

Abstract
The S. cerevisiae Cdc13 is a multifunctional protein with key roles in regulation of telomerase, telomere end protection, and conventional telomere replication, all of which are cell cycle-regulated processes. Given that phosphorylation is a key mechanism for regulating protein function, we identified sites of phosphorylation using nano liquid chromatography-tandem mass spectrometry (nanoLC-MS/MS). We also determined phosphorylation abundance on both wild type (WT) and a telomerase deficient form of Cdc13, encoded by the cdc13-2 allele, in both G1 phase cells, when telomerase is not active, and G2/M phase cells, when it is. We identified 21 sites of in vivo phosphorylation, of which only five had been reported previously. In contrast, phosphorylation of two in vitro targets of the ATM-like Tel1 kinase, S249 and S255, was not detected. This result helps resolve conflicting data on the importance of phosphorylation of these residues in telomerase recruitment. multiple residues showed differences in their cell cycle pattern of modification. For example, phosphorylation of S314 was significantly higher in the G2/M compared to the G1 phase and in WT versus mutant Cdc13, and a S314D mutation negatively affected telomere length. Our findings provide new targets in a key telomerase regulatory protein for modulation of telomere dynamics.

Keywords: telomere regulation, Cdc13, phosphorylation, quantitative proteomics, mass spectrometry
INTRODUCTION
The telomere is a nucleoprotein structure that caps the end of linear chromosomes and is essential for preserving genome integrity. Saccharomyces cerevisiae telomeric DNA is composed of ~300 bp of degenerate TG1–3 repeats, which are maintained by the enzyme telomerase.1 The minimal components for in vitro telomerase activity are Est2, the reverse transcriptase subunit,2 and TLC1, the RNA template for G-rich strand synthesis.3 Three other proteins are also required for in vivo telomerase action: Cdc13, the telomeric single strand (ss) DNA binding protein, 4,5 Est1, the recruiter-activator of telomerase,6–8 and Est3, an accessory subunit of unknown function.7
Telomere replication is regulated by both the cell cycle and telomere length. Semiconservative replication of telomeric DNA and telomere lengthening via telomerase occur only during the late S-G2 phase.9,10 Since telomerase activity is present throughout most of the cell cycle, current models propose that substrate accessibility is the rate-liming step for telomerase action. Telomerase recruitment to telomeres in the late S/G2 phase depends on a specific interaction between Cdc13 and Est1.11–14 Telomeric C-strand degradation, which generates long ss G-tails, the presumed substrates for telomerase action, is also limited to late S/G2 phase.1 As in mammals, short telomeres are preferentially elongated.15 Est2 and Est1 both bind preferentially to short telomeres.16–18
As for many cellular processes, telomere replication is regulated at least in part by phosphorylation. Cyclin-dependent kinase 1 (CDK1) is required for cell cycle regulated C-strand degradation19,20 while telomere length is strongly dependent on the kinase activity of Tel1, the yeast homologue of ataxia-telangiectasia mutated (ATM) (and to a lesser extent on Mec1, the homologue of AT-related or ATR).21,22 Tel1 itself binds preferentially to short telomeres, and its activity is important for preferential lengthening of short telomeres.16,23,24 An important step in determining mechanisms of telomere length control is to identify the telomere relevant targets of these kinases.
Cdc13, which binds to telomeric ssDNA with high affinity and sequence specificity,4,5,25 is a crucial player in regulating telomere replication. It is essential for telomerase action in vivo, as a single amino acid mutation at position 252, the cdc13-2 allele, can lead to a telomerase null phenotype.5 It is also required for telomere end protection, as removing Cdc13 leads to rapid telomere degradation and activation of a DNA damage checkpoint that arrests cells in the G2/M phase.26 In addition to binding telomeric ssDNA, Cdc13 forms a homodimer27,28 and associates with a number of nuclear proteins important in maintaining telomere structure and telomerase activity. Cdc13 binding to Est1 through a genetically and biochemically defined telomerase recruitment domain (RD) is essential for G-strand elongation by telomerase.12–14 The interaction between Cdc13 and Pol1, the largest subunit of DNA polymerase α, is implicated in C-strand synthesis by the conventional replication machinery.29,30 Cdc13 also forms a complex with Stn1 and Ten1, proteins essential for telomere end protection.31–33 Genetic evidence suggests that these interactions compete with each other for the execution of different functions of Cdc13. For example, mutations that reduce Cdc13-Pol129 or Cdc13-Stn131,33,34 interaction lead to long telomeres, while overexpression or telomere-tethering of STN1 result in telomere shortening.35
The importance of Cdc13 in telomere dynamics makes it an ideal target for phosphorylation-dependent regulation. Cdc13 can be phosphorylated in vitro and in vivo by Tel1, Mec1, and CDK1 kinases.36–38 Simultaneous mutation of two of the in vitro targets of Tel1/Mec1 within the RD of Cdc13, S249A, and S255A eliminates telomerase mediated telomere lengthening.36 These observations suggest that phosphorylation of Cdc13 by Tel1/Mec1 at these residues is critical for telomerase recruitment in vivo. However, a different disruption in the Tel1/Mec1 motifs (Q250A Q256A) does not render the cell telomerase deficient,39 raising the concern that S249 and S255 may not be in vivo targets of Tel1/Mec1 kinases. Mutation of the CDK1 target, T308A, leads to modestly shorter telomeres and reduces Est1 telomere binding.37,38 Mec1 phosphorylation of S306 does not affect telomere length but suppresses de novo telomere addition at DNA double strand breaks (DSBs).40 To date, it is not known if Cdc13 is subject to modification by other kinases.
Here we report a comprehensive analysis of Cdc13 phosphorylation using mass spectrometric and genetic approaches. In addition to confirming previously reported sites, we identified 16 novel phosphorylation sites on Cdc13. To determine the importance of these modifications, we employed a label-free quantitative approach to identify residues that are differentially phosphorylated as a function of cell cycle stage and/ or between wild type (WT) Cdc13 and the telomerase defective mutant protein encoded by cdc13-2. These studies reveal a novel phosphorylation site that was enriched in the G2 phase on WT Cdc13 and that negatively modulated telomere length in vivo.
METHODS
Protein Purification
WT and the E252K mutant of Cdc13, as well as protein fragments, were overexpressed in S. cerevisiae and purified as described.14 For cell cycle-arrested cultures, cells were induced for Cdc13 overexpression first for ~8 h before addition of α factor (20 ng/mL) or nocodazole (Sigma-Aldrich, 15 µg/mL). Cells were grown for another 3.5 h before harvesting. Cell cycle distributions were determined by flow cytometry.
