

Abstract
The N-terminal stretch of human frataxin (hFXN) intermediate (residues 42–80) is not conserved throughout evolution and, under defined experimental conditions, behaves as a random-coil. Overexpression of hFXN56–210 in Escherichia coli yields a multimer, whereas the mature form of hFXN (hFXN81–210) is monomeric. Thus, cumulative experimental evidence points to the N-terminal moiety as an essential element for the assembly of a high molecular weight oligomer. The secondary structure propensity of peptide 56–81, the moiety putatively responsible for promoting protein–protein interactions, was also studied. Depending on the environment (TFE or SDS), this peptide adopts α-helical or β-strand structure. In this context, we explored the conformation and stability of hFXN56–210. The biophysical characterization by fluorescence, CD and SEC-FPLC shows that subunits are well folded, sharing similar stability to hFXN90–210. However, controlled proteolysis indicates that the N-terminal stretch is labile in the context of the multimer, whereas the FXN domain (residues 81–210) remains strongly resistant. In addition, guanidine hydrochloride at low concentration disrupts intermolecular interactions, shifting the ensemble toward the monomeric form. The conformational plasticity of the N-terminal tail might impart on hFXN the ability to act as a recognition signal as well as an oligomerization trigger. Understanding the fine-tuning of these activities and their resulting balance will bear direct relevance for ultimately comprehending hFXN function.
Keywords: Frataxin, Helical propensity, Protein stability, Oligomerization, Folding
Graphical abstract
Highlights
-
•
hFXN56–210 is well-folded and shares similar stability to hFXN90–210.
-
•
The oligomeric form of hFXN56–210 can be disassembled and reassembled in vitro.
-
•
Proteolysis leads to the oligomer disassembly: subunits are abridged to hFXN81–210.
-
•
Isolated peptide hFXN56–81 acquires structure in TFE and SDS solutions.
-
•
The N-terminal tail is structurally malleable and triggers oligomerization.
1. Introduction
Macromolecular recognition is probably one of the most important subjects in biochemistry. Intriguing links exist among molecular recognition, motions, stability, aggregation and biological function. Moreover, there is a large number of examples that point out the importance of structure flexibility, local unfolding and the presence of intermediate states in different biologically relevant processes, particularly those involving protein–ligand and protein–protein interactions [1–4]. In this context, it is very relevant to understand how these interactions control the dynamics, ultimately affecting the nature of the final ensemble responsible for the tertiary and quaternary structure leading to protein function.
In this regard, frataxin (FXN, a member of CyaY protein family), a ∼130 residue protein with α/β structure becomes a very interesting model. FXN acts as an iron chaperone and is crucially implicated in iron homeostasis in prokaryotes and in the mitochondria of eukaryotes. FXN is able to interact and transfer this metal to specific proteins, such as mitochondrial aconitase [5] and ferrochelatase [6,7], the latter being the key enzyme catalyzing the last step of heme biosynthesis. In its monomeric form, human FXN (hFXN) binds six iron atoms per protein molecule [8]. The deficiency of hFXN function is the cause of an autosomal recessive neurodegenerative disorder termed Friedreich's Ataxia (FRDA), which leads to metabolic disorders, extensive cell damage by radical species, and impaired assembly of Fe–S clusters [9,10].
The 3D structure of hFXN has been determined by X-ray crystallography [11] and by NMR [12] and its backbone dynamics has been thoroughly analyzed by NMR [13]. Furthermore, thermodynamic stability has been extensively investigated by temperature and chemical unfolding, proving that hFXN is a highly cooperative protein, with a stability of ∼7–8 kcal mol−1 [14,15]. However, stability of the FXN domain varies significantly among different organisms [16].
It has been established previously that yeast frataxin (yFXN) has an N-terminal tail which finely modulates both the conformation of the yFXN monomer and the assembly of its quaternary structure, consisting of 48 subunits. The assembly of the biologically competent homo-multimeric form is induced by iron and results in a particle storing nearly 2400 iron atoms [17]. However, the ability of an oligomerization-deficient variant of yFXN to function in vivo suggests that oligomerization-induced iron storage, is not a critical function of this protein [18]. The role of the N-terminal region of hFXN is not clear. Mitochondrial maturation of the human variant occurs via a mechanism involving at least two-steps, the first yielding the intermediate hFXN42–210 which further matures to hFXN81–210 [19]. Variant hFXN56–210 was also detected in actively dividing cells by high-resolution SDS–PAGE and Western blotting. In addition, it has been shown that hFXN56–210 can be generated by proteolysis when the primary 80–81 site is blocked [20,21]. The biological role of variants hFXN42–210 and 56–210 is not obvious and remains unknown. FXN interacts with components of the Fe–S cluster biosynthesis machinery, such as proteins NFS1, ISCU and ISD11. No difference in the interaction with ISCU and NFS1 was detected between the intermediate (GST-hFXN42–210) and the mature (GST-hFXN81–210) forms of frataxin in pull-down experiments using mitochondria enriched extracts, indicating that the site of interaction with the complex is fully comprised within the sequence of the mature form [19]. However, some aspects of these interactions remain largely unclear. Very recently, it has been suggested that the oligomeric form might play a role in binding to the isolated components of the complex [17]. Two opposing views could be set in this scenario. Under particular metabolic conditions, a longer N-terminus – that present in variants starting at residue 42 or at residues 55–56 – might render FXN forms endowed with the capacity to oligomerize. This would result in an enhanced iron detoxification or iron storage activity in the cell. On the other hand, as hFXN aggregation may take place via the N-terminal peptide – bringing about an impairment of FXN function – cleavage at position 80 would have been selected along evolution.
It is worthy of note that two novel FXN isoforms (II and III) lacking the mitochondrial signal peptide were detected and identified. Variants II and III are specifically expressed in cerebellum and heart tissues, respectively. Both proteins seem to be functionally relevant and may be implicated in FRDA. Interestingly, isoform III lacks the 47 amino acid stretch between positions 9 and 55, a feature that resembles it to hFXN56–210. Furthermore, based on the expression profile, it has been suggested that hFXN56–210 and isoform III share the same iron-self processing mechanism [22].
The N-terminal sequence (residues 56–78) of hFXN is not conserved throughout evolution. NMR experiments in solution suggest that this segment is unstructured in the monomeric form, and that its presence does not bear any effect on the protein structure [23]. However, during overexpression of hFXN56–210 in Escherichia coli, oligomerization is clearly observed. Approximately 60–70% of hFXN5–210 adopts a stable homo polymer conformation of high molecular weight [23,24]. Moreover, supramolecular assembled forms of hFXN have been isolated from human heart mitochondria [21]. On the other hand, the recombinant variants hFXN80–210 and hFXN90–210 lack the capability of oligomerization in physiological buffers, and their hydrodynamic behavior corresponds to that expected for the monomer. Thus, the whole experimental evidence, points to the N-terminal segment (residues 56–78) as a structural element critical for the assembly of the high molecular weight multimer.
The origin of hFXN self recognition as well as the physicochemical conditions which favor the assembly process remain unknown. In this context, we undertook the study of the homo-oligomerization of hFXN, inquiring on the importance of the protein surfaces involved [25,26], whether this process is cooperative and if protomers share features of the native state. In this paper, we focus on the specific role of the N-terminal region in the in vitro homo-oligomerization process. We suggest that the N-terminal tail may participate in the stabilization of a competent state for oligomerization, establishing crucial interactions removable by limited proteolysis or by the addition of a chaotropic agent. In so doing, data presented herein also shed light on the folding process of variant hFXN56–210.
2. Results
Recombinant hFXN56–210 and hFXN90–210 variants were purified as described in Section 4. Their masses (17,253 ± 2 and 13,603 ± 2, respectively) differ in less than 3 Da by comparison to the expected masses deduced from the hFXN sequence. Variant hFXN90–210 conserves its N-terminal Met residue, whereas hFXN56–210 loses it during expression in E. coli. Both proteins are expressed at high level and mostly partition in the soluble fraction after cell disruption and centrifugation (data not shown). Proteolytic damage is evident only in the case of variant hFXN56–210 in E. coli lysates. Neither hFXN90–210 nor hFXN56–210 seem to contain significant iron after purification, as judged by the 1,10-phenanthroline method.
2.1. Oligomerization state of hFXN variants inferred from the hydrodynamic behavior
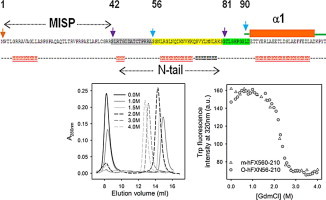
To study the oligomerization state of recombinant hFXN56–210 we investigated the hydrodynamic behavior of both proteins using SEC-FPLC (Fig. 1). Variant hFXN56–210 elutes in the exclusion volume (VO) of the columns (Sephacryl S200 and Superose S200), indicating the high molecular weight of the soluble multimer form. On the other hand, the behavior of hFXN90–210 in aqueous solution corresponds to that of an exclusively monomeric species, as it was similarly observed for the mature form hFXN81–210 [23]. In addition, at 100 mM NaCl the elution volume of the shorter variant is slightly lower than that expected from its molecular weight (data not shown). This effect is corrected at higher salt concentrations (e. g. 1.0 M NaCl). We suggest that the electrostatic effect due to the acidic nature of the surface of this protein may add to the exclusion of the protein from the matrix. This behavior has been observed in our lab for other acidic proteins, like fogrin [27].
Fig. 1.
