

Abstract
Groupers of the family Epinephelidae are a diverse and economically valuable group of reef fishes. To investigate the evolution of their mitochondrial genomes we characterized and compared these genomes among 22 species, 17 newly sequenced. Among these fishes we identified three distinct genome organizations, two of them never previously reported in vertebrates. In 19 of these species, mitochondrial genomes followed the typical vertebrate canonical organization with 13 protein-coding genes, 22 tRNAs, two rRNAs, and a non-coding control region. Differing from this, members of genus Variola have an extra tRNA-Ile between tRNA-Val and 16S rRNA. Evidence suggests that this evolved from tRNA-Val via a duplication event due to slipped strand mispairing during replication. Additionally, Cephalopholis argus has an extra tRNA-Asp in the midst of the control region, likely resulting from long-range duplication of the canonical tRNA-Asp through illicit priming of mitochondrial replication by tRNAs. Along with their gene contents, we characterized the regulatory elements of these mitochondrial genomes’ control regions, including putative termination-associated sequences and conserved sequence blocks. Looking at the mitochondrial genomic constituents, rRNA and tRNA are the most conserved, followed by protein-coding genes, and non-coding regions are the most divergent. Divergence rates vary among the protein-coding genes, and the three cytochrome oxidase subunits (COI, II, III) are the most conserved, while NADH dehydrogenase subunit 6 (ND6) and the ATP synthase subunit 8 (ATP8) are the most divergent. We then tested the phylogenetic utility of this new mt genome data using 12 protein-coding genes of 48 species from the suborder Percoidei. From this, we provide further support for the elevation of the subfamily Epinephelinae to family Epinephelidae, the resurrection of the genus Hyporthodus, and the combination of the monotypic genera Anyperodon and Cromileptes to genus Epinephelus , and Aethaloperca to genus Cephalopholis .
Introduction
The Perciform family Epinephelidae (previously subfamily Epinephelinae in family Serranidae), commonly known as groupers, is an assemblage of reef fishes comprising more than 160 species in 16 genera [1]. These commercially important fishes inhabit coastal areas of the tropics and subtropics, with species frequently sharing small geographic ranges. Their evolutionary biology has been of continuing interest due in part to their biological diversity and apparent rapid sympatric speciation [1–3]. While many areas of their evolutionary biology are under active investigation, current understanding of the structure and evolution of the groupers’ mitochondrial (mt) genome is limited as few have been sequenced [4,5] and none have yet to be comprehensively described. This is of particular interest as the mt genome presents a simpler system than the nuclear genome for studying the molecular dynamics and mechanisms of rearrangements that underlie variations in the genome.
While organization of the fish mt genome was long thought to be highly conserved, there is increasing appreciation that it may be considerably more plastic than originally believed. The canonical fish mt genome contains 13 protein-coding genes (ATP8, ATP6, COI-III, Cyt b, ND1-6 and ND4L), two ribosomal RNA genes (12S rRNA and 16S rRNA), and 22 transfer RNA genes (tRNAs). Additionally, there are two major non-coding regions involved in mtDNA replication and transcription, the light-strand (L-strand) origin of replication (OL) and the control region (CR) [6]. However, recent work has shown that the fish mt genomes can differ from this canonical organization in both gene content and order, with several recognized hotspots of reorganization. These include the tRNA-WANCY cluster (W, Trp; A, Ala; N, Asn; C, Cys; Y, Tyr) located between ND2 and COI [7,8], the tRNA-IQM cluster (I, Ile; Q, Gln; M, Met) located between ND1 and ND2 [9,10], and the CR, usually involved in the translocation of ND6 and tRNA-Glu to CR [11,12]. In addition to these hotspots, large-scale reorganizations of the mt genome have also been observed in some fishes [13,14]. Our interest in this study is first in comparing the mt genome arrangement and sequence among the groupers for insight into the evolutionary dynamics and molecular mechanisms that have shaped them. Beyond this though, these fully sequenced mt genomes provide a broadly useful molecular resource for these fishes.
Among its other uses, several features of the mtDNA make it particularly attractive as a tool for studying evolutionary biology [15]. These include its small size, cellular abundance, maternal inheritance, compact gene arrangement, and high rate of evolution [16]. Thus, mtDNA is generally considered a good molecular marker for phylogenetic analyses among fish taxa. However, short mt gene fragments exhibit limitations in resolving complicated phylogenetic relationships in many fish lineages [17]. The additional informative sites from longer DNA sequences (e.g. mt genomes) allow these deeper branches and higher-level relationships to be more fully resolved [18]. Hence, the mt genomes provided in this study may help resolve the evolutionary relationships among the groupers [1,19,20] where recent studies have led to suggested taxonomic revisions.
Additionally, the mt genomes from these fishes can be applied towards species identification and in aid of conservation efforts for this commercially important group. mtDNA has been used for DNA barcoding allowing for identification on the level of species or population. This is particularly useful for groupers where similarities in color patterns, a lack of morphological variation, and ontogenetic changes combine to cause taxonomic confusion [21]. Such improved means of identification are important for future efforts to manage grouper fisheries with 33 grouper species already assessed as ‘endangered’, ‘vulnerable’, or ‘near threatened’ on the ICUN (International Union for Conservation of Nature and Natural Resources) Red List [22].
To better understand the structure of the grouper mt genome and its evolution we took a detailed view of its organization, gene content, and functional regions in 22 species, including representatives from nine of the 16 genera in family Epinephelidae (Table 1). Not only does this provide new insight into novel mt genome organization among these fishes, but the additional informative sites available from these fully sequenced mt genomes allows us to test recently proposed taxonomic revisions. Finally, this work also provides important new molecular resources for the species identification, fishery management, and conservation biology of groupers.
Table 1. Genome sizes and nucleotide compositions for the mitochondrial genomes of 22 grouper species analyzed in this study and sampling locations of the 17 newly sequenced ones.
| Species | Sampling location | Genome size (bp) | Base compositions (%) | AT-skew | GC-skew | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | T | G | C | |||||||||||||||||
| Aethaloperca rogaa | HongKong, China | 16538 | 29.4 | 27.3 | 15.9 | 27.4 | 0.04 | -0.27 | ||||||||||||
| Anyperodon leucogrammicus | — | 16616 | 28.7 | 26.9 | 15.7 | 28.7 | 0.03 | -0.29 | ||||||||||||
| Cephalopholis argus | Taiwan | 16767 | 29.2 | 27.7 | 16.2 | 26.9 | 0.03 | -0.25 | ||||||||||||
| Cephalopholis sonnerati | HongKong, China | 16587 | 29.6 | 26.2 | 15.9 | 28.3 | 0.06 | -0.28 | ||||||||||||
| Cromileptes altivelis | Hainan, China | 16504 | 29.1 | 26.3 | 15.7 | 29.0 | 0.05 | -0.30 | ||||||||||||
| Epinephelus akaara | Fujian, China | 16795 | 28.7 | 27.3 | 16.2 | 27.8 | 0.03 | -0.26 | ||||||||||||
| Epinephelus areolatus | Hainan, China | 16965 | 28.7 | 27.1 | 16.3 | 28.0 | 0.03 | -0.26 | ||||||||||||
| Epinephelus awoara | Fujian, China | 16802 | 28.5 | 27.3 | 16.5 | 27.7 | 0.02 | -0.25 | ||||||||||||
| Epinephelus bruneus | — | 16692 | 28.5 | 26.6 | 16.2 | 28.7 | 0.03 | -0.28 | ||||||||||||
| Epinephelus coioides | Fujian, China | 16418 | 28.7 | 26.5 | 15.8 | 28.9 | 0.04 | -0.29 | ||||||||||||
| Epinephelus epistictus | Hainan, China | 16927 | 29.2 | 27.0 | 15.7 | 28.1 | 0.04 | -0.28 | ||||||||||||
| Epinephelus fuscoguttatus | Hainan, China | 16648 | 29.2 | 26.9 | 15.6 | 28.3 | 0.04 | -0.29 | ||||||||||||
| Epinephelus lanceolatus | — | 16714 | 29.5 | 26.5 | 15.1 | 28.8 | 0.05 | -0.31 | ||||||||||||
| Epinephelus moara | — | 16696 | 28.6 | 26.5 | 16.1 | 28.9 | 0.04 | -0.28 | ||||||||||||
| Epinephelus trimaculatus | Hainan, China | 16777 | 29.0 | 27.1 | 15.9 | 28.0 | 0.03 | -0.27 | ||||||||||||
| Hyporthodus octofasciatus | Fujian, China | 16545 | 28.6 | 27.4 | 16.3 | 27.8 | 0.02 | -0.26 | ||||||||||||
| Hyporthodusseptemfasciatus | — | 16558 | 28.6 | 26.8 | 16.3 | 28.3 | 0.03 | -0.27 | ||||||||||||
| Plectropomus areolatus | HongKong, China | 16770 | 28.8 | 27.6 | 16.3 | 27.2 | 0.02 | -0.25 | ||||||||||||
| Plectropomus leopardus | Fujian, China | 16753 | 29.1 | 28.0 | 16.2 | 26.7 | 0.02 | -0.24 | ||||||||||||
| Triso dermopterus | Hainan, China | 16605 | 28.2 | 25.7 | 16.7 | 29.4 | 0.05 | -0.28 | ||||||||||||
| Variola albimarginata | Taiwan | 16768 | 28.4 | 27.7 | 16.4 | 27.6 | 0.01 | -0.26 | ||||||||||||
| Variola louti | Hainan, China | 16770 | 28.4 | 27.8 | 16.3 | 27.5 | 0.01 | -0.26 | ||||||||||||
The species with listed sampling locations were sequenced in this study. These sampling locations are for fish markets where specimens were purchased. In China fresh groupers are usually only available in fish markets that are supplied by local fishermen, making these representative of the local fish fauna.