MS and MS/MS Analysis
Purified Cdc13 proteins were treated with 5 mM dithiothreitol (DTT) at 51 °C for 1 h to reduce disulfide bonds. The cysteines were then alkylated by treating with 14 mM iodoacetamide in the dark at room temperature for 45 min, as previously described.41 The resulting proteins were enzymatically digested using either trypsin (Promega, 1:100 weight) or LysC (Roche, 1:5 weight) and desalted prior to mass spectrometry (MS) analysis as previously described.42 Alternatively, purified proteins were subject to thiol reduction, alkylation, and trypsin digestion according to the FASP methodology.43
Approximately 5 µg of sample was loaded by an autosampler (AS-2; Eksigent Technologies Inc., CA, USA) onto a 75 µm fused silica capillary column with an ESI tip that was hand-packed with 130 mm of C18 reverse phase resin (5 µm particles, 200 Å pore size). Samples were chromatographically resolved using a 110 min 1–100% buffer B gradient (buffer A = 0.1 mol/L acetic acid, buffer B = 95% acetonitrile in 0.1 mol/L acetic acid) at a flow rate of approximately 200–300 nL/min controlled by an HPLC pump (1200 series; Agilent, Santa Rosa, CA, USA). The HPLC effluent was coupled directly to an LTQ-Orbitrap XL hybrid mass spectrometer (ThermoFisher Scientific, Carlsbad, CA, USA) with a resolution of 30,000 for full MS followed by seven data-dependent and targeted MS/MS acquisitions, alternating between CID and ETD fragmentation (ETD reaction time of 100 ms, ETD activation time of 100 ms, activation Q of 0.25).
Method for Phosphorylation Site Identification and Relative Quantitation
A traditional “shotgun” nanoLC-MS/MS approach based on identification and subsequent quantification of PTMs would not be appropriate for the purpose of this experiment. Most of the phosphorylation sites on Cdc13 are present on peptides that can be modified on more than one residue, and tandem mass spectra of these modified isoforms share a significant number of fragment ion peaks, resulting in a large number of false positive identifications by traditional tandem MS search algorithms. In addition, the relative concentrations of modified isoforms can span orders of magnitudes (e.g., 100 copies of site 1 phosphorylation are present for every copy of site 2 phosphorylation). Therefore, tandem MS is often not available for lower abundance isoforms, since MS2 events are often triggered by the largest signals in data-dependent MS2 acquisition, creating false negativity for the low abundance phosphorylation events due to undersampling.
In order to address these problems, we developed an algorithm that incorporates MS, MS/MS, and chromatographic information into the PTM identification and quantitation. The advantage of this approach is that we can confidently assign a signal to a particular PTM based on its LC retention time and MS isotopic distribution even when MS/MS data are not available. Low abundance PTMs identified in this fashion were subsequently targeted for MS2 fragmentation in later runs to confirm the assignments. In addition, all MS2 identifications were extensively manually validated to ensure site localization accuracy (tandem MS annotations for the phosphorylation sites identified are provided in Supporting Information Figure 1). As summarized in Figure 1, our method uses a mathematical model that simultaneously identifies and quantitates all modified isoforms of a single peptide sequence by maximizing (1) the fit between the experimental and theoretical isotopic distributions of each modified isoform in the MS and (2) the number of observed fragment ion peaks that support the modified isoform in any available MS2. This objective function is solved with respect to constraints that restrict modified isoform assignments to be chromatographically feasible. Specifically, under our acidic mobile phase conditions, phosphorylation of a residue on a peptide generally results in that modified isoform becoming more hydrophobic than its nonphosphorylated counterpart. Thus, all identifications are required to satisfy these temporal relationships based on their relative chemicophysical properties. The model also enforces the constraint that multiple charge states of the same modified isoform should coelute. Lastly, because several phosphorylation sites were in close proximity of arginines and lysines clusters (i.e., “RK”, “KRKRK”, “KK”, etc.) and we often observed alternative cleavage patterns with Lys-C and trypsin, missed cleavages were allowed in the in silico digest of the Cdc13 protein sequence.
Figure 1.

Schematic illustration of the LC-MS/MS experimental and computational approach for phosphorylation identification and quantitation.
To illustrate the concept behind the proposed method, consider the theoretical peptide presented in the lower left of Figure 1, which has two possible sites of phosphorylation (denoted by the gray circles). We see that two peaks in the MS potentially correspond to the modified isoform with two phoshorylations, three peaks in the MS potentially correspond to the modified isoform with one phoshorylation, two peaks potentially correspond to the unmodified state of the peptide (i.e., unphosphorylated). The goal of the proposed model is to assign one (or none) of these MS peaks to each modified form. When considering the assignment of a MS peak to a particular modified peptide, it is important that the observed experimental MS isotopic distribution (i.e., monoisotopic peak, +1 isotope, and +2 isotope) of the peptide resembles its theoretical isotopic distribution, which can be approximated by the Euclidean distance between the two profiles. Also, if a tandem MS is available for any of the potential candidate MS peaks, its fragment ions should support the amino acid sequence and postulated sites of phosphorylation of the modified form under consideration. The combined evidence that the experimental MS isotopic distribution and tandem MS of a given MS peak explain a particular modified form is simultaneously balanced against the constraint that the final MS peaks assigned to these three modified peptides must be chromatographically consistent (i.e., the unmodified peptide elutes first, the monophosphorylated peptide elutes second, and the diphosphorylated peptide elutes last, as illustrated in Figure 1). By formulating this problem as an optimization problem, we are able to simultaneously consider these modes of information when assigning MS peaks to the possible modified forms.
The MS signal was used to estimate the relative abundance of the each modified form. Specifically, for a given modified form, we integrated the MS signal over all isotopic peaks (i.e., monoisotopic, +1 and +2 isotopes) and across all observed charge states, including the contributions from exogenous modifications introduced during sample processing, such as oxidized methionine and carbamidomethylation of cysteine. This abundance was then normalized to the 100% relative signal by dividing the summation of the abundances for all modified forms of the same peptide. Several technical and biological replicates were examined to provide quantitative estimates of cell cycle specific changes in phosphorylation.
To examine the quantitation consistency of our approach, we applied this method to two nonphosphorylated Cdc13 peptides, 23-DFEGYPSK-30 and 567-LELNEGFK-574. These peptides are present in +2 and +3 charge states and come from different domains of the Cdc13 sequence. The results are presented in Supporting Information Figure 2, where it is observed that the average ratio between these two peptides across several replicates is consistently close to 0.4 for asynchronous and G1 and G2/M phase cells.
Figure 2.

(A) Schematic illustration of Cdc13 domain organization. (B) Phosphatase treatment of Cdc13. 1.5 µg of purified Cdc13 was incubated with 40 U of λ protein phosphatase (NEB, lanes 2 and 3) and 1 mM MnCl2 (lanes 3 and 4) in 1X vendor-supplied buffer at 30 °C for 30 min. Samples were resolved on 8% SDS-PAGE gel and stained with Coomassie blue.
