

Abstract
In fragment-based drug discovery, the weak affinities exhibited by fragments pose significant challenges for screening. Biophysical techniques are used to address this challenge, but there is no clear consensus on which cascade of methods is best suited to identify fragment hits that ultimately translate into bound X-ray structures and provide bona fide starting points for synthesis. We have benchmarked an integrated biophysical approach for fragment screening and validation against Mycobacterium tuberculosis pantothenate synthetase. A primary screen of 1,250 fragments library was performed by thermal shift, followed by secondary screen using one-dimensional NMR spectroscopy (water ligand observed gradient spectroscopy and saturation transfer difference binding experiments) and ultimate hit validation by isothermal titration calorimetry and X-ray crystallography. Our multibiophysical approach identified three distinct binding sites for fragments and laid a solid foundation for successful structure-based elaboration into potent inhibitors.
Keywords: drug design, hot spots, protein–ligand interactions
Fragment-based small molecule design is now established in many industrial and academic laboratories, but the paradigm for the discovery of lead compounds for drug development and of new chemical tools for biology is still evolving. The approach involves the identification of low molecular weight “fragment” compounds, typically <250–300 Da (1), which bind to the target protein of interest, and their further elaboration into high-affinity small molecules (2). The rationale of fragment-based strategies and examples in which they have been successfully applied in industry and academia over the past decade have been extensively reviewed (3–8). One of the reasons for the successes and broad impact of fragment-based approaches is the growing consensus that fragments generally represent more attractive and synthetically tractable starting points for medicinal chemistry compared with the larger, functionally more complex structures of many hits identified by high-throughput screening and that fragment growing or linking provides a more efficient approach to exploring chemical space, especially for novel classes of target proteins. Therefore, a crucial aspect of any fragment-based program is the identification of a number of fragments that bind noncovalently to the protein target and the elucidation of their binding properties to make informed decisions as to which “privileged” structures to use for subsequent chemical elaboration. However, the weak affinities displayed by fragments (usually in the high micromolar to millimolar range) make it challenging not only to screen and detect fragment binding but also to characterize reliably their binding interaction with the protein target. Over the past few years, a wide range of methods has been explored and applied, often individually, toward fragment screening (9, 10). These include experimental techniques such as ligand-observed and protein-observed NMR spectroscopy (11, 12), X-ray crystallography (11, 13, 14), mass spectrometry (MS) (15), surface plasmon resonance (SPR) (16), high-concentration bioassays (17), and isothermal titration calorimetry (ITC) (18, 19). There are also studies describing computational screening to enrich hits from virtual fragment libraries (16, 20). Each of these approaches has different observables, detection, and sensitivity capabilities (21). Although several methods—for example, NMR, MS, and ITC—are conducted in solution, others require protein crystals (X-ray crystallography) or immobilization to a surface (SPR). Some of these methods can be applied with significant automation and throughput. Because of the broad differences among the techniques that are used for fragment screening and their complementary advantages and disadvantages, there is a lack of consensus as to which strategy should be chosen for screening fragment libraries. It is becoming apparent that a combination of techniques should be used for fragment screening and validation to increase success in translating the identified hits into bound X-ray structures and to inform as broadly as possible decision making for further optimization of fragments by medicinal chemistry.
In this article, we demonstrate the successful application of a cascade of biophysical techniques for fragment screening, validation, and characterization against Mycobacterium tuberculosis pantothenate synthetase (Pts). Tuberculosis is a highly dangerous infectious disease for which there is an urgent need to develop new medicines (22). Pts has been suggested as a possible target for developing novel antitubercular drugs (23, 24). The enzyme performs the condensation of pantoate and β-alanine, by using ATP as cofactor, to form the vitamin pantothenate (25–28). Several approaches have been used to develop inhibitors, including high-throughput screening (29, 30), mimicking the reaction intermediate (31, 32), and more recently, we have demonstrated the application of dynamic combinatorial chemistry (33) as well as fragment growing and fragment linking (34, 35). These further justify the choice of Pts as a model system to benchmark our biophysical fragment screening strategy.
Results
Biophysical Screening of the Library.
A systematic screening strategy was devised that involved a primary screen using fluorescence-based thermal shift (36, 37) of a rule-of-three compliant fragment library (1,250 fragments; Fig. S1 and Table S1), followed by a secondary screen using one-dimensional 1H NMR spectroscopy experiments: water ligand observed gradient spectroscopy (WaterLOGSY) (38) and saturation transfer difference (STD) (39). Pts has an average thermal unfolding value of 38.1 ± 0.2 °C, which remains unaltered when in the presence of up to 10% vol/vol DMSO. For all experiments a positive control was used, namely addition of 1 mM ATP, for which an increased unfolding temperature is observed of 43.2 ± 0.9 °C, corresponding to a shift (ΔTm) of 5.1 ± 0.9 °C. Fragments were screened at 10 mM concentration, and hits were identified as stabilizing the enzyme by at least 0.5 °C (Fig. S2). The choice for this cutoff was based on the screening thermal window used for detecting fragment binding in this assay, effectively representing the possible minimal error. Based on this threshold value, 39 compounds were identified as hits (Fig. S3), corresponding to a hit rate of 3.1%. A similar hit rate was also observed for a fragment-based, thermal-shift screening campaign of the ankyrin domain of Notch-1 receptor (3.2%) (40) and for a mutant of p53, Y220C (2.4%) (41). This hit rate includes both true and false hits. A low hit rate does not reflect a limitation of the technique, but rather the potential druggability and stability of the target being investigated (42) or even the design of the library used (43).
