled complex of T4 DNA replication helicase (gp41) and polymerase (gp43) can perform rapid and processive DNA strand-displacement synthesis. Proc Natl Acad Sci U S A. 1996;93:14456–61. doi: 10.1073/pnas.93.25.14456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donmez I, Patel SS. Mechanisms of a ring shaped helicase. Nucleic Acids Res. 2006;34:4216–24. doi: 10.1093/nar/gkl508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont S, Cheng W, Serebrov V, Beran RK, Tinoco I, Jr, Pyle AM, Bustamante C. RNA translocation and unwinding mechanism of HCV NS3 helicase and its coordination by ATP. Nature. 2006;439:105–8. doi: 10.1038/nature04331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enemark EJ, Joshua-Tor L. On helicases and other motor proteins. Curr Opin Struct Biol. 2008;18:243–57. doi: 10.1016/j.sbi.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essevaz-Roulet B, Bockelmann U, Heslot F. Mechanical separation of the complementary strands of DNA. Proc Natl Acad Sci U S A. 1997;94:11935–40. doi: 10.1073/pnas.94.22.11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan HF, Li HW. Studying RecBCD helicase translocation along Chi-DNA using tethered particle motion with a stretching force. Biophys J. 2009;96:1875–83. doi: 10.1016/j.bpj.2008.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer CJ, Lohman TM. ATP-dependent translocation of proteins along single-stranded DNA: models and methods of analysis of pre-steady state kinetics. J Mol Biol. 2004;344:1265–86. doi: 10.1016/j.jmb.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Fischer CJ, Maluf NK, Lohman TM. Mechanism of ATP-dependent translocation of E.coli UvrD monomers along single-stranded DNA. J Mol Biol. 2004;344:1287–309. doi: 10.1016/j.jmb.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Ha T. Need for speed: mechanical regulation of a replicative helicase. Cell. 2007;129:1249–50. doi: 10.1016/j.cell.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Ha T, Rasnik I, Cheng W, Babcock HP, Gauss GH, Lohman TM, Chu S. Initiation and re-initiation of DNA unwinding by the Escherichia coli Rep helicase. Nature. 2002;419:638–41. doi: 10.1038/nature01083. [DOI] [PubMed] [Google Scholar]
- Hamdan SM, Johnson DE, Tanner NA, Lee JB, Qimron U, Tabor S, van Oijen AM, Richardson CC. Dynamic DNA helicase-DNA polymerase interactions assure processive replication fork movement. Mol Cell. 2007;27:539–49. doi: 10.1016/j.molcel.2007.06.020. [DOI] [PubMed] [Google Scholar]
- Hamdan SM, Loparo JJ, Takahashi M, Richardson CC, van Oijen AM. Dynamics of DNA replication loops reveal temporal control of lagging-strand synthesis. Nature. 2009;457:336–9. doi: 10.1038/nature07512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa N, Bianco PR, Baskin RJ, Kowalczykowski SC. Direct visualization of RecBCD movement reveals cotranslocation of the RecD motor after chi recognition. Mol Cell. 2005;17:745–50. doi: 10.1016/j.molcel.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Henn A, Medalia O, Shi SP, Steinberg M, Franceschi F, Sagi I. Visualization of unwinding activity of duplex RNA by DbpA, a DEAD box helicase, at single-molecule resolution by atomic force microscopy. Proc Natl Acad Sci U S A. 2001;98:5007–12. doi: 10.1073/pnas.071372498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilbert M, Karow AR, Klostermeier D. The mechanism of ATP-dependent RNA unwinding by DEAD box proteins. Biol Chem. 2009;390:1237–50. doi: 10.1515/BC.2009.135. [DOI] [PubMed] [Google Scholar]
- Honda M, Park J, Pugh RA, Ha T, Spies M. Single-Molecule Analysis Reveals Differential Effect of ssDNA-Binding Proteins on DNA Translocation by XPD Helicase. Molecular Cell. 2009;35:694–703. doi: 10.1016/j.molcel.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfner KP, Michaelis J. Mechanisms of nucleic acid translocases: lessons from structural biology and single-molecule biophysics. Curr Opin Struct Biol. 2007;17:87–95. doi: 10.1016/j.sbi.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Jankowsky E, Gross CH, Shuman S, Pyle AM. The DExH protein NPH-II is a processive and directional motor for unwinding RNA. Nature. 2000;403:447–51. doi: 10.1038/35000239. [DOI] [PubMed] [Google Scholar]