Telomere Length Determination
Mutations were introduced into the Cdc13 ORF using the Quick-Change site-directed mutagenesis kit (Strategene). The mutations and ORF integrity were verified by DNA sequencing and then subcloned into a pRS314::CDC1329 (for Southern blot analysis) or a pRS314::Cdc13-myc9 plasmid (for Western blot analysis). Cells heterozygous for CDC13 (YPH501 cdc13Δ::HIS3/+) were transformed with pRS314::CDC13 or the mutant plasmids, sporulated, and dissected, and genotypes were determined by biosynthetic markers. At least two independent isolates of each mutation were chosen for telomere length analysis using Southern blotting and/or protein expression using Western blot analysis against the myc9 epitope. Freshly cleaved spores were struck three or more consecutive times on rich medium (YEPD) to reach the equilibrium telomere length. Genomic DNAs were digested with PstI and XhoI, separated on 0.8% agarose gels, transferred to Hybond N+ nylon membrane (GE Healthcare), and blotted with a randomly primed 32P-labled telomere probe isolated from pCT300.44
RESULTS
Cdc13 Is Phosphorylated in Vivo
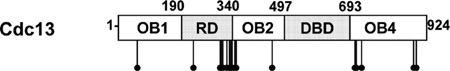
Structural, genetic, and biochemical data have identified functional domains within Cdc13. In addition to the RD, Cdc13 consists of four oligosaccharide-oligonucleotide binding (OB) folds (Figure 2A). The N-terminal OB1 (aa 14–225) is required for homodimerization,27,28 as well as for interaction with Pol1 (the largest subunit of DNA Polα) and Sir4 (a silencing protein).28–30 OB3 (aa 497–693) is the canonical DNA binding domain (DBD).25,45 Functions of OB2 (aa 323–485) and OB4 (aa 712–924) are unclear, but OB4 is implicated in Stn1 binding, and its deletion results in telomere hyperelongation.31,34 The RD (aa 190–340) is located between the OB1 and OB2 domains.
Previously, we purified full-length Cdc13 from its native host, S. cerevisiae, for functional studies and showed that yeast derived Cdc13 is highly active in ssDNA and Est1 binding.14 This purified Cdc13 migrated in SDS-PAGE gel as multiple bands (Figure 2B, lane 1). Treating purified Cdc13 with both λ protein phosphatase and its metal cofactor MnCl2, but not with either one alone, converted all bands to a single, faster-migrating species (lanes 2–4), confirming that purified Cdc13 is phosphorylated. Since Cdc13 phosphorylation has been implicated in telomerase regulation,36–38 we decided to characterize its sites of phosphorylation in more detail.
Characterization of Cdc13 Phosphorylation Revealed Previously Unreported Sites of Modification
There are 145 serines and threonines spread throughout the 924-amino acid Cdc13 ORF that are candidate sites for phosphorylation. We first identified Cdc13 phosphorylation sites by nano liquid chromatography tandem mass spectrometry (nanoLC-MS/MS) using protein affinity-purified from asynchronously growing cells (see Figure 1 and Methods for experimental design). Prior to nanoLC-MS/MS, purified Cdc13 was digested with either trypsin or LysC protease. Some post-translational modifications (PTMs), including phosphorylation, are relatively labile, which can result in prompt loss during collision-induced dissociation (CID) fragmentation without further informative fragmentation of the modified peptide. Electron transfer dissociation (ETD) is a complementary alternative to CID in that it is a radical-based nonergotic process and does not convey levels of excess energy to the peptide as in CID, thereby minimizing prompt loss of labile species. However, due to Coulombic forces, this approach increases in efficiency, with longer peptide sequences bearing a higher total positive charge. Thus, we performed separate tryptic and LysC digests to obtain peptides of varying size and charge. Both digests were analyzed using either CID or ETD based on the decision-tree algorithm46 (see Figure 1) to obtain a sufficient sampling of tandem MS for each fragmentation process. It was discovered that ETD fragmentation for tryptic and LysC digests did not result in sufficient fragmentation for phosphorylation site localization, perhaps due to the relatively small size and charge states of the peptides examined, so we consolidated the workflow to only tryptic digestion followed by CID fragmentation for the remainder of the study.
In total, we identified 21 phosphorylation sites on full-length Cdc13 (Figure 3, Table 1). Five of these 21 sites (S225, S306, T308, S333, S336) were reported previously.36–38,40,47 Of these, one (S225) had not been confirmed to be present in vivo; and only two (S306 and T308) have been characterized for their biological function.37,38,40 When compared to the secondary structure of Cdc13 previously determined by X-ray crystallography or structural predictions,28 all phosphorylation sites map to positions on or immediately next to a loop, which are usually more flexible regions compared to α helices or β strands.
Figure 3.
Summary of Cdc13 phosphorylation sites with respect to domain structure.
Table 1.
Summary of Cdc13 Phosphorylation Sites Identified from Full-Length, WT Proteina
| position | flanking sequence | telomere length | G1/G2 abundance | WT/MT abundance | putative kinase |
|---|---|---|---|---|---|
| S56 | CLLGFSNFER | ND | ND | ND | |
| S225 | QKVLSQKSK | unchangedb | ND | ND | Tel1/Mec1b |
| S306 | KSYIQSQTPE | unchanged | unchanged | unchanged | Mec1c |
| T308 | KSYIQSQTPE | T308A: slightly shortc | moderate increase in G2/M |
unchanged | CDK1c |
| S314 | RKTSVPNNW | S314A: slightly longer in cdc13-2 background |
substantial increase in G2/M |
drastic increase in WT in G2/M | |
| S314D: slightly short | |||||
| S324 | DDDSGSKR | S324A, S324D: unchanged | substantial increase in G2/M |
drastic increase in WT in G2/M | |
| S333 | KLSFHSPNAS | ND | unchanged | drastic increase in WT in G1 | |
| S336 | KLSFHSPNAS | S336A: unchangedc | unchanged | drastic increase in WT in G1 | CDK1c |
| S340 | FHSPNASSIR | ND | ND | ND | |
| S341 | SSIRKAISY | ND | moderate increase in G2/M |
unchanged | |
| S347 | SSIRKAISY | ND | unchanged | moderate decrease in WT | |
| S352 | LSLASVGSVE | ND | ND | ND | |
| S460 | DKTASPGMA | ND | unchanged | unchanged | CDK/MAPK (not CDK1)c |
| S708 | VKHEPKKSPT | S708A: slightly longer | moderate increase in G1 |
unchanged | CDK/MAPK (not CDK1)c |
| S710 | SPTTPALAE | ND | unchanged | unchanged | |
| S711 | SPTTPALAE | T711A: unchanged | unchanged | unchanged | CDK/MAPK (not CDK1c) |
| S726 | LNADVSSFD | ND | ND | ND | |
| S727 | LNADVSSFD | ND | ND | ND | |
| S883 | PDSGSLDCAI | ND | unchanged | moderate decrease in WT in G1 but moderate increase in WT in G2/M |
|
| S885 | PDSGSLDCAI | ND | unchanged | unchanged | |
| T893 | NATVNPLRLL | T893A, T893D: unchanged | ND | ND |
The table indicates the position of phosphorylation, effect on telomere lengths, and change of abundance between cell cycle phases, and that between WT Cdc13 and the Cdc13-E252K mutant (MT). Sites from different domains are separated by dashed lines. The change in phosphorylation abundance for each site was derived from the most consistently observed peptide. Moderate, substantial, and drastic reflect a 2–5-fold, 5–10-fold, and more than 10-fold change in abundance. ND: not determined.