Results and Discussion
Canonical genome organization and composition
The mt genomes of the 22 analyzed groupers range in size from 16,418 to 16,965 bp and show three distinct organizations. While Variola albimarginata , V . louti , and Cephalopholis argus have novel mt genome organization, the 19 remaining species share the canonical mt gene content and arrangement (Figure 1A and Table 2). The canonical mt genomes of the 19 species contain 37 genes encoding 13 proteins, 22 tRNAs, and two rRNAs. As in many teleosts, most of the mt genes are located on the heavy-strand (H-strand) with the exceptions of ND6 and eight tRNA (tRNA-Gln, tRNA-Ala, tRNA-Asn, tRNA-Cys, tRNA-Tyr, tRNA-Ser(UCN), tRNA-Glu, tRNA-Pro), which are encoded on the L-strand. The overall genome nucleotide identity between these 19 mt genomes which show identical synteny is 83.9%.
Figure 1. Gene maps for mitochondrial genomes.

Genes encoded on the heavy and light strand are shown outside and inside the circle, respectively. The inner grey ring indicates the GC content. This genome map was constructed via OrganellarGenomeDRAW [81] with manual modifications. (A) The mt gemone organization of Aethaloperca rogaa, Anyperodon leucogrammicus, Cephalopholis sonnerati, Cromileptes altivelis, Epinephelus akaara, E. areolatus, E. awoara, E. bruneus, E. coioides, E. epistictus, E. fuscoguttatus, E. lanceolatus, E. moara, E. trimaculatus, Hyporthodus octofasciatus, H. septemfasciatus, Plectropomus areolatus, P. leopardus, and Triso dermopterus. (B) The mt gemone organization of genera Variola, V. albimarginata and V. louti. (C) The mt gemone organization of Cephalopholis argus.
Table 2. Gene content of the mitochondrial genomes of 19 grouper species with canonical genome organization (genome structure shown in Figure 1A).
| Gene or region | Size | Codon | Intergenic nucleotides* | Strand | ||
|---|---|---|---|---|---|---|
| Nucleotide (bp) | Amino acid | Start | Stop | |||
| tRNA-Phe | 69 to 71 | 0 | H | |||
| 12S rRNA | 952 to 961 | 0 | H | |||
| tRNA-Val | 70 to 73 | -1 to 2 | H | |||
| 16S rRNA | 1695 to 1722 | 0 to 20 | H | |||
| tRNA-Leu(UUR) | 73 to 76 | 0 | H | |||
| ND1 | 975 | 324 | ATG | TAA/TAG | 0 | H |
| tRNA-Ile | 69 to 70 | 4 to 6 | H | |||
| tRNA-Gln | 71 | -2 to 0 | L | |||
| tRNA-Met | 69 to 70 | -1 to 0 | H | |||
| ND2 | 1045 to 1048 | 348 to 349 | ATG | T/TA/TAA/TAG | 0 | H |
| tRNA-Trp | 70 to 72 | -2 to 4 | H | |||
| tRNA-Ala | 69 | 1 to 2 | L | |||
| tRNA-Asn | 73 to 75 | -2 to 1 | L | |||
| OL | 33 to 43 | -1 to 0 | - | |||
| tRNA-Cys | 66 to 68 | 0 to 27 | L | |||
| tRNA-Tyr | 70 to 71 | 0 to 1 | L | |||
| COI | 1551 | 516 | GTG | TAA/TAG | 1 | H |
| tRNA-Ser(UCN) | 71 | 0 to 3 | L | |||
| tRNA-Asp | 72 to 73 | 2 to 8 | H | |||
| COII | 691 | 230 | ATG | T | 0 to 8 | H |
| tRNA-Lys | 72 to 74 | 0 to 1 | H | |||
| ATPase8 | 168 | 55 | ATG | TAA | 0 to 1 | H |
| ATPase6 | 684 | 227 | ATG/CTG/TTG | TAA | -10 | H |
| COIII | 785 to 786 | 261 | ATG | TA/TAA | -1 | H |
| tRNA-Gly | 70 to 73 | -1 to 0 | H | |||
| ND3 | 349 | 116 | ATG | T | -2 to 0 | H |
| tRNA-Arg | 69 | 0 | H | |||
| ND4L | 297 | 98 | ATG | TAA | 0 | H |
| ND4 | 1381 | 460 | ATG | T | -7 | H |
| tRNA-His | 69 to 73 | 0 | H | |||
| tRNA-Ser(AGY) | 70 to 82 | 0 to 10 | H | |||
| tRNA-Leu(CUN) | 73 to 74 | 0 to 10 | H | |||
| ND5 | 1836 to 1839 | 611 to 612 | ATG | TAA/TAG | -2 to 0 | H |
| ND6 | 522 | 173 | ATG | TAA/TAG | -4 | L |
| tRNA-Glu | 69 to 70 | 0 | L | |||
| Cytb | 1141 to 1144 | 380 | ATG | T | 2 to 9 | H |
| tRNA-Thr | 72 to 74 | 0 to 1 | H | |||
| tRNA-Pro | 70 to 71 | -1 to 4 | L | |||
| Control Region | 721 to 1264 | 0 | - | |||
* Negative numbers indicate overlapping nucleotides between adjacent genes.
The nucleotide compositions of the 22 mt genomes are shown in Table 1. Strand asymmetry of nucleotide composition is usually described by AT and GC skews. The mt genome AT-skews of these groupers are barely above zero, while the GC-skews are all negative. These results indicate that the A content is only slightly higher than T, whereas C is considerably more prevalent than G. Such skews towards a particular nucleotide are attributed to differential mutational pressures imposed on the L- and H-strands [23], resulting from the asymmetric replication of mtDNA [24,25].
Novel genome organizations and their plausible evolutionary mechanisms
Two novel types of mt genome organizations were found among the groupers, each containing an additional tRNA. Both species of genus Variola have an additional intergenic spacer of around 100 bp between tRNA-Val and 16S rRNA. This spacer can fold into the conventional cloverleaf structure of tRNA and includes an anticodon of tRNA-Ile. This additional tRNA-Ile shares no meaningful sequence similarity with the canonical tRNA-Ile between ND1 and tRNA-Gln. Furthermore, it has an anticodon AAU different from the GAU of canonical tRNA-Ile. However, the additional tRNA-Ile shares a high sequence identity with the adjacent tRNA-Val (83% in V . albimarginata and 76% in V . louti ), and in both species it is encoded by the H-strand. Thus, we speculate the new tRNA-Ile may have evolved from tRNA-Val via a duplication event due to slipped strand mispairing during mtDNA replication. Nucleotide mutations of the anticodon from TAC to AAT then switched the redundant tRNA-Val (anticodon UAC) to a new tRNA-Ile (anticodon AAU). This novel mt genome organization is a genus-specific characteristic of Variola and distinct from other grouper genera (Figure 1B).
Similar intergenic sequences between tRNA-Val and 16S rRNA have also been observed in another clade of Serranidae fishes, fairy basslets in subfamily Anthiinae [26]. Although the intergenic sequences of Anthiinae do not reveal cloverleaf secondary structure of tRNA, they still can be folded into stem-loop structures and were also thought to have been retained from the ancestral tRNA-Val [26]. Therefore, this can be regarded as a case of convergent evolution in which similar mt genome rearrangements have occurred independently in two separate lineages of Percoidei fishes.
The other novel mt genome organization was observed in C . argus . This species has an additional tRNA-Asp on the L-strand inserted in the middle of the CR (Figure 1C). This tRNA gene shares 97% sequence identity and the same anticodon GUC with the canonical H-strand coded tRNA-Asp found between tRNA-Ser (UCN) and COII. This additional copy is therefore likely the result of a long-range duplication event, which could be explained by illicit priming of mitochondrial replication by tRNAs and the resultant integration of tRNA genes in the CR [27,28]. Although the canonical tRNA-Asp is distant from the H-strand replication origin (OH) in the CR, at the start of replication the tRNA encoded by this gene could illegitimately prime DNA synthesis of the H-strand which would lead to an extra copy of the tRNA gene within the CR on the L-strand.