The hits were mostly fused 5–6 membered rings (15 in total) and six membered rings (seven in total). The fused 5–6 membered and six membered rings represent 12.2% and 13% of the fragment library. A prominent feature for 19 of the 39 resulting hits was the presence of a carboxylate group. At this stage, we attributed this to possible interactions with positively charged His44, His47, Lys160, and Arg198 residues that are involved in binding the triphosphate backbone of ATP. One of the larger recorded ΔTm was for fragment 1 (2.5 °C, Fig. 1). Interestingly, this fragment belongs to a scaffold overrepresented in the library, the benzodioxoles/benzoxoles, with 31 compounds in total (∼3% of the library). Four of these fragments showed a ΔTm in the range 0.5–2.5 °C.
Fig. 1.

The hits identified in the primary thermal shift screen and validated by secondary NMR spectroscopy screen, with Kd determined by ITC. The fragments exhibited a varied range in both ΔTm (from 0.5 to 3.0 °C) and affinities (Kd from 480 μM to 17.3 mM).
About 17.2% of the fragments (215 in total) displayed a ΔTm between 0 and 0.5 °C. Present in this range were fragments belonging to scaffolds with a ΔTm higher than the threshold used for this study. The majority of the fragments, 73.2% (915 in total), displayed negative ΔTm values. This could be attributed to fragments stabilizing the unfolded state of the protein, or fragments aggregating and causing early destabilization and unfolding of the protein (37). Some fragments that possessed scaffolds identified as hits in this primary screen were members of this group displaying negative ΔTm values. Not all compounds were amenable to analysis by this assay, as 45 fragments consistently failed to produce a typical thermal unfolding curve. These fragments were not part of a distinctive class and could not be grouped according to their structures. Some of the fragments had fluorescence properties that might interfere in the assay.
The 39 hits identified by the primary thermal shift screen were further analyzed using one-dimensional 1H NMR spectroscopy as a secondary screen. Both WaterLOGSY and STD NMR experiments were performed to test for binding of fragments to Pts by comparing the proton signals of the fragments in the presence of the enzyme with those recorded from a sample under identical conditions but in which the enzyme was absent (Fig. S4). This follow-up screen validated 17 of the 39 hits (a 56% attrition rate) identified by thermal shift (Fig. 1). The remaining compounds did not show any evidence of binding by NMR spectroscopy at the concentrations used. This success rate in validating thermal shift hits by NMR is larger than typical hit rates from NMR screens of random fragment libraries (5–10%, depending on the target) and in particular was significantly larger than the hit rate found by performing a WaterLOGSY screen of 52 randomly selected fragments on Pts. The latter experiment yielded one validated hit, 5-methoxyindole (34), corresponding to a hit rate of 2%. This analysis demonstrates the successful enrichment of fragment hits achieved by prescreening the entire library using thermal shift. Interestingly, 5-methoxyindole showed a ΔTm of 0.0 °C, suggesting that it would have been missed (a false negative) from the thermal shift screen had it been present in the initial library.
The binding specificity of the fragments that were positive in the NMR screen was investigated by monitoring the changes in the proton signals of the fragments upon addition of a saturating concentration of ATP, Kd of 10 μM (32). Compounds 1–6, 8, and 14 showed a significant decrease in their LOGSY and STD signals, suggesting almost complete displacement of the fragments by ATP. Most of the remaining fragments (compounds 7 and 9–17) exhibited little or no change in their NMR signals upon addition of ATP, indicating that they are not significantly displaced. This observation suggests these fragments bind at another site, possibly the pantoate pocket. To address this question, pantoate was added to a sample containing the fragment being investigated, ATP, and enzyme. The goal was to form in situ pantoyladenylate, the intermediate in the reaction catalyzed by Pts (27). There is no report on the affinity of pantoyladenylate to Pts, however an isosteric sulfonamide analog has a reported Kd determined by ITC of 125 nM (32). Once the pantoyladenylate intermediate is formed, it would be tightly bound to Pts, occupying both the adenine and pantoate pocket and effectively competing with the fragment for the pantoate pocket. As a result of the addition of pantoate, a decrease in the proton signals of both ATP and pantoate ligands was observed by NMR spectroscopy, consistent with the formation of pantoyladenylate preventing the two substrates binding to the enzyme active site. Upon formation of pantoyladenylate, the NMR signals of fragments 7 and 9–16 reverted back to control level, consistent with their displacement by the reaction intermediate, suggesting they were originally binding most probably in the pantoate pocket. One fragment, 17, was not displaced by either ATP or the pantoyladenylate intermediate, suggesting either binding at a third site or a nonspecific interaction. Fragments that are selected at the end of this stringent screening process were then evaluated in terms of their ligand efficiency and binding mode.
Validation of Fragment Binding by ITC and X-Ray Crystallography.
