

Abstract
Myopia is an extremely common eye disorder but the pathogenesis of its isolated form, which accounts for the overwhelming majority of cases, remains poorly understood. There is strong evidence for genetic predisposition to myopia, but determining myopia genetic risk factors has been difficult to achieve. We have identified Mendelian forms of myopia in four consanguineous families and implemented exome/autozygome analysis to identify homozygous truncating variants in LRPAP1 and CTSH as the likely causal mutations. LRPAP1 encodes a chaperone of LRP1, which is known to influence TGF-β activity. Interestingly, we observed marked deficiency of LRP1 and upregulation of TGF-β in cells from affected individuals, the latter being consistent with available data on the role of TGF-β in the remodeling of the sclera in myopia and the high frequency of myopia in individuals with Marfan syndrome who characteristically have upregulation of TGF-β signaling. CTSH, on the other hand, encodes a protease and we show that deficiency of the murine ortholog results in markedly abnormal globes consistent with the observed human phenotype. Our data highlight a role for LRPAP1 and CTSH in myopia genetics and demonstrate the power of Mendelian forms in illuminating new molecular mechanisms that may be relevant to common phenotypes.
Main Text
Myopia, or short-sightedness, is a disorder of ocular refraction in which the eye presents the image anterior to, rather than exactly at, the plane of the retina, which results in blurred vision.1 This refractive error is extremely common in humans with an estimated prevalence of 25% although it can reach 70% or higher in some Asian populations.2,3 Although myopia is usually a benign disorder that can be easily managed with optical means, e.g., glasses and contact lenses, individuals with high myopia are at increased risk of other eye pathologies, most notably retinal degeneration or even detachment.4 Most cases of myopia are isolated but there are syndromic forms that are important to consider clinically, e.g., Stickler syndrome.5
The etiology of the common isolated form of myopia is poorly understood but is believed to encompass complex interaction between genetic and environmental factors, as is typical of other common phenotypes.5 Myopia can be induced in animal models by inducing excessive accommodation.6 In addition, epidemiological data suggest increasing variability in refraction as young children grow to become teenagers and young adults, i.e., that myopia is a developmental process.7 These observations are the basis of the “form-deprivation” model of myopia, i.e., projection of blurred images stimulate the elongation of axial length of the globe during childhood and results in a myopic eye.8 There is also evidence that limited outdoor activity also increases risk of myopia, an effect that seems to be mediated by light intensity as suggested by epidemiological data and animal models.9,10 These important environmental factors must be influenced by genetic factors because myopia is characterized by very high heritability, estimated at 90% by some, and displays strong familial clustering.11
As is typical of other common phenotypes, genetic risk factors of myopia have been very difficult to study because of their complex nature. Conventional linkage analysis has produced several loci for nonsyndromic myopia, usually autosomal dominant, but did not reveal causative mutations.12 Similarly, candidate gene association studies have highlighted a number of genes involved in extracellular matrix (ECM) remodeling but the biased nature of this approach limits its utility. More recently, several GWAS signals have been identified but their effect size is small.5,13,14 An attractive complementary approach would be to study Mendelian forms of myopia because these tend to be much more tractable and this approach has proven effective in elucidating novel disease mechanisms for other common diseases.15,16 Two recent studies from China implemented next-generation sequencing in families with an apparently autosomal-dominant form of myopia and identified a variant each in ZNF644 (MIM 614159) and CCDC111, highlighting them as candidate genes involved in myopia pathogenesis, although definitive loss-of-function alleles in these genes have not been identified.17,18 One major limiting factor in this approach is the availability of such cases because large effect size variants (causal in the case of true Mendelian forms) tend to be rare in the general population.19
We hypothesized that our highly consanguineous population will be enriched for the otherwise rare occurrence of Mendelian forms of myopia.20 Therefore, we set out to search for families in which consanguineous healthy parents have children with extreme myopia. We specifically searched for extreme myopia because Mendelian forms tend to be more severe and to minimize the risk of recruiting families with the expected familial clustering of the common form of myopia. We show that this approach led to the identification of several families in which extreme myopia is indeed an autosomal-recessive trait probably caused by fully penetrant inactivating mutations in two genes, LRPAP1 (MIM 104225) and CTSH (MIM 116820).
All subjects were Saudi children (2–16 years old) clinically diagnosed to have extreme myopia (spherical equivalent of −17 diopters or greater) that was not related to crystalline lens subluxation and was not part of conditions known to be associated with high myopia (e.g., Stickler syndrome, premature birth). Written informed consent was obtained from all subjects prior to their enrollment in this IRB-approved research protocol. Four families were identified in which healthy consanguineous parents had children with extreme myopia (Figures 1 and 2). Best-corrected visual acuity was subnormal; this decreased visual acuity may have been from amblyopia and/or chorioretinal atrophy. Clinical findings and biometric data are summarized in the Table 1.