The gene contents of fish mt genomes are generally stable with variation typically the result of gene duplications [29,30]. What mt genome rearrangements have been seen in fishes are typically gene translocations [13,31,32], resulting from tandem duplication of gene regions. These are then often followed by deletions of the functionally redundant duplicated gene; however, there are examples of tRNAs retained in the mt genome after duplication. Beyond the additional tRNA-Ile and tRNA-Asp described here, the non-canonical tRNAs observed thus far in fish mt genomes include the extra tRNA-Ser downstream of ND5 in seabass Morone saxatilis [33], the extra tRNA-Met in the tRNA-IQM cluster in Pampus species [10] and the pseudo tRNA-Met in the same position in parrotfish Chlorurus sordidus [9], the additional tRNA-Asn and pseudo tRNA-Ala in the tRNA-WANCY cluster in polar cod Boreogadus saida [7], the duplicated tRNA-Thr and tRNA-Pro in the CR of Antarctic notothenioids [12]. While new tRNAs appear to be scattered through different mt regions, most in fact remain located at the rearrangement hotspots (tRNA clusters and CR) in the fish mt genome.
Protein-coding genes
The cumulative lengths of the groupers’ mt protein coding genes range from 11,408 to 11,417 bp. This accounts for 69.5% to 67.3% of the mt genome’s total length. Animal mtDNA is generally very compact and contains some overlapping regions between the adjacent protein-coding genes. Among the 13 protein-coding genes in these groupers, we found a 10 bp overlap between ATP8 and ATP6, 7 bp between ND4L and ND4, 4 bp between ND5 and ND6, and 1 bp between ATP6 and COIII (Table 2).
Most of the grouper mt protein-coding genes begin with the typical start codon ATG. As in many other metazoans [16], the grouper COI gene differs in that it begins with GTG. Additionally, while most teleost fishes retain ATG as the ATP6 start codon, this is CTG or TTG in groupers outside of the basal genus Plectropomus [19,34,35]. This latter difference is not common among teleosts, and could be considered a linage-specific characteristic of the more derived epinephelid clade (clade A in Figure 2) with the exceptions of E . bruneus , E . moara , and H . septemfasciatus .
Figure 2. Phylogenetic tree of 22 groupers in family Epinephelidae and 29 representatives from other families in suborder Percoidei.

A species from the suborder Labrodei family Labridae, Pseudolabrus sieboldi, was selected as outgroup. Congruent tree topology was inferred from partitioned Maximum likelihood and Bayesian analyses using the concatenated nucleotide sequences of 12 mitochondrial protein-coding genes (excluding ND6). The Bayesian posterior probability values (top) and bootstrap values (bottom) are labeled at branch nodes. Branch length information from the Bayesian tree is shown. NCBI RefSeq or GenBank accession number of each species was listed on the right of the species name. Clade A indicates the derived epinephelid clade whose ATP6 start codon is not ATG but CTG or TTG, different from most other teleosts and basal groupers.
Unlike the start codon, variation is more common in the termination codons of mt genes [16]. Among the groupers this was seen in the protein-coding genes COII, ND2, ND3, ND4, and Cyt b all of which terminate with the incomplete stop codon T or TA (Table 2). These stop codons are completed with the addition of 3' adenine residues to the mRNA by post-transcriptional polyadenylation [36,37].
Calculating the nucleotide base frequency at each codon position across all 13 mt protein-coding genes (Table 3), T was found over-represented at the second codon position (P < 0.01). Since the triplet codons with T at the second positions all code for hydrophobic residues this observed bias is explained by the high proportion of hydrophobic residues among mt transmembrane proteins. The 12 protein genes encoded on H-strand share a strong anti-G bias (lower than 8%, P < 0.01) at the third codon positions typical in vertebrates [38]. However, ND6 encoded on the L-strand exhibits the opposite trend with a strong anti-C bias (lower than 10%, P < 0.01) at the third codon positions. This may suggest that the well-known mitochondrial anti-G bias is actually strand-specific rather than a common feature for all mt protein genes. Moreover, among groupers this bias is primarily at the third codon positions, likely due to the less stringent selective pressure on synonymous mutations at this position.
Table 3. Nucleotide compositions of different regions in grouper mitochondrial genomes.
| Strand | Gene/Region | Position | Nucleotide composition (%) |
||||
|---|---|---|---|---|---|---|---|
| A | T | G | C | A+T | |||
| H | Protein-coding gene | 1st codon | 26.3±0.4 | 21.0±0.4 | 24.8±0.4 | 27.9±0.5 | 47.3±0.8 |
| 2nd codon | 18.3±0.1 | 40.5±0.2 | 13.2±0.1 | 28.0±0.2 | 58.8±0.3 | ||
| 3rd codon | 35.5±1.3 | 24.4±2.5 | 6.1±1.1 | 34.3±2.4 | 59.9±3.8 | ||
| tRNA gene | 30.6±0.6 | 25.6±0.4 | 21.1±0.6 | 22.7±0.6 | 56.2±1.0 | ||
| rRNA gene | 32.2±0.5 | 21.5±0.5 | 20.9±0.3 | 25.4±0.7 | 53.7±1.0 | ||
| Control region | 34.5±1.7 | 32.9±2.0 | 13.8±1.1 | 18.8±2.5 | 67.4±3.7 | ||
| L | Protein-coding gene (ND6) | 1st codon | 15.1±1.3 | 30.5±1.5 | 42.9±1.8 | 11.5±1.2 | 45.6±2.8 |
| 2nd codon | 11.8±0.7 | 42.4±1.6 | 23.3±1.1 | 22.5±1.7 | 54.2±2.2 | ||
| 3rd codon | 21.8±4.9 | 41.8±3.8 | 27.8±3.0 | 8.6±2.8 | 63.6±8.7 | ||
| tRNA gene | 25.6±0.6 | 31.9±0.6 | 25.7±0.7 | 16.9±0.4 | 57.5±1.2 | ||
Numbers in the table are means and standard deviations of 22 grouper species.
Ribosomal and transfer RNA genes
Comparing rRNA among groupers, 12S rRNAs range in length from 952 to 961 bp, and 16S rRNAs from 1,695 to 1,722 bp. The two rRNAs, both encoded by the H-strand, indicate moderate nucleotide compositional bias with A% > C% > T% > G%(P < 0.01) (Table 3). This compositional bias is similar to, but not as strong as, that observed in H-strand coded protein-coding genes. Thus, this confirms that the biased nucleotide composition is a strand-specific characteristic of mtDNA. Similar phenomena have been found in the mt genomes of other fishes [39] and mammals [40], which is thought to be correlated with asymmetric replication of the L- and H-strand [40]. The reduced compositional bias of rRNA compared to protein-coding genes is likely due to its structural constraints, particularly in stem regions [41].
Except for the two Variola species and C . argus , the mt genomes of the remaining 19 grouper species all contain 22 tRNAs, 14 of which are encoded by the H-strand and eight encoded by L-strand. Among the 22 tRNAs, two forms of tRNA-Leu (UUR and CUN) and tRNA-Ser (UCN and AGY) were observed in all groupers (Table 2). These tRNAs range in size from 66 to 76 bp, and in some their end sequences overlap with neighboring tRNA or rRNA. All tRNAs except tRNA-Ser (AGY) could be folded into the typical clover-leaf secondary structure as determined by tRNAscan-SE. In all groupers tRNA-Ser (AGY) was found to lack the entire dihydrouridine arm, reducing its secondary structure to a 'truncated cloverleaf', resembling that of most metazoans [42].
Non-coding regions
The non-coding regions in the grouper mtDNA include the CR, the OL, and several short intergenic spacers that range in size from 1 to 27 bp. Located between tRNA-Pro and tRNA-Phe, CR is the largest of these non-coding regions. It is AT rich (average 67.4%) and ranges in size among groupers from 721 to 1264 bp. The grouper CR consists of several termination-associated sequences (TAS) comprising the extended termination-associated sequences (ETAS) region. This is followed by six conserved sequence blocks (CSB-F to CSB-A) making up the central conserved domain (CCD), then three additional conserved sequence blocks (CSB-1 to CSB-3) located downstream from the central conserved domain (Figure 3). However, none of these sequence elements are found in the CR of C . argus which shares no significant sequence similarity with other grouper species and contains an extra tRNA-Asp insertion in the middle of the region.
Figure 3. Schematic structures of mitochondrial control regions.
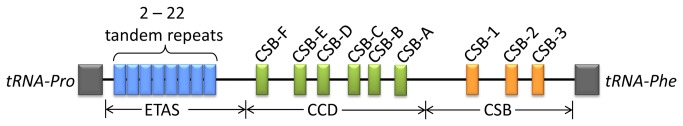
ETAS, extended termination associated sequences; CCD, central conserved domain; CBSs, conserved sequence blocks. This figure does not show the structure for the CR of Cephalopholis argus.