To elucidate the respective binding affinities and binding modes of the fragments, thermodynamic and structural studies were conducted using ITC and X-ray crystallography. An accurate characterization of the binding affinity for the interaction using ITC under low c-value (ratio of macromolecule concentration to dissociation constant < 1) experimental design can be achieved provided that (i) the binding enthalpy is sufficiently large (typically |ΔH| > 5 kcal⋅mol−1) to allow for the instrument sensitivity and (ii) a large proportion of the binding site, ideally >80%, is saturated with ligand at the end of the titration (18, 44). Under the experimental conditions used, the latter should be true for dissociation constants around or below 5 mM. We were able to characterize reliably the affinity for 14 out of the 17 fragments studied, which exhibited a Kd in the 0.5–17 mM range, with an average ΔG of –3.4 ± 0.3 kcal⋅mol−1 and a ligand efficiency in the range 0.18–0.37 kcal⋅mol−1 nonhydrogen atom−1 (Table 1 and Fig. S5). All fragment binding by ITC was found to be enthalpically driven, with ΔH values ranging from –5.3 to –21 kcal⋅mol−1, resulting in unfavorable binding entropy in all cases. The wide range of enthalpy values across the series could reflect entropy/enthalpy compensation effects. However, ΔH values returned from data fitting of hyperbolic ITC titrations under low c-value conditions should be interpreted with caution, as the binding stoichiometry cannot be measured under these conditions and it is not always possible to achieve complete saturation of the binding site at the end of the titration. Two fragments, 11 and 16, were among those postulated to bind at the pantoate pocket based on the NMR data. These two fragments showed an increase in affinity of 1.8 and 12 times, respectively, when titrated against the enzyme in the presence of saturating amounts of ATP. These results are consistent with a model of fragments binding at the pantoate pocket in a cooperative fashion with ATP binding. In addition, fragments binding to the Pts–ATP complex showed a significantly smaller binding enthalpy, resulting in these cases in a favorable binding entropy. Inspection of the crystal structure of ATP bound to Pts [retrieved data from the Protein Data Bank (PDB) ID code 2A84] shows that the α-phosphate is engaged in key hydrogen bonds with Met40 and His47, stabilizing a loop at the pantoate pocket that is usually disordered.
Table 1.
Thermodynamic binding parameters determined by ITC
| Fragment | ΔH, kcal/mol | ΔG, kcal/mol | Kd, mM | LE |
| 1 | −13.9 ± 0.1 | −4.0 ± 0.1 | 1.2 ± 0.01 | 0.29 |
| 2 | −10.1 ± 0.2 | −4.5 ± 0.3 | 0.5 ± 0.01 | 0.30 |
| 3 | −11.6 ± 0.2 | −3.1 ± 0.3 | 5.1 ± 0.2 | 0.26 |
| 6 (racemate) | −10.3 ± 0.05 | −4.2 ± 0.1 | 0.9 ± 0.04 | 0.32 |
| 6 (R-enantiomer) | −7.9 ± 0.1 | −4.1 ± 0.1 | 0.9 ± 0.02 | 0.32 |
| 6 (S-enantiomer) | −12.3 ± 0.2 | −4.3 ± 0.2 | 0.7 ± 0.02 | 0.33 |
| 7 | −9.4 ± 0.1 | −3.3 ± 0.1 | 3.5 ± 0.04 | 0.28 |
| 8 | −7.51 ± 0.1 | −4.2 ± 0.1 | 0.7 ± 0.02 | 0.32 |
| 10 | −7.2 ± 0.1 | −3.7 ± 0.1 | 1.9 ± 0.03 | 0.37 |
| 11 | −16.2 ± 0.8 | −3.4 ± 0.9 | 3.2 ± 0.2 | 0.31 |
| 11 (in the presence of ATP) | −1.5 ± 0.05 | −3.8 ± 0.1 | 1.7 ± 0.08 | 0.34 |
| 12 | −21.2 ± 1.0 | −2.7 ± 1.0 | 10.9 ± 0.6 | 0.27 |
| 13 | −10.7 ± 0.3 | −3.1 ± 0.3 | 4.9 ± 0.2 | 0.26 |
| 14 | −8.7 ± 0.2 | −4.0 ± 0.2 | 1.0 ± 0.01 | 0.33 |
| 15 | −12.3 ± 0.7 | −2.3 ± 0.7 | 11.5 ± 0.8 | 0.18 |
| 16 | −11.3 ± 0.02 | −2.6 ± 0.1 | 17.3 ± 0.8 | 0.22 |
| 16 (in the presence of ATP) | −1.8 ± 0.4 | −3.8 ± 0.4 | 1.4 ± 0.02 | 0.32 |
| 17 | −5.3 ± 0.2 | −3.1 ± 0.2 | 4.5 ± 0.2 | 0.34 |
Fragment 6, present in the library as a racemic mixture, showed a Kd of 880 μM against Pts. The individual enantiomers of 6 showed comparable binding affinities by ITC, the S enantiomer exhibiting slightly higher affinity (Kd = 670 μM; Table 1 and Fig. S6). Three fragments, 4, 5, and 9, could not be analyzed by ITC. The fragments were soluble in the conditions used for analysis (buffer with 2–5% DMSO added). The ITC was repeated at a lower temperature to explore the possibility that the enthalpic contribution associated to fragment binding may change, however no signal could be detected. This could be due to the very weak affinities these compounds might exhibit.
The 17 fragment hits were then moved forward to protein X-ray crystallography. Pts can be readily crystallized as a dimeric apoenzyme, yielding high-resolution diffraction data (27, 28, 32). To locate the fragment binding sites and gain insight into the nature of protein–ligand interactions, fragments were soaked in Pts crystals overnight, at concentrations ranging from 50 to 200 mM, depending on the solubility of the fragments.
Briefly, the active site of Pts can be divided into three distinct regions: (i) the adenosine binding site (comprising an adenine region and a ribose region), (ii) the triphosphate binding site (adjacent to the β-alanine site), and (iii) the pantoate binding site (Fig. S7). If more than one fragment is found to bind at the same binding site, a fragment merging approach would be considered. Alternatively, if fragments are found to bind in adjacent pockets, they offer the opportunity to explore a fragment linking approach. We were able to identify reliably electron density corresponding to the soaked compound for eight out of the 17 fragments studied (∼47% of the total), with resolutions for the final refined structures in the 1.63–2.33 Å range (Table S2).
Fragments Validated to Bind at the Adenine Pocket.