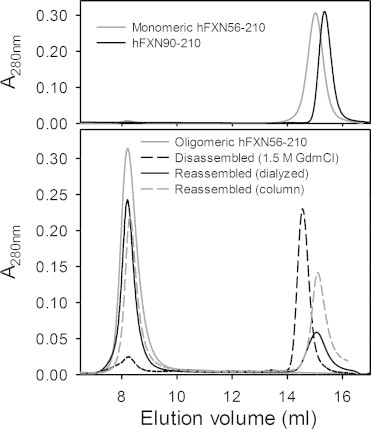
Hydrodynamic behavior of protein constructs studied by size exclusion chromatography (SEC). Lower panel: protein hFXN56–210 in native condition (the oligomer, grey line) or after incubation in 1.5 M GdmCl and subsequent dialysis (black line). In addition, a protein sample was injected after incubation in 1.5 M GdmCl without the dialysis step and the column was equilibrated in the presence of 1.5 M GdmCl (black dashed line) or in the absence of denaturant (grey dashed line). Upper panel: as control samples, hFXN90–210 and monomeric hFXN56–210 were also run. In all cases buffer was 20 mM Tris–HCl, 100 mM NaCl, 1 mM EDTA, pH 7.0 and runs were performed at room temperature.
Then, we asked ourselves whether a low concentration of chaotropic agent may induce the disassembly of the multimer. When hFXN56–210 was incubated in 1.5 M GdmCl and injected into the SEC-FPLC system equilibrated in buffer at the same concentration of GdmCl, essentially all the molecules turned into a slightly expanded monomeric conformation, as revealed by the somewhat lower exclusion volume measured for this form as compared with that expected. In addition, to test if, after removal of the chaotrope, reassembly occurs into an oligomeric conformation in vitro, we incubated hFXN56–210 for 16 h in 1.5 M GdmCl, followed by exhaustive dialysis for 36 h at 4 °C (Fig. 1). After this treatment, re-association of hFXN56–210 into a soluble multimer of high molecular weight is evident (Fig. 1). However, a fraction of molecules remains as monomeric species, pointing to the complexity and lack of completeness of the oligomerization process. Similarly, when hFXN56–210 was incubated in 1.5 M GdmCl for 16 h, but injected into the SEC-FPLC system equilibrated in buffer without GdmCl, both monomeric (RS = 20.4 ± 0.5 Å) and oligomeric forms (RS > 90 Å) are present in significant amounts. Despite the fast dilution experienced by the sample, under these circumstances close to 60% of the protein is able to achieve the oligomeric state in vitro (Fig. 1). The remaining monomeric fraction elutes similarly to the monomeric form of variant hFXN56–210 purified from E. coli (upper panel in Fig. 1). Mass spectrometry confirms that protein samples do not undergo proteolysis at any step along these treatments. This is particularly important to note, because of the known lability of this variant in the monomeric state.
Taken together, these results indicate that the N-tail of the protein contains in its sequence the information to promote those intermolecular interactions necessary for the assembly of the oligomeric form. Thus, oligomerization is not the result of recombinant over-expression of the protein.
2.2. Optical characterization of the homo-oligomeric form of hFXN
To investigate secondary and tertiary structure content of the recombinant oligomeric form of hFXN56–210, we carefully compared the circular dichroism spectra corresponding to hFXN56–210 in its oligomeric form with that of monomeric hFXN90–210 (Fig. 2). CD main signals in the far-UV region of the variants are very similar, although at 230 nm dichroic absorbance of aromatic residues might contribute to the spectrum of hFXN90–210 (Fig. 2A). Correspondingly, the near-UV CD spectrum of the shorter variant shows an enhanced fine-structured pattern of aromatic bands (Fig. 2B). The decrease in intensity of the signals in this range for hFXN56–210 in the oligomeric form might reflect changes in the close environment of the aromatic side-chains or conformational diversity among subunits. Interestingly, these results might be compatible with changes in tertiary structure of the frataxin domain, including the stabilization of a molten-globule like conformation in the oligomeric context.
Fig. 2.
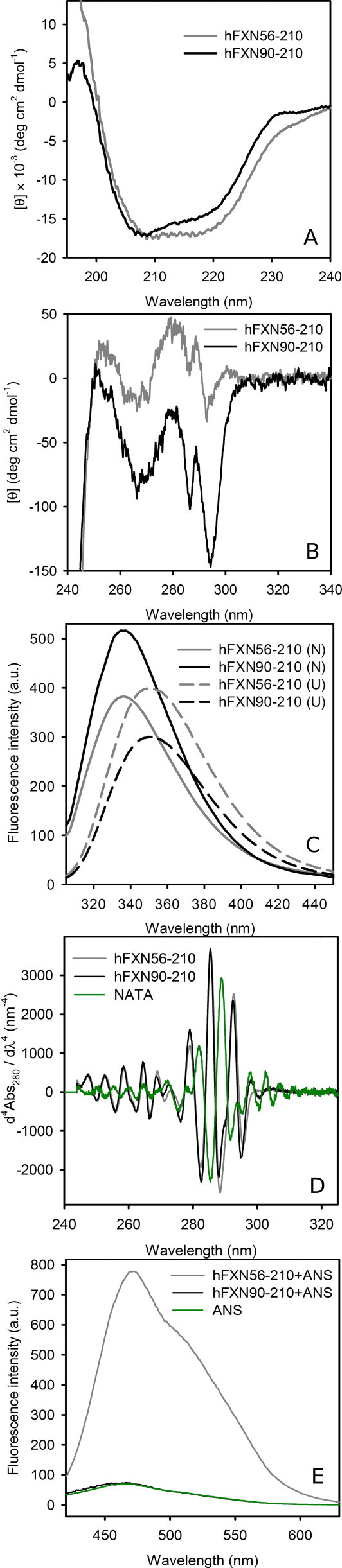
Spectroscopic characterization of hFXN variants. (A) Far-UV CD; (B) near-UV CD; (C) Trp fluorescence emission (the excitation wavelength is 295 nm); (D) fourth-derivative UV-absorption; and (E) ANS fluorescence emission (the excitation wavelength is 350 nm). Both proteins hFXN90–210 and hFXN56–210 are in buffer 20 mM Tris–HCl, 100 mM NaCl, 1 mM EDTA, pH 7.0 at 20 °C.
As mentioned previously, although variant hFXN56–210 expressed in E. coli yields mainly the oligomeric form, nearly 30% of the protein remains as soluble monomer [24]. Taking into account this fact, we purified the latter form to compare its spectral features with those of the oligomer and hFXN90–210 (Fig. S1). It is evident that monomeric hFXN56–210 yields a far UV CD spectrum which is practically superimposable to that observed for hFXN90–210, including the contribution of the aromatic band at 230 nm. In the same fashion, the near-UV CD spectrum of the monomer bears more resemblance to that of variant hFXN90–210 than to hFXN56–210 (data not shown). Remarkably, the calculated spectral difference (Soligomer – Smonomer) suggests the acquisition of additional secondary structure in the context of the oligomer. In addition, the lower signal measured below 208 nm for monomeric hFXN56–210 by comparison with the oligomer points to the contribution of a random-coil conformation of the N-tail to the spectrum of the former.
The fluorescence spectra of hFXN56–210 and hFXN90–210 show a similar maximal wavelength of emission (Fig. 2C). Nevertheless, the former exhibits enhanced quenching, probably related to its multimeric state. Likewise, both forms show almost superimposable fourth-derivative absorption spectra (Fig. 2D). Particularly, bands corresponding to Trp are slightly red-shifted: 292.8 and 285.7 nm for hFXN56–210, and 292.6 and 285.5 nm for hFXN90–210. By comparison, N-acetyl-tryptophanamide (NATA) – a model compound of full solvent exposure for Trp – shows bands at 289.1 and 282.1 nm. For both variants, these data indicate a definite apolar environment for this aromatic residue.
The existence of solvent exposed apolar surfaces in the oligomeric state of hFXN56–210 was revealed by the fluorophore ANS. Fig. 2E shows that – although ANS does not attach to hFXN90–210 – the probe is able to bind hFXN56–210 in the oligomeric form. Binding of this dye to accessible native surfaces has been well documented, e.g. as in the case of BSA [28] and FABP [29]. Nevertheless, binding to a partially structured N-tail in hFXN56–210 or to sites present in a less compact molten globule like state cannot be underestimated.
2.3. Oligomer disassembly by proteolysis: the C-terminal domain of hFXN56–210 is resistant to proteolysis
The lower magnitude of signals in the near-UV CD region observed for the oligomer, by comparison with variant hFXN90–210, and the presence of significant binding of ANS to the former might be attributed to a less rigid environment around the aromatic chromophores and the appearance of exposed hydrophobic patches to the aqueous solvent in the quaternary assembly. However, multimeric hFXN56–210 proves to be soluble and resistant to proteolysis when expressed in E. coli. This fact led us to hypothesize that both the N-terminal tail and the C-terminal domain of this form remain well-folded and/or inaccessible to proteases in the supramolecular assembly. Thus, it becomes of key importance to investigate the extent of structure in the oligomer.
Limited proteolysis in vitro followed by SDS–PAGE and SEC-FPLC shows that upon incubation with trypsin (1:100, mass ratio, protease to protein) the oligomer hFXN56–210 disassembles, as hFXN56–210 molecules become shortened to the form hFXN81–210. Proteolysis does not proceed beyond the latter (Fig. 3). In all likelihood, this fact indicates that the FXN domain is well-folded. Most significantly, resistance to proteolysis is a distinguishing feature of native state hFXN90–210 (Fig. 3, top right panel). This evidence suggests that the N-terminal moiety of hFXN56–210 is only partially structured, therefore, to some extent exposed to protease action. Similarly, challenging variant hFXN90–210 with chymotrypsin reveals limited susceptibility to proteolysis, as evidenced by a single point cut at residue Tyr 205 after incubation for 4 h at 20 °C [15].