The repeat-rich ETAS region is the most variable part of the CR, with 2 to 22 tandem repeats depending on the species. This region also includes repetitive elements that range in length from 15 to 134 bp, each containing a conserved TAS motif TACAT and the reversed complement cTAS motif ATGTA. The TAS motif can pair with the cTAS motif, leading to formation of stable hairpin loops which presumably function as sequence-specific signals for termination of mtDNA replication [43]. Repeats in the ETAS region occur in diverse fish taxa [44–46], and the distribution of number of repeated copies suggests that they are randomly added and deleted. The variation in tandem repeats observed in the ETAS region likely evolved via the process of illegitimate elongation [46] or imprecise termination during the replication of the circular mtDNA, and are probably mediated by the mechanism of slipped-strand mispairing [47].
We aligned the conserved regions of CRs in the 21 grouper species, and identified six conserved sequence blocks (CSB-F to CSB-A) with high sequence similarity to the CSBs of other fishes [48]. While five conserved sequence blocks (CSB-F to CSB-B) in the central conserved domain have been reported in mammalian mt CRs [49], only three (CSB-F to CSB-D) are typically found in teleosts [5,50,51]. Looking at these blocks in more detail, first CSB-F demarcates the boundary of ETAS region and central conserved domain. Although this has little sequence similarity with the mammalian CSB-F, both are equivalently positioned within the mt genome CR. Next, the grouper CSB-E has the identifying GTGGG-box of teleost CSB-E [50]. Both CSB-F and CSB-E share 85% sequence identity among these groupers. The CSB-D has a critical role in maintaining the proper regulatory functions of CR and is recognized as the most universally conserved CR segment among teleost fishes [50], which was also seen among the groupers (95% sequence identity). The remaining three CSBs, CSB-C, CSB-B, and CSB-A, are the least conserved (81%–84%), although they can still be identified by the consensus sequences.
We determined the consensus sequences of the six CSBs in the central conserved domain of grouper CR (Figure S1), which are:
CSB-F, GCACAGTAAGAACCTACCAA;
CSB-E, GACAATAATTGTGGGGGT;
CSB-D, TATTCCTGGCATTTGGTTCCTACTTCAGGGCCA;
CSB-C, CTTTCATTGACGCTTGCATAAGTTAATG;
CSB-B, CATTCGACTCGTTACCCA;
CSB-A, TCCAGAGGGTAGGGGGTT.
In addition, we also identified three conserved sequence blocks downstream from the central conserved domain of grouper CR (Figure S1 and Table 4). Comparing these to available CSB sequences from representative teleost fishes (Table 4) we identified the following key features: CSB-1 has an A/T rich region followed by a conserved GATACA motif. Unlike those of higher vertebrates, teleost CSB-2 is the most conserved among the three downstream CSBs and is characterized by a poly-C stretch separated by TA or TTA. CSB-3 contains a sequence of three or four As followed by a poly-C stretch. These CSBs are thought to be involved in the formation of proper RNA primer for mtDNA replication and play a role in the switch from RNA to DNA synthesis that commences at OH [52,53].
Table 4. Conserved sequence blocks in the mitochondrial control region of some teleost representatives.
| Species | CSB-1 | CSB-2 | CSB-3 | reference |
|---|---|---|---|---|
| three Branchiostegus species | TT-CTTAATGCATACTCTTATTGA-GGTG | A-AAACCCCCCTACCCCC | GAAAACCCCCC-G-AAACA | [68] |
| Chanos chanos | NA | AAACCCCCCCCTCCCCCCA | TGTTAAACCCCCAAAACCA | [69] |
| Collichthysniveatus | AT-TACTGTATTTTAGTGCATAA | TAGACCCCCCTACCCCCC | T-AA-C-CCTAAAAACA | [70] |
| Crossostoma lacustre | CTATGTATGTAGAATGAGCATAA | ACAAACCCCCCTACCCCCCT | TGCTCAAACCCCGAAACCA | [71] |
| Cyprinids (different ploidy level) | ATT-AATTAATG-T-GCAGGACA-TA | CAAA-CCCCCCTACCCCC | TGTCAAACCCGAAACCA | [51] |
| Cyprinella spiloptera | AGGTTAATGATTATAAGACATAA | CAAACCCCCTTACCCCC | TGTCAAACCCCGAAAGCA | [72] |
| Dascyllus trimaculatus | CACTACTGTCTTCCCGGACATACA | TAAAACCCCCCTACCCCCCT | TGAAACCCCCCGAAAACA | [73] |
| Dicentrarchus labrax | TTTATCGTAAGTGACATAGGTAA | AATTTCCCCCCCTACCCCCCC | TGCTCAAAATAAATCCCCCTAAGAAAAAGC | [74] |
| Larimichthys crocea , L . polyactis | ATTT-AAGTATTC-AGTGCATTA | TAGACCCCCCTACCCCCC | TAA--CCC-TAAAAACA | [75] |
| Oryzias latipes | ATACAGACCTTGTTGACAAG | CAAACCCCCCTACCCCC | TGCAAACCCCCCGGAAACA | [76] |
| Pampus sp., Trichiurus japonicas | ATAACTGATATCAAGAGCATAA | TA--CCCCCCTACCCCCC | T--AAACCC-----AAACA | [77] |
| Pleuragramma antarcticum | TTCTGGGGCATAA | AACCCCCCCCCACCCCC | NA | [12] |
| Rastrelliger brachysoma | ATATAAGGATATCATGAGCATAA | TAAACCCCCCTACCCCC | TGCAAACCCCCCGGAAA | [78] |
| Rivulus marmoratus | ATAACTGATATCACGGGCATATC | TAGACCCCCCTACCCCCCT | CATTACAAACTTAAACCA | [79] |
| Scomber colias , S. japonicas | ACATTTTCCTCGCATAA | GTCAAACCCCCCCACCCCCC | CTGCAAACCCCCCGGGAAACA | [80] |
| 22 groupers in family Epinephelidae | CATAACTGATTTCAAGAACATAA | TAAACCCCCCTACCCCCC | TGTAAACCCCCCGGAAACA | this study |
Dashed lines indicate the unconserved nucleotides, i.e. substitutions, insertions, or deletions.
The OL non-coding region in groupers is formed on the H-strand located in a cluster of five tRNA genes (Trp, W; Ala, A; Asn, N; Cys, C; Tyr, Y), the so called WANCY region. The OL region region is a short stretch of 33 to 43 nucleotides with a predicted stable stem-loop secondary structure featuring a GC-rich stem and T-rich loop. It is priming of this latter poly-T site that is thought to initiate L-strand synthesis [54]. Groupers share two common features of the vertebrate OL, a pyrimidine-rich 5’ flanking region of the stem, and a conserved motif (5’-GCCGG-3’) found at the base of the stem within the tRNA-Cys. Both may be associated with accuracy and efficiency of DNA replication at OL as suggested by study of the human mt genome [54].
Sequence variation and evolutionary rates of different grouper mt genome regions
While the mt genome as a whole generally evolves faster than the nuclear genome, the extent of interspecific sequence variation differs among mt genes. To compare the sequence variability of different regions in grouper mt genomes we calculated the mean of the pairwise sequence identities for each mt gene type and non-coding region among the 22 species. Overall, the most conserved gene types are rRNA (89.2±4.2%) and tRNA (88.4±3.8%), protein-coding genes present moderate variation (82.6±3.7%), and as expected, the non-coding CR (59.1±9.1%) are the most divergent (P < 0.01).
Except for tRNA-Val and tRNA-Ser (AGY), rRNAs and the remaining tRNAs show high levels of sequence similarity (sequence identity > 83%) among the investigated species (Figure 4A). The conservation of rRNAs and tRNAs is presumably required for the formation of their secondary structures and tertiary interactions. This functional constraint would explain their high sequence identities despite the high mutation rate in mtDNA. The metazoan tRNA-Ser (AGY) generally exhibits considerable sequence variability [42], which may be explained by taking into account that its dihydrouridine arm is replaced by a variable loop, likely weakening structural constraints.
Figure 4. Sequence variations among mitochondrial genes.

Genes were ranked by their sequence identity percentages from low to high (left to right). (A) Sequence identities of 22 tRNA genes. (B) NT% (dark grey) and deduced AA% (light grey) of 13 protein-coding genes. Genes were ranked by the AA%.
Sequence identity comparisons of both nucleotides (NT %) and their deduced amino acids (AA %) (Figure 4B) from the 13 protein-coding genes show these to be less strictly conserved than the rRNA and tRNA sequences. Divergence varied among these genes, with the three cytochrome oxidase subunits, COI, II, and III, being the most conserved (AA > 96%, NT > 85%), while NADH dehydrogenase subunit 6 (ND6) of complex I and ATP synthase subunit 8 (ATP8) are the most divergent (AA< 81%, NT < 80%). The difference in divergence among mt protein-coding genes can be explained by the presence of all the functional groups of complex IV in the mt electron transport chain, including COI, II, and III, are mitochondrially encoded, whereas most of the complex I and ATP synthase subunits are instead encoded by nuclear genes [27,55].