Analysis of the crystal structures indicated that, among the eight fragment hits for which X-ray structures were obtained, fragments 1, 2, 3, and 8 bound at the adenine binding region, the binding site of 5-methoxyindole, a previously identified fragment hit for Pts (34). This observation suggests that this pocket is a “hot-spot” for fragment binding at the enzyme active site. Fragment 3 (benzodioxole-5-carboxylic acid; Fig. 2 A and B) engages deep in the adenine pocket, where one of the oxygen atoms in the dioxole moiety is interacting through a hydrogen bond with the main-chain NH of Val187. The fragment further engages Pts through hydrogen bonds between the carboxylic acid moiety and the side-chain residues of His44 and Lys160. Interestingly, a water molecule is positioned in the β-phosphate spot, in close proximity to the carboxylate. This water molecule further mediates a hydrogen bond bridge between the fragment and the main-chain NH of Ser197. Fragment 5 (2,3-dihydrobenzofuran-5-carboxylic acid) is structurally related to fragment 3. The ΔTm value registered for 5 was identical to that of 3, however no crystal structure was obtained and we were unable to measure its Kd by ITC. Inspection of the crystal structure of 3 highlights that only one of the oxygens from the benzodioxole ring forms a hydrogen bond with the protein. As fragment 5 also contains an oxygen at this position, we would expect it to bind in a similar fashion. Fragment 1 (benzodioxole-5-propanoic acid; Fig. 2 C and D) recapitulates all of the important interactions described for 3. The water coordinating the interaction with 3 is now displaced, and the carboxylate makes a hydrogen bond with Ser196 and Ser197. In addition, Lys160 adopts a different conformation, to capture a hydrogen bond with the carboxylate.
Fig. 2.

X-ray crystal structures of fragments found to bind the Pts active site at the adenine pocket. Crystal structures of bound fragments 3 (A and B, 1.75 Å of resolution), 1 (C and D, 1.94 Å of resolution), 2 (E and F, 2.07 Å of resolution), and 8 (G and H, 1.63 Å of resolution) are shown. The Fo–Fc omit electron density maps are shown as a yellow mesh contoured at 3σ around the fragments. B, D, F, and H depict the amino acids found to be participating in hydrogen bonds with the fragments, represented by dashed red lines. Carbon atoms are depicted by gray, oxygen by red, and nitrogen by blue colors. Small red spheres represent water molecules coordinating binding between the fragments and Pts. Images rendered by Pymol v0.99 (49).
One of the hits with the highest ΔTm observed from the screening campaign was fragment 2 (6-methoxybenzofuran carboxylic acid). This fragment has a distinctive methoxy group, also present in 5-methoxyindole (34). Indeed, the crystal structure of 2 bound to the enzyme (Fig. 2 E and F) shows the methoxy moiety interacting through a hydrogen bond to main-chain NH of Val187, exhibiting the same role as the oxygen from the benzodioxole/benzoxole scaffolds (fragments 1, 3, and 5) and overlaying exactly the same group of 5-methoxyindole. This key interaction effectively recapitulates one of the hydrogen bonds formed by the adenine ring of ATP at this site (Fig. S7A). This type of interaction is also seen for fragments binding at the ATP binding sites of protein kinases, where most fragments reproduce two of the hydrogen bonds formed by the adenine moiety of ATP at the hinge region of the kinase active site (14). The carboxylate group of fragment 2 extends further toward the exit of the pocket, interacting with Lys160 and Ser196 through hydrogen bonds and recapitulating the position of the carboxylate of fragment 1, forming key interactions also observed for the β-phosphate of ATP (27, 28) and for the bound sulfate ion from the crystallization buffer (34). Fragment 8 retains the key interaction observed for the previous fragments (Fig. 2 G and H). In this case, it is the nitrogen atom of the pyrazole moiety instead of the oxygen atom, observed for fragments 1, 2 and 3, in a hydrogen bond interaction with Val187. The carboxylic acid moiety of fragment 8 is engaging Pts via a water molecule, to the main-chain carbonyl of Pro38. Interestingly, the crystal structure shows an ethylene glycol molecule occupying the same position as the carboxylate group of fragments 1 and 2.
All fragments identified to bind at the adenine site reside in the same plane. This plane deviates by 35° from the adenine ring plane of ATP. It is noteworthy that across all of the structures with fragments bound in the adenine pocket, there are no significant changes in protein conformation, apart from a slight rearrangement of the side chain of Lys160. Overall, two hydrogen bond acceptor hot-spots were successfully identified on the ATP binding site, one for the methoxy moiety of 2, another for the carboxylate/sulfate anions. Fragments 1, 2, and 3 were fully occupied in both pockets of Pts dimer and with identical binding mode, whereas 8 binds at only one pocket, fully occupied.
Fragments Validated to Bind at the Pantoate Pocket.