Fig. 3.
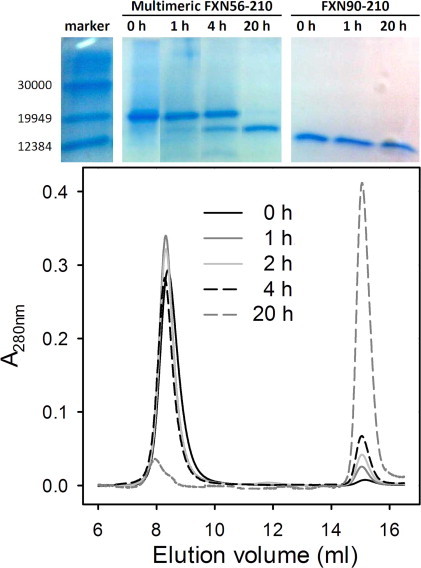
Limited proteolysis of hFXN variants. Both hFXN90–210 and hFXN56–210 were incubated with trypsin 1:100 (protein to protease mass ratio) at 20 °C, in 20 mM Tris–HCl, 100 mM NaCl, 1 mM EDTA, pH 7.0. Aliquots were separated at different times (0, 1, 2, 4 and 20 h) and the reaction was immediately stopped by the addition of 2× SDS–PAGE sample buffer. Samples were kept at −20 °C until loaded into the gel (upper panel). Products were also analyzed by SEC-FPLC (lower panel) and the integrity of the covalent structure of the protein samples was checked by ESI-MS.
2.4. Incubation of hFXN56–210 with GdmCl triggers the disassembly of the oligomer
To further investigate the conformation of variant hFXN56–210, we tested its chemically induced unfolding (Fig. 4). In particular, we evaluated the cooperativity of the transition observed by (i) the magnitude observed for the parameter mNU, that discloses the dependence of the free energy difference of unfolding with denaturant concentration, and (ii) the extrapolated ΔG°NU, H20 value, measuring the stability of hFXN56–210 by comparison with the hFXN90–210 variant.
Fig. 4.
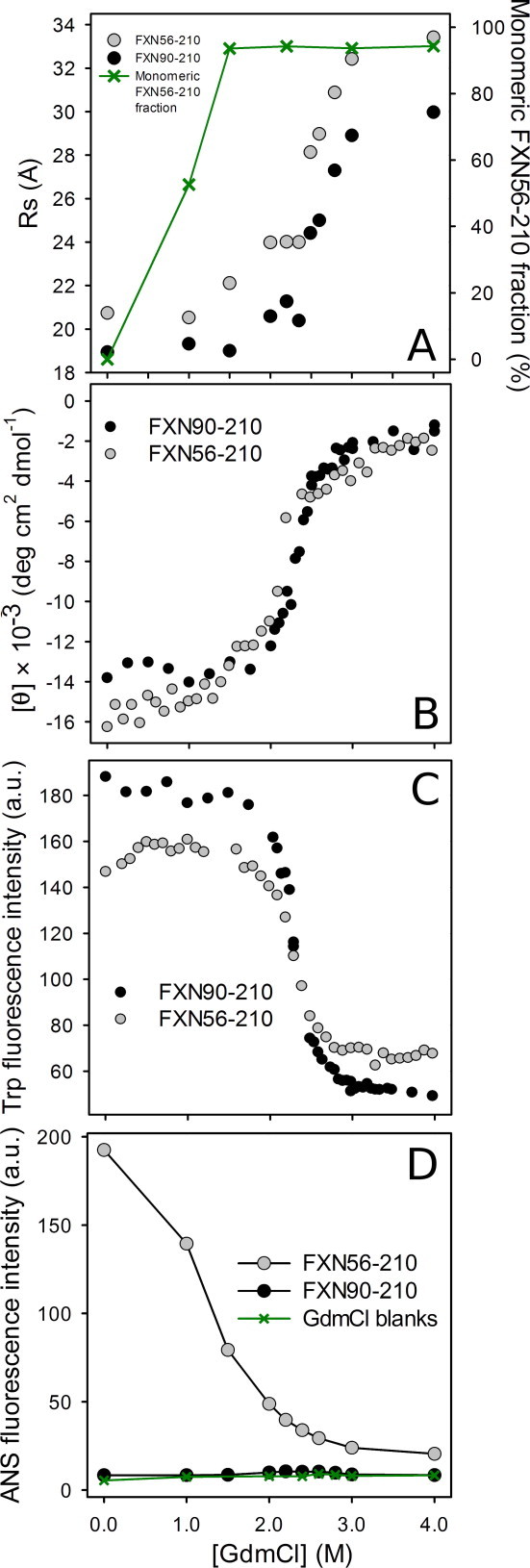
Chemical unfolding of hFXN variants followed by (A) SEC-FPLC, (B) CD signal at 220 nm, (C) Trp fluorescence emission intensity at 320 nm, and (D) ANS fluorescence emission intensity at 480 nm. Variant hFXN90–210 is indicated in open symbols (○) and hFXN56–210, in filled symbols (•). Unfolding of hFXN56–210 was achieved by incubation for 16 h at room temperature. Buffer was 20 mM Tris–HCl, 100 mM NaCl, 1 mM EDTA, pH 7.0 and measurements were performed at 20 °C. For the RS estimations by SEC (Sephacryl S-200), a calibration curve (log RS vs. Ve) with proteins of known RS was carried out and NaCl solutions of different ionic strength were assayed (from 100 mM to 1.0 M, data not shown). Unfolding data corresponding to CD and Trp fluorescence for variant hFXN90-210 was extracted from [15].
The SEC experiment allowed us to distinguish the occurrence of two separate phenomena, namely, (i) the disassembly of the oligomer, most likely not a fully reversible process, as judged by partial re-association after dialysis, from (ii) the subsequent reversible unfolding of the monomeric species (see below). Regarding the hydrodynamic behavior of hFXN (Fig. 4), at 1.0 M GdmCl, more than 50% of the oligomer dissociates into a monomer characterized by a RS = 20.5 ± 0.1 Å, essentially identical to that expected for a globular species of the molecular mass corresponding to that of monomeric FXN56–210 (RS = 20.3 Å, according to Uversky [30], Figs. 4A and S2), and slightly larger than that measured for hFXN90–210 (RS = 18.9 ± 0.1 Å). In addition, at 1.0 M GdmCl a slight but significant increase in the Trp fluorescence emission and a red shift of the maximum occurs (Figs. 4C and S3A), probably indicating a perturbation in the closed-packed environment around these aromatic residues. This is not the case for hFXN90–210 (Fig. S3D). Naturally, at this GdmCl concentration range, observations might be governed by the changes in the oligomeric state of the protein. In this regard, at 1.5 M GdmCl ∼100% of molecules populate a monomeric conformation (Fig. 4A). The presence of oligomeric and/or eventually partially folded species in the conformational ensemble of hFXN56–210 are revealed by the binding of ANS. By contrast, at all GdmCl concentrations assayed hFXN90–210 fails to bind this fluorophore (Fig. 4D).
In a different vein, between 1 and 2 M GdmCl a significant increase in the elution volume – compatible with the expansion of the monomeric form – is observed for hFXN56–210: the RS values derived are 22.6 ± 0.2 and 24.0 ± 0.1 Å, for 1.5 and 2.2 M, respectively. As the perturbation in the elution volume at this GdmCl concentration range is much less evident in the case of hFXN90–210, we suggest that the changes observed might be related to the unfolding and the concomitant increased solvation of the 34-residue N-terminal stretch, although more experiments should be designed to clarify this point. The integrity of all species was ascertained by ESI-MS after incubation for 16 h in 0, 1.0, 1.5, and 2.0 M GdmCl solutions. Proteolysis was not detected in either of these conditions.
At higher GdmCl concentrations (2–3 M), a major change in the elution volume is observed for both variants hFXN56–210 and hFXN90–210 (Fig. 4A). This represents a progressive evolution in the elution volume giving rise to very sharp peaks, and values of the hydrodynamic radius that do not correspond to either that expected for the native or the completely unfolded states: e.g. at 2.6 M, the RS values for hFXN56–210 and hFXN90–210 are 29.0 ± 0.1, and 25.0 ± 0.1 Å, respectively (Fig. S2). On the other hand, the RS observed for hFXN56–210 and hFXN90–210 at 4.0 M GdmCl are 33.4 ± 0.1, and 30.0 ± 0.1 Å, respectively. This result is consistent with the GdmCl-induced unfolded state of hFXN co-existing in fast equilibrium with the native state. The conformational transitions observed for each variant between 2 and 3 M GdmCl superimpose well, exhibiting common trends: decreases in the far-UV CD signal and in the Trp fluorescence intensity, the latter also associated to an important red shift of the wavelength of maximum emission (Fig. 4B and C). This result is compatible with a major conformational change in tertiary and secondary structures. The global interpretation of the SEC and spectroscopic data in the range 2–3 M GdmCl points to a transition dominated by the unfolding of the monomeric form of hFXN56–210 (Figs. 4, S2 and S3).