Looking at the divergence rates of these genes allows us to compare their utility for DNA barcoding. Though COI is commonly used for DNA barcoding [56], the low divergence rate seen in this gene among groupers suggests it may not be able to clearly distinguish between closely related species. As the optimal mt genes for DNA barcoding can vary between taxonomic groups, our analysis of pairwise sequence identities of the 13 protein-coding grouper mt genes leads us to suggest ND2 as a better candidate for barcoding identification in groupers. ND2 has a higher percentage of variable sites (52.2% among 22 grouper species) than COI (36.9%) and can therefore better discriminate between newly derived species. Additionally, the smaller size of ND2 (1046 bp) compared to COI (1551 bp) makes it easier to completely sequence using only a pair of primers. Thus, a complete gene instead of a partial gene can be used for barcoding.
In order to analyze the level of selective pressures imposed on different protein-coding genes, we calculated the ratio of the rate of non-synonymous substitutions (Ka) to the rate of synonymous substitutions (Ks) (Figure 5). Our results show that all 13 genes have the pairwise comparative Ka/Ks ratios smaller than 1, indicating a signal of strong purifying selection against deleterious mutations on all mt protein genes in these groupers. However, the average Ka/Ks ratio (0.019 to 0.26) differs substantially among individual genes, suggesting varying functional constraints. Among these genes the ratio is highest in ND6 (0.26), implying this is under the least purifying selective pressure.
Figure 5. Average Ka /Ks ratios of 13 protein-coding genes.

Ka /Ks is the ratio of non-synonymous substitutions rate (Ka) to synonymous substitutions substitution rate (Ks).
Phylogenetic analyses
Recent phylogenetic analyses based on several mitochondrial and nuclear genes have challenged traditional views of grouper systematics [19,20]. To test these revised classifications on the mt genome level, we performed partition Bayesian and maximum likelihood (ML) analyses using the concatenated nucleotide sequences (10,963 bp) of the 12 H-strand coded mt protein genes. We excluded the ND6 gene as its rapidly evolving sequence may have led to multiple substitutions at some sites and such homoplasy would reduce the resolution of our phylogenetic analysis.
Both phylogenetic analyses yielded congruent tree topologies with strong support on all nodes of concern (Figure 2). They uniformly agreed on the monophyly of Epinephelinae with a bootstrap value of 100% on the ML tree and a posterior probability of 1 on the Bayesian tree. While groupers were traditionally considered as the subfamily Epinephelinae belonging to the family Serranidae, a large-scale phylogenetic study of percomorph assemblages suggested elevating the monophyletic grouper species (traditional Epinephelinae) to familial status as they were not allied with the remaining members of the polyphyletic Serranidae [20]. In our study, the Serranidae species Hypoplectrus gemma clusters together with the family Percidae and they together comprise a sister clade to the grouper species with high support values (100% for Baysian and 81% for ML) (Figure 2), lending further corroboration to the elevation of subfamily Epinephelinae to family Epinephelidae.
In terms of the relationships within the family Epinephelidae, our analyses provide high support (100% for both methods) for the initial divergence of genus Plectropomus (Figure 2). Although only two species are available from the genus Plectropomus , the monophyletic nature of this group [19] allows us to use these species to draw inference for the entire genus. Therefore, our study is indicative of a basal evolutionary status of Plectropomus in the Epinephelidae lineage in agreement with previous studies of molecular and morphological phylogeny [19,34,35]. Craig and Hastings (2007) [19] proposed a taxonomic change placing a clade of Epinephelus species into a new genus designated Hyporthodus based on a set of distinct morphologically characters. This is corroborated by our phylogenetic analyses (100% support for both methods) with the two representatives of the Hyporthodus genus, H . septemfasciatus and H . octofasciatus , branching from the remaining Epinephelus species and grouping with T . dermopterus (Figure 2). Furthermore, our resulting trees show that two monotypic genera Anyperodon and Cromileptus ( A . leucogrammicus and C . altivelis ) are deeply nested within the clade of the genus Epinephelus , and another monotypic genus Aethaloperca ( A . rogaa ) grouped with the species of genus Cephalopholis (Figure 2), which is consistent with prior studies using different genetic markers [19,57]. Although these monotypic genera have their unique morphological features, such derived traits are phylogenetically uninformative for tracing the evolution along a lineage. Our results agree with the taxonomic revision to synonymize Anyperodon and Cromileptus within the genus Epinephelus , and Aethaloperca within genus Cephalopholis , reflecting the monophyletic taxon [19].
Short Mt gene fragments exhibit limitations in resolving complicated phylogenetic relationships in many fish lineages [17]. The added informative sites from longer DNA sequences allow these deeper branches and higher-level relationships to be more fully resolved [18]. Accordingly, more genetic data (e.g. mt genomes) along with extensive taxon sampling is necessary to fully elucidate the phylogenetic relationships among groupers and their higher-level affinities to other percomorph lineages.
Conclusion
We characterized and compared the complete mt genomes of 22 groupers including 17 newly sequenced species. In studying the evolution of their mt genomes we identified three distinct genome organizations, two of which have never previously been reported. These include the addition of a second tRNA-Ile in genus Variola and a second tRNA-Asp in C . argus .
Looking at the constituents of the mt genome in more detail, we identified a lineage-specific modification of the ATP6 start codon to CTG or TTG in the more derived epinephelid clade. Examining the non-coding mt genome CR, all species outside C . argus contained six conserved sequence blocks rather than the three typically found in teleosts. Comparison of the divergence rates among constituents of the mt genome identified rRNA and tRNA as the most conserved gene types, followed by protein-coding genes, and the non-coding OL and CR as the most divergent. Among the protein-coding genes divergence rates varied with COI, II, and III being the most conserved while ND6 and ATP8 are the most divergent.
Applying these new mt genomic sequences to test recent suggested taxonomic reorganization of the groupers, our phylogenetic analyses corroborated the rearrangement of grouper lineages. The results of this work, besides offering insight into the evolution of the grouper mt genome, provides important resources for the further study of grouper species identification, conservation genetics, speciation and other evolutionary biology studies.
Materials and Methods
Sample collection, PCR amplification, and sequencing
Specimens of 17 grouper species were obtained from local fish markets in coastal areas of mainland China and Taiwan. Species identifications were performed according to FAO Groupers of the World [1,21] and tissue samples (mostly dorsal muscle) were collected. All experiments were conducted in accordance with the guidelines approved by the Institutional Animal Care and Use Committee at the Xiamen University. Total genomic DNA was then isolated using standard phenol–chloroform extraction and ethanol precipitation methods. Contiguous and overlapping segments of the grouper mt genomes were amplified using a set of 16 primer pairs and then sequenced. The primers, PCR conditions, and sequencing reactions were based on the previously described methods in Zhuang et al. [58] with slight modifications.
Mitochondrial genome sequence assembly and gene annotation
Sequencing results were first manually proofread and edited using ChromasPro v.1.42 (Technelysium) then the 17 mt genomes were assembled using Sequencher v.4.7 (Gene Codes Corp.). These complete mt genome sequences have been deposited in GenBank under the Accession numbers listed in Figure 2 and Table S1. Protein-coding and rRNA genes were identified by BLAST searches (http://www.blast.ncbi.nlm.nih.gov/Blast.cgi) and annotated based on alignments with the mt genomes of closely related species found in the GenBank database. Most of the tRNA genes and their secondary structures could be predicted with tRNAscan-SE [59]. However, both the tRNA-Ser (AGY) gene and V . louti ’s extra copy of tRNA-Ile gene instead were identified by inspecting anti-codon sequences and their tRNA-like secondary structure because they cannot form perfect clover-leaf structure and therefore were not detected by the computer program. These sequencing and annotation results were then combined to map the organization of each species’ mt genome (Figure 1).
Sequence analyses
Mt genomes were analyzed for 22 grouper species, the 17 sequenced in this study, and 5 species available from GenBank (Accession numbers in Figure 2 and Table S1). Nucleotide compositions were calculated using the program DNASTAR (DNASTAR Inc.) while AT and GC-skew were each computed using the formulas (A-T)/ (A+T) and (G-C)/ (G+C) [38]. Tandem Repeat Finder was used to identify the repetitive sequences in the control region [60]. Pairwise sequence identities for entire mt genomes and each gene type and Ka/Ks ratios for protein-coding genes were calculated using the program MEGA 5.0 [61]. We tested for significant differences in sequence variability of different regions using a one-way analysis of variance (ANOVA).
Phylogenetic analyses
At the time of this study, complete mt genome sequences were available in GenBank from representatives of 27 families in the suborder Percoidei. We utilized all available species from the families Epinephelidae, Percidae and Serranidae, and selected one species from each remaining family in our phylogenetic analyses. In total, 51 Percoidei species (Figure 2) and the outgroup species Pseudolabrus sieboldi (Labrodei, Labridae; GenBank Accession Number: AP006019) were included.