The binding of fragments 14, 6, and 7 to Pts was all confirmed by ITC with Kd in the range 0.88–3.5 mM. These fragments share a common carboxylate group and showed some level of competition with ATP in the NMR binding experiments. X-ray crystallographic studies revealed that all three fragments bind at the pantoate binding site (Fig. 3). The X-ray crystal structure for fragment 14 (benzofuran carboxylate) revealed that the carboxylate group is directed toward the adenine site, forming hydrogen bonds with His47 side chain and with main-chain NH of Met40 (Fig. 3 A and B). Comparison with the crystal structure of ATP bound to Pts shows that the carboxylate group overlaps well with the α-phosphate of ATP, thus providing a structural basis for the competitive binding with ATP observed by NMR (Fig. S4A). There remains uncertainty as to the position of the oxygen on the furan moiety of 14. Whichever position the oxygen assumes, no hydrogen bond to any close protein atom could be identified. An X-ray crystal structure for the racemate mixture of fragment 6 (benzodioxane carboxylate) was obtained. This compound has a chiral center on the carbon atom adjacent to the carboxyl group. From the X-ray crystal structure it is not possible to ascertain which enantiomer is predominantly bound; as such, both the S and R enantiomers were modeled. The compound engages Pts at the pantoate pocket in a fashion similar to the benzofuran (Fig. 3 C and D), however the fragment is now at a distance of <3.5 Å to Gln172, making a key hydrogen bond with its side-chain amide group. The X-ray crystal structure does not provide a clear explanation as to why the S enantiomer is bound with a slightly higher affinity. A conserved water molecule located in the vicinity (3 Å) is engaged in hydrogen bonds with the main chain carbonyl groups of Pro38 and Phe156 and could be mediating an interaction with 6. Fragment 7 (benzothiadiazole-5-carboxylate) was also found to bind at the pantoate pocket, and to recapitulate the hydrogen bonds with Met40 and His47 (Fig. 3 E and F).
Fig. 3.

X-ray crystal structures of fragments found to bind the Pts active site at the pantoate pocket. Crystal structures of bound fragments 14 (A and B, 2.4 Å of resolution, retrieved data from the PDB ID code 3IME) (34), 6 (C and D, 2.25 Å of resolution), and 7 (E and F, 1.85 Å of resolution) are shown. The Fo–Fc omit electron density maps are shown as a yellow mesh contoured at 3σ around the fragments.
A Fragment Validated to Bind Deep in the Pantoate Pocket.
The thiophene sulfonamide fragment 11 was not displaced by ATP in the NMR competition experiment but only upon forming the pantoyl adenylate intermediate (Fig. S4B). ITC showed fragment 11 binds with a Kd = 3.2 mM. The preliminary binding data suggested that fragment 11 was binding at a different site to the previous fragments. This was found to be the case, as X-ray crystallography shows it binds deeply into the pantoate pocket (Fig. 4 A and B). Further analysis of the crystal structure revealed loss of electron density for loop Gln72–Asp80. This suggests a displacement of this loop upon fragment binding, an unexpected observation, not least because it was assumed this conformational change would be energetically unfavorable and it would seem difficult to induce it with a weakly binding fragment. Nevertheless, fragment 11 binds to Pts with good ligand efficiency (LE) (Kd = 3.2 mM, LE = 0.31 in the absence of ATP; Kd = 1.7 mM, LE = 0.34 in the presence of ATP).
Fig. 4.

X-ray crystal structures of fragment 11 found to bind deeper in the pantoate pocket of Pts. Crystal structures of fragment 11 bound to Pts in the absence (A and B) and presence of ATP (C and D). The Fo–Fc omit electron density maps are shown as a yellow mesh contoured at 3σ around the ligands.
To investigate further the binding of fragment 11, we obtained a crystal structure of it bound in the presence of ATP (Fig. 4 C and D). The structure confirmed that fragment 11 retains the same binding mode in the absence and presence of ATP. In addition, no density is observed for the loop 72–80, as was the case for the binary structure. This suggests that the change of thermodynamic signature observed is not due to significant changes in the fragment-binding mode. This fragment could potentially be exploited in the generation of inhibitors that could “trap” the enzyme into an inactive state by displacing the catalytically competent conformation required for the Gln72 loop. To our knowledge, crystal structure of a fragment-like small molecule bound next to ATP at the active site of an enzyme is to date unprecedented.
Fragments Validated to Bind Pts but for Which an X-Ray Crystal Structure Was Not Obtained.
A set of fragments was shown to bind Pts by NMR spectroscopy, but for which it was not possible to obtain an X-ray crystal structure. Of these, fragments 9 and 16 are of particular interest. These fragments share a benzoisoxazole scaffold, which has been described (29) as part of novel inhibitors for Pts. The authors relied on docking results to predict how the inhibitor would engage Pts, however the predicted poses showed no preference for a particular orientation. Fragment 16 was validated to bind in the pantoate pocket by NMR and ITC. Both WaterLOGSY and STD NMR spectra showed the signal for the fragment returning only upon addition of both ATP and pantoate. The titration of this fragment with Pts–ATP mixture resulted in a Kd of 1.4 mM (LE = 0.32). This suggests this fragment is binding deep in the pantoate pocket, as described above for fragment 11. An ITC experiment for fragment 9 failed to produce any binding isotherm. Almost all of the fragments that failed to produce an X-ray crystal structure had Kd greater than 3 mM. The only exception was fragment 8, with a Kd of 720 μM. It was also not possible to acquire an X-ray crystal structure of fragment 17 bound to Pts. This fragment bound with a Kd of 4.5 mM, however due to its small size (137 Da), its LE is 0.37. NMR experiments showed fragment 17 was not displaced competitively by either ATP or pantoyladenylate. It is possible that this fragment binds at a site away from the active site.
Discussion
Our multiapproach for fragment screening resulted in the discovery of several hits from a library of 1,250 fragments. The use of several different biophysical techniques was essential for the validation of the hits proceeding from the thermal shift analysis. Thermal shift provided a fast way to screen the entire library, and NMR allowed the subsequent validation of the resulting hits. Thermal shift provided a >50% rate of false positives that were later verified not to bind by NMR, however it significantly enriched the hit rate of the secondary NMR screen. Most fragments validated by NMR and ITC were shown by X-ray crystallography to bind to Pts at the adenine pocket, competitively with ATP, and pantoate pocket, competitively with pantoyladenylate. One fragment was identified to bind deeper in the pantoate pocket, noncompetitively with ATP, inducing a conformational change in a protein loop, a feature that to our knowledge has not been previously observed. One of the fragments described in this study has already been elaborated to provide potent inhibitors (34). The unique fragments described here will bolster efforts to optimize the current inhibitors and to develop other chemical series. Work toward these objectives is underway in our laboratories and will be disclosed in future publications.