The interpretation of this body of results can be achieved in the simplest fashion by assuming a model for the dissociation/unfolding where the species present are the oligomer, the monomer and the unfolded state. Unfortunately, the partial irreversibility observed for the oligomer disassembly process taking place in the range 0 to ∼1.0 M GdmCl precluded a rigorous thermodynamic treatment of this part of the data. One should also take into account the fact that the observed changes associated to the major transition (that occurring in the range 1.5–4.0 M GdmCl) comply with full reversibility and involve only monomeric and unfolded states. Thus, the derived thermodynamic parameters for variants hFXN56–210 and hFXN90–210 were calculated and summarized in Table 1. Remarkably, the unfolding curve corresponding to the monomer of hFXN56–210, as purified from E. coli extracts in the monomeric form, is essentially superimposable to that observed for the variant purified as the oligomeric species (Fig. S4). Regardless of its source, once hFXN56–210 adopts a monomeric state, it unfolds with a similar difference in free energy as hFXN90–210. In regard to this point, the mass of the monomeric form was checked by ESI-MS to check the integrity of the protein (data not shown).
Table 1.
GdmCl-induced unfolding parameters for the hFXN variants.a
| hFXN | Fluorescence |
Circular dichroism |
||||
|---|---|---|---|---|---|---|
| ΔG°NU H2O | mNU | CmNU | ΔG°UN H2O | mNU | CmNU | |
| 90–210 | 6.7 ± 0.2 | 2.9 | 2.3 ± 0.1 | 6.7 ± 0.2 | 2.9 | 2.3 ± 0.1 |
| O-56–210b | 6.4 ± 0.1 | 2.8 ± 0.3 | 2.3 ± 0.2 | 6.1 ± 0.8 | 2.8 ± 0.4 | 2.2 ± 0.3 |
| m-56–210c | 6.5 ± 0.5 | 2.9 ± 0.3 | 2.2 ± 0.4 | 6.3 ± 0.1 | 2.9 ± 0.4 | 2.2 ± 0.3 |
A two-state N ↔ U model is assumed. Parameter mNU is the slope of the linear dependence of free energy of unfolding (ΔG°NU) on denaturant concentration, and ΔG°NU H2O represents the extrapolated value of ΔG°NU at zero denaturant concentration. In the fittings corresponding to variant hFXN90–210, we used the predicted mNU value for GdmCl-induced unfolding, considering a globular protein of identical molecular weight: 2.9 kcal mol−1 M−1 for hFXN90–210. The units of mNU are kcal mol−1 M−1. Cm and free energy are in M and kcal mol−1, respectively. The parameters for hFXN56–210 were calculated by non-linear least-square fitting of the model to the data shown in Fig. 5, as described in Section 4 and fitting for hFXN90–210 is the same as in Ref. [15].
Variant hFXN56–210 expressed in E. coli yields mainly the oligomeric form (O-hFXN56–210). In this case this fraction was purified from E. coli extracts and analyzed.
When hFXN56–210 is expressed in E. coli, nearly 30% of the recombinant protein remains as soluble monomer (m-hFXN56–210). In this case this fraction was purified and analyzed.
2.5. Isolated peptide hFXN56–81 acquires structure in TFE and SDS solutions
As secondary structure predictions by Jpred3 [31] carried out on the sequence of hFXN uncovers a moderate tendency to form α-helical (residues 59–70) and β-strand (residues 72–79) structures (Fig. S5A), we decided to study the propensity of the N-terminal tail of hFXN (represented by peptide hFXN56–81) to acquire secondary structure in isolation.
As judged by the shape of the far-UV CD spectrum, the synthetic peptide hFXN56–81 is unstructured in its isolated form (Fig. 5A). However, after incubation with the co-solvent 2,2,2-trifluoroethanol (TFE), the far-UV CD spectrum becomes compatible with the stabilization of α-helical structure, as indicated by the appearance of the negative bands at 208 and 220 nm, Fig. 5A. The ellipticity observed in 30% TFE solution is approximately 37% of the total signal expected for a full α-helix. Accordingly, the AGADIR algorithm [32,33] predicts a somewhat stable α-helical structure in the N-tail (Fig. S5B). Remarkably, when sequence 56–90 was submitted to the I-TASSER server, an integrated platform for automated protein structure prediction [34], the stretch 59–70 was also predicted to fold as an α-helix in all five 3D models presented as the output (data not shown).
Fig. 5.
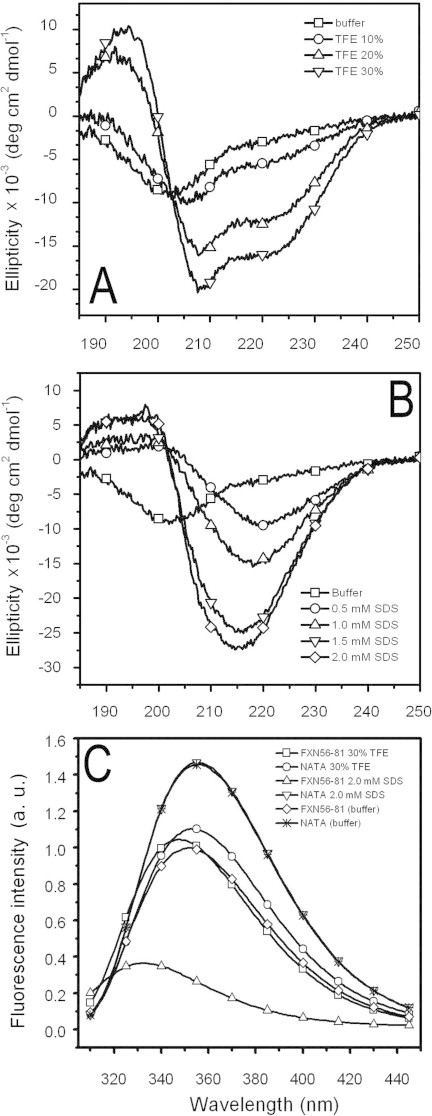
Peptide hFXN56–81 acquires structure in the presence of co-solvents. Synthetic peptide was incubated with TFE (A, 0, 10, 20, or 30%, v/v) or SDS (B, 0, 0.5, 1.0, 1.5 and 2.0 mM). Far-UV CD (A and B) and Trp fluorescence (C) spectra were acquired. NATA (N-acetyl tryptophanamide) was included as a reference compound. Excitation wavelength was 295 nm and buffer was 20 mM Tris–HCl and 100 mM NaCl, 1 mM EDTA, pH 7.0.
In addition, we analyzed whether the detergent sodium dodecyl sulfate (SDS) is able to stabilize a structured conformation. Strikingly, in the presence of SDS (0.5–2 mM), spectra are characterized by a single negative band at 216 nm, a feature compatible with the existence of β-strands (Fig. 5B). It is noteworthy that in the case of the synthetic peptide corresponding to the amphipathic C-terminal α-helix of hFXN (residues 181–195), SDS is indeed able to stabilize the helical conformation [35].
Taking advantage of the presence of the intrinsic probe Trp66 in hFXN56–81, we also studied the fluorescence emission in TFE or SDS to establish whether this reporter residue is able to sense changes in the chemical environment (Fig. 5C). In the absence of co-solvent, emission shows λMAX = 351 nm, indicating a polar milieu around the fluorophore. On the other hand, both co-solvents TFE and SDS produce significant blue shifts (4 and 19 nm, respectively). As a control sample, we studied the fluorescence emission of N-acetyl- l-tryptophanamide (NATA) under the same experimental conditions. Clearly, the change in the environment of Trp66 depends on the polypeptide context of hFXN56–81, since hardly any change occurs for NATA. Significantly, the presence of SDS not only results in the shift, but also causes a dramatic quenching effect. This behavior may result primarily as the consequence of SDS/Trp side-chain interaction, not excluding an environmental alteration brought about by an SDS-induced change in the conformation of the peptide. These results agree well with the notion that the N-terminal tail might acquire a structured conformation upon interaction with protein apolar surfaces of hFXN56–210, or even with other protein partners inside the cell.
2.6. Interaction between peptide hFXN56–81 and hFXN
To establish whether peptide hFXN56–81 is able to interact with hFXN, and whether this interaction guides the folding of the N-terminal peptide, and assuming that residues 81–89 are most likely unstructured [23], we evaluated changes in the far-UV CD spectrum upon mixing the above peptide with protein hFXN90–210. After incubation for 1 h at room temperature, the spectrum of this mixture was compared with the calculated sum of the spectra of each component in isolation (15 μM final concentration). Under these conditions, no change was detected in the CD signals (not shown), a result that agrees with NMR evidence by Prischi et al. [23], where chemical shifts compatible with a random coil are observed for residues 56–80 in the context of monomeric hFXN56–210. Interestingly, when peptide hFXN56–81 and hFXN90–210 are incubated (at a 10:1 peptide:protein molar ratio) with the dye ANS, an enhancement of fluorescence at 480 nm was observed (Fig. S6A). On the other hand, the incubation of peptide (50 μM) or protein hFXN90–210 (5 μM) in isolated form does not yield significant change in the ANS fluorescence spectrum. These results may put forward the notion that peptide and protein may indeed interact, giving rise to the emergence of new exposed apolar surfaces. However, the absence of ANS binding at a low peptide:protein ratio suggests the weak nature of the interaction between both moieties (data not shown). In this context, one should note that hFXN56–210 as a monomer shares the capability with the complex of binding this fluorescent dye (Fig. S6B). One could speculate that the conformation of residues 56–81 may be constrained by the presence of the hFXN domain, particularly when the N-terminal peptide is covalently bound to the rest of the protein.