The concatenated sequences of 12 protein-coding genes on mt H-stand were aligned with codon constraint using CLUSTAL X [62] with minor manual adjustments (Figure S2). Individual gene sequences were also aligned and jModelTest was used to infer the best evolution model of nucleotide substitution [63]. Likelihood ratio tests were conducted to compare different models and the best selected based on both Akaike information criterion (AIC) and Bayesian information criterion (BIC). Among the 12 genes, GTR+I+G was selected as the best model for ND1, ND2, ND4, ND4L, ND5, ATP6, and ATP8, TVM+I+G was selected for COI, COIII, and Cytb, and TrN+I+G was selected for COII and ND3. The 12 genes were split into three model-defined partitions which were used in the following phylogenetic analyses.
A partitioned Bayesian phylogenetic analysis was performed using MrBayes v. 3.1.2 [64]. Two independent analyses were run with Markov chain Monte Carlo (MCMC) sampling with four chains. Trees and parameters were sampled every 250 generations over a total of three million generations, with the first 10% of the samples discarded as burn-in. Acceptable convergence to the stationary distribution was checked using Tracer 1.4.1 [65]. A majority consensus tree with Bayesian posterior probabilities (BPP) was computed. Partitioned maximum likelihood phylogenetic analysis was performed with GARLI v.0.951 [66]. Each analysis was run with 100 replicates using a random starting tree. Search replicates were evaluated by likelihood score, and only that with the best score was retained. Node supports of the best tree were evaluated with 1000 bootstrap replications, which were then processed by Sumtree [67] to generate a majority consensus tree with bootstrap values.
Supporting Information
Sequence alignment of the central conserved domains and the conserved sequence blocks in the mitochondrial control regions. Dashed lines indicate gaps introduced by the alignment. Asterisks indicate nucleotide identity in the column. The conserved sequence blocks are shown in red and boxed. This excludes the grouper species Cephalopholis argus .
(PDF)
Sequence alignment of the 12 mitochondrial protein-coding genes (excluding ND6) used for phylogenetic analyses. Multiple sequence alignment was generated using CLUSTAL X [62] with minor manual adjustments. The manual modifications are highlighted in yellow.
(DOCX)
GenBank accession numbers for mitochondrial genome sequences of the Percoidei species used in phylogenetic analysis.
(DOCX)
Acknowledgments
We are grateful to Kevin Bilyk, Lauren Fields and two anonymous reviewers for their constructive comments on this manuscript.
Funding Statement
This study was supported by Program for New Century Excellent Talents in University 2012 and National Basic Research Special Foundation of China (2013FY110700). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Craig MT, Sadovy YJ, Heemstra PC (2011) Groupers of the World: A Field and Market Guide. Grahamstown, South Africa: National Inquiry Services Centre. [Google Scholar]
- 2. Gilles A, Miquelis A, Quignard J-P, Faure É (2000) Molecular phylogeography of western Mediterranean dusky grouper Epinephelus marginatus . C R Académie SciencesIIISciences Vie 323: 195-205. PubMed: 10763438. [DOI] [PubMed] [Google Scholar]
- 3. Han J, Lv F, Cai H (2011) Detection of species-specific long VNTRs in mitochondrial control region and their application to identifying sympatric Hong Kong grouper (Epinephelus akaara) and yellow grouper (Epinephelus awoara). Mol Ecol Resour 11: 215-218. doi:10.1111/j.1755-0998.2010.02911.x. PubMed: 21429126. [DOI] [PubMed] [Google Scholar]
- 4. Zhu ZY, Yue GH (2008) The complete mitochondrial genome of red grouper Plectropomus leopardus and its applications in identification of grouper species. Aquaculture 276: 44-49. doi:10.1016/j.aquaculture.2008.02.008. [Google Scholar]
- 5. Liu M, Li JL, Ding SX, Liu ZQ (2013) Epinephelus moara: a valid species of the family Epinephelidae (Pisces: Perciformes). J Fish Biol 82: 1684-1699. doi:10.1111/jfb.12112. PubMed: 23639162. [DOI] [PubMed] [Google Scholar]
- 6. Boore JL (1999) Animal mitochondrial genomes. Nucleic Acids Res 27: 1767-1780. doi:10.1093/nar/27.8.1767. PubMed: 10101183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Breines R, Ursvik A, Nymark M, Johansen SD, Coucheron DH (2008) Complete mitochondrial genome sequences of the Arctic Ocean codfishes Arctogadus glacialis and Boreogadus saida reveal oriL and tRNA gene duplications. Polar Biol 31: 1245-1252. doi:10.1007/s00300-008-0463-7. [Google Scholar]
- 8. Ponce M, Infante C, Jiménez-Cantizano RM, Pérez L, Manchado M (2008) Complete mitochondrial genome of the blackspot seabream, Pagellus bogaraveo (Perciformes: Sparidae), with high levels of length heteroplasmy in the WANCY region. Gene 409: 44-52. doi:10.1016/j.gene.2007.11.004. PubMed: 18191919. [DOI] [PubMed] [Google Scholar]
- 9. Mabuchi K, Miya M, Satoh TP, Westneat MW, Nishida M (2004) Gene rearrangements and evolution of tRNA pseudogenes in the mitochondrial genome of the parrotfish (Teleostei: Perciformes: Scaridae). J Mol Evol 59: 287-297. doi:10.1007/s00239-004-2621-z. PubMed: 15553084. [DOI] [PubMed] [Google Scholar]
- 10. Cui Z, Liu Y, Li CP, Chu KH (2011) Species delineation in Pampus (Perciformes) and the phylogenetic status of the Stromateoidei based on mitogenomics. Mol Biol Rep 38: 1103-1114. doi:10.1007/s11033-010-0207-y. PubMed: 20574712. [DOI] [PubMed] [Google Scholar]
- 11. Inoue JG, Miya M, Tsukamoto K, Nishida M (2001) Complete mitochondrial DNA sequence of the Japanese anchovy Engraulis japonicus. Fish Sci 67: 828-835. doi:10.1046/j.1444-2906.2001.00329.x. [Google Scholar]
- 12. Zhuang X, Cheng CH (2010) ND6 gene "lost" and found: evolution of mitochondrial gene rearrangement in Antarctic notothenioids. Mol Biol Evol 27: 1391-1403. doi:10.1093/molbev/msq026. PubMed: 20106908. [DOI] [PubMed] [Google Scholar]
- 13. Inoue JG, Miya M, Tsukamoto K, Nishida M (2003) Evolution of the deep-sea gulper eel mitochondrial genomes: large-scale gene rearrangements originated within the eels. Mol Biol Evol 20: 1917-1924. doi:10.1093/molbev/msg206. PubMed: 12949142. [DOI] [PubMed] [Google Scholar]
- 14. Miya M, Satoh TP, Nishida M (2005) The phylogenetic position of toadfishes (order Batrachoidiformes) in the higher ray-finned fish as inferred from partitioned Bayesian analysis of 102 whole mitochondrial genome sequences. Biol J Linn Soc 85: 289-306. doi:10.1111/j.1095-8312.2005.00483.x. [Google Scholar]
- 15. Moritz C, Dowling T, Brown W (1987) Evolution of animal mitochondrial DNA: relevance for population biology and systematics. Annu Rev Ecol Syst 18: 269-292. doi:10.1146/annurev.es.18.110187.001413. [Google Scholar]
- 16. Wolstenholme DR (1992) Animal mitochondrial DNA: structure and evolution. Int Rev Cytol 141: 173-216. doi:10.1016/S0074-7696(08)62066-5. PubMed: 1452431. [DOI] [PubMed] [Google Scholar]
- 17. Stepien CA, Kocher TD (1997) Molecules and morphology in studies of fish evolution. Mol Sys Fishes: 1-11. [Google Scholar]
- 18. Miya M, Nishida M (2000) Use of mitogenomic information in teleostean molecular phylogenetics: a tree-based exploration under the maximum-parsimony optimality criterion. Mol Phylogenet Evol 17: 437-455. doi:10.1006/mpev.2000.0839. PubMed: 11133198. [DOI] [PubMed] [Google Scholar]
- 19. Craig MT, Hastings PA (2007) A molecular phylogeny of the groupers of the subfamily Epinephelinae (Serranidae) with a revised classification of the Epinephelini. Ichthyol Res 54: 1-17. doi:10.1007/s10228-006-0367-x. [Google Scholar]
- 20. Smith WL, Craig MT (2007) Casting the Percomorph Net Widely: The Importance of Broad Taxonomic Sampling in the Search for the Placement of Serranid and Percid Fishes. Copeia 1: 35-55. [Google Scholar]
- 21. Heemstra PC, Randall JE (1993) FAO species catalogue. v. 16: Groupers of the world (Family Serranidae, Subfamily Epinephelinae). An annotated and illustrated catalogue of the grouper, Rockcod, Hind, Coral grouper and Lyretail species known to date: FAO.