Figure 1.
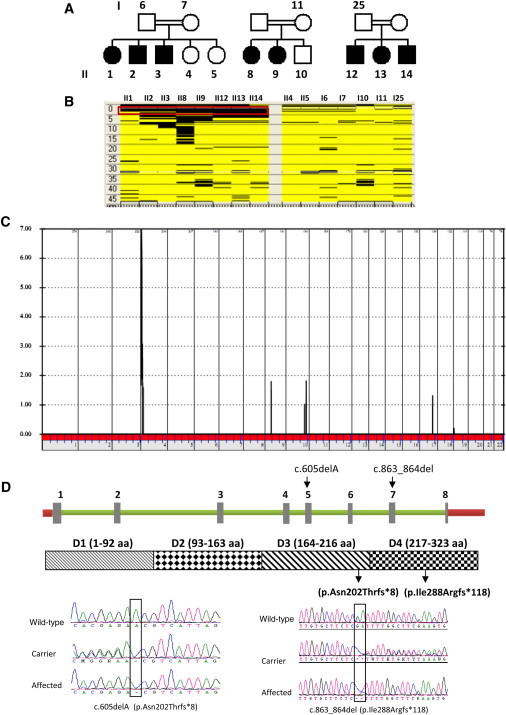
Identification of LRPAP1 Mutations in a Mendelian Form of Myopia
(A) Pedigrees of three consanguineous families in which extreme myopia appears to follow an autosomal-recessive mode of inheritance.
(B) Autozygome analysis shows a single block of autozygosity (boxed in red, the coordinates are 1,725,469 to 4,136,325) on chromosome 4 that is exclusively shared by the affected members of the three families (columns represent individual cases and rows represent individual SNP calls; black is homozygous and yellow is heterozygous).
(C) Linkage analysis shows one peak on chromosome 4 with a LOD score of 7.
(D) Schematic of LRPAP1 and the protein it encodes with the sites of the two truncating mutations shown. DNA chromatograms are shown for the mutations.
Figure 2.
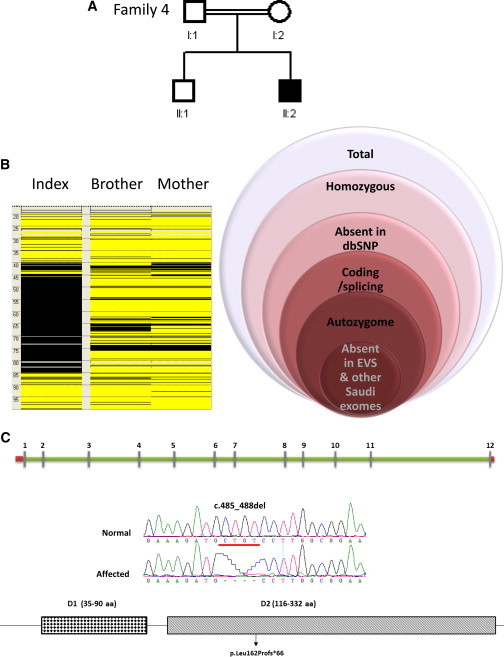
Identification of a CTSH Mutation in a Mendelian Form of Myopia
(A) Pedigree of a consanguineous family with one child with extreme myopia.
(B) Autozygome analysis shows several blocks of autozygosity but one block is shown that harbors CTSH and next to it is a schematic of the filtration strategy used to highlight the CTSH mutation.
(C) Schematic of CTSH and the protein it encodes with the sites of the two truncating mutations shown. DNA chromatograms for the reported mutation are shown.
Table 1.