We proceeded to investigate whether peptide hFXN56–81 is able to disassemble the quaternary structure of hFXN56–210 -perhaps by interacting with the N-terminal tail of the protomers in the context of the oligomer. Surprisingly, when peptide hFXN56–81 was added to a solution of the soluble multimer form of hFXN56–210, the aggregation into an insoluble species of this protein was triggered, as judged by the onset of turbidity and the quantitative assessment by centrifugation followed by SDS–PAGE (Fig. 6). This result demonstrates that peptide hFXN56–81 is capable of satisfying key contacts with the oligomer leading to the assembly of large aggregates. In the same fashion, when hFXN56–210 was submitted to the server PASTA (Prediction of Amyloid Structure Aggregation [36]) we found that the stretch of 14 residues 65–78 (Fig. S7) would form a seed for oligomerization, adopting a parallel β-strand conformation like that occurring in a fibril core.
Fig. 6.
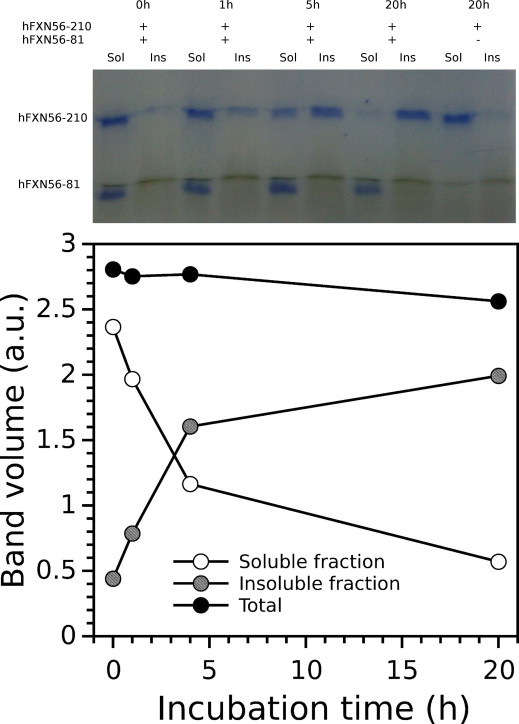
Aggregation of hFXN56–210 induced by the N-terminal peptide hFXN56–81. Variants hFXN90–210 and hFXN56–210 were incubated at 37 °C in the presence or in the absence of peptide hFXN56–81 (1:10, protein:peptide molar ratio) in buffer 20 mM Tris–HCl, 100 mM NaCl, 1 mM EDTA, pH 7.0. Aliquots were taken at different incubation times (0, 1, 5 and 20 h). Samples were centrifuged for 10 min at 4 °C, the soluble and the insoluble fractions were isolated and the reaction was immediately stopped by addition of 4× SDS–PAGE sample buffer. The samples were finally loaded onto a 16% gel. Separated bands (upper panel) and their quantification after Coomassie Brilliant Blue staining (lower panel) at each indicated time are shown. Errors were estimated at less than 10%.
Finally, we explored whether hFXN56–210 in the monomeric state (6.0 μM protein concentration) may alter its propensity to aggregate upon addition of 20% TFE. If this were the case – and hFXN90–210 did not aggregate under the same experimental conditions – we might correlate the tendency of hFXN to aggregate with the presence of the N-tail and the conformational change that might be taking place in this region of the protein. Interestingly, TFE does not produce significant changes in the far-UV CD spectrum of hFXN90–210 and the protein remains completely soluble (data not shown), whereas after addition of TFE variant hFXN56–210 aggregates into an insoluble form, as judged by UV absorption (data not shown). Taking together, these results are compatible with the existence of a conformational change involving the N-tail of the protein that could alter the dynamics and the quaternary structure of hFXN.
3. Discussion
NMR studies showed that the N-terminal chain of hFXN56–210 adopts a random-coil state, as indicated by the analysis of the chemical shift of signals assigned to this part of the backbone [23]. Most importantly, these observations were gathered on the monomer species of this variant, where the hFXN domain shows chemical shifts close to those of native hFXN90–210. Likewise, the analysis of the amino acid sequence 56–80 shows low secondary structure propensity. However, this could represent a common attribute of several polypeptide chains that acquire structure upon interacting with their cognate partners, as is the case of several intrinsically disordered proteins (IDPs). In this scenario, an in-depth structural study of this part of the protein is needed to reach a thorough understanding of the molecular details of this key biological phenomenon underlying FRDA. Particularly revealing are the electrostatic properties of the N-terminal sequence of hFXN56–210. A net charge of +5 indicates that this peptide might interact with negatively charged segments located further down along the sequence, like the ones existing at the iron binding site (α-helix 1, loop1 and β-strand 1, Fig. S5). Suggestively, Kondapalli and co-workers [37] showed that for residues 54, 58, 60, 62, 66, 69 and 70, chemical shift values appear to be dispersed in the HSQC spectrum, even though a defined kind of secondary structure cannot be assigned to this section of the backbone. In the same fashion, the analysis of mutual information in the CyaY protein family points to the co-evolution of several residue positions belonging to this same stretch, a fact that might indicate a tendency to acquire local structure (positions 23, 51, 52, 53, 55, 58, 61, 62, 63, 64, 65, 66 and 70, plus 196, cluster B in Fig. S8).
It is noteworthy that residues 80–90 of the mature form of hFXN are predicted as random coil, in accord with the analysis of chemical shifts, most likely indicating that this segment is unstructured [37], [23]. In this context, we believe that this amino acid stretch may act as a linker exposing a site for proteolysis at position 81 (Fig. S5).
The biophysical characteristics of hFXN56–210 explored in this work – including UV absorption, CD and fluorescence spectroscopies, and the hydrodynamic behavior – indicate that the multimeric form is indeed structured and its constituent monomers display a cooperative unfolding transition that differ only marginally from hFXN90–210 ( are 6.3 and 6.7 kcal mol−1, respectively), with a value for the mNU parameter (2.8 ± 0.3 kcal mol−1 M−1) closely resembling that corresponding to the hFXN domain. This suggests that in the range of 1.5–3 M GdmCl concentrations corresponding to the unfolding transition, the N-terminal tail does not pack well onto the folded FXN domain, because typically the predicted mNU for a globular protein of this size should be ∼3.8 kcal mol−1 M−1, a value considerably larger than that experimentally measured here.
The existence of solvent exposed apolar surfaces in the oligomeric state of hFXN56–210 is revealed by the binding of the fluorophore ANS. This phenomenon points to the presence of partially folded conformations in the context of the oligomer. In the same way, the decreased intensity of the near-UV CD signals for hFXN56–210 in the oligomeric form might reflect conformational heterogeneity among subunits with molten globule-like dynamics. However, hFXN56–210 unfolds in a cooperative fashion showing similar stability to the observed for variant hFXN90–210, pointing to the fact that conformational differences between oligomer subunits and the monomer should not be drastic. This is also in agreement with the reduced sensitivity to proteolysis observed for the subunits of the oligomer. This feature clearly distinguishes its nature from that of a typical molten-globule, or even from other destabilized native-like states of hFXN, as is the case for the C-terminal truncated variant of human frataxin hFXN90–195 [15].
The oligomerization process of hFXN shows distinctive features: (i) in contrast to the yFXN, where this phenomenon is triggered by iron, purified monomeric fractions of forms hFXN56–210 [24] and hFXN42–210 [37] do not appear to assemble into oligomers in vitro under native conditions; (ii) it was shown that large oligomers of hFXN can be irreversibly disassembled into stable monomers by the addition of SDS [38]; (iii) we show herein that monomeric species of hFXN56–210 stabilized by incubation at moderate concentrations of denaturant (1.5 M GdmCl) can reassemble into the oligomeric form upon dialysis or by removal of GdmCl by gel filtration (SEC). Taken together, these results allowed us to draw the following picture. Limited conformational changes on the native-state facilitated by the chaotropic agent, promote oligomer assembly from the monomeric form of hFXN56–210, which becomes competent for oligomerization (monomerOC).
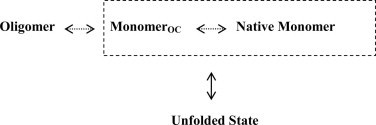
3.1. Unfolded state
In this scenario, the N-terminal tail of hFXN56–210 seems to be an essential key conferring the protein the capability to oligomerize. Moreover, the oligomeric form of hFXN56–210 may disassemble into native monomers by a protease treatment removing the N-terminal extension, while maintaining intact the rest of the protein. This fact supports the idea that the N-terminal region mediates the interaction among subunits, and indicates that this moiety is most likely exposed to the solvent, and – as such – to protease action. The latter indicates that sequence 56–81 must be covalently bound to the rest of the protein to promote the oligomerization of the hFXN domain. Independently, the non-covalent interaction between the N-terminal tail and the rest of the protein is also possible, as revealed by the ANS binding to the complex hFXN90–210/hFXN56–81 and the onset of aggregation of hFXN56–210 triggered by the addition of excess amounts of peptide hFXN56–81.
Regarding the homologue of frataxin yFXN, studies performed by Karadaghi and co-workers on the mutant Y73A showed that, by contrast to wild-type yFXN, this protein can spontaneously assemble into larger oligomeric species in the absence of iron [39]. Thus, the oligomerization behavior of yFXN depends on fine details of the amino acid sequence of the N-tail. Remarkably, data from the same group indicate that the N-terminus of this protein is also highly flexible [40]. In our case, although the N-tail of hFXN is predicted as an intrinsically disordered segment [23], residues 66–75 display a lower tendency to form disordered regions, by comparison to residues 45–61 and 76–90 (IUPRED [41] prediction shown in Fig. S9). We envision that the N-terminal tail of hFXN may acquire a non-random conformation upon binding to other proteins inside the cell. The notion that the N-terminal tail of hFXN56–210 might contain short interaction-prone segments which could experience order/disorder transitions upon specific binding is reinforced after taking into account the capability of peptide hFXN56–81 to acquire secondary structure in the presence of co-solvents like TFE or SDS. The stabilization of two different conformations in each of these solvent conditions would also be compatible with one of the distinctive features of IDPs, namely, their ability to attain alternative structures in a template-dependent fashion. This capability is attractively illustrated by the C-terminal binding region of p53 [42,43].