Result reported in a previously published work.36
Strikingly, 11 of the 21 phosphorylation sites identified from the full-length Cdc13 were within a 128 aa region (S225 to S352) that overlaps both the RD and OB2 domains. In contrast, only two phosphorylation sites were mapped to OB1, and none was present within the DBD. The C-terminal OB4 domain was also phosphorylation-rich, as it contained eight phosphorylation sites in two clusters (S708, T710, T711, S726, and S727 in one cluster and S883, S885, and T893 in the other). Despite repeated analytical attempts and exhaustive search of the resulting raw MS data, phosphorylation of the highly conserved RD core (N234 to D287),39 including the two previously identified in vitro Tel1/ Mec1 targets, S249 and S255,36 was not detected.
Identification of Phosphorylation Sites from Cdc13 Fragments
We also expressed two Cdc13 fragments in yeast: a.a. 1–340 (OB1-RD) and a.a. 445–697 (DBD) and determined phosphorylation sites within these fragments by nanoLC-MS/ MS similarly as described for full-length Cdc13. From the OB1-RD fragment, we could detect phosphorylation on all sites that were detected in the full-length Cdc13. We also detected two additional phosphorylation sites, T3 and S170, which were not found in the full-length protein (Figure 3, Table 1). From the DBD fragment, we detected five additional sites that were not observed in the full-length Cdc13 (T458, S467, S472, T373, T507). Because three of the five sites identified from the DBD fragment are located in regions predicted to be in a β strand,28 we suspect that these five residues are buried within the interior of Cdc13 in the context of full-length protein; thus, their phosphorylation is unlikely to be biologically relevant. T3 is located at the distal N terminus, and S170 maps to a disordered loop on the protein surface of the Cdc13 OB1 crystal structure.28 Hence, both sites should be surface accessible in the OB1-RD fragment. Although we do not think this phosphorylation is biologically meaningful, the absence of S170 phosphorylation in the context of full-length protein suggests that the surface containing S170 may be involved in a long-range interaction with another domain of Cdc13.
Methods for Analyzing the Frequency of Cdc13 Phosphorylation as a Function of Cell Cycle Progression
Since telomerase action is strictly cell cycle regulated,9,10 key Cdc13 phosphorylation events might be cell cycle regulated. To test this possibility, we prepared Cdc13 from cells at two specific points in the cell cycle. Cells were arrested in α factor (late G1 phase) when telomerase is not active or in nocodazole (G2/M phase) when telomerase is active.9,48 Cdc13 purified from specific cell cycle phases was subjected to enzymatic digestion and nanoLC-MS/MS analyses to measure quantitatively changes in phosphorylation abundance between the two cell cycle stages. We then used the algorithm described in the Methods section to determine sites of phosphorylation and perform relative quantitation between different modified forms of the same peptide. A graphical summary of the phosphorylation levels for wild type Cdc13 is presented (Figure 4). We quantified all alternatively cleaved peptides and graphed them separately, with the most consistently observed one listed first in each of the panels.
Figure 4.

Cell cycle analysis of phospho-peptide levels from WT Cdc13. Results are presented as averages from at least two independent measurements. Error bars represent 1 standard deviation. Peptides that showed significant difference in abundance between G1 and G2/M phases are indicated by an asterisk.
Cdc13 Phosphorylation as a Function of Cell Cycle Position
Out of all the peptides containing S306 and T308 (Figure 4A), the peptide 302-SYIQSQTPER-311 was the most consistently observed. MS/MS analysis confirmed that the site of monophosphorylation on this peptide was preferentially T308, not S306 (data not shown). Monophosphorylation of this peptide was significantly higher in G2/M cells compared to G1 cells, a result consistent with previous reports that CDK1-dependent T308 phosphorylation occurs in the S/G2 phase.37,38 The diphosphorylated form of this peptide was also higher in G2/M cells compared to G1 cells, but this increase was not statistically significant.
Of the monophosphorylated peptides containing S314 and S324 (Figure 4B), S314 was the dominant phosphorylation site. However, we also observed low levels of S324 monophosphorylation within the 313-TSVPNNWHDDDSGSKR-328 peptide. Phosphorylation of both S314 and S324 was significantly higher in G2/M cells compared to G1 cells.
There is a strong grouping of potential phosphorylation sites (S333, S336, S340, S341) within the same peptides 331-KLSFHSPNASSIR-343 and 332-LSFHSPNASSIR-343 (Figure 4C). S336 was the most abundant monophosphorylation mark, and its phosphorylation was relatively constant across the cell cycle. S341 is a novel site of Cdc13 phosphorylation that was only observed in peptide 331-KLSFHSPNASSIR-343 in conjunction with phosphorylation of both S333 and S336. This triphosphorylated peptide was not particularly abundant, but its abundance was significantly higher in protein from G2/M phase than from G1 phase cells.
S347 was the major monophosphorylation site on peptide 345-AISYEQLSLASVGSVER-361 (Figure 4D). Monophosphorylation of S352 was also detected, but its abundance was an order of magnitude lower than that of S347.
Peptides 707-KSPTTPALAEHIPDLNADVSSFDVK-731 and 708-SPTTPALAEHIPDLNADVSSFDVK-731 (Figure 4E) contained a cluster of phosphorylation sites (S708, T710, T711, S726, S727). NanoLC-MS/MS revealed that the most abundant modification in this region was monophosphorylation of S708, and this modification was modestly higher in the G1 phase. Diphosphorylated versions of this peptide were modified at S708 and either T710 or T711.
Lastly, low but consistent phosphorylation was observed on S883 on peptide 877-VAAAPDSGSLDCAINATVNPLR-898 (Figure 4F). This phosphorylation did not change significantly across the cell cycle.
It is important to note that, for peptides containing more than one site of phosphorylation, there are two possible issues that can complicate isoform quantitation. The first is that the phosphopeptide isoforms are not chromatographically resolvable, resulting in a “mixed” tandem mass spectrum containing different percentages of each of the isobaric, coeluting isoforms. Upon careful inspection of the annotated tandem mass spectra (Supporting Information Figure 1), it was determined that this was not a limiting issue in our analysis. In the second scenario, the peptides are chromatographically resolvable, resulting in two or more MS peaks in the chromatogram corresponding to unique isoforms. We manually inspected the raw data for the existence of multiple chromatographic peaks consistent with distinct isobaric phosphopeptide isoforms, but we did not find any in this study.
Phosphorylation of Cdc13-E252K, a Separation-of-Function Mutant That Does Not Support Telomerase Action
The high density of phosphorylation events in the RD/OB2 boundary was particularly interesting given the key role of the RD in telomerase regulation. To determine if any of the detected phosphorylated sites are associated with defects in telomerase recruitment, we purified full-length Cdc13-E252K and analyzed it by nanoLC-MS/MS. All of the 21 phosphorylation sites seen in the full-length WT Cdc13 were also detected in the mutant protein. Moreover, no new sites of phosphorylation were detected in Cdc13-E252K.