- 22. IUCN (2012) IUCN 2012. IUCN Red List of Threatened Species. Version 2012.2. Accessed: 31 January 2013: Iucn. [Google Scholar]
- 23. Hassanin A, LéGer N, Deutsch J (2005) Evidence for Multiple Reversals of Asymmetric Mutational Constraints during the Evolution of the Mitochondrial Genome of Metazoa, and Consequences for Phylogenetic Inferences. Syst Biol 54: 277-298. doi:10.1080/10635150590947843. PubMed: 16021696. [DOI] [PubMed] [Google Scholar]
- 24. Clayton DA (1982) Replication of animal mitochondrial DNA. Cell 28: 693–705. doi:10.1016/0092-8674(82)90049-6. PubMed: 6178513. [DOI] [PubMed] [Google Scholar]
- 25. Tanaka M, Ozawa T (1994) Strand asymmetry in human mitochondrial DNA mutations. Genomics 22: 327-335. doi:10.1006/geno.1994.1391. PubMed: 7806218. [DOI] [PubMed] [Google Scholar]
- 26. Smith WML, Smith KR, Wheeler WC (2009) Mitochondrial intergenic spacer in fairy basslets (Serranidae: Anthiinae) and the simultaneous analysis of nucleotide and rearrangement data. Am Mus Novit: 1-10.
- 27. Cantatore P, Gadaleta MN, Roberti M, Saccone C, Wilson AC (1987) Duplication and remoulding of tRNA genes during the evolutionary rearrangement of mitochondrial genomes. Nature 329: 853-855. doi:10.1038/329853a0. PubMed: 3670390. [DOI] [PubMed] [Google Scholar]
- 28. Jacobs HT, Asakawa S, Araki T, Miura K, Smith MJ et al. (1989) Conserved tRNA gene cluster in starfish mitochondrial DNA. Curr Genet 15: 193-206. doi:10.1007/BF00435506. PubMed: 2766382. [DOI] [PubMed] [Google Scholar]
- 29. San Mauro D, Gower DJ, Zardoya R, Wilkinson M (2006) A hotspot of gene order rearrangement by tandem duplication and random loss in the vertebrate mitochondrial genome. Mol Biol Evol 23: 227-234. PubMed: 16177229. [DOI] [PubMed] [Google Scholar]
- 30. Bernt M, Braband A, Schierwater B, Stadler PF (2012) Genetic aspects of mitochondrial genome evolution. Mol Phylogenet Evol (. (2012)) PubMed: 23142697. [DOI] [PubMed] [Google Scholar]
- 31. Kong X, Dong X, Zhang Y, Shi W et al. (2009) A novel rearrangement in the mitochondrial genome of tongue sole, Cynoglossus semilaevis: control region translocation and a tRNA gene inversion. Genome 52: 975-984. doi:10.1139/G09-069. PubMed: 19953125. [DOI] [PubMed] [Google Scholar]
- 32. Miya M, Nishida M (1999) Organization of the mitochondrial genome of a deep-sea fish, Gonostoma gracile (Teleostei: Stomiiformes): first example of transfer RNA gene rearrangements in bony fishes. Mar Biotechnol 1: 416-426. doi:10.1007/PL00011798. PubMed: 10525676. [DOI] [PubMed] [Google Scholar]
- 33. Williams EP, Peer AC, Miller TJ, Secor DH, Place AR (2012) A phylogeny of the temperate seabasses (Moronidae) characterized by a translocation of the mt‐nd6 gene. J Fish Biol 80: 110-130. doi:10.1111/j.1095-8649.2011.03158.x. PubMed: 22220893. [DOI] [PubMed] [Google Scholar]
- 34. Leis JM (1986) Larval development in four species of Indo-Pacific coral trout Plectropomus (Pisces Serranidae Epinephelinae) with an analysis of the relationships of the genus. Bull Mar Sci 38: 525-552. [Google Scholar]
- 35. Ding S, Zhuang X, Guo F, Wang J, Su Y et al. (2006) Molecular phylogenetic relationships of China Seas groupers based on cytochrome b gene fragment sequences. Sci China C 49: 235-242. doi:10.1007/s11427-006-0235-y. PubMed: 16856492. [DOI] [PubMed] [Google Scholar]
- 36. Coucheron DH, Nymark M, Breines R, Karlsen BO, Andreassen M et al. (2011) Characterization of mitochondrial mRNAs in codfish reveals unique features compared to mammals. Curr Genet 57: 213-222. doi:10.1007/s00294-011-0338-2. PubMed: 21484258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ojala D, Montoya J, Attardi G (1981) tRNA punctuation model of RNA processing in human mitochondria. Nature 290: 470-474. doi:10.1038/290470a0. PubMed: 7219536. [DOI] [PubMed] [Google Scholar]
- 38. Perna NT, Kocher TD (1995) Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol 41: 353-358. doi:10.1007/BF01215182. PubMed: 7563121. [DOI] [PubMed] [Google Scholar]
- 39. Ortí G, Petry P, Porto JI, Jégu M, Meyer A (1996) Patterns of nucleotide change in mitochondrial ribosomal RNA genes and the phylogeny of piranhas. J Mol Evol 42: 169-182. doi:10.1007/BF02198843. PubMed: 8919869. [DOI] [PubMed] [Google Scholar]
- 40. Reyes A, Gissi C, Pesole G, Saccone C (1998) Asymmetrical directional mutation pressure in the mitochondrial genome of mammals. Mol Biol Evol 15: 957-966. doi:10.1093/oxfordjournals.molbev.a026011. PubMed: 9718723. [DOI] [PubMed] [Google Scholar]
- 41. Wang HY, Lee SC (2002) Secondary structure of mitochondrial 12S rRNA among fish and its phylogenetic applications. Mol Biol Evol 19: 138-148. doi:10.1093/oxfordjournals.molbev.a004066. PubMed: 11801742. [DOI] [PubMed] [Google Scholar]
- 42. Garey JR, Wolstenholme DR (1989) Platyhelminth mitochondrial DNA: evidence for early evolutionary origin of a tRNA ser AGN that contains a dihydrouridine arm replacement loop, and of serine-specifying AGA and AGG codons. J Mol Evol 28: 374-387. doi:10.1007/BF02603072. PubMed: 2545889. [DOI] [PubMed] [Google Scholar]
- 43. Saccone C, Pesole G, Sbisá E (1991) The main regulatory region of mammalian mitochondrial DNA: structure-function model and evolutionary pattern. J Mol Evol 33: 83-91. doi:10.1007/BF02100199. PubMed: 1909377. [DOI] [PubMed] [Google Scholar]
- 44. Broughton RE, Dowling TE (1997) Evolutionary dynamics of tandem repeats in the mitochondrial DNA control region of the minnow Cyprinella spiloptera . Mol Biol Evol 14: 1187-1196. doi:10.1093/oxfordjournals.molbev.a025728. PubMed: 9402730. [DOI] [PubMed] [Google Scholar]
- 45. Faber JE, Stepien CA (1998) Tandemly Repeated Sequences in the Mitochondrial DNA Control Region and Phylogeography of the Pike-Perches Stizostedion . Mol Phylogenet Evol 10: 310-322. doi:10.1006/mpev.1998.0530. PubMed: 10051384. [DOI] [PubMed] [Google Scholar]
- 46. Buroker NE, Brown JR, Gilbert TA, O’Hara PJ, Beckenbach AT et al. (1990) Length heteroplasmy of sturgeon mitochondrial DNA: an illegitimate elongation model. Genetics 124: 157-163. PubMed: 1968410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fumagalli L, Taberlet P, Favre L, Hausser J (1996) Origin and evolution of homologous repeated sequences in the mitochondrial DNA control region of shrews. Mol Biol Evol 13: 31-46. doi:10.1093/oxfordjournals.molbev.a025568. PubMed: 8583904. [DOI] [PubMed] [Google Scholar]
- 48. Zhang Y, Zhang H, Gao T, Miao Z (2011) Structure of mitochondrial DNA control region and molecular phylogenetic relationship among three flounders of genus Pleuronectes . Biochem Syst Ecol 39: 627-634. doi:10.1016/j.bse.2011.05.008. [Google Scholar]
- 49. Southern ŠO, Southern PJ, Dizon AE (1988) Molecular characterization of a cloned dolphin mitochondrial genome. J Mol Evol 28: 32-42. doi:10.1007/BF02143495. PubMed: 3148740. [DOI] [PubMed] [Google Scholar]
- 50. Lee WJ, Conroy J, Howell WH, Kocher TD (1995) Structure and evolution of teleost mitochondrial control regions. J Mol Evol 41: 54-66. PubMed: 7608989. [DOI] [PubMed] [Google Scholar]
- 51. Guo X, Liu S, Liu Y (2003) Comparative analysis of the mitochondrial DNA control region in cyprinids with different ploidy level. Aquaculture 224: 25-38. doi:10.1016/S0044-8486(03)00168-6. [Google Scholar]
- 52. Clayton DA (1991) Nuclear gadgets in mitochondrial DNA replication and transcription. Trends Biochem Sci 16: 107-111. doi:10.1016/0968-0004(91)90043-U. PubMed: 2057998. [DOI] [PubMed] [Google Scholar]
- 53. Shadel GS, Clayton DA (1997) Mitochondrial DNA maintenance in vertebrates. Annu Rev Biochem 66: 409-435. doi:10.1146/annurev.biochem.66.1.409. PubMed: 9242913. [DOI] [PubMed] [Google Scholar]