Regarding hFXN81–210, the mature form of hFXN, is preponderantly found under physiological conditions, the excision of the N-terminal tail in hFXN may be an important step precluding oligomer assembly of hFXN in vivo.
Very recently, some clues provided evidence for the existence of a post-translational regulation mechanism of the hFXN level mediated by the ubiquitin–proteasome system (UPS), thus opening the possibility to hamper hFXN degradation by increasing its bioavailability in FRDA [44]. However, in light of the results presented herein, it will be essential to evaluate the level of intermediate states of hFXN and the ability of these forms (and of the precursor) to trigger oligomerization in vivo, in the presence of small molecules such as those used to inhibit UPS-mediated degradation [44]. Finally, the extra mitochondrial isoform III of hFXN – lacking a 47 amino acid stretch (9–55) – seems to be functionally relevant and might be implicated in FRDA. This variant shares the same iron-catalyzed self-processing mechanism of hFXN56–210 [45], perhaps preventing the aggregation of this isoform mediated by its N-terminal tail. On the other hand, isoform III might still keep the capability to interact with other cytoplasmic proteins through its N-terminal tail.
The presence of the conformationally plastic N-terminal tail might impart hFXN the ability to act as a recognition signal as well as an oligomerization trigger. Understanding the fine tuning of these activities and the resulting balance will ultimately be relevant for understanding hFXN function in the cell.
4. Materials and methods
4.1. Peptide synthesis and purification
Peptide hFXN56–81, of sequence SSNQRGLNQIWNVKKQSVYLMNLRKS, was synthesized and partially purified by GenScript Corp (Piscataway, NJ). The purification was completed by HPLC (Rainin Dynamax, NY) using a reverse phase C18 semi-preparative column (Vydac) equilibrated in 0.05% aqueous TFA. The peptide was eluted with a linear gradient from 30% to 45% aqueous acetonitrile (ACN), 0.05% TFA. The peptide typically elutes at 31% ACN. Fractions containing >98.0% pure peptide were pooled and lyophilized. Peptide concentration was calculated by measuring absorbance in the near-UV region, using an extinction coefficient: ε280nm = 6990 M−1 cm−1. The molecular mass of peptide (theoretical value: 3092.55 Da) was checked after HPLC by ESI-MS. As a control sample, we had peptide FXN181–195 prepared of sequence SLHELLAAELTKALKY (1800.11 Da), corresponding to the amphipathic α-helix 2 of hFXN. In this case, a C-terminal Tyr tag was added to allow the concentration measurement by absorbance in the near-UV region, using the extinction coefficient: ε280nm = 1490 M−1 cm−1. AGADIR prediction [33,46,47] based on the sequence of peptide hFXN181–195 yields a low value of helical propensity: 5.8% helicity at 25 °C.
4.2. Protein expression and purification
Human frataxin cDNA was kindly provided by Dr. Hèléne Puccio from IGBMC (Strasbourg, France). To prepare hFXN90–210, primers NdeI FXN90, tttaagaaggagatatacatatgctagatgagaccacctatgaa and XbaI FXN210 gcatggatcctcaagcatcttttccggaataggc were used to amplify the cDNA corresponding to the truncated version 90–210 of hFXN. The PCR product was sub-cloned into a pET9b plasmid vector, and the identity of the insert was confirmed by DNA sequencing. E. coli BL21 (DE3) cells were then electroporated with this construction. Bacteria cultures (2–3 L Terrific Broth, pH 7.2) were grown at 37 °C and 280 rpm. hFXN90–210 was induced at OD = 1.0 by the addition of 1.0 mM IPTG to the culture medium (37 °C, 280 rpm). After induction for 3.5 h, bacteria were centrifuged at 6000 rpm and the pellet was stored at −20 °C until rupture using a French press. Soluble and insoluble fractions were separated by centrifugation at 10,000 rpm (20 min). The soluble fraction was carefully loaded onto a preparative Sephadex G-100 chromatography (SEC, 93 cm × 2.7 cm) column, previously equilibrated with 20 mM Tris–HCl at pH 7.0 and 100 mM NaCl, 1 mM EDTA. Mass was confirmed by ESI-MS (theoretical molecular mass value: 13,605.1 Da, considering the presence of the N-terminal Met). To measure protein concentration, the extinction coefficient used for hFXN90–210 was ε280nm = 26,930 M−1 cm−1 (A280nm = 1.98 for a 1 mg/mL solution).
To prepare hFXN56–210, primers NdeIFXN56 of sequence tttaagaaggagatatacatatgagttcgaaccaacgtggcctcaa and XbaI FXN210 were used to amplify the cDNA corresponding to this variant. Sub-cloning into pET9b and expression steps were identical to those previously described for variant hFXN90–210. To purify hFXN56–210, we took advantage of the increase in stability of the soluble aggregated form of this variant by comparison to its monomeric form: the latter being much more sensitive to protease action along expression and purification. In this case, an ionic exchange chromatography (IE) was used to clarify the soluble fraction (particularly, to reduce the DNA content). At pH 7.0, the hFXN56–210 does not bind to the DE52 matrix. Then, we performed a second IE in the same matrix at pH 8.0, a condition in which hFXN56–210 binds, thus allowing its straightforward purification. Finally, a step of size exclusion chromatography (SEC) using preparative Sephadex G-100 chromatography (SEC, 93 cm × 2.7 cm) column, previously equilibrated with 20 mM Tris–HCl at pH 7.0 and 100 mM NaCl, 1 mM EDTA was carried out (as in the case of hFXN90–210). In this case, fractions eluting at Vo, containing 95% pure hFXN56–210, were pooled and conserved at 4 °C until later use. The extinction coefficient used for this variant to measure protein concentration was ε280nm = 33,920 M−1 cm−1 (A280nm = 1.97 for a 1 mg/mL solution); the molecular mass of hFXN56–210 was confirmed by ESI-MS (theoretical molecular mass value: 17,255.3 Da, without considering the N-terminal Met).
4.3. Fluorescence measurements
Steady-state fluorescence measurements were performed in a Jasco FP-6500 spectrofluorometer operating in the ratio mode and equipped with a thermostated cell holder connected to a circulating water bath set at 20 °C. To this end, a 1.0 cm path length cell sealed with a Teflon cap was used. When the intrinsic fluorescence of proteins was measured, excitation wavelength was 295 nm and emission data were collected in the range 310–450 nm. The spectral slit-width was set to 3 nm for both monochromators.
For ANS-binding experiments, hFXN variants (5 μM) in 20 mM Tris–HCl, 100 mM NaCl, pH 7.0 were incubated at room temperature (5 min), with the dye ANS (8-anilino-1-naphthalene sulfonic acid) at a final concentration of 50 μM. A control sample consisting of ANS (50 μM) in buffer solution was prepared. The excitation wavelength was 350 nm and the emission spectra were collected between 400 and 600 nm. The bandwidth used was 3.0 nm for both excitation and emission. In order to estimate the concentration of the dye, an extinction coefficient value of 4,950 M−1 cm−1 at 350 nm was used [48].
4.4. Circular dichroism spectroscopy
Ellipticity of protein samples was evaluated using a Jasco 810 spectropolarimeter. Far-UV CD spectra were recorded in the range between 185 and 250 nm. A cell of 0.1 cm was used and protein concentration was 10 μM. For near-UV CD spectra, the wavelength range was 240–340 nm, protein concentration was 20 μM, and the path length was 1.0 cm. In all cases data was acquired at a scan speed of 20 nm/min and at least 3 scans were averaged for each sample. Blank scans were subtracted from the spectra and values of ellipticity were expressed in units of deg cm2 dmol−1, unless otherwise indicated in the text. Theoretical calculation of ellipticity for a 100% helical peptide was performed as explained by Luo and Baldwin [49–51].
4.5. Limited trypsinolysis
hFXN variants were incubated with trypsin 1:100 (protein:protease, mass ratio) at 20 °C in 20 mM Tris–HCl, 100 mM NaCl, 1 mM EDTA, pH 7.0. Aliquots were separated at different times (0, 15, 30, and 45 min, 1, 2, and 16 h) and the reaction was immediately stopped by addition of the sample buffer for SDS–PAGE. Samples were kept at −20 °C until loaded into the gel. Proteolysis products were also analyzed by RP-HPLC followed by ESI-MS.
4.6. Protein unfolding and oligomer disassembly experiments
Isothermal unfolding experiments were carried out by incubating the hFXN variants with 0–6 M GdmCl in buffer solution for 16 h at room temperature, followed by far UV CD analysis, tryptophan fluorescence measurements, and SEC hydrodynamic radii determination. To calculate thermodynamic parameters, a two-state unfolding mechanism was assumed, i.e. only native (N) and unfolded (U) species exist at equilibrium. In addition, a three-state model, considering native (N), partially folded (I) and unfolded (U) species was also applied. Theoretical functions corresponding to each model were independently fit to each data set [52]. GdmCl concentration was measured by refractometry.