We also investigated whether phosphorylation of Cdc13-E252K was cell cycle regulated in the same manner as WT Cdc13. We purified Cdc13-E252K from cells arrested in the G1 phase with α factor or in the G2/M phase with nocodazole and subjected the purified proteins to enzymatic digestion and quantitative nanoLC-MS/MS analysis as described for WT Cdc13 (see Figure 5 for a graphical summary of the WT-tomutant ratio for each modified isoform).
Figure 5.

Comparison of phospho-peptide quantification between WT Cdc13 and Cdc13-E252K mutant (MT). Results are presented as the Log2 value of the WT/MT ratio, and the error bar represents 1 standard deviation. Peptides that showed significant difference in abundance between WT and mutant are indicated by an asterisk.
There were several significant differences in the pattern of cell cycle regulated phosphorylation in mutant versus WT protein. First, peptide 312-KTSVPNNWHDDDSGSK-327 diphosphorylated on both S314 and S324 was >4,000-fold more abundant in WT protein from G2/M phase cells compared to G2/M phase Cdc13-E252K (Figure 5B). Second, peptide 332-LSFHSPNASSIR-343 diphosphorylated on S333 and S336 was 6.8-fold more abundant in WT protein from G1 phase cells compared to mutant protein from the same cell cycle phase (Figure 5C). We also observed a 129- and 56-fold decrease in the unphosphorylated form of peptide 331-KLSFHSPNASSIR-343 in WT protein compared to mutant protein in G1 and G2/M cell phases, respectively (Figure 5C). In addition, we identified a small but significantly lower monophosphorylation of S347 in WT compared to peptides derived from the mutant protein across the cell cycle (Figure 5D). Additionally, S883 monophosphorylation was modestly downregulated in WT Cdc13 in G1 but upregulated in the G2/M phase of the cell cycle (Figure 5F).
Cdc13 S314 Phosphorylation Negatively Affects Telomere Lengths
Mutation of S225, S306, and S336 to a nonphosphorylatable residue was shown previously to have no effect on telomere length37–39 while a T308A mutation leads to modest reductions in both telomere length and Est1 telomere binding.37,38 Since phosphorylation of S314 and S324 was significantly higher in WT relative to Cdc13-E252K in the G2/M phase, we decided to investigate the role of S314 and S324 phosphorylation in telomere length regulation. Using site directed mutagenesis, we changed S314 and S324 to either alanine (A), which cannot be phosphorylated, or aspartate (D), a change that mimics constitutive phosphorylation. Mutant alleles were expressed from the Cdc13 promoter on a CEN plasmid in cells in which the mutant protein was the only version of Cdc13. Cells were grown for at least three consecutive restreaks, and genomic DNA was extracted for telomere length analysis. Strains expressing Cdc13-S314A, Cdc13-S324A, or Cdc13-S324D had WT length telomeres (Figure 6A, telomeres are on average 15 ± 3, 5 ± 1, 5 ± 1 bp longer than WT, respectively, n = 4). However, the S314D mutation led to stable but slightly shorter telomeres (37 ± 17 bp shorter than WT, n = 3) (Figure 6A, Table I). Moreover, the S314A S324A double mutant showed a synergistic effect that lead to telomeres 82 ± 30 bp longer than WT. The S314D S324D mutant, however, maintained telomeres 47 ± 14 bp shorter than WT, a length similar to that in the S314D mutant.
Figure 6.

Telomere length analysis of Cdc13 phosphorylation mutants. cdc13Δ cells carrying WT or mutant Cdc13 on a CEN TRP1 plasmid were analyzed for their telomere lengths as described in Methods. Mutants analyzed are (A) S314A, S314D, S324A, S324D, S314A S324A, and S314D S324D, and (B) cdc13-2 and cdc13-2 S314A mutants. (C) Western blot analysis of myc9-tagged Cdc13 mutants expressed from Cdc13 promoter on a CEN TRP1 plasmid (upper panel). The same blot was probed with anti-α tubulin as loading control (lower panel). UT: untagged.
To provide more evidence that phosphorylation at S314 affects telomere length, we introduced mutations at this residue into the cdc13-2 allele to generate double mutants. Although the cdc13-2, S314A double mutant still senesced, its telomeres were on average 77 ± 21 bp longer compared to cdc13-2 alone (n = 3, Figure 6B). These differences were not due to effects of these mutations on protein abundance (Figure 6C). These results suggest that while phosphorylation of S324 is not important for telomere length, phosphorylation of S314A negatively regulates telomere length.
DISCUSSION
Misregulation of telomerase is associated with aging and cancer.49 In budding yeast, telomerase is strictly controlled, occurring only in the late S/G2 phase and acting preferentially on short telomeres. Cdc13 is a multifunctional telomere binding protein that acts as a type of molecular switch with its binding partners determining if it forms a protective cap (Stn1, Ten1) or promotes telomerase elongation (Est1) or C-strand resynthesis (Pol α). Given its multiple binding partners and functions, Cdc13 is a good candidate for regulation by phosphorylation.
We determined in vivo phosphorylation sites on both WT and the telomerase defective Cdc13-E252K protein in asynchronous, G1 arrested (telomerase inactive), and G2/M arrested (telomerase active) cells using a new label-free nanoLC-MS/ MS approach to quantitate relative levels of phosphorylation at individual residues. This approach provides an alternative to methods that use internal peptide standards to provide the absolute abundance of a given modification.50 Our approach is useful when, as with the project described here, the sites of phosphorylation are not known, since our label-free nanoLC-MS/MS method allows the simultaneous identification and relative abundance determination of phosphorylation sites. The relative abundance of a given modification was determined by comparing across samples (i.e., between WT and mutant Cdc13 or WT from G1 versus WT from G2/M phase cells) but never between different peptides. Estimating changes in relative abundances is valid, since the differences in ionization efficiency resulting from different PTMs on the same peptide sequence should be consistent across different runs. Indeed, our quantitative data were highly reproducible with small differences in relative abundances of modified residues across multiple technical and biological replicates (Figure 4 and 5).
Because our analysis was able to document modified isoforms that were present over a wide range of concentrations (Figure 4), this approach should be generally useful for analysis of other hypermodified proteins. While identifications based on shotgun tandem MS and blind database searching only would have missed many lower abundance modifications, our method was able to identify these sites based on their relative retention time and MS isotopic distribution. If we had used only a traditional tandem MS search algorithm, we likely would have identified a large number of additional false sites of modification on the same peptide sequences because modified isoforms share a significant number of fragment ion peaks in tandem MS. Since our method simultaneously uses MS, MS2, and retention time to identify sites and degrees of phosphorylation, the false positive rate was low and even allowed discovery of sites in the absence of tandem MS. It should be noted here that all modified isoforms identified without available tandem MS were later targeted for MS2 in subsequent nanoLC-MS/MS runs to confirm their assignments.