Clinical Characteristics of Individuals with Autosomal-Recessive Mendelian Phenocopies of Myopia
| ID | Age | Sex | Axial | Retinoscopy | BCVA | Mutation | Comments |
|---|---|---|---|---|---|---|---|
| II-8 | 15 | F | 35.35 | −25.00, −3.00×060 | 20/70 | LRPAP1: c.605delA (p.Asn202Thrfs∗8) | none |
| 35.37 | −27.00, −3.00×030 | 20/70 | |||||
| II-9 | 6 | F | 32.44 | −18.50, −5.00×150 | 20/60 | LRPAP1: c.605delA (p.Asn202Thrfs∗8) | diaphragm surgery soon after birth |
| 31.89 | −14.50, −5.00×160 | 20/60 | |||||
| II-12 | 16 | M | 37.65 | −23.00, −1.00×010 | 20/70 | LRPAP1: c.863_864del (p.Ile288Argfs∗118) | left esotropia |
| 37.45 | −24.75 | 20/100 | |||||
| II-13 | 15 | F | 35.20 | −23.75, −0.75×005 | 20/100 | LRPAP1: c.863_864del (p.Ile288Argfs∗118) | esotropia |
| 35.81 | −23.50, −1.75×150 | 20/100 | |||||
| II-14 | 4 | M | 31.19 | −20.00 | CSM | LRPAP1: c.863_864del (p.Ile288Argfs∗118) | exotropia |
| 30.82 | −20.00 | CSM | |||||
| II-1 | 7 | F | 31.55 | −26.00 | 20/100 | LRPAP1: c.863_864del (p.Ile288Argfs∗118) | esotropia |
| 30.95 | −26.00 | 20/100 | |||||
| II-1 | 4 | M | 29.64 | −23.50 | CSM | LRPAP1: c.863_864del (p.Ile288Argfs∗118) | none |
| 29.80 | −23.00 | CSM | |||||
| II-3 | 2 | M | NA | −19.00 | CSM | LRPAP1: c.863_864del (p.Ile288Argfs∗118) | intermittent |
| NA | −19.00 | CSUM | esotropia | ||||
| F4 | 1 | M | 27.60 | −17.00 | CSM | CTSH: c.485_488del | phthisis bulbi in the other eye, dysmorphia |
Where relevant for a given individual, first row refers to right eye and second row to left eye. Age is given in years. Sex is indicated as “M” for male and “F” for female. Retinoscopy in given in diopters after cyclopentolate 1% (first number is the sphere and second number is the cylinder [indicating astigmatism] followed by the axis). Abbreviations are as follows: BCVA, best-corrected visual acuity; NA, not able to be performed because of young age; CSM, central steady and maintained fixation; CSUM, central steady and unmaintained fixation.
We mostly recruited multiplex cases with extreme myopia whose parents are consanguineous in order to enrich for the possibility of an autosomal-recessive Mendelian phenocopy of myopia that is tractable by autozygome analysis.15,20 Therefore, DNA samples extracted from the cases and their unaffected siblings and parents were searched for genome-wide runs of homozygosity (ROH) >2 Mb in length and >107 SNPs in density, which were used as surrogates of autozygosity by performing genome-wide SNP genotyping on Axiom platform (Affymetrix) that includes >550,000 SNPs followed by autoSNPa analysis.21
For families 1–3, autozygome analysis revealed only one autozygous interval that is exclusively shared among all eight affected members, which was later confirmed by linkage analysis (Figure 1). Although these three families are not known to be related to one another, haplotype analysis of the critical autozygous interval in families 2 and 3 was identical but different from that in family 1. Therefore, we proceeded with exome sequencing of one affected member from family 2 and another from family 1. The only gene that harbored a coding/splicing variant within the critical autozygous interval that is not reported in dbSNP build 135 was LRPAP1. Consistent with the haplotype analysis result, exome sequencing revealed two different mutations in family 1 (RefSeq accession number NM_002337.3; c.605delA) and family 2 (c.863_864del), and the latter was subsequently found in family 3 by direct sequencing (Figure 1). Both mutations are truncating in nature (p.Asn202Thrfs∗8 and p.Ile288Argfs∗118), fully segregated with the extreme myopia phenotype in both families and absent in 210 in-house Saudi exome files and publically available SNP databases including the 1000 Genomes Project (Integrated Phase 1 Release) and the Exome Variant Server (ESP6500SI-V2).
In family 4, autozygome analysis of the index, as expected, revealed several autozygous regions that were used to filter the resulting exome variants (Figure 2). Interestingly, a single variant survived this and the other filters we applied: a 4 bp deletion (RefSeq NM_004390.1; c.485_488del) in CTSH that predicts frameshift and premature truncation (Figure 2). This variant was found heterozygous in the healthy brother and parents and was absent in 210 in-house Saudi exome files and publically available SNP databases including the 1000 Genomes Project (Integrated Phase 1 Release) and the Exome Variant Server (ESP6500SI-V2). Immunoblot analysis of LRPAP1 revealed absence of the normal protein in affected individuals with LRPAP1 mutations, whereas RT-PCR showed marked reduction of the abundance of the mutant transcription cells from the affected individual with CTSH mutation compared to controls (90%) when quantified by real-time RT-PCR (Figure 3), most probably as a result of NMD. Taken together, our data show that the mutations in LRPAP1 and CTSH are probably loss-of-function mutations.
Figure 3.