4.7. Hydrodynamic behavior
Size exclusion chromatography. Changes in hydrodynamic volumes were monitored by chromatography on a SEC-FPLC system equipped with a 280 nm UV detector and a Superose 12 or a Superdex S-200 HR 10/30 columns (Pharmacia Biotech, Sweden), equilibrated at room temperature in buffer solution, 20 mM Tris–HCl and 100 mM NaCl, 1 mM EDTA, pH 7.0. This type of chromatographic resin enabled us to confidently test for the presence of soluble aggregates in the samples. The flow rate was 0.2 or 0.5 mL/min and the injection volume was 100–200 μL. Samples were centrifuged at 14,000 rpm before loading into the column that was previously calibrated with appropriate molecular weight markers. The hydrodynamic radius (RS) of each species was derived from the elution volume (Ve) considering for that a calibration curve according Uversky [30].
4.8. Spectrophotometric determination of iron
Iron concentration was determined using the method of 1,10-phenanthroline [53]. Briefly, 1,10-phenanthroline was prepared in 0.1 N HCl and a standard iron solution (18 mM) was prepared in 0.47 mM sulfuric acid for calibration curves. Ascorbic acid (100 μL, 10%) and sodium citrate (100 μL, 10%) were mixed with the protein sample and water (up to a final volume of 700 μL). After that, 1,10-phenanthroline solution (100 μL) was added and mixed. After 15 min incubation, the sample was centrifuged in a 1.5 mL Eppendorf tube at maximum speed for 5 min. This centrifugation step is important to remove protein aggregates that disperse light, thus leading to incorrect estimates. Next, the absorbance at 512 nm was read.
4.9. Aggregation of hFXN56–210 induced by peptide hFXN56–81
Variants hFXN90–210 and hFXN56–210 were incubated at 37 °C in the presence or in the absence of peptide hFXN56–81, 1:10 (protein:peptide, molar ratio) in buffer 20 mM Tris–HCl and 100 mM NaCl, 1 mM EDTA, pH 7.0. Aliquots of the reactions were taken at different incubation times (0, 1, 2, 3, 5 and 20 h). Samples were centrifuged for 10 min at 4 °C to isolate the soluble from the insoluble fractions and the reaction was immediately stopped by addition of 4-fold concentrated sample buffer for SDS–PAGE. Then, samples were kept at −20 °C until loaded into the gel. To evaluate the effect of TFE on oligomerization and aggregation of hFXN56–210, protein samples (6.0 μM) were incubated at room temperature in buffer 20 mM Tris–HCl, 100 mM NaCl, 1 mM EDTA, pH 7.0 in the presence or in the absence of 20% (v/v) TFE. After a short incubation time (5 min) the samples were centrifuged (15 min at 4 °C) and the supernatants were analyzed by UV absorption to determine protein concentration and by circular dichroism to evaluate the secondary structure content. As running experiments with TFE on the full proteins can be cumbersome, e.g. TFE at relatively low concentration (20–25%) triggers aggregation of a number of proteins, in this experiment hFXN90–210 was included as a sample control.
4.10. Bioinformatics, secondary structure, aggregation propensity and mutual information analysis
Frataxin sequences (948) were obtained from PFAM and aligned using SeaView (muscle algorithm). From the global alignment (MSA948), 165 sequences containing N-terminal extensions were selected and a new MSA was performed (MSA165). Mutual information and conservation scores were calculated for both MSAs as in references [54–56]. Secondary structure predictions were calculated using Jpred [31,57] on a MSA of 30 mammalian sequences fished by BLAST by using hFXN1–210 as input. Helical stability was predicted using the AGADIR algorithm [32,33]. Aggregation propensity was predicted using the PASTA algorithm [36]. Prediction of intrinsically disordered segments along the sequence of hFXN using IUPRED [41].
Acknowledgments
This work was supported by Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) and Universidad de Buenos Aires (UBACyT). We specially thank Dr. Hélène Puccio for kindly providing us with the hFXN gene.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Contributor Information
José M. Delfino, Email: delfino@qb.ffyb.uba.ar.
Javier Santos, Email: jsantos@qb.ffyb.uba.ar.
Supplementary material
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.fob.2013.07.004.
Appendix. Supplementary materials
Supplementary materials for "The role of the N-terminal tail for the oligomerization, folding and stability of human frataxin".
References
- 1.Schrank T.P., Bolen D.W., Hilser V.J. Rational modulation of conformational fluctuations in adenylate kinase reveals a local unfolding mechanism for allostery and functional adaptation in proteins. Proc Natl Acad Sci U S A. 2009;106:16984–16989. doi: 10.1073/pnas.0906510106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang Y., Liu Z. Kinetic advantage of intrinsically disordered proteins in coupled folding-binding process: a critical assessment of the “fly-casting” mechanism. J Mol Biol. 2009;393:1143–1159. doi: 10.1016/j.jmb.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 3.Turjanski A.G., Gutkind J.S., Best R.B., Hummer G. binding-induced folding of a natively unstructured transcription factor. PLoS Comput Biol. 2008;4:e1000060. doi: 10.1371/journal.pcbi.1000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turjanski A.G., Hummer G., Gutkind J.S. How mitogen-activated protein kinases recognize and phosphorylate their targets: a QM/MM study. J Am Chem Soc. 2009;131:6141–6148. doi: 10.1021/ja8071995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bulteau A.L., O’Neill H.A., Kennedy M.C., Ikeda-Saito M., Isaya G., Szweda L.I. Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science. 2004;305:242–245. doi: 10.1126/science.1098991. [DOI] [PubMed] [Google Scholar]
- 6.Bencze K.Z., Yoon T., Millan-Pacheco C., Bradley P.B., Pastor N., Cowan J.A., Stemmler T.L. Human frataxin: iron and ferrochelatase binding surface. Chem Commun (Camb) 2007:1798–1800. doi: 10.1039/b703195e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He Y., Alam S.L., Proteasa S.V., Zhang Y., Lesuisse E., Dancis A., Stemmler T.L. Yeast frataxin solution structure, iron binding, and ferrochelatase interaction. Biochemistry. 2004;43:16254–16262. doi: 10.1021/bi0488193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoon T., Cowan J.A. Iron–sulfur cluster biosynthesis. Characterization of frataxin as an iron donor for assembly of [2Fe-2S] clusters in ISU-type proteins. J Am Chem Soc. 2003;125:6078–6084. doi: 10.1021/ja027967i. [DOI] [PubMed] [Google Scholar]
- 9.Pandolfo M., Pastore A. The pathogenesis of Friedreich ataxia and the structure and function of frataxin. J Neurol. 2009;256(Suppl. 1):9–17. doi: 10.1007/s00415-009-1003-2. [DOI] [PubMed] [Google Scholar]
- 10.Bencze K.Z., Kondapalli K.C., Cook J.D., McMahon S., Millan-Pacheco C., Pastor N., Stemmler T.L. The structure and function of frataxin. Crit Rev Biochem Mol Biol. 2006;41:269–291. doi: 10.1080/10409230600846058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dhe-Paganon S., Shigeta R., Chi Y.I., Ristow M., Shoelson S.E. Crystal structure of human frataxin. J Biol Chem. 2000;275:30753–30756. doi: 10.1074/jbc.C000407200. [DOI] [PubMed] [Google Scholar]
- 12.Musco G., Stier G., Kolmerer B., Adinolfi S., Martin S., Frenkiel T., Gibson T., Pastore A. Towards a structural understanding of Friedreich's ataxia: the solution structure of frataxin. Structure. 2000;8:695–707. doi: 10.1016/s0969-2126(00)00158-1. [DOI] [PubMed] [Google Scholar]
- 13.Correia A.R., Pastore C., Adinolfi S., Pastore A., Gomes C.M. Dynamics, stability and iron-binding activity of frataxin clinical mutants. FEBS J. 2008;275:3680–3690. doi: 10.1111/j.1742-4658.2008.06512.x. [DOI] [PubMed] [Google Scholar]
- 14.Correia A.R., Adinolfi S., Pastore A., Gomes C.M. Conformational stability of human frataxin and effect of Friedreich's ataxia-related mutations on protein folding. Biochem J. 2006;398:605–611. doi: 10.1042/BJ20060345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roman E.A., Faraj S.E., Gallo M., Salvay A.G., Ferreiro D.U., Santos J. Protein stability and dynamics modulation: the case of human frataxin. PLoS One. 2012;7:e45743. doi: 10.1371/journal.pone.0045743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adinolfi S., Nair M., Politou A., Bayer E., Martin S., Temussi P., Pastore A. The factors governing the thermal stability of frataxin orthologues: how to increase a protein's stability. Biochemistry. 2004;43:6511–6518. doi: 10.1021/bi036049+. [DOI] [PubMed] [Google Scholar]
- 17.Gakh O., Bedekovics T., Duncan S.F., Smith D.Y.T., Berkholz D.S., Isaya G. Normal and Friedreich ataxia cells express different isoforms of frataxin with complementary roles in iron–sulfur cluster assembly. J Biol Chem. 2010;285:38486–38501. doi: 10.1074/jbc.M110.145144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aloria K., Schilke B., Andrew A., Craig E.A. Iron-induced oligomerization of yeast frataxin homologue Yfh1 is dispensable in vivo. EMBO Rep. 2004;5:1096–1101. doi: 10.1038/sj.embor.7400272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmucker S., Argentini M., Carelle-Calmels N., Martelli A., Puccio H. The in vivo mitochondrial two-step maturation of human frataxin. Hum Mol Genet. 2008;17:3521–3531. doi: 10.1093/hmg/ddn244. [DOI] [PubMed] [Google Scholar]