We identified 21 phosphorylation sites on the 924-amino acid Cdc13 (summarized in Figure 3, Table 1). Of these 21 sites, 17 had not been detected previously in vivo (one of the 17 sites, S225, was seen on protein phosphorylated in vitro36). All of the phosphorylation sites are located at residues predicted to be in flexible regions of Cdc13 that are more likely to be solvent accessible.28 In contrast, phosphorylation sites detected only in the Cdc13 DBD (S467, S472, T473) were not on exposed surfaces and probably are not relevant to intact protein. However, S170, a residue that was only phosphorylated in OB1-RD, is on a disordered loop,28 indicating it is on the protein surface in the context of OB1-RD but not in the context of fulllength Cdc13. Therefore, the S170-containing surface may be responsible for a long-range interaction within the Cdc13 protein.
The phosphorylated residues were not spread uniformly throughout the protein. To our surprise, regions that make critical biological contacts, such as the core of the DBD and OB1, and the highly conserved N-terminal half of RD that interacts with Est1, were devoid of detectable phosphorylation (Figure 3, Table 1). Perhaps the phosphorylation sites surrounding these interaction interfaces do not regulate these functions as an on-off switch, as do the phosphorylation sites on the CDKs.51,52 Rather, we suggest they are present to modulate and coordinate these interactions, or mediate long-range interactions between Cdc13 domains.
There was a cluster of ten phosphorylation sites within a 47 amino acid region at the boundary of the RD/OB2 domains (aa 306 to 352; Figure 3). This cluster includes the two previously identified phosphorylation sites (S306 and T308) and the one identified here (S314) whose phosphorylation affects telomere length or telomerase action (Figure 6). All three of these residues are located within the C-terminal half of the RD. The extension of this phosphorylation cluster into the OB2 domain suggests that OB2 may have a previously unidentified role in affecting telomerase recruitment in addition to its previously suggested roles as an effector of the affinity and sequence specificity of the DBD.53
Of particular interest, phosphorylation of two in vitro identified Tel1/Mec1 targets, S249 and S255 within the RD,36 was not detected despite repeated attempts. The lack of detectable phosphorylation on these two residues was probably not due to false negativity or low sensitivity from the search algorithms, since we detected phosphorylation at two other Tel1/Mec1 sites, S225 and S306, as well as modifications present in ~0.01% of the sample (as in phosphorylation of 331-KLSFHSPNASSIR-343, Figure 4C). A lack of in vivo phosphorylation at these residues provides an explanation for seemingly contradictory results from earlier studies. S249 and S255 are both phosphorylated by Tel1 and Mec1 in vitro, and changing both residues to alanine confers a telomerase defective phenotype.36 Nonetheless, a cdc13-Q250A Q256A allele is not telomerase deficient.39 Our results support the interpretation that these residues are not Tel1/Mec1 substrates in vivo. Since these two SQ motifs are extremely conserved in Cdc13 from closely related fungi,28,39 we propose that they play a structural, rather than a signaling, role. In this model, the telomerase-null phenotype of the cdc13-S249A S255A allele is explained by a local loss of structural integrity rather than a lack of phosphorylation. These results emphasize the importance of determining sites of modification in vivo even when genetic data are available.
To gain information about the importance of phosphorylation of given residues, we determined if modifications were correlated with cell cycle phase (Figure 4) and/or WT versus telomerase deficient protein (Figure 5). There were no differences between mutant and WT protein in terms of sites of phosphorylation. This finding may indicate that regulated phosphorylation occurs upstream of telomerase recruitment. Alternatively, quantitative rather than qualitative differences in phosphorylation may explain functional differences, as there were six sites that showed different levels of phosphorylation between WT and mutant Cdc13 (S314, S324, S333, S336, S347, S883). In addition, there were five residues (T308, S314, S324, S341, S708) that showed cell cycle-specific differences. In both cases, these differences could be substantial. For example, phosphorylation of S314, S324 on the peptide 311-KTSVPNNWHDDDSGSK-327 was 6-fold more abundant in WT protein in the G2/M versus the G1 phase (Figure 4B) and >4,000-fold less frequent in mutant Cdc13 (Figure 5B). As the abundance of S314 phosphorylation was affected both by the cdc13-2 mutation and by the cell cycle, we mutated this site to alanine or aspartate. Although S314 mutations conferred effects on telomere length, consistent with phosphorylation at this residue having a negative effect on telomerase (Figure 6), the effects were subtle, similar to what has been seen previously for mutation of the Cdk1 target T308.37,38 Thus, individual phosphorylation events may act redundantly to influence telomere length, and multiple mutations may be needed for large effects. Consistent with this view, we observed that the S314A S324A double mutation resulted in longer telomeres while either mutation alone had no effect (Figure 6A). Our analysis provides many more candidates for residues that can be mutated singly and in combination with other mutations for effects on telomerase recruitment and telomere structure.
CDK/MAPKs phosphorylate S/TP motifs and ATM/ATR kinases phosphorylate S/TQ motifs. There are seven S/TP and ten S/TQ motifs in Cdc13. Five of the seven S/TP motifs were phosphorylated in vivo, but only two were CDK1 targets in vitro.37,38 Four of the ten S/TQ motifs can be phosphorylated by Tel1 or Mec1 in vitro,36 but only two (S225, S306) appeared to be phosphorylated in vivo. Clearly, factors other than sequence specificity contribute to substrate recognition and phosphorylation in vivo. The three S/TP motifs that are not direct CDK1 targets in vitro but were phosphorylated in vivo may be phosphorylated by another kinase with the same consensus motif. Indeed, the kinases responsible for most of the detected phosphorylation events (17 out of 21) could not be determined by the sequence context of the modified residue. However, a recent high-throughput study identified three kinases, Cak1, Hsl1, and Vhs1, that affect cell cycle progression and that also interact with Cdc13.54 All three are excellent candidates to explain phosphorylation of residues for which there is, as yet, no evident responsible kinase.
This study has provided a wealth of knowledge about Cdc13 phosphorylation, but there is still much to learn. Although our study identified 17 novel phosphorylation sites, the biological functions of most of these are unknown. A full understanding of their importance will likely involve analysis of multiply modified proteins to circumvent biological redundancy. By using information on the abundance of phosphorylation events as a function of the cell cycle and in vivo function, we provide a first step for a rational next stage analysis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Andrew Taggart and Yasumasa Tsukamoto for their initial work on Cdc13 phosphorylation and Saw Kyin of Princeton Mass Spec Facility for the initial work with mass spectrometry analysis. This work was supported by U.S. National Institute of Health Grant GM43265 to V.A.Z. B.A.G. acknowledges funding support from a National Science Foundation (NSF) Early Faculty CAREER award, an NSF grant (CBET-0941143), the American Society for mass spectrometry research award sponsored by the Waters Corporation, and a grant supported by Award Number DP2OD007447 from the Office Of The Director, National Institutes of Health. Y.W. was supported in part by postdoctoral fellowship DRG-1943-07 from the Damon Runyon-Robert Black Cancer Research Foundation, and P.A.D. was supported by NIH Kirschstein-NSRA F32 GM093490.