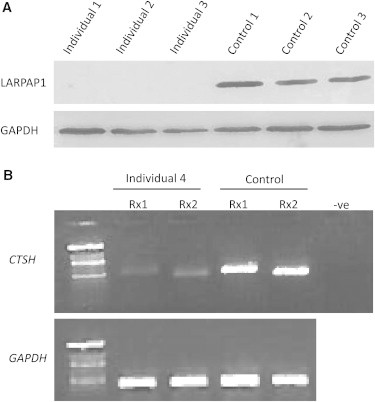
LRPAP1 and CTSH Mutations Are Probably Loss of Function
(A) Immunoblot analysis with LRPAP1 antibody shows nearly complete loss of LRPAP1 in the index of each of families 1–3 compared to controls.
(B) RT-PCR of two cDNA fragments from CTSH (Rx1 and Rx2) shows severe instability of the mutant transcript in the index in family 4 compared to control.
LRPAP1 is a widely expressed gene that encodes Low Density Lipoprotein Receptor-Related Protein-Associated Protein 1, a 357 amino acid protein that is thought to act as a chaperone that binds and protects the lipoprotein receptor-related proteins LRP1 and LRP2.22,23 It contains four independently folded domains, D1, D2, D3, and D4, which encompass residues 1–92, 93–163, 164–216, and 217–323, respectively.24 Because the truncating mutations we report are predicted to severely truncate the D4 domain (Figure 1), which is the domain shown experimentally to be responsible for binding to LRP, we sought to test whether these mutations may abolish the chaperone activity and result in increased degradation of LRP1. Indeed, immunoblot analysis revealed that cells from affected individuals have marked reduction in LRP1 compared to controls (Figure 4).
Figure 4.
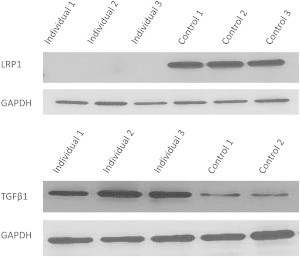
LRPAP1 Deficiency Is Associated with Severe Depletion of LRP1 and Upregulation of TGFB1
Top: immunoblot analysis with LRP1 antibody showing marked reduction of the band corresponding to LRP1 in affected individuals compared to controls (GAPDH is shown for loading control).
Bottom: immunoblot analysis with TGF-β antibody showing >2-fold increase in TGF-β in affected individuals compared to controls (GAPDH is shown for loading control).
LRP1 is identical to TGF-βR(V) but mice deficient in LRP1 have paradoxically activated TGF-β signaling.25 This is not surprising because mutations in TGF-β2, TGF-βR(I), and TGF-βR(II) are also known to result in a paradoxical activation of TGF-β, giving rise to a Marfan-like phenotype (Marfan syndrome itself is characterized by increased TGF-β).26–29 Therefore, we wanted to test whether TGF-β is increased in cells with LRPAP1 truncation. Indeed, more than 2-fold increase in TGF-β level was observed (Figure 4). Interestingly, TGF-β is one of the most reproducibly dysregulated genes in the study of myopia development and is thought to exert an effect through modulating ECM of the sclera, thereby allowing the eye to increase in axial length in response to “form deprivation.”30
CTSH encodes 1 of 11 papain-like cysteine proteases, known as cysteine-cathepsins, existing in humans.31 Active cathepsin H (CTSH) is a 37 kD protein that possesses both endo- and exopeptidase function.32,33
Although Ctsh has a ubiquitous expression pattern, there has been recent interest in exploring a potential nonredundant role in specific cell and tissue types. Gene targeting of Ctsh in mice established roles for this protease in the processing of the surfactant-glycoprotein SP-B in pneumocytes, in coactivation of granzyme B in lymphocytes, in N-terminal trimming of pituitary gland neuropeptides, as well as in tumor growth and angiogenesis in neuroendocrine pancreatic tumors.34–38 However, no severe general phenotype was observed in these Ctsh knockout mice and the eye has not been studied so far. Therefore, we asked whether CTSH deficiency recapitulates the eye-specific phenotype we observed in the affected individuals. Remarkably, Ctsh−/− eyes displayed markedly abnormal posterior chamber that assumes a “<” configuration compared to the rounded appearance in wild-type littermates, a pattern suggestive of abnormal lengthening, similar to that observed in the affected individuals (Figure 5).
Figure 5.
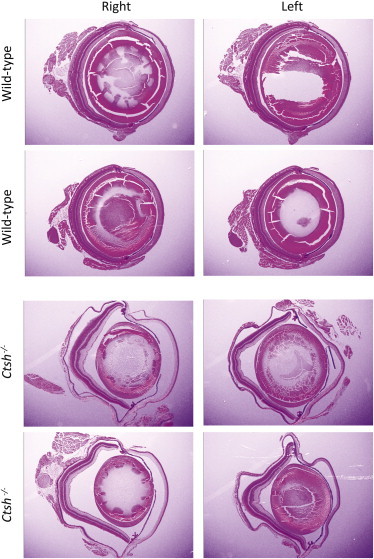
CTSH Deficiency in Mouse Results in Abnormal Globe Development
A panel of comparable eye H&E-stained sections from Ctsh−/− and their wild-type littermate (four animals are shown, two eyes each for a total of eight sections). Note the grossly abnormal globes in the knockout mouse that assume a “<” shape compared to the rounded appearance in controls, most probably indicative of elongated axial length.