Methods
Protein Expression, Purification, and Crystallization.
Expression, purification, tag removal, and crystallization of Pts were as described (27, 28, 32). Final Pts buffer was 50 mM Hepes pH 7.6, 50 mM NaCl, and 5 mM MgCl2.
Thermal Shift.
The thermal shift assay was performed as described previously (32). Briefly, a stock solution containing 4 μM Pts, 10× Sypro Orange (Invitrogen), and 1× Pts buffer was prepared, from which a volume of 90 μL was dispensed into each of a 96-well plate. Fragments (Maybridge) were added, from a master stock of 100 mM, to each plate to obtain a final concentration of 10 mM. Quadruplicate controls were used for all experiments, where 10% vol/vol DMSO (reference) and 1 mM ATP (positive control) were used instead of fragment. The resulting data (fluorescence intensity vs. temperature) were fitted to obtain the denaturing temperature Tm (point of sigmoidal inflection) as the maximum of each curve’s derivative. This analysis was performed by using an in-house developed Microsoft Office Excel macro. The reference unfolding temperature of Pts in 10% (vol/vol) DMSO 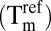 was subtracted from the values in the presence of fragment
was subtracted from the values in the presence of fragment 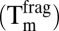 to obtain thermal shifts,
to obtain thermal shifts, 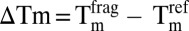 . Fragments were considered to be hits when
. Fragments were considered to be hits when 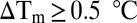 .
.
NMR Spectroscopy.
The 1H NMR experiments were performed at 298 K on a 500 MHz or 700 MHz Bruker NMR spectrometers equipped with a 5 mm Triple Resonance Inverse (TXI) cryoprobe with z gradients and a Bruker Automatic Sample Changer (BACS-60). Samples (700 µL) comprising 0.5 mM fragment in the absence and presence of 10 µM His6–Pts were prepared in 25 mM Tris⋅HCl buffer at pH 8.0 with 5 mM MgCl2, 10% (vol/vol) D2O, and 0.5% (vol/vol) DMSO-d6 and loaded into 528-PP-8 NMR tubes (Wilmad-LabGlass). Deuterated (Trimethylsilyl)–propionic acid–d4 (20 μM) was present in all samples for calibration purposes. For displacement experiments, 0.5 mM ATP was added; in samples that showed no displacement with ATP, pantoate was added to a final concentration of 1 mM. WaterLOGSY experiments used a 20-ms selective Gaussian 180° shaped pulse at the water signal frequency and an NOE mixing time of 1 s. STD experiments used a 40-ms selective Gaussian 180° shaped pulse at a frequency alternating between on-resonance (0.8 ppm) and off-resonance (40 ppm) after each scan. Water signal suppression was achieved using a W5 Watergate gradient spin-echo pulse sequence. The resulting spectra were analyzed with Bruker TopSpin software.
ITC.
ITC experiments were performed using a VP-ITC or ITC200 instruments (Microcal, GE). Fragments were solubilized in either 5% or 2% vol/vol DMSO, at a range of concentration of 10–20 mM. Protein solutions were dialyzed overnight in Pts buffer. The solution containing the fragment was titrated against Pts and the thermodynamic characterization and affinity determined through the use of the Origin software by using a single ligand binding model.
X-Ray Crystallography.
Fragments were soaked onto Pts crystals at concentrations between 50 and 200 mM overnight or longer. For fragment 8, a powder soak was performed instead. For the ternary complex fragment 11–Pts–ATP, fragment 11 and ATP were soaked at 50 and 5 mM, respectively. To facilitate the accessibility of the pocket to the fragments, the sulfate ion bound at the active site had to be back-washed (34). To achieve this, a solution identical to the crystallization condition was used but where Li2SO4 was replaced by LiCl. Crystals were then flash-frozen in liquid nitrogen and stored for data collection at the synchrotron. All derived data were indexed and scaled using either the HKL suite (45) or MOSFLM (46). After data collection, indexing, and scaling, crystallographic refinement was carried out, using the graphical interface of the CCP4 suite, running REFMAC 5.0 (47), with the apo structure of Pts as a starting model (PDB ID code 3COV) (32). After this initial step, further refinement was performed under Refmac 5, interspersed with model building with the fragment and Pts, by COOT (48). Table S2 summarizes the X-ray crystallography data collection and refinement statistics.
Supplementary Material
Acknowledgments
We thank Dr. Dimitri Y. Chirgadze (Crystallographic X-Ray Facility at the Department of Biochemistry, University of Cambridge) and are grateful for the technical support at the European Synchrotron Radiation Facility (ESRF), Swiss Light Source (SLS), and Diamond Light Source (DLS) Facilities for their assistance. This research was supported by the Bill & Melinda Gates Foundation [“Integrated Methods for TB Drug Development (IMTB)” Accelerator Grant], the UK Biotechnology and Biological Sciences Research Council (Grant BB/D006104/1), the Fundação para a Ciência e Tecnologia (PhD sponsorship to H.L.S.), and Homerton College (Junior Research Fellowship to A.C.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. D.R. is a guest editor invited by the Editorial Board.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4EF6, 4DDH, 4DDK, 4G5F, 4FZJ, 4DDM, 4EFK, and 4G5Y).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1304045110/-/DCSupplemental.