Footnotes
Supporting Information
Histogram of the ratio of 23-DFEGYPSK-30 to 567-LELNEGFK-574, and annotated CID tandem mass spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
REFERENCES
- 1.Wellinger RJ, Wolf AJ, Zakian VA. Saccharomyces telomeres acquire single-strand TG1–3 tails late in S phase. Cell. 1993;72:51–60. doi: 10.1016/0092-8674(93)90049-v. [DOI] [PubMed] [Google Scholar]
- 2.Lingner J, Hughes TR, Shevchenko A, Mann M, Lundblad V, Cech TR. Reverse transcriptase motifs in the catalytic subunit of telomerase. Science. 1997;276:561–567. doi: 10.1126/science.276.5312.561. [DOI] [PubMed] [Google Scholar]
- 3.Singer MS, Gottschling DE. TLC1: template RNA component of Saccharomyces cerevisiae telomerase. Science. 1994;266:404–409. doi: 10.1126/science.7545955. [DOI] [PubMed] [Google Scholar]
- 4.Lin JJ, Zakian VA. The Saccharomyces CDC13 protein is a single-strand TG1–3 telomeric DNA-binding protein in vitro that affects telomere behavior in vivo. Proc. Natl. Acad. Sci. U.S.A. 1996;93:13760–13765. doi: 10.1073/pnas.93.24.13760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nugent CI, Hughes TR, Lue NF, Lundblad V. Cdc13p: a single-strand telomeric DNA-binding protein with a dual role in yeast telomere maintenance. Science. 1996;274:249–252. doi: 10.1126/science.274.5285.249. [DOI] [PubMed] [Google Scholar]
- 6.Lundblad V, Szostak JW. A mutant with a defect in telomere elongation leads to senescence in yeast. Cell. 1989;57:633–643. doi: 10.1016/0092-8674(89)90132-3. [DOI] [PubMed] [Google Scholar]
- 7.Lendvay TS, Morris DK, Sah J, Balasubramanian B, Lundblad V. Senescence mutants of Saccharomyces cerevisiae with a defect in telomere replication identify three additional EST genes. Genetics. 1996;144:1399–412. doi: 10.1093/genetics/144.4.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taggart AK, Teng SC, Zakian VA. Est1p as a cell cycle-regulated activator of telomere-bound telomerase. Science. 2002;297:1023–1026. doi: 10.1126/science.1074968. [DOI] [PubMed] [Google Scholar]
- 9.Diede SJ, Gottschling DE. Telomerase-mediated telomere addition in vivo requires DNA primase and DNA polymerases alpha and delta. Cell. 1999;99:723–733. doi: 10.1016/s0092-8674(00)81670-0. [DOI] [PubMed] [Google Scholar]
- 10.Marcand S, Brevet V, Mann C, Gilson E. Cell cycle restriction of telomere elongation. Curr. Biol. 2000;10:487–490. doi: 10.1016/s0960-9822(00)00450-4. [DOI] [PubMed] [Google Scholar]
- 11.Evans SK, Lundblad V. Est1 and Cdc13 as comediators of telomerase access. Science. 1999;286:117–120. doi: 10.1126/science.286.5437.117. [DOI] [PubMed] [Google Scholar]
- 12.Pennock E, Buckley K, Lundblad V. Cdc13 delivers separate complexes to the telomere for end protection and replication. Cell. 2001;104:387–396. doi: 10.1016/s0092-8674(01)00226-4. [DOI] [PubMed] [Google Scholar]
- 13.Bianchi A, Negrini S, Shore D. Delivery of yeast telomerase to a DNA break depends on the recruitment functions of Cdc13 and Est1. Mol. Cells. 2004;16:139–146. doi: 10.1016/j.molcel.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 14.Wu Y, Zakian VA. The telomeric Cdc13 protein interacts directly with the telomerase subunit Est1 to bring it to telomeric DNA ends in vitro. Proc. Natl. Acad. Sci. U. S. A. 2011;108:20362–20369. doi: 10.1073/pnas.1100281108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teixeira MT, Arneric M, Sperisen P, Lingner J. Telomere length homeostasis is achieved via a switch between telomerase-extendible and -nonextendible states. Cell. 2004;117:323–335. doi: 10.1016/s0092-8674(04)00334-4. [DOI] [PubMed] [Google Scholar]
- 16.Sabourin M, Tuzon CT, Zakian VA. Telomerase Tel1p preferentially associate with short telomeres in S. cerevisiae. Mol. Cells. 2007;27:550–561. doi: 10.1016/j.molcel.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bianchi A, Shore D. Increased association of telomerase with short telomeres in yeast. Genes Dev. 2007;21:1726–1730. doi: 10.1101/gad.438907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bianchi A, Shore D. Early replication of short telomeres in budding yeast. Cell. 2007;128:1051–1062. doi: 10.1016/j.cell.2007.01.041. [DOI] [PubMed] [Google Scholar]
- 19.Frank CJ, Hyde M, Greider CW. Regulation of telomere elongation by the cyclin-dependent kinase CDK1. Mol. Cells. 2006;24:423–432. doi: 10.1016/j.molcel.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 20.Vodenicharov MD, Wellinger RJ. DNA degradation at unprotected telomeres in yeast is regulated by the CDK1 (Cdc28/Clb) cell-cycle kinase. Mol. Cells. 2006;24:127–137. doi: 10.1016/j.molcel.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 21.Greenwell PW, Kronmal SL, Porter SE, Gassenhuber J, Obermaier B, Petes TD. TEL1 a gene involved in controlling telomere length in Scerevisiae, is homologous to the human ataxia telangiectasia gene. Cell. 1995;82:823–829. doi: 10.1016/0092-8674(95)90479-4. [DOI] [PubMed] [Google Scholar]
- 22.Ritchie KB, Mallory JC, Petes TD. Interactions of TLC1 (which encodes the RNA subunit of telomerase), TEL1 MEC1 in regulating telomere length in the yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 1999;19:6065–6075. doi: 10.1128/mcb.19.9.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hector RE, Shtofman RL, Ray A, Chen BR, Nyun T, Berkner KL, Runge KW. Tel1p preferentially associates with short telomeres to stimulate their elongation. Mol. Cells. 2007;27:851–858. doi: 10.1016/j.molcel.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 24.Arneric M, Lingner J. Tel1 kinase and subtelomere-bound Tbf1 mediate preferential elongation of short telomeres by telomerase in yeast. EMBO Rep. 2007;8:1080–1085. doi: 10.1038/sj.embor.7401082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hughes TR, Weilbaecher RG, Walterscheid M, Lundblad V. Identification of the single-strand telomeric DNA binding domain of the Saccharomyces cerevisiae Cdc13 protein. Proc. Natl. Acad. Sci. U. S. A. 2000;97:6457–6462. doi: 10.1073/pnas.97.12.6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garvik B, Carson M, Hartwell L. Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol. Cell. Biol. 1995;15:6128–6138. doi: 10.1128/mcb.15.11.6128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitchell MT, Smith JS, Mason M, Harper S, Speicher DW, Johnson FB, Skordalakes E. Cdc13 N-terminal dimerization, DNA binding telomere length regulation. Mol. Cell. Biol. 2010;30:5325–5334. doi: 10.1128/MCB.00515-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun J, Yang Y, Wan K, Mao N, Yu TY, Lin YC, DeZwaan DC, Freeman BC, Lin JJ, Lue NF, Lei M. Structural bases of dimerization of yeast telomere protein Cdc13 and its interaction with the catalytic subunit of DNA polymerase alpha. Cell Res. 2011;21:258–274. doi: 10.1038/cr.2010.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qi H, Zakian VA. The Saccharomyces telomere-binding protein Cdc13p interacts with both the catalytic subunit of DNA polymerase alpha and the telomerase-associated est1 protein. Genes Dev. 2000;14:1777–1788. [PMC free article] [PubMed] [Google Scholar]