- Janscak P, Garcia PL, Hamburger F, Makuta Y, Shiraishi K, Imai Y, Ikeda H, Bickle TA. Characterization and mutational analysis of the RecQ core of the bloom syndrome protein. J Mol Biol. 2003;330:29–42. doi: 10.1016/s0022-2836(03)00534-5. [DOI] [PubMed] [Google Scholar]
- Johnson DS, Bai L, Smith BY, Patel SS, Wang MD. Single-molecule studies reveal dynamics of DNA unwinding by the ring-shaped T7 helicase. Cell. 2007;129:1299–309. doi: 10.1016/j.cell.2007.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo C, Balci H, Ishitsuka Y, Buranachai C, Ha T. Advances in single-molecule fluorescence methods for molecular biology. Annu Rev Biochem. 2008;77:51–76. doi: 10.1146/annurev.biochem.77.070606.101543. [DOI] [PubMed] [Google Scholar]
- Karow AR, Klostermeier D. A conformational change in the helicase core is necessary but not sufficient for RNA unwinding by the DEAD box helicase YxiN. Nucleic Acids Res. 2009;37:4464–71. doi: 10.1093/nar/gkp397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JB, Hite RK, Hamdan SM, Xie XS, Richardson CC, van Oijen AM. DNA primase acts as a molecular brake in DNA replication. Nature. 2006;439:621–4. doi: 10.1038/nature04317. [DOI] [PubMed] [Google Scholar]
- Lee JY, Yang W. UvrD helicase unwinds DNA one base pair at a time by a two-part power stroke. Cell. 2006;127:1349–60. doi: 10.1016/j.cell.2006.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden MH, Hartmann RK, Klostermeier D. The putative RNase P motif in the DEAD box helicase Hera is dispensable for efficient interaction with RNA and helicase activity. Nucleic Acids Res. 2008;36:5800–11. doi: 10.1093/nar/gkn581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionnet T, Dawid A, Bigot S, Barre FX, Saleh OA, Heslot F, Allemand JF, Bensimon D, Croquette V. DNA mechanics as a tool to probe helicase and translocase activity. Nucleic Acids Res. 2006;34:4232–44. doi: 10.1093/nar/gkl451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionnet T, Spiering MM, Benkovic SJ, Bensimon D, Croquette V. Real-time observation of bacteriophage T4 gp41 helicase reveals an unwinding mechanism. Proc Natl Acad Sci U S A. 2007;104:19790–5. doi: 10.1073/pnas.0709793104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Putnam A, Jankowsky E. ATP hydrolysis is required for DEAD-box protein recycling but not for duplex unwinding. Proc Natl Acad Sci U S A. 2008;105:20209–14. doi: 10.1073/pnas.0811115106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohman TM, Bjornson KP. Mechanisms of helicase-catalyzed DNA unwinding. Annu Rev Biochem. 1996;65:169–214. doi: 10.1146/annurev.bi.65.070196.001125. [DOI] [PubMed] [Google Scholar]
- Lohman TM, Tomko EJ, Wu CG. Non-hexameric DNA helicases and translocases: mechanisms and regulation. Nat Rev Mol Cell Biol. 2008;9:391–401. doi: 10.1038/nrm2394. [DOI] [PubMed] [Google Scholar]
- Lucius AL, Maluf NK, Fischer CJ, Lohman TM. General methods for analysis of sequential “n-step” kinetic mechanisms: application to single turnover kinetics of helicase-catalyzed DNA unwinding. Biophys J. 2003;85:2224–39. doi: 10.1016/s0006-3495(03)74648-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucius AL, Vindigni A, Gregorian R, Ali JA, Taylor AF, Smith GR, Lohman TM. DNA unwinding step-size of E. coli RecBCD helicase determined from single turnover chemical quenched-flow kinetic studies. J Mol Biol. 2002;324:409–28. doi: 10.1016/s0022-2836(02)01067-7. [DOI] [PubMed] [Google Scholar]
- Maluf NK, Fischer CJ, Lohman TM. A Dimer of Escherichia coli UvrD is the active form of the helicase in vitro. J Mol Biol. 2003;325:913–35. doi: 10.1016/s0022-2836(02)01277-9. [DOI] [PubMed] [Google Scholar]
- Manosas M, Spiering MM, Zhuang Z, Benkovic SJ, Croquette V. Coupling DNA unwinding activity with primer synthesis in the bacteriophage T4 primosome. Nat Chem Biol. 2009;5:904–12. doi: 10.1038/nchembio.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden S, Nardelli M, Linder P, McCarthy JE. Unwinding single RNA molecules using helicases involved in eukaryotic translation initiation. J Mol Biol. 2006;361:327–35. doi: 10.1016/j.jmb.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Moffitt JR, Chemla YR, Smith SB, Bustamante C. Recent advances in optical tweezers. Annu Rev Biochem. 2008;77:205–28. doi: 10.1146/annurev.biochem.77.043007.090225. [DOI] [PubMed] [Google Scholar]