References
- 1.Congreve M, Murray CW, Carr R, Rees DC. Fragment-based lead discovery. Annu Rep Med Chem. 2007;42:431–448. [Google Scholar]
- 2.Ciulli A, Abell C. Fragment-based approaches to enzyme inhibition. Curr Opin Biotechnol. 2007;18(6):489–496. doi: 10.1016/j.copbio.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rees DC, Congreve M, Murray CW, Carr R. Fragment-based lead discovery. Nat Rev Drug Discov. 2004;3(8):660–672. doi: 10.1038/nrd1467. [DOI] [PubMed] [Google Scholar]
- 4.Erlanson DA. Fragment-based lead discovery: A chemical update. Curr Opin Biotechnol. 2006;17(6):643–652. doi: 10.1016/j.copbio.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 5.Hajduk PJ, Greer J. A decade of fragment-based drug design: Strategic advances and lessons learned. Nat Rev Drug Discov. 2007;6(3):211–219. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- 6.Congreve M, Chessari G, Tisi D, Woodhead AJ. Recent developments in fragment-based drug discovery. J Med Chem. 2008;51(13):3661–3680. doi: 10.1021/jm8000373. [DOI] [PubMed] [Google Scholar]
- 7.Chessari G, Woodhead AJ. From fragment to clinical candidate—A historical perspective. Drug Discov Today. 2009;14(13-14):668–675. doi: 10.1016/j.drudis.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 8.Murray CW, Rees DC. The rise of fragment-based drug discovery. Nat Chem. 2009;1(3):187–192. doi: 10.1038/nchem.217. [DOI] [PubMed] [Google Scholar]
- 9.Erlanson DA, McDowell RS, O’Brien T. Fragment-based drug discovery. J Med Chem. 2004;47(14):3463–3482. doi: 10.1021/jm040031v. [DOI] [PubMed] [Google Scholar]
- 10.Ciulli A, Blundell TL, Abell C. In: Computational and Structural Approaches to Drug Discovery: Ligand-Protein Interactions. Stroud RM, Finer-Moore J, editors. London: Royal Society of Chemistry; 2008. pp. 293–318. [Google Scholar]
- 11.Jhoti H, Cleasby A, Verdonk M, Williams G. Fragment-based screening using X-ray crystallography and NMR spectroscopy. Curr Opin Chem Biol. 2007;11(5):485–493. doi: 10.1016/j.cbpa.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 12.Pellecchia M, et al. Perspectives on NMR in drug discovery: A technique comes of age. Nat Rev Drug Discov. 2008;7(9):738–745. doi: 10.1038/nrd2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blundell TL, Jhoti H, Abell C. High-throughput crystallography for lead discovery in drug design. Nat Rev Drug Discov. 2002;1(1):45–54. doi: 10.1038/nrd706. [DOI] [PubMed] [Google Scholar]
- 14.Hartshorn MJ, et al. Fragment-based lead discovery using X-ray crystallography. J Med Chem. 2005;48(2):403–413. doi: 10.1021/jm0495778. [DOI] [PubMed] [Google Scholar]
- 15.Erlanson DA, et al. Site-directed ligand discovery. Proc Natl Acad Sci USA. 2000;97(17):9367–9372. doi: 10.1073/pnas.97.17.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teotico DG, et al. Docking for fragment inhibitors of AmpC β-lactamase. Proc Natl Acad Sci USA. 2009;106(18):7455–7460. doi: 10.1073/pnas.0813029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barker J, Courtney S, Hesterkamp T, Ullmann D, Whittaker M. Fragment screening by biochemical assay. Expert Opin Drug Discov. 2006;1(3):225–236. doi: 10.1517/17460441.1.3.225. [DOI] [PubMed] [Google Scholar]
- 18.Ciulli A, Williams G, Smith AG, Blundell TL, Abell C. Probing hot spots at protein-ligand binding sites: A fragment-based approach using biophysical methods. J Med Chem. 2006;49(16):4992–5000. doi: 10.1021/jm060490r. [DOI] [PubMed] [Google Scholar]
- 19.Taylor JD, Gilbert PJ, Williams MA, Pitt WR, Ladbury JE. Identification of novel fragment compounds targeted against the pY pocket of v-Src SH2 by computational and NMR screening and thermodynamic evaluation. Proteins. 2007;67(4):981–990. doi: 10.1002/prot.21369. [DOI] [PubMed] [Google Scholar]
- 20.Chen Y, Shoichet BK. Molecular docking and ligand specificity in fragment-based inhibitor discovery. Nat Chem Biol. 2009;5(5):358–364. doi: 10.1038/nchembio.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pröll F, Fechner P, Proll G. Direct optical detection in fragment-based screening. Anal Bioanal Chem. 2009;393(6-7):1557–1562. doi: 10.1007/s00216-008-2477-y. [DOI] [PubMed] [Google Scholar]
- 22.Janin YL. Antituberculosis drugs: Ten years of research. Bioorg Med Chem. 2007;15(7):2479–2513. doi: 10.1016/j.bmc.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 23.Sambandamurthy VK, et al. A pantothenate auxotroph of Mycobacterium tuberculosis is highly attenuated and protects mice against tuberculosis. Nat Med. 2002;8(10):1171–1174. doi: 10.1038/nm765. [DOI] [PubMed] [Google Scholar]
- 24.Sambandamurthy VK, et al. Mycobacterium tuberculosis DeltaRD1 DeltapanCD: A safe and limited replicating mutant strain that protects immunocompetent and immunocompromised mice against experimental tuberculosis. Vaccine. 2006;24(37-39):6309–6320. doi: 10.1016/j.vaccine.2006.05.097. [DOI] [PubMed] [Google Scholar]
- 25.Zheng R, Blanchard JS. Steady-state and pre-steady-state kinetic analysis of Mycobacterium tuberculosis pantothenate synthetase. Biochemistry. 2001;40(43):12904–12912. doi: 10.1021/bi011522+. [DOI] [PubMed] [Google Scholar]