Deciphering the genetics of myopia is complicated by the same set of challenges that are usually seen in other multifactorial disorders.39 Despite these challenges, research continues into myopia genetics because it is hoped that identifying genetic risk factors will illuminate the molecular pathogenesis of this extremely common disorder and make it possible to consider potential targets for prevention or therapy. Although Mendelian forms of multifactorial disorders are rare, they lend themselves readily to the same powerful tools of gene mapping that made the field of Mendelian genetics very successful. More importantly, by identifying a single lesion in a genetically homogeneous cohort, one can assign causality more confidently and direct resources to the understanding of the underlying mechanism that may inform our understanding of the pathogenesis of the multifactorial counterpart. Indeed, we show that pursuit of rare Mendelian forms of myopia identified two genes previously unsuspected as playing a role in axial length determination.
Our study suggests a model wherein LRPAP1 deficiency leads to deficiency of LRP1, which we propose leads to perturbation of TGF-β regulation and may result in abnormal ECM remodeling in the developing eye. This model is supported by the observation that myopia caused by increased axial length is one of the characteristic features of Marfan syndrome, which is also characterized by increased TGF-β.40,41 Lack of other Marfan features in the study individuals can be due to several factors including divergence in other aspects of the molecular pathogenesis. Although it remains to be seen how generalizable our findings are to common myopia, they provide supportive evidence of the role of TGF-β signaling in myopia development in humans. Various manifestations of Marfan syndrome have been shown to respond to treatment with antagonists of TGF-β but it remains to be seen whether myopia in these individuals responds to this therapeutic strategy, especially if initiated early before unfavorable ECM remodeling takes place.41 If it does, it will be of interest to consider this approach in individuals with LRPAP1-related myopia.
Although the level of evidence is less robust when compared to LRPAP1 because we have only one human mutation, our study strongly supports a causal link between CTSH deficiency and severe myopia development and reveals a role for a cathepsin in the axial length determination. First, the mutation we identified is the only sequence variant that survived the various filters we applied in the interpretation of the thousands of exome-generated variants.42 Second, the mutation is truncating in nature and our RNA analysis supports severe deficiency as a result. Third, we show that mice deficient in its ortholog have markedly abnormal globe suggestive of increased axial length. The mechanism by which this protease modifies axial length developmentally remains unknown but is likely to involve modification of the ECM. We note here that mutations in another protease, PRSS56, cause an almost mirror image of myopia with severe shortening of the axial length in a condition known as posterior microphthalmos.43–45
How much, if at all, does variation in CTSH and LRPAP1 influence the common form of myopia? The LRPAP1 locus has not been highlighted in any GWAS on myopia in the past. On the other hand, we note that CTSH is in remarkable proximity to RASGRF1 (45 kb away), which was reported recently as the source of a major association signal in a GWAS (and replicated by two others) on myopia even though the mouse phenotype is not consistent with its presumed role in myopia pathogenesis.13,14,46 We have sequenced CTSH and LRPAP1 in a panel of 100 individuals with myopia of ≥6 D but did not find any evidence of increased load of rare variants in these individuals compared to 100 similarly screened controls (data not shown). Thus, it remains to be seen whether these genes, especially CTSH, are directly involved in the pathogenesis of the common form of myopia although the pathways involved may be logical targets for future studies.
In summary, we present two recessive Mendelian forms of myopia that are solved at the gene level. The nature of the two genes identified in this study suggests a role of ECM remodeling in myopia development through TGF-β signaling and protease-mediated pathways. Although we could not find evidence for increased load of rare or common variants in LRPAP1 or CTSH in individuals with the common form of myopia, future studies are needed to fully explore a potential role their pathways may play in the pathogenesis of common myopia.