- Myong S, Bruno MM, Pyle AM, Ha T. Spring-loaded mechanism of DNA unwinding by hepatitis C virus NS3 helicase. Science. 2007;317:513–6. doi: 10.1126/science.1144130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myong S, Cui S, Cornish PV, Kirchhofer A, Gack MU, Jung JU, Hopfner KP, Ha T. Cytosolic viral sensor RIG-I is a 5′-triphosphate-dependent translocase on double-stranded RNA. Science. 2009;323:1070–4. doi: 10.1126/science.1168352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myong S, Rasnik I, Joo C, Lohman TM, Ha T. Repetitive shuttling of a motor protein on DNA. Nature. 2005;437:1321–5. doi: 10.1038/nature04049. [DOI] [PubMed] [Google Scholar]
- Niedziela-Majka A, Chesnik MA, Tomko EJ, Lohman TM. Bacillus stearothermophilus PcrA monomer is a single-stranded DNA translocase but not a processive helicase in vitro. J Biol Chem. 2007;282:27076–85. doi: 10.1074/jbc.M704399200. [DOI] [PubMed] [Google Scholar]
- Pandey M, Syed S, Donmez I, Patel G, Ha T, Patel SS. Coordinating DNA replication by means of priming loop and differential synthesis rate. Nature. 2009 doi: 10.1038/nature08611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SS, Donmez I. Mechanisms of helicases. J Biol Chem. 2006;281:18265–8. doi: 10.1074/jbc.R600008200. [DOI] [PubMed] [Google Scholar]
- Perkins TT, Li HW, Dalal RV, Gelles J, Block SM. Forward and reverse motion of single RecBCD molecules on DNA. Biophys J. 2004;86:1640–8. doi: 10.1016/S0006-3495(04)74232-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips R, Kondev J, Theriot J. Garland Science. 2008. Physical Biology of the Cell. [Google Scholar]
- Pugh RA, Lin Y, Eller C, Leesley H, Cann IK, Spies M. Ferroplasma acidarmanus RPA2 facilitates efficient unwinding of forked DNA substrates by monomers of FacXPD helicase. J Mol Biol. 2008;383:982–98. doi: 10.1016/j.jmb.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Pyle AM. Translocation and unwinding mechanisms of RNA and DNA helicases. Annu Rev Biophys. 2008;37:317–36. doi: 10.1146/annurev.biophys.37.032807.125908. [DOI] [PubMed] [Google Scholar]
- Ramanathan SP, van Aelst K, Sears A, Peakman LJ, Diffin FM, Szczelkun MD, Seidel R. Type III restriction enzymes communicate in 1D without looping between their target sites. Proc Natl Acad Sci U S A. 2009;106:1748–53. doi: 10.1073/pnas.0807193106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasnik I, Myong S, Ha T. Unraveling helicase mechanisms one molecule at a time. Nucleic Acids Res. 2006;34:4225–31. doi: 10.1093/nar/gkl452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliff GC, Erie DA. A novel single-molecule study to determine protein--protein association constants. J Am Chem Soc. 2001;123:5632–5. doi: 10.1021/ja005750n. [DOI] [PubMed] [Google Scholar]
- Rothenberg E, Trakselis MA, Bell SD, Ha T. MCM forked substrate specificity involves dynamic interaction with the 5′-tail. J Biol Chem. 2007;282:34229–34. doi: 10.1074/jbc.M706300200. [DOI] [PubMed] [Google Scholar]
- Roy R, Hohng S, Ha T. A practical guide to single-molecule FRET. Nat Methods. 2008;5:507–16. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrock RD, Alberts B. Processivity of the gene 41 DNA helicase at the bacteriophage T4 DNA replication fork. J Biol Chem. 1996;271:16678–82. doi: 10.1074/jbc.271.28.16678. [DOI] [PubMed] [Google Scholar]
- Serebrov V, Beran RK, Pyle AM. Establishing a mechanistic basis for the large kinetic steps of the NS3 helicase. J Biol Chem. 2009;284:2512–21. doi: 10.1074/jbc.M805460200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serebrov V, Pyle AM. Periodic cycles of RNA unwinding and pausing by hepatitis C virus NS3 helicase. Nature. 2004;430:476–80. doi: 10.1038/nature02704. [DOI] [PubMed] [Google Scholar]