- 26.Zheng R, Dam TK, Brewer CF, Blanchard JS. Active site residues in Mycobacterium tuberculosis pantothenate synthetase required in the formation and stabilization of the adenylate intermediate. Biochemistry. 2004;43(22):7171–7178. doi: 10.1021/bi049676n. [DOI] [PubMed] [Google Scholar]
- 27.Wang S, Eisenberg D. Crystal structures of a pantothenate synthetase from M. tuberculosis and its complexes with substrates and a reaction intermediate. Protein Sci. 2003;12(5):1097–1108. doi: 10.1110/ps.0241803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang S, Eisenberg D. Crystal structure of the pantothenate synthetase from Mycobacterium tuberculosis, snapshots of the enzyme in action. Biochemistry. 2006;45(6):1554–1561. doi: 10.1021/bi051873e. [DOI] [PubMed] [Google Scholar]
- 29.Velaparthi S, et al. 5-tert-butyl-N-pyrazol-4-yl-4,5,6,7-tetrahydrobenzo[d]isoxazole-3-carboxamide derivatives as novel potent inhibitors of Mycobacterium tuberculosis pantothenate synthetase: Initiating a quest for new antitubercular drugs. J Med Chem. 2008;51(7):1999–2002. doi: 10.1021/jm701372r. [DOI] [PubMed] [Google Scholar]
- 30.White EL, et al. A novel inhibitor of Mycobacterium tuberculosis pantothenate synthetase. J Biomol Screen. 2007;12(1):100–105. doi: 10.1177/1087057106296484. [DOI] [PubMed] [Google Scholar]
- 31.Tuck KL, Saldanha SA, Birch LM, Smith AG, Abell C. The design and synthesis of inhibitors of pantothenate synthetase. Org Biomol Chem. 2006;4(19):3598–3610. doi: 10.1039/b609482a. [DOI] [PubMed] [Google Scholar]
- 32.Ciulli A, et al. Inhibition of Mycobacterium tuberculosis pantothenate synthetase by analogues of the reaction intermediate. ChemBioChem. 2008;9(16):2606–2611. doi: 10.1002/cbic.200800437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scott DE, Dawes GJ, Ando M, Abell C, Ciulli A. A fragment-based approach to probing adenosine recognition sites by using dynamic combinatorial chemistry. ChemBioChem. 2009;10(17):2772–2779. doi: 10.1002/cbic.200900537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hung AW, et al. Application of fragment growing and fragment linking to the discovery of inhibitors of Mycobacterium tuberculosis pantothenate synthetase. Angew Chem Int Ed Engl. 2009;48(45):8452–8456. doi: 10.1002/anie.200903821. [DOI] [PubMed] [Google Scholar]
- 35.Sledz P, et al. Optimization of the interligand Overhauser effect for fragment linking: Application to inhibitor discovery against Mycobacterium tuberculosis pantothenate synthetase. J Am Chem Soc. 2010;132(13):4544–4545. doi: 10.1021/ja100595u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lo M-C, et al. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem. 2004;332(1):153–159. doi: 10.1016/j.ab.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 37.McDonnell PA, et al. Assessing compound binding to the Eg5 motor domain using a thermal shift assay. Anal Biochem. 2009;392(1):59–69. doi: 10.1016/j.ab.2009.05.044. [DOI] [PubMed] [Google Scholar]
- 38.Dalvit C, Fogliatto G, Stewart A, Veronesi M, Stockman B. WaterLOGSY as a method for primary NMR screening: Practical aspects and range of applicability. J Biomol NMR. 2001;21(4):349–359. doi: 10.1023/a:1013302231549. [DOI] [PubMed] [Google Scholar]
- 39.Mayer M, Meyer B. Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew Chem Int Ed. 1999;38(12):1784–1788. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1784::AID-ANIE1784>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 40.Abdel-Rahman N, Martinez-Arias A, Blundell TL. Probing the druggability of protein-protein interactions: Targeting the Notch1 receptor ankyrin domain using a fragment-based approach. Biochem Soc Trans. 2011;39(5):1327–1333. doi: 10.1042/BST0391327. [DOI] [PubMed] [Google Scholar]
- 41.Basse N, et al. Toward the rational design of p53-stabilizing drugs: Probing the surface of the oncogenic Y220C mutant. Chem Biol. 2010;17(1):46–56. doi: 10.1016/j.chembiol.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 42.Barelier S, Pons J, Gehring K, Lancelin J-M, Krimm I. Ligand specificity in fragment-based drug design. J Med Chem. 2010;53(14):5256–5266. doi: 10.1021/jm100496j. [DOI] [PubMed] [Google Scholar]
- 43.Chen I-J, Hubbard RE. Lessons for fragment library design: Analysis of output from multiple screening campaigns. J Comput Aided Mol Des. 2009;23(8):603–620. doi: 10.1007/s10822-009-9280-5. [DOI] [PubMed] [Google Scholar]
- 44.Turnbull WB, Daranas AH. On the value of c: Can low affinity systems be studied by isothermal titration calorimetry? J Am Chem Soc. 2003;125(48):14859–14866. doi: 10.1021/ja036166s. [DOI] [PubMed] [Google Scholar]
- 45.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 46.Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography. 1992;26 10.1073/pnas.1114128109. [Google Scholar]
- 47.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 48.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 49.DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific LLC; 2008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.