Acknowledgments
We thank the affected individuals and their families for their enthusiastic participation. We are indebted to the Genomic and Sequencing Core Facilities at KFSHRC for their technical help. F.S.A. is supported by KACST grant 08MED and DHFMR Collaborative Research Grant. T.R. is supported by the Deutsche Forschungsgemeinschaft SFB 850/B7.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Curtin B.J. Harper & Row; Philadelphia: 1985. The Myopias: Basic Science and Clinical Management. [Google Scholar]
- 2.Sperduto R.D., Seigel D., Roberts J., Rowland M. Prevalence of myopia in the United States. Arch. Ophthalmol. 1983;101:405–407. doi: 10.1001/archopht.1983.01040010405011. [DOI] [PubMed] [Google Scholar]
- 3.Lin L.L., Chen C.J., Hung P.T., Ko L.S. Nation-wide survey of myopia among schoolchildren in Taiwan, 1986. Acta Ophthalmol. Suppl. 1988;185:29–33. doi: 10.1111/j.1755-3768.1988.tb02657.x. [DOI] [PubMed] [Google Scholar]
- 4.Morgan I.G., Ohno-Matsui K., Saw S.M. Myopia. Lancet. 2012;379:1739–1748. doi: 10.1016/S0140-6736(12)60272-4. [DOI] [PubMed] [Google Scholar]
- 5.Wojciechowski R. Nature and nurture: the complex genetics of myopia and refractive error. Clin. Genet. 2011;79:301–320. doi: 10.1111/j.1399-0004.2010.01592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raviola E., Wiesel T.N. An animal model of myopia. N. Engl. J. Med. 1985;312:1609–1615. doi: 10.1056/NEJM198506203122505. [DOI] [PubMed] [Google Scholar]
- 7.Siegwart J.T., Jr., Norton T.T. Binocular lens treatment in tree shrews: Effect of age and comparison of plus lens wear with recovery from minus lens-induced myopia. Exp. Eye Res. 2010;91:660–669. doi: 10.1016/j.exer.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lawrence M.S., Azar D.T. Myopia and models and mechanisms of refractive error control. Ophthalmol. Clin. North Am. 2002;15:127–133. doi: 10.1016/s0896-1549(01)00002-5. [DOI] [PubMed] [Google Scholar]
- 9.Ashby R., Ohlendorf A., Schaeffel F. The effect of ambient illuminance on the development of deprivation myopia in chicks. Invest. Ophthalmol. Vis. Sci. 2009;50:5348–5354. doi: 10.1167/iovs.09-3419. [DOI] [PubMed] [Google Scholar]
- 10.Sherwin J.C., Hewitt A.W., Coroneo M.T., Kearns L.S., Griffiths L.R., Mackey D.A. The association between time spent outdoors and myopia using a novel biomarker of outdoor light exposure. Invest. Ophthalmol. Vis. Sci. 2012;53:4363–4370. doi: 10.1167/iovs.11-8677. [DOI] [PubMed] [Google Scholar]
- 11.Lyhne N., Sjølie A.K., Kyvik K.O., Green A. The importance of genes and environment for ocular refraction and its determiners: a population based study among 20-45 year old twins. Br. J. Ophthalmol. 2001;85:1470–1476. doi: 10.1136/bjo.85.12.1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abbott D., Li Y.J., Guggenheim J.A., Metlapally R., Malecaze F., Calvas P., Rosenberg T., Paget S., Zayats T., Mackey D.A. An international collaborative family-based whole genome quantitative trait linkage scan for myopic refractive error. Mol. Vis. 2012;18:720–729. [PMC free article] [PubMed] [Google Scholar]
- 13.Kiefer A.K., Tung J.Y., Do C.B., Hinds D.A., Mountain J.L., Francke U., Eriksson N. Genome-wide analysis points to roles for extracellular matrix remodeling, the visual cycle, and neuronal development in myopia. PLoS Genet. 2013;9:e1003299. doi: 10.1371/journal.pgen.1003299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verhoeven V.J., Hysi P.G., Wojciechowski R., Fan Q., Guggenheim J.A., Höhn R., MacGregor S., Hewitt A.W., Nag A., Cheng C.-Y., Consortium for Refractive Error and Myopia (CREAM) Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Research Group. Wellcome Trust Case Control Consortium 2 (WTCCC2) Fuchs’ Genetics Multi-Center Study Group Genome-wide meta-analyses of multiancestry cohorts identify multiple new susceptibility loci for refractive error and myopia. Nat. Genet. 2013;45:314–318. doi: 10.1038/ng.2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Al-Mayouf S.M., Sunker A., Abdwani R., Abrawi S.A., Almurshedi F., Alhashmi N., Al Sonbul A., Sewairi W., Qari A., Abdallah E. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat. Genet. 2011;43:1186–1188. doi: 10.1038/ng.975. [DOI] [PubMed] [Google Scholar]