- Singleton MR, Dillingham MS, Wigley DB. Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- Singleton MR, Wigley DB. Modularity and specialization in superfamily 1 and 2 helicases. J Bacteriol. 2002;184:1819–26. doi: 10.1128/JB.184.7.1819-1826.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatter AF, Thomas CD, Webb MR. PcrA helicase tightly couples ATP hydrolysis to unwinding double-stranded DNA, modulated by the initiator protein for plasmid replication, RepD. Biochemistry. 2009;48:6326–34. doi: 10.1021/bi900101h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spies M, Amitani I, Baskin RJ, Kowalczykowski SC. RecBCD enzyme switches lead motor subunits in response to chi recognition. Cell. 2007;131:694–705. doi: 10.1016/j.cell.2007.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spies M, Bianco PR, Dillingham MS, Handa N, Baskin RJ, Kowalczykowski SC. A molecular throttle: the recombination hotspot chi controls DNA translocation by the RecBCD helicase. Cell. 2003;114:647–54. doi: 10.1016/s0092-8674(03)00681-0. [DOI] [PubMed] [Google Scholar]
- Stano NM, Jeong YJ, Donmez I, Tummalapalli P, Levin MK, Patel SS. DNA synthesis provides the driving force to accelerate DNA unwinding by a helicase. Nature. 2005;435:370–3. doi: 10.1038/nature03615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Wei KJ, Zhang B, Zhang XH, Dou SX, Li M, Xi XG. Impediment of E. coli UvrD by DNA-destabilizing force reveals a strained-inchworm mechanism of DNA unwinding. Embo J. 2008;27:3279–87. doi: 10.1038/emboj.2008.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner NA, Hamdan SM, Jergic S, Loscha KV, Schaeffer PM, Dixon NE, van Oijen AM. Single-molecule studies of fork dynamics in Escherichia coli DNA replication. Nat Struct Mol Biol. 2008;15:170–6. doi: 10.1038/nsmb.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner NA, Loparo JJ, Hamdan SM, Jergic S, Dixon NE, van Oijen AM. Real-time single-molecule observation of rolling-circle DNA replication. Nucleic Acids Res. 2009;37:e27. doi: 10.1093/nar/gkp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theissen B, Karow AR, Kohler J, Gubaev A, Klostermeier D. Cooperative binding of ATP and RNA induces a closed conformation in a DEAD box RNA helicase. Proc Natl Acad Sci U S A. 2008;105:548–53. doi: 10.1073/pnas.0705488105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomko EJ, Fischer CJ, Niedziela-Majka A, Lohman TM. A nonuniform stepping mechanism for E. coli UvrD monomer translocation along single-stranded DNA. Mol Cell. 2007;26:335–47. doi: 10.1016/j.molcel.2007.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velankar SS, Soultanas P, Dillingham MS, Subramanya HS, Wigley DB. Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell. 1999;97:75–84. doi: 10.1016/s0092-8674(00)80716-3. [DOI] [PubMed] [Google Scholar]
- Wickersham CE, Cash KJ, Pfeil SH, Bruck I, Kaplan DL, Plaxco KW, Lipman EA. Tracking a molecular motor with a nanoscale optical encoder. NanoLetters. 2010 doi: 10.1021/n1904192m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi J, Zhang Z, Zhuang Z, Yang J, Spiering MM, Hammes GG, Benkovic SJ. Interaction between the T4 helicase loading protein (gp59) and the DNA polymerase (gp43): unlocking of the gp59-gp43-DNA complex to initiate assembly of a fully functional replisome. Biochemistry. 2005a;44:7747–56. doi: 10.1021/bi047296w. [DOI] [PubMed] [Google Scholar]
- Xi J, Zhuang Z, Zhang Z, Selzer T, Spiering MM, Hammes GG, Benkovic SJ. Interaction between the T4 helicase-loading protein (gp59) and the DNA polymerase (gp43): a locking mechanism to delay replication during replisome assembly. Biochemistry. 2005b;44:2305–18. doi: 10.1021/bi0479508. [DOI] [PubMed] [Google Scholar]
- Yang Y, Dou SX, Ren H, Wang PY, Zhang XD, Qian M, Pan BY, Xi XG. Evidence for a functional dimeric form of the PcrA helicase in DNA unwinding. Nucleic Acids Res. 2008;36:1976–89. doi: 10.1093/nar/gkm1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yodh JG, Stevens BC, Kanagaraj R, Janscak P, Ha T. BLM helicase measures DNA unwound before switching strands and hRPA promotes unwinding reinitiation. Embo J. 2009;28:405–16. doi: 10.1038/emboj.2008.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Dillingham MS, Thomas CD, Allen S, Roberts CJ, Soultanas P. Directional loading and stimulation of PcrA helicase by the replication initiator protein RepD. J Mol Biol. 2007;371:336–48. doi: 10.1016/j.jmb.2007.05.050. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Spiering MM, Trakselis MA, Ishmael FT, Xi J, Benkovic SJ, Hammes GG. Assembly of the bacteriophage T4 primosome: single-molecule and ensemble studies. Proc Natl Acad Sci U S A. 2005;102:3254–9. doi: 10.1073/pnas.0500327102. [DOI] [PMC free article] [PubMed] [Google Scholar]