- 20.Condo I., Ventura N., Malisan F., Rufini A., Tomassini B., Testi R. In vivo maturation of human frataxin. Hum Mol Genet. 2007;16:1534–1540. doi: 10.1093/hmg/ddm102. [DOI] [PubMed] [Google Scholar]
- 21.O’Neill H.A., Gakh O., Isaya G. Supramolecular assemblies of human frataxin are formed via subunit-subunit interactions mediated by a non-conserved amino-terminal region. J Mol Biol. 2005;345:433–439. doi: 10.1016/j.jmb.2004.10.074. [DOI] [PubMed] [Google Scholar]
- 22.Xia H., Cao Y., Dai X., Marelja Z., Zhou D., Mo R., Al-Mahdawi S., Pook M.A., Leimkuhler S., Rouault T.A. Novel frataxin isoforms may contribute to the pathological mechanism of friedreich ataxia. PLoS One. 2012;7:e47847. doi: 10.1371/journal.pone.0047847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prischi F., Giannini C., Adinolfi S., Pastore A. The N-terminus of mature human frataxin is intrinsically unfolded. FEBS J. 2009;276:6669–6676. doi: 10.1111/j.1742-4658.2009.07381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cavadini P., O’Neill H.A., Benada O., Isaya G. Assembly and iron-binding properties of human frataxin, the protein deficient in Friedreich ataxia. Hum Mol Genet. 2002;11:217–227. doi: 10.1093/hmg/11.3.217. [DOI] [PubMed] [Google Scholar]
- 25.Dayhoff J.E., Shoemaker B.A., Bryant S.H., Panchenko A.R. Evolution of protein binding modes in homooligomers. J Mol Biol. 2010;395:860–870. doi: 10.1016/j.jmb.2009.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schulz G.E. The dominance of symmetry in the evolution of homo-oligomeric proteins. J Mol Biol. 2010;395:834–843. doi: 10.1016/j.jmb.2009.10.044. [DOI] [PubMed] [Google Scholar]
- 27.Noguera M.E., Primo M.E., Sosa L.N., Risso V.A., Poskus E., Ermacora M.R. Biophysical characterization of the membrane-proximal ectodomain of the receptor-type protein-tyrosine phosphatase phogrin. Protein Pept Lett. 2012 doi: 10.2174/0929866511320090007. [DOI] [PubMed] [Google Scholar]
- 28.Celej M.S., Dassie S.A., Freire E., Bianconi M.L., Fidelio G.D. Ligand-induced thermostability in proteins: thermodynamic analysis of ANS–albumin interaction. Biochim Biophys Acta. 2005;1750:122–133. doi: 10.1016/j.bbapap.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 29.Kurian E., Kirk W.R., Prendergast F.G. Affinity of fatty acid for (r)rat intestinal fatty acid binding protein: further examination. Biochemistry. 1996;35:3865–3874. doi: 10.1021/bi952589y. [DOI] [PubMed] [Google Scholar]
- 30.Uversky V.N. Use of fast protein size-exclusion liquid chromatography to study the unfolding of proteins which denature through the molten globule. Biochemistry. 1993;32:13288–13298. doi: 10.1021/bi00211a042. [DOI] [PubMed] [Google Scholar]
- 31.Cole C., Barber J.D., Barton G.J. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36:W197–201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fisinger S., Serrano L., Lacroix E. Computational estimation of specific side chain interaction energies in alpha helices. Protein Sci. 2001;10:809–818. doi: 10.1110/ps.34901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lacroix E., Viguera A.R., Serrano L. Elucidating the folding problem of alpha-helices: local motifs, long-range electrostatics, ionic-strength dependence and prediction of NMR parameters. J Mol Biol. 1998;284:173–191. doi: 10.1006/jmbi.1998.2145. [DOI] [PubMed] [Google Scholar]
- 34.Roy A., Kucukural A., Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roman E.A, Rosi P., Gonzalez L., Ebrero M.C., Wuilloud R., Gonzalez F., lecha F.L., Delfino J.M., Santos J. Gain of local structure in an amphipathic peptide does not require a specific tertiary framework. Proteins. 2010;78:2757–2768. doi: 10.1002/prot.22789. [DOI] [PubMed] [Google Scholar]
- 36.Trovato A., Seno F., Tosatto S.C. The PASTA server for protein aggregation prediction. Protein Eng Des Sel. 2007;20:521–523. doi: 10.1093/protein/gzm042. [DOI] [PubMed] [Google Scholar]
- 37.Kondapalli K.C., Bencze K.Z., Dizin E., Cowan J.A., Stemmler T.L. NMR assignments of a stable processing intermediate of human frataxin. Biomol NMR Assign. 2010;4:61–64. doi: 10.1007/s12104-010-9209-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Neill H.A., Gakh O., Park S., Cui J., Mooney S.M., Sampson M., Ferreira G.C., Isaya G. Assembly of human frataxin is a mechanism for detoxifying redox-active iron. Biochemistry. 2005;44:537–545. doi: 10.1021/bi048459j. [DOI] [PubMed] [Google Scholar]
- 39.Karlberg T., Schagerlof U., Gakh O., Park S., Ryde U., Lindahl M., Leath K., Garman E., Isaya G., Al-Karadaghi S. The structures of frataxin oligomers reveal the mechanism for the delivery and detoxification of iron. Structure. 2006;14:1535–1546. doi: 10.1016/j.str.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 40.Soderberg C.A., Shkumatov A.V., Rajan S., Gakh O., Svergun D.I., Isaya G., Al-Karadaghi S. Oligomerization propensity and flexibility of yeast frataxin studied by X-ray crystallography and small-angle X-ray scattering. J Mol Biol. 2011;414:783–797. doi: 10.1016/j.jmb.2011.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dosztanyi Z., Csizmok V., Tompa P., Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21:3433–3434. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- 42.Xue B., Dunker A.K., Uversky V.N. Retro-MoRFs: identifying protein binding sites by normal and reverse alignment and intrinsic disorder prediction. Int J Mol Sci. 2010;11:3725–3747. doi: 10.3390/ijms11103725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uversky V.N. Multitude of binding modes attainable by intrinsically disordered proteins: a portrait gallery of disorder-based complexes. Chem Soc Rev 2011 Mar;40:1623-34 [DOI] [PubMed]
- 44.Rufini A., Fortuni S., Arcuri G., Condo I., Serio D., Incani O., Malisan F., Ventura N., Testi R. Preventing the ubiquitin–proteasome-dependent degradation of frataxin, the protein defective in Friedreich's ataxia. Hum Mol Genet. 2011;20:1253–1261. doi: 10.1093/hmg/ddq566. [DOI] [PubMed] [Google Scholar]
- 45.Yoon T., Dizin E., Cowan J.A. N-terminal iron-mediated self-cleavage of human frataxin: regulation of iron binding and complex formation with target proteins. J Biol Inorg Chem. 2007;12:535–542. doi: 10.1007/s00775-007-0205-2. [DOI] [PubMed] [Google Scholar]
- 46.Munoz V., Serrano L. Elucidating the folding problem of helical peptides using empirical parameters. Nat Struct Biol. 1994;1:399–409. doi: 10.1038/nsb0694-399. [DOI] [PubMed] [Google Scholar]
- 47.Munoz V., Serrano L. Elucidating the folding problem of helical peptides using empirical parameters. III. Temperature and pH dependence. J Mol Biol. 1995;245:297–308. doi: 10.1006/jmbi.1994.0024. [DOI] [PubMed] [Google Scholar]
- 48.Daniel E., Weber G. Cooperative effects in binding by bovine serum albumin. I. The binding of 1-anilino-8-naphthalenesulfonate. Fluorimetric titrations. Biochemistry. 1966;5:1893–1900. doi: 10.1021/bi00870a016. [DOI] [PubMed] [Google Scholar]
- 49.Luo P., Baldwin R.L. Mechanism of helix induction by trifluoroethanol: a framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water. Biochemistry. 1997;36:8413–8421. doi: 10.1021/bi9707133. [DOI] [PubMed] [Google Scholar]
- 50.Luo P., Baldwin R.L. Interaction between water and polar groups of the helix backbone: an important determinant of helix propensities. Proc Natl Acad Sci U S A. 1999;96:4930–4935. doi: 10.1073/pnas.96.9.4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luo P., Baldwin R.L. Origin of the different strengths of the (i,i+4) and (i,i+3) leucine pair interactions in helices. Biophys Chem. 2002;96:103–108. doi: 10.1016/s0301-4622(02)00010-8. [DOI] [PubMed] [Google Scholar]
- 52.Santoro M.M., Bolen D.W. A test of the linear extrapolation of unfolding free energy changes over an extended denaturant concentration range. Biochemistry. 1992;31:4901–4907. doi: 10.1021/bi00135a022. [DOI] [PubMed] [Google Scholar]
- 53.Marczenko Z. Wiley; New York: 1976. Spectrophotometric determination of elements Chalmer, R. A. [Google Scholar]
- 54.Aguilar D., Oliva B., Marino B., Uslje C. Mapping the mutual information network of enzymatic families in the protein structure to unveil functional features. PLoS One. 2012;7:e41430. doi: 10.1371/journal.pone.0041430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marino Buslje C, Teppa E., Di Domenico T., Delfino J.M., Nielsen M. Networks of high mutual information define the structural proximity of catalytic sites: implications for catalytic residue identification. PLoS Comput Biol. 2010;6:e1000978. doi: 10.1371/journal.pcbi.1000978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Teppa E., Wilkins A.D., Nielsen M., Marino Buslje C. Disentangling evolutionary signals: conservation, specificity determining positions and coevolution. Implication for catalytic residue prediction. BMC Bioinformatics. 2012;13:235. doi: 10.1186/1471-2105-13-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cuff J.A., Barton G.J. Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins. 2000;40:502–511. doi: 10.1002/1097-0134(20000815)40:3<502::aid-prot170>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials for "The role of the N-terminal tail for the oligomerization, folding and stability of human frataxin".