- 54. Hixson JE, Wong TW, Clayton DA (1986) Both the conserved stem-loop and divergent 5'-flanking sequences are required for initiation at the human mitochondrial origin of light-strand DNA replication. J Biol Chem 261: 2384-2390. PubMed: 3944140. [PubMed] [Google Scholar]
- 55. Zardoya R, Garrido-Pertierra A, Bautista JM (1995) The complete nucleotide sequence of the mitochondrial DNA genome of the rainbow trout, Oncorhynchus mykiss . J Mol Evol 41: 942-951. PubMed: 8587139. [DOI] [PubMed] [Google Scholar]
- 56. Hebert PD, Cywinska A, Ball SL, deWaard JR (2003) Biological identifications through DNA barcodes. Proc Biol Sci 270: 313-321. doi:10.1098/rspb.2002.2218. PubMed: 12614582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Craig MT, Pondella DJ, Franck JPC, Hafner JC (2001) On the status of the serranid fish genus Epinephelus: evidence for paraphyly based upon 16S rDNA sequence. Mol Phylogenet Evol 19: 121-130. doi:10.1006/mpev.2000.0913. PubMed: 11286497. [DOI] [PubMed] [Google Scholar]
- 58. Zhuang X, Ding S, Wang J, Wang Y, Su Y (2009) A set of 16 consensus primer pairs amplifying the complete mitochondrial genomes of orange-spotted grouper (Epinephelus coioides) and Hong Kong grouper (Epinephelus akaara). Mol Ecol Resour 9: 1551-1553. doi:10.1111/j.1755-0998.2009.02716.x. PubMed: 21564956. [DOI] [PubMed] [Google Scholar]
- 59. Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25: 0955. doi:10.1093/nar/25.5.955. PubMed: 9023104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27: 573-580. doi:10.1093/nar/27.2.573. PubMed: 9862982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tamura K, Peterson D, Peterson N, Stecher G, Nei M et al. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731-2739. doi:10.1093/molbev/msr121. PubMed: 21546353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25: 4876-4882. doi:10.1093/nar/25.24.4876. PubMed: 9396791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Posada D (2008) jModelTest: phylogenetic model averaging. Mol Biol Evol 25: 1253-1256. doi:10.1093/molbev/msn083. PubMed: 18397919. [DOI] [PubMed] [Google Scholar]
- 64. Huelsenbeck JP, Mark P, Ronquist F (2001) MrBayes: Bayesian inference of phylogenetic trees. 3.1. 2. Bioinformatics 17: 754-755. doi:10.1093/bioinformatics/17.8.754. PubMed: 11524383. [DOI] [PubMed] [Google Scholar]
- 65. Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7: 214. doi:10.1186/1471-2148-7-214. PubMed: 17996036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zwickl DJ (2006) Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. Ph.D. dissertation, the University of Texas at Austin. [Google Scholar]
- 67. Sukumaran J, Holder MT (2010) DendroPy: a Python library for phylogenetic computing. Bioinformatics 26: 1569-1571. doi:10.1093/bioinformatics/btq228. PubMed: 20421198. [DOI] [PubMed] [Google Scholar]
- 68. Oh DJ, Oh BS, Jung MM, Jung YH (2010) Complete mitochondrial genome of three Branchiostegus (Perciformes, Malacanthidae) species: Genome description and phylogenetic considerations. Mitochondr DNA 21: 151-159. doi:10.3109/19401736.2010.503241. PubMed: 20958223. [DOI] [PubMed] [Google Scholar]
- 69. Ravago RG, Monje VD, Juinio-Meñez MA (2002) Length and sequence variability in mitochondrial control region of the milkfish, Chanos chanos . Mar Biotechnol 4: 40-50. doi:10.1007/s10126-001-0076-4. PubMed: 14961287. [DOI] [PubMed] [Google Scholar]
- 70. Cheng J, Ma G-q, Song N, Gao T-x (2012) Complete mitochondrial genome sequence of bighead croaker Collichthys niveatus (Perciformes, Sciaenidae): A mitogenomic perspective on the phylogenetic relationships of Pseudosciaeniae. Gene 491: 210-223. doi:10.1016/j.gene.2011.09.020. PubMed: 21989484. [DOI] [PubMed] [Google Scholar]
- 71. Tzeng CS, Hui CF, Shen SC, Huang PC (1992) The complete nucleotide sequence of the Crossostoma lacustre mitochondrial genome: conservation and variations among vertebrates. Nucleic Acids Res 20: 4853-4858. doi:10.1093/nar/20.18.4853. PubMed: 1408800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Broughton RE, Dowling TE (1994) Length variation in mitochondrial DNA of the minnow Cyprinella spiloptera . Genetics 138: 179-190. PubMed: 8001785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chen CA, Ablan MCA, McManus JW, Bell JD, Tuan VS et al. (2004) Variable Numbers of Tandem Repeats (VNTRs), Heteroplasmy, and Sequence Variation of the Mitochondrial Control Region in the Threespot Dascyllus, Dascyllus trimaculatus (Perciformes: Pomacentridae). Zool Stud 43: 803-821. [Google Scholar]
- 74. Cecconi F, Giorgi M, Mariottini P (1995) Unique features in the mitochondrial D-loop region of the European seabass Dicentrarchus labrax . Gene 160: 149-155. doi:10.1016/0378-1119(95)00232-U. PubMed: 7642088. [DOI] [PubMed] [Google Scholar]
- 75. Cui Z, Liu Y, Li CP, You F, Chu KH (2009) The complete mitochondrial genome of the large yellow croaker, Larimichthys crocea (Perciformes, Sciaenidae): unusual features of its control region and the phylogenetic position of the Sciaenidae. Gene 432: 33-43. doi:10.1016/j.gene.2008.11.024. PubMed: 19100818. [DOI] [PubMed] [Google Scholar]
- 76. Hirayama M, Mukai T, Miya M, Murata Y, Sekiya Y et al. (2010) Intraspecific variation in the mitochondrial genome among local populations of Medaka Oryzias latipes . Gene 457: 13-24. doi:10.1016/j.gene.2010.02.012. PubMed: 20193748. [DOI] [PubMed] [Google Scholar]
- 77. Cui Z, Liu Y, Chu KH (2010) Broader pattern of tandem repeats in the mitochondrial control region of Perciformes. Chin J Oceanol Limnol 28: 785-794. doi:10.1007/s00343-010-9091-5. [Google Scholar]
- 78. Jondeung A, Karinthanyakit W (2010) The complete mitochondrial DNA sequence of the short mackerel (Rastrelliger brachysoma), and its phylogenetic position within Scombroidei, Perciformes. Mitochondr DNA 21: 36-47. doi:10.3109/19401731003622529. PubMed: 20331328. [DOI] [PubMed] [Google Scholar]
- 79. Lee JS, Miya M, Lee YS, Kim CG, Park EH et al. (2001) The complete DNA sequence of the mitochondrial genome of the self-fertilizing fish Rivulus marmoratus (Cyprinodontiformes, Rivulidae) and the first description of duplication of a control region in fish. Gene 280: 1-7. doi:10.1016/S0378-1119(01)00765-X. PubMed: 11738812. [DOI] [PubMed] [Google Scholar]
- 80. Catanese G, Manchado M, Infante C (2010) Evolutionary relatedness of mackerels of the genus Scomber based on complete mitochondrial genomes: strong support to the recognition of Atlantic Scomber colias and Pacific Scomber japonicus as distinct species. Gene 452: 35-43. doi:10.1016/j.gene.2009.12.004. PubMed: 20035845. [DOI] [PubMed] [Google Scholar]
- 81. Lohse M, Drechsel O, Bock R (2007) OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet 52: 267-274. doi:10.1007/s00294-007-0161-y. PubMed: 17957369. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequence alignment of the central conserved domains and the conserved sequence blocks in the mitochondrial control regions. Dashed lines indicate gaps introduced by the alignment. Asterisks indicate nucleotide identity in the column. The conserved sequence blocks are shown in red and boxed. This excludes the grouper species Cephalopholis argus .
(PDF)
Sequence alignment of the 12 mitochondrial protein-coding genes (excluding ND6) used for phylogenetic analyses. Multiple sequence alignment was generated using CLUSTAL X [62] with minor manual adjustments. The manual modifications are highlighted in yellow.
(DOCX)
GenBank accession numbers for mitochondrial genome sequences of the Percoidei species used in phylogenetic analysis.
(DOCX)