- 16.Alangari A., Alsultan A., Adly N., Massaad M.J., Kiani I.S., Aljebreen A., Raddaoui E., Almomen A.K., Al-Muhsen S., Geha R.S., Alkuraya F.S. LPS-responsive beige-like anchor (LRBA) gene mutation in a family with inflammatory bowel disease and combined immunodeficiency. J. Allergy Clin. Immunol. 2012;130:481–488. doi: 10.1016/j.jaci.2012.05.043. e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi Y., Li Y., Zhang D., Zhang H., Li Y., Lu F., Liu X., He F., Gong B., Cai L. Exome sequencing identifies ZNF644 mutations in high myopia. PLoS Genet. 2011;7:e1002084. doi: 10.1371/journal.pgen.1002084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao F., Wu J., Xue A., Su Y., Wang X., Lu X., Zhou Z., Qu J., Zhou X. Exome sequencing reveals CCDC111 mutation associated with high myopia. Hum. Genet. 2013 doi: 10.1007/s00439-013-1303-6. Published online April 12, 2013. [DOI] [PubMed] [Google Scholar]
- 19.Baird P.N., Schäche M., Dirani M. The GEnes in Myopia (GEM) study in understanding the aetiology of refractive errors. Prog. Retin. Eye Res. 2010;29:520–542. doi: 10.1016/j.preteyeres.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 20.Alkuraya F.S. Autozygome decoded. Genet. Med. 2010;12:765–771. doi: 10.1097/GIM.0b013e3181fbfcc4. [DOI] [PubMed] [Google Scholar]
- 21.Carr I.M., Flintoff K.J., Taylor G.R., Markham A.F., Bonthron D.T. Interactive visual analysis of SNP data for rapid autozygosity mapping in consanguineous families. Hum. Mutat. 2006;27:1041–1046. doi: 10.1002/humu.20383. [DOI] [PubMed] [Google Scholar]
- 22.Willnow T.E., Armstrong S.A., Hammer R.E., Herz J. Functional expression of low density lipoprotein receptor-related protein is controlled by receptor-associated protein in vivo. Proc. Natl. Acad. Sci. USA. 1995;92:4537–4541. doi: 10.1073/pnas.92.10.4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Willnow T.E., Rohlmann A., Horton J., Otani H., Braun J.R., Hammer R.E., Herz J. RAP, a specialized chaperone, prevents ligand-induced ER retention and degradation of LDL receptor-related endocytic receptors. EMBO J. 1996;15:2632–2639. [PMC free article] [PubMed] [Google Scholar]
- 24.Medved L.V., Migliorini M., Mikhailenko I., Barrientos L.G., Llinás M., Strickland D.K. Domain organization of the 39-kDa receptor-associated protein. J. Biol. Chem. 1999;274:717–727. doi: 10.1074/jbc.274.2.717. [DOI] [PubMed] [Google Scholar]
- 25.Boucher P., Li W.P., Matz R.L., Takayama Y., Auwerx J., Anderson R.G., Herz J. LRP1 functions as an atheroprotective integrator of TGFbeta and PDFG signals in the vascular wall: implications for Marfan syndrome. PLoS ONE. 2007;2:e448. doi: 10.1371/journal.pone.0000448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boileau C., Guo D.C., Hanna N., Regalado E.S., Detaint D., Gong L., Varret M., Prakash S.K., Li A.H., d’Indy H., National Heart, Lung, and Blood Institute (NHLBI) Go Exome Sequencing Project TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet. 2012;44:916–921. doi: 10.1038/ng.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mizuguchi T., Matsumoto N. Recent progress in genetics of Marfan syndrome and Marfan-associated disorders. J. Hum. Genet. 2007;52:1–12. doi: 10.1007/s10038-006-0078-1. [DOI] [PubMed] [Google Scholar]
- 28.Mizuguchi T., Collod-Beroud G., Akiyama T., Abifadel M., Harada N., Morisaki T., Allard D., Varret M., Claustres M., Morisaki H. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat. Genet. 2004;36:855–860. doi: 10.1038/ng1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lindsay M.E., Schepers D., Bolar N.A., Doyle J.J., Gallo E., Fert-Bober J., Kempers M.J., Fishman E.K., Chen Y., Myers L. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 2012;44:922–927. doi: 10.1038/ng.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jobling A.I., Nguyen M., Gentle A., McBrien N.A. Isoform-specific changes in scleral transforming growth factor-beta expression and the regulation of collagen synthesis during myopia progression. J. Biol. Chem. 2004;279:18121–18126. doi: 10.1074/jbc.M400381200. [DOI] [PubMed] [Google Scholar]