- 30.Hsu CL, Chen YS, Tsai SY, Tu PJ, Wang MJ, Lin JJ. Interaction of Saccharomyces Cdc13p with Pol1p, Imp4p, Sir4p and Zds2p is involved in telomere replication, telomere maintenance and cell growth control. Nucleic Acids Res. 2004;32:511–521. doi: 10.1093/nar/gkh203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chandra A, Hughes TR, Nugent CI, Lundblad V. Cdc13 both positively and negatively regulates telomere replication. Genes Dev. 2001;15:404–414. doi: 10.1101/gad.861001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grandin N, Reed SI, Charbonneau M. Stn1, a new Saccharomyces cerevisiae protein, is implicated in telomere size regulation in association with Cdc13. Genes Dev. 1997;11:512–527. doi: 10.1101/gad.11.4.512. [DOI] [PubMed] [Google Scholar]
- 33.Puglisi A, Bianchi A, Lemmens L, Damay P, Shore D. Distinct roles for yeast Stn1 in telomere capping telomerase inhibition. EMBO J. 2008 doi: 10.1038/emboj.2008.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hang LE, Liu X, Cheung I, Yang Y, Zhao X. SUMOylation regulates telomere length homeostasis by targeting Cdc13. Nat. Struct. Mol. Biol. 2011;18:920–926. doi: 10.1038/nsmb.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grandin N, Damon C, Charbonneau M. Cdc13 cooperates with the yeast Ku proteins and Stn1 to regulate telomerase recruitment. Mol. Cell. Biol. 2000;20:8397–8408. doi: 10.1128/mcb.20.22.8397-8408.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tseng SF, Lin JJ, Teng SC. The telomerase-recruitment domain of the telomere binding protein Cdc13 is regulated by Mec1p/ Tel1p–dependent phosphorylation. Nucleic Acids Res. 2006;34:6327–6336. doi: 10.1093/nar/gkl786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li S, Makovets S, Matsuguchi T, Blethrow JD, Shokat KM, Blackburn EH. Cdk1-dependent phosphorylation of Cdc13 coordinates telomere elongation during cell-cycle progression. Cell. 2009;136:50–61. doi: 10.1016/j.cell.2008.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tseng SF, Shen ZJ, Tsai HJ, Lin YH, Teng SC. Rapid Cdc13 turnover and telomere length homeostasis are controlled by Cdk1-mediated phosphorylation of Cdc13. Nucleic Acids Res. 2009;37:3602–3611. doi: 10.1093/nar/gkp235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao H, Toro TB, Paschini M, Braunstein-Ballew B, Cervantes RB, Lundblad V. Telomerase recruitment in Saccha-romyces cerevisiae is not dependent on Tel1-mediated phosphorylation of Cdc13. Genetics. 2010;186:1147–1159. doi: 10.1534/genetics.110.122044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang W, Durocher D. De novo telomere formation is suppressed by the Mec1-dependent inhibition of Cdc13 accumulation at DNA breaks. Genes Dev. 2010;24:502–515. doi: 10.1101/gad.1869110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Villen J, Gygi SP. The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat. Protoc. 2008;3:1630–1638. doi: 10.1038/nprot.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rappsilber J, Ishihama Y, Mann M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 2003;75:663–670. doi: 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- 43.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat. Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 44.Bourns BD, Alexander MK, Smith AM, Zakian VA. Sir proteins, Rif proteins, and Cdc13p bind Saccharomyces telomeres in vivo. Mol. Cell. Biol. 1998;18:5600–5608. doi: 10.1128/mcb.18.9.5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anderson EM, Halsey WA, Wuttke DS. Delineation of the high-affinity single-stranded telomeric DNA-binding domain of Saccharomyces cerevisiae Cdc13. Nucleic Acids Res. 2002;30:4305–4313. doi: 10.1093/nar/gkf554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swaney DL, McAlister GC, Coon JJ. Decision tree-driven tandem mass spectrometry for shotgun proteomics. Nat. Methods. 2008;5:959–964. doi: 10.1038/nmeth.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chi A, Huttenhower C, Geer LY, Coon JJ, Syka JE, Bai DL, Shabanowitz J, Burke DJ, Troyanskaya OG, Hunt DF. Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2007;104:2193–2198. doi: 10.1073/pnas.0607084104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Osterhage JL, Talley JM, Friedman KL. Proteasome-dependent degradation of Est1p regulates the cell cycle-restricted assembly of telomerase in Saccharomyces cerevisiae. Nat. Struct. Mol. Biol. 2006;13:720–728. doi: 10.1038/nsmb1125. [DOI] [PubMed] [Google Scholar]
- 49.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 50.Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. Regulatory roles of cyclin dependent kinase phosphorylation in cell cycle control. Proc. Natl. Acad. Sci. U. S. A. 2003;100:6940–6945. doi: 10.1073/pnas.0832254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lew DJ, Kornbluth S. Regulatory roles of cyclin dependent kinase phosphorylation in cell cycle control. Curr. Opin. Cell. Biol. 1996;8:795–804. doi: 10.1016/s0955-0674(96)80080-9. [DOI] [PubMed] [Google Scholar]
- 52.Berry LD, Gould KL. Regulation of Cdc2 activity by phosphorylation at T14/Y15. Prog. Cell Cycle Res. 1996;2:99–105. doi: 10.1007/978-1-4615-5873-6_10. [DOI] [PubMed] [Google Scholar]
- 53.Zappulla DC, Roberts JN, Goodrich KJ, Cech TR, Wuttke DS. Inhibition of yeast telomerase action by the telomeric ssDNA-binding protein, Cdc13p. Nucleic Acids Res. 2009;37:354–367. doi: 10.1093/nar/gkn830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fasolo J, Sboner A, Sun MG, Yu H, Chen R, Sharon D, Kim PM, Gerstein M, Snyder M. Diverse protein kinase interactions identified by protein microarrays reveal novel connections between cellular processes. Genes Dev. 2011;25:767–778. doi: 10.1101/gad.1998811. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.