- 31.Rawlings N.D., Barrett A.J., Bateman A. MEROPS: the peptidase database. Nucleic Acids Res. 2010;38(Database issue):D227–D233. doi: 10.1093/nar/gkp971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baudys M., Meloun B., Gan-Erdene T., Fusek M., Mares M., Kostka V., Pohl J., Blake C.C. S-S bridges of cathepsin B and H from bovine spleen: a basis for cathepsin B model building and possible functional implications for discrimination between exo- and endopeptidase activities among cathepsins B, H and L. Biomed. Biochim. Acta. 1991;50:569–577. [PubMed] [Google Scholar]
- 33.Kirschke H., Langner J., Wiederanders B., Ansorge S., Bohley P., Hanson H. Cathepsin H: an endoaminopeptidase from rat liver lysosomes. Acta Biol. Med. Ger. 1977;36:185–199. [PubMed] [Google Scholar]
- 34.Bühling F., Kouadio M., Chwieralski C.E., Kern U., Hohlfeld J.M., Klemm N., Friedrichs N., Roth W., Deussing J.M., Peters C., Reinheckel T. Gene targeting of the cysteine peptidase cathepsin H impairs lung surfactant in mice. PLoS ONE. 2011;6:e26247. doi: 10.1371/journal.pone.0026247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ueno T., Linder S., Na C.L., Rice W.R., Johansson J., Weaver T.E. Processing of pulmonary surfactant protein B by napsin and cathepsin H. J. Biol. Chem. 2004;279:16178–16184. doi: 10.1074/jbc.M312029200. [DOI] [PubMed] [Google Scholar]
- 36.D’Angelo M.E., Bird P.I., Peters C., Reinheckel T., Trapani J.A., Sutton V.R. Cathepsin H is an additional convertase of pro-granzyme B. J. Biol. Chem. 2010;285:20514–20519. doi: 10.1074/jbc.M109.094573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gocheva V., Chen X., Peters C., Reinheckel T., Joyce J.A. Deletion of cathepsin H perturbs angiogenic switching, vascularization and growth of tumors in a mouse model of pancreatic islet cell cancer. Biol. Chem. 2010;391:937–945. doi: 10.1515/BC.2010.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu W.D., Funkelstein L., Toneff T., Reinheckel T., Peters C., Hook V. Cathepsin H functions as an aminopeptidase in secretory vesicles for production of enkephalin and galanin peptide neurotransmitters. J. Neurochem. 2012;122:512–522. doi: 10.1111/j.1471-4159.2012.07788.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldstein D.B. Common genetic variation and human traits. N. Engl. J. Med. 2009;360:1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 40.Loeys B.L., Dietz H.C., Braverman A.C., Callewaert B.L., De Backer J., Devereux R.B., Hilhorst-Hofstee Y., Jondeau G., Faivre L., Milewicz D.M. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 2010;47:476–485. doi: 10.1136/jmg.2009.072785. [DOI] [PubMed] [Google Scholar]
- 41.Brooke B.S., Habashi J.P., Judge D.P., Patel N., Loeys B., Dietz H.C., 3rd Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N. Engl. J. Med. 2008;358:2787–2795. doi: 10.1056/NEJMoa0706585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alkuraya F.S. Discovery of rare homozygous mutations from studies of consanguineous pedigrees. Curr. Protocols Hum. Genet. 2012 doi: 10.1002/0471142905.hg0612s75. Unit 6, 12. [DOI] [PubMed] [Google Scholar]
- 43.Gal A., Rau I., El Matri L., Kreienkamp H.J., Fehr S., Baklouti K., Chouchane I., Li Y., Rehbein M., Fuchs J. Autosomal-recessive posterior microphthalmos is caused by mutations in PRSS56, a gene encoding a trypsin-like serine protease. Am. J. Hum. Genet. 2011;88:382–390. doi: 10.1016/j.ajhg.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nair K.S., Hmani-Aifa M., Ali Z., Kearney A.L., Ben Salem S., Macalinao D.G., Cosma I.M., Bouassida W., Hakim B., Benzina Z. Alteration of the serine protease PRSS56 causes angle-closure glaucoma in mice and posterior microphthalmia in humans and mice. Nat. Genet. 2011;43:579–584. doi: 10.1038/ng.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nowilaty S.R., Khan A.O., Aldahmesh M.A., Tabbara K.F., Al-Amri A., Alkuraya F.S. Biometric and molecular characterization of clinically diagnosed posterior microphthalmos. Am. J. Ophthalmol. 2013;155:361–372. doi: 10.1016/j.ajo.2012.08.016. e7. [DOI] [PubMed] [Google Scholar]
- 46.Hysi P.G., Young T.L., Mackey D.A., Andrew T., Fernández-Medarde A., Solouki A.M., Hewitt A.W., Macgregor S., Vingerling J.R., Li Y.J. A genome-wide association study for myopia and refractive error identifies a susceptibility locus at 15q25. Nat. Genet. 2010;42:902–905. doi: 10.1038/ng.664. [DOI] [PMC free article] [PubMed] [Google